

Abstract
Chronic administration of drugs leads to the activation of compensatory mechanisms that may inhibit some of their activity and induce unwanted toxicity. These mechanisms are an obstacle for maintaining a sustainable effect for many chronic medications. Pathways that adapt to the burden induced by chronic drugs, whether or not related to the underlying disease, can lead to a partial or complete loss of effect. Variability characterizes many biological systems and manifests itself as large intra- and inter-individual differences in the response to drugs. Circadian rhythm-based chronotherapy is further associated with variability in responses noted among patients. This paper reviews current knowledge regarding the loss of effect of chronic medications and the range of variabilities that have been described in responses and loss of responses. Establishment of a personalized platform for overcoming these prohibitive mechanisms is presented as a model for ensuring long-term sustained medication effects. This novel platform implements personalized variability signatures and individualized circadian rhythms for preventing and opposing the prohibitive effect of the compensatory mechanisms induced by chronic drug administration.
Keywords: drug resistance, compensatory mechanisms, chronotherapy, chronobiology, variability
Graphical Abstract
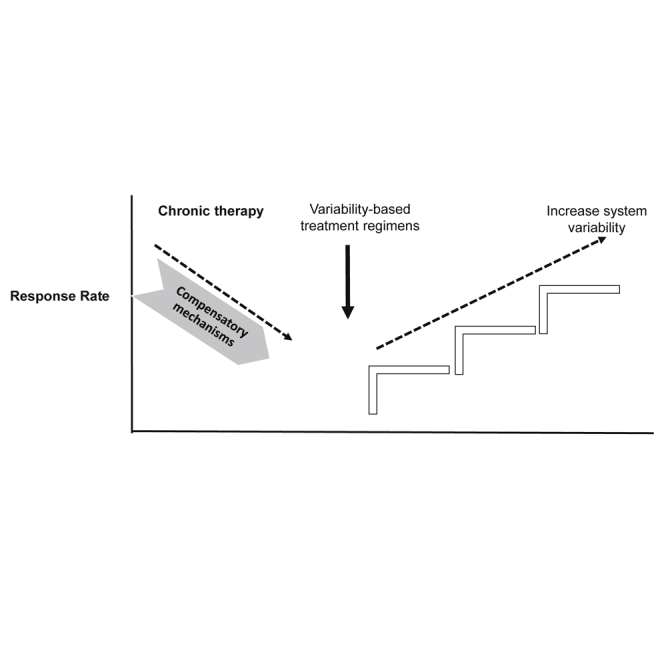
Chronic drug administration leads to the activation of compensatory mechanisms that inhibit some of their activity and induce toxicity. Establishment of a personalized platform for overcoming these prohibitive mechanisms is presented as a model for ensuring long-term sustained medication effects using personalized variability signatures and individualized circadian rhythms.
Main Text
Compensatory mechanisms are activated toward many chronically administered medications and are obstacles for maintaining sustainable effects of these drugs. While the body may initially respond to a drug in a beneficial way, the intrinsic adaptation of biological systems to the burden induced by a drug, whether or not related to the underlying disease, can lead to a partial or complete loss of effect. The de novo intrinsic resistance to a drug is attributed, in some cases, to the activation of compensatory opposing signaling pathways or other cellular pathways. Many of the currently used small-molecule drugs are aimed at activation or inhibition of a specific pathway. These drugs may engage compensatory mechanisms that lead to drug tolerance or to a partial or complete resistance to the drug.1 In some cases, compensatory mechanisms activate opposing pathways that promote exacerbation of the underlying disorder or cause unwanted side effects. The use of biological agents, including antibodies, recombinant hormones, and gene transfer-based or cell-based technologies, was expected to overcome these obstacles.1 However, drug resistance or tolerance can also be developed toward these modalities. Combinations of treatments that target multiple pathways simultaneously have been proposed as a method to overcome these issues. Treatment-induced toxicity of these combinations and an inability to use continuous pharmacodynamically effective doses of many targeted treatments require intermittent drug regimens.2 This paper reviews the data on the potential deleterious effects of these compensatory pathways and the role of variability and chronotherapy. The establishment of a platform to overcome these compensatory mechanisms in an individualized way is presented.
The Compensatory Response to Chronic Medications Can Lead to a Partial or Complete Loss of Their Effects and Is Associated with Increased Toxicity
Partial or complete loss of response to drugs is a result of compensatory opposing mechanisms that are activated upon their chronic administration. One-third of patients with rheumatoid arthritis (RA) show an inadequate primary response to anti-tumor necrosis factor (TNF)-based drugs.3 Several mechanisms were proposed to underlie this non-responsiveness.4 Some studies report a loss of efficacy in up to 48% of patients.5,6 Anti-TNF drug survival in patients with RA is 47 months.5 The overall 10-year retention rate of first-line anti-TNF agents is 23%.7,8 A primary loss of response toward anti-TNFs occurs in up to 40% of patients with inflammatory bowel disease.9 A secondary loss of response, following an initial effect, occurs in 25%–61% of these patients.10, 11, 12
Inhibitors of angiotensin-converting enzyme and angiotensin II type 1 receptor are used for the treatment of hypertension; however, they induce compensatory mechanisms that increase plasma renin activity.13 In patients with chronic heart failure, the modest effect of renin-angiotensin system inhibitors is attributed to the ability of renin-angiotensin activity to escape from suppression via potentiation of endogenous opposing vasoactive peptides.14 Compensatory mechanisms are similarly associated with neuro-endocrine circuits associated with obesity.15 Peroxisome proliferator-activated receptor gamma (PPARγ) is the target of thiazolidinediones (TZDs), which improve insulin sensitivity by reducing de novo lipogenesis in the liver. The hepatocyte-specific effects of PPARγ counterbalance the positive therapeutic actions of systemic delivery of TZDs.16
Compensatory mechanisms are also relevant to the degree of effect of neurological drugs. GABAA receptors mediate a “tonic” form of signaling that depends on the detection of GABA by extrasynaptic receptors. This process in dentate granule cells is modulated by endogenous neurosteroids, which undergo changes related to hormonal status and stress. However, tonic currents also exert a paradoxical excitatory role via depolarization of neurons or via a network effect, leading to polarization of thalamocortical neurons. Tonic currents are increased in models of focal epilepsy, due to a compensatory change that prevents seizure generation. Drugs that increase GABA potentiate the tonic currents, leading to an antiepileptic effect.17 The imaging features of drug-resistant epilepsy in idiopathic generalized tonic-clonic seizure show that hippocampal functional connectivity is impaired and this correlates with disease duration. In contrast, the hippocampal functional connectivity in drug-sensitive patients shows a compensatory enhancement, which may be used as a marker to identify and predict drug resistance.18 Dopamine depletion in the putamen is linked to changes in network functional connectivity in Parkinson’s disease, and while attributed to the pathology of the disease, it is also part of a compensatory mechanism. Patients with mild-moderate Parkinson’s disease studied after an overnight dopamine replacement therapy washout showed increased putamen functional connectivity, both in the cerebellum and in the primary motor cortex (M1). Increased cerebellar functional connectivity in this study correlated with improved motor performance, and increased M1 connectivity predicted poorer motor performance. Following the administration of a standard dose of levodopa, motor performance improved, along with reduced putamen-cerebellar connectivity. This increase in M1 functional connectivity reflects a pathological change that is harmful to motor performance, while the increased putamen-cerebellar connectivity reflects a compensatory mechanism in the brain.19
Compensatory mechanisms toward anti-malignant drugs may be associated with the development of resistance. Tumor cells adapt to therapeutic regimens by activating alternative survival and growth pathways. Current methods for overcoming resistance use targeting multiple non-overlapping pathways in an attempt to generate synergism.20 Cancer drug resistance is associated with a patient’s genetic background or, alternatively, it can be acquired by tumors that are more sensitive to anti-malignant drugs. This acquired resistance is attributed, in part, to the micro-clonality/micro-genetic heterogeneity of the tumors.21
Protein kinase-directed cancer therapies show only a transient effect in most patients. Resistance toward these drugs develops within 1 or 2 years, due to secondary mutations or other changes within the targeted kinase or due to feedback loops that bypass and compensate for the inhibited kinases.22 Protein kinase signaling is associated with feedback regulation and compensatory signaling. The emergence of resistance in leukemia, melanoma, and lung and breast tumors is associated with similar secondary compensatory mutations. In addition to mutations, resistance results from alternative/compensatory signaling pathways that are being activated.23 Drug efficacy is attenuated by alterations of the drug target, including steric interference, compensatory activity, and conformational changes. It can also be attenuated by compensatory signaling associated with bypass mechanisms and phenotype switching.24 The compensatory mechanisms within the phosphatidylinositol 3-kinase (PI3K)/phosphatase and tensin homolog (PTEN)/AKT signaling network that afford resistance to receptor tyrosine kinase (RTK) inhibition by anti-HER2 monoclonal antibodies (mAbs) has been modeled. This dynamic signaling pathway shows a sensitivity-to-resistance transition. The model has been tested in human ovarian carcinoma cell lines, using combinatorial inhibition of RTK, PTEN, and PI3K enzymes as a method for overcoming resistance.25
Current methods to overcome resistance to tyrosine kinase inhibitors include the use of second- and third-generation inhibitors that overcome new mutations, drug combinations that simultaneously block the primary oncogenic pathway and compensatory routes, and mAbs.22 A combined dose of PIK-75, an inhibitor of the PI3K/AKT pathway, and vemurafenib, which inhibits both the PI3K/AKT and mitogen-activated protein kinase (MAPK) pathways, overcomes the compensatory pathways in resistant, advanced melanomas. miR-126 has a synergistic role in overcoming resistance when included in a triple combination alongside PIK-75 and vemurafenib.26 The receptor tyrosine kinase, MET, is upregulated or over-activated in glioblastoma (GBM). Resistance to anti-MET drugs frequently occurs. Several proteins are altered in MET inhibitor-resistant GBM, including mammalian target of rapamycin (mTOR), fibroblast growth factor receptor 1 (FGFR1), epidermal growth factor receptor (EGFR), signal transducer and activator of transcription 3 (STAT3), and cyclooxygenase 2 (COX-2). Simultaneous inhibition of MET and one of these upregulated proteins promotes cell death and inhibits cell proliferation in resistant cells.27 A combination of three drugs, cetuximab, trastuzumab (an anti-HER2 mAb), and osimertinib, prevents the onset of resistance to osimertinib.28 However, these combinations may be associated with the development of multiple drug resistance.
Anti-cancer drugs targeting single metabolic pathways show limited efficacy due to the tumor’s ability to compensate by using other metabolic pathways to meet its energy and growth demands. Combinations of metabolism-targeting drugs are used to improve therapeutic efficacy in the face of compensatory response mechanisms. The combination of propranolol and dichloroacetate attenuates tumor cell metabolism and mTOR signaling, inhibits proliferation and colony formation, and induces apoptosis.29 Tumor resistance to antiangiogenic drugs is associated with the upregulation of compensatory/alternative pathways for angiogenesis, leading to tumor growth in the midst of an anti-angiogenic environment.30,31 A relative redundancy in angiogenic signaling is associated with the activation of alternative proangiogenic factors upon treatment with anti-VEGF agents. Dose intensity and schedules impact the development of resistance. Higher doses of these agents are associated with greater stimulation of compensatory proangiogenic pathways, which decreases the efficacy of these drugs, leading to escape mechanisms.32 It has been proposed that computational modeling, based on circulating biomarkers and pharmacodynamics (PD), may be utilized to attenuate this resistance.32
Pancreatic ductal adenocarcinoma (PDAC) shows chemoresistance due to the upregulation of alternative compensatory pathways.33 The effect of nanoliposomal irinotecan in the treatment of gemcitabine-refractory metastatic pancreatic cancer is limited by ATP-binding cassette G2 (ABCG2) transporter-mediated irinotecan efflux from tumor cells. It has been suggested that benzoporphyrin derivative-based photodynamic therapy (PDT), a photochemical cytotoxic modality, can activate the apoptotic pathway and reduce ABCG2 expression, thereby increasing intracellular irinotecan levels in the tumor. In animal models, the combination of PDT and a subclinical dose of nanoliposomal irinotecan synergistically inhibit tumor growth.20 The development of de novo or adaptive resistance to abiraterone limits its efficacy in patients with metastatic castration-resistant prostate cancer. Cabozantinib enhances the efficacy of abiraterone by blocking multiple compensatory survival mechanisms, including IGFIR activation.34
Compensatory mechanisms, along with the activation of major pathways unrelated to the disease, may be associated with unwanted and sometimes serious toxicity. The calcineurin inhibitors cyclosporine and tacrolimus are immunosuppressive drugs that induce nephrotoxicity. The resulting renal damage is due to tubular cell death, pro-fibrotic effects, and increased renal inflammation. Several pro-inflammatory pathways are activated by these drugs, including JAK2/STAT3 and TAK1/JNK/AP-1 pathways, TLR4/Myd88/IRAK signaling, and the unfolded protein response, which can promote nuclear factor κB (NF-κB) activation and pro-inflammatory gene expression.35
Antipsychotic drugs can cause type 2 diabetes by inhibiting insulin signaling pathways in muscle cells, hepatocytes, and adipocytes and by increasing free fatty acid levels and inflammation, thus inducing direct damage and apoptosis of β cells.36 Antipsychotic-induced movement disorders, including antipsychotic-induced parkinsonism and tardive dyskinesia, are well-described effects of these drugs. Genes associated with susceptibility to these side effects are modifier genes that determine the clinical expression of motor sub-phenotypes of the disease, disease severity, and the rate of disease progression via their impact on compensatory mechanisms for striatal dopamine loss.37
Overall, these examples suggest a deleterious impact of the compensatory effects to chronic medication use.
Inter- and Intra-patient Variability in Compensatory Pathways Impact the Clinical Effect of Chronically Administered Drugs
Variability is a characteristic of many biological systems and is also seen in the compensatory effects toward chronic medications.38, 39, 40 The unpredictable response to drugs remains a major challenge for designing effective therapeutic regimens. The variability in compensatory pathways may impact the clinical efficacy of chronic drug administration. Inter- and intra-patient variability in the mechanisms of drug resistance development can explain patient-to-patient differences in the clinical effects of the drug.24 Differences in compensatory mechanisms may have a greater impact than pharmacogenomics on the pharmacodynamics of drugs.
Diversity in drug responses and in drug toxicity is associated with ethnic variances, which manifest as alterations in the pharmacokinetics (PK) and pharmacodynamics of drugs. Pharmacogenomics underlie some of these differences.41 Inter-individual differences in drug responses are associated with variability in the expression of drug-metabolizing enzymes, transporters at sites of absorption, and in drug distribution, leading to an unpredictable exposure and tissue distribution of drugs, which then impacts efficacy and toxicity.42 Marked inter-individual variability in the analgesic response to nonsteroidal anti-inflammatory drugs has been described. This variability represents the associations between the mechanisms of pain and differences in patient phenotypes and genotypes.43,44 In a clinical trial examining pharmacodynamics according to the genotype at two COX SNPs, the inhibitory effects of celecoxib on COX-2 induction were associated with the COX-2 genotype, with rs689466 having been associated with variability in the response to COX-2 inhibition.45
P-glycoprotein is an efflux transporter expressed at the blood-brain barrier (BBB), which limits the delivery of drugs to the brain. The variability in its function at the BBB is associated with unpredictability in the response to and toxicity of drugs that target the brain.46 Similarly, albuminuria-lowering drugs have a marked variability among patients. A previous study aimed to determine whether inter-individual variability in the albuminuria response after therapy reflects a random variability or a true response variation to treatment. No correlation between on- and off-treatment albuminuria changes were observed in the placebo arm, while associations between the on- and off-treatment responses were shown for patients treated with albuminuria-lowering drugs. There was a correlation between responses when the same individual was re-exposed to the same drug at the same dose, suggesting that the effect on albuminuria with medication varies between patients, but is independent of the type of drug used.47
Cardiovascular drugs are characterized by wide inter-individual variability in dose, plasma concentration, and response (therapeutic and/or toxic) relationships. This variability is attributed to differences in pharmacokinetics and/or pharmacodynamics. Genetics, sex, age, disease state, environmental factors (e.g., smoking and diet), drug-drug interactions, and race all contribute to this variability.48 The marked inter-individual variability in the pro-arrhythmic toxicity of these drugs has been modeled by quantification of the repolarization reserve.49 Variability was simulated by randomizing model parameters in a human ventricular myocyte model, and parameters associated with action potentials were shown to impact the variability, suggesting that this nonintuitive behavior can be attributed to ionic currents that prolong action potentials, but decrease drug-induced action potential prolongation.49 The heterogeneity of treatment effects and side effects in patients with hypertension is attributed to age, race/ethnicity, mutations, and multiple polymorphisms within the components of the renin-angiotensin-aldosterone system.50
Healthy volunteers tested following a single oral administration of diclofenac showed multiple peaks in individual plasma concentration profiles. The data showed that the release of the drug from the tablets was variable and dependent on the applied stress during gastrointestinal transit.51 Intra-patient variability, in addition to inter-patient variability, also contributes to the differences in response to chronically administered medication. An assessment of the plasma levels of lamotrigine (LTG), levetiracetam (LEV), and topiramate (TPM) after the substitution of a stable brand-name drug regimen with a generic drug in patients with epilepsy has been reported. The proportion of patients showing an intra-patient change in plasma drug concentration greater than ±20% was high and was similar in the brand name and the generic group for LTG (22% versus 33%), LEV (44% versus 38%), and TPM (41% versus 6%). A significant inter-day variability in intra-patient plasma concentrations was noted, even in patients stabilized with the same brand name product over time.52
Marked intra-patient variability in etanercept trough levels, following regular dosing, decreases the feasibility of therapeutic drug monitoring. The median intra-patient variability of etanercept trough concentrations is 33% and ranges from 8% to 155%. A higher variability is correlated with lower trough levels and with non-responsiveness.53 The intra-patient variability in tacrolimus levels was studied in 432 renal transplant patients during a 4-year period. Patients with high variability had an increased risk of rejection during the first post-transplant year and showed impaired glomerular filtration rates post-transplant compared to patients with low variability.54 In contrast, the intra-patient variability in tacrolimus exposure had no effect on the outcome of heart transplants.55 Intra-patient variation in the area under the curve for plasma etoposide concentration versus time following intravenous drug administration is minimal, but increases following oral administration, where high variation in drug toxicity is seen. Mean etoposide bioavailability was 64% in this study, showing both inter- and intra-patient variability. The inter-patient variability was found to be 3-fold greater than the within-patient variability.56
Marked variability is a characteristic of many intracellular processes.39,40,57,58 Drug-target interactions in the microenvironment of the cell do not always follow the rules of simple diffusion and chemical reactions. Multiple non-specific interactions of drugs with macromolecules in cells are not measured by simple pharmacodynamics methods. Non-specific drug-target interactions are associated with the slow incorporation of DNA-binding drugs. The variable interactions in different cellular compartments contribute to the variability in intracellular drug kinetics.59 Many of the functions of the microtubules manifest as distinct variability patterns.39,60 Ongoing dynamic changes in the strength of synaptic connections in excitatory neurons occur when calcium ions Ca2+ bind the Ca2+ sensor, calmodulin (CaM), thus contributing to synaptic plasticity. Modeling the activation of downstream Ca2+/CaM-dependent binding proteins has shown that the model output is robust to parameter variability. While variations in the expression of the CaM-binding protein neurogranin decrease Ca2+/CaM-dependent kinase II activation, the overall level of receptor phosphorylation is preserved, suggesting that the compensatory response to variability is part of a regulatory process in these dynamic systems.61 Variability between cells is a characteristic of the innate immune response. Transcriptionally diverging genes that control immune functions, including the production of cytokines and chemokines, differ between cells and have distinct promoter structures.62,63 A wide intra-subject variability has been shown in the expression of intracellular nitric oxide in immune cells.64 This variability has been shown to predict an individual’s immune response to influenza vaccination and is associated with altered antibody levels.65
Studies on the compensatory effects of drugs are based on the responses of patients to regular dosing, based on persistent therapeutic regimens. Regular treatment regimens may be associated with a more profound activation of prohibitive compensatory mechanisms and, as such, may reduce the beneficial effects of drugs.38, 39, 40 Overall, these data suggest that the compensatory response to a drug is characterized by both intra- and inter-patient variability, which determine the overall response rate to the drug and the drug’s toxicity. The unpredictability of these responses makes is difficult to improve the efficacy of drugs and to overcome drug resistance using standard methods of “a single solution for all.”
A Role for Chronotherapy in Improving Efficacy and Reducing Toxicity
Chronotherapy, which is drug administration based on chronobiology, has been suggested to increase efficacy and reduce toxicity. The circadian clock synchronizes and regulates many genes and metabolic and physiological pathways, based on daily light and dark cycles.66 The master clock for this regulation is located in the suprachiasmatic nucleus of the brain and it synchronizes functions based on inputs continuously received from the retina and other tissues. In addition, most cells in the body contain molecular clocks that drive circadian rhythms from the level of chromatin to proteins.66 The synchronizations regulated by the master oscillator and by the tissue oscillators are required for the normal function of cells and organs.67 Disruption of these clocks is associated with various pathologies, and several diseases display marked circadian variation in their symptoms and severity.68,69 Shift workers have an increased incidence of chronic diseases, including diabetes, obesity, and cardiovascular mortality.70,71
Information regarding cellular molecular clocks can assist in identifying ideal times for the administration of drugs to improve both efficacy and safety.68,69 Adjusting the timing of medical treatment depending on the patient’s circadian state is applied in the treatment of nocturnal asthma, arthritis, neurological disorders, cancer, and hypertension.72 An analysis of the effectiveness of chronotherapy in more than 40 clinical trials has supported its use in the majority of cases.73 However, a lack of efficacy of chronotherapy has been shown for the use of statins to treat hyperlipidemia and in several small, short-term trials that lacked data on clinically relevant outcomes.74
The cardiovascular system is affected by the 24-h rhythm, showing altered platelet aggregability during the morning, which is associated with an increased incidence of cardiovascular events. Chronotherapy with low-dose aspirin shows a greater decrease in early morning platelet activity after evening intake compared with morning intake.75 A high-amplitude circadian rhythm of the renin-angiotensin-aldosterone system is activated during sleep, which suggests that the administration of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers at bedtime has a greater effect than when they are administered in the morning. Bedtime chronotherapy with conventional hypertension medication results in a 61% reduction in cardiovascular events compared with morning administration.76 Over-activation of the renin-angiotensin-aldosterone system underlies the pathophysiology of congestive heart failure. A pharmacokinetic-pharmacodynamic model has been developed to assess the kinetics of angiotensin-converting enzyme inhibitors in correlation with the time-dependent changes in systemic renin-angiotensin-aldosterone biomarkers. This model has shown the beneficial effect of bedtime dosing.77 Several trials have shown that mean blood pressure during sleep is an independent and more reliable predictor of cardiovascular disease risk than either mean blood pressure when awake or 24-h mean blood pressure. Sleep-time hypertension is prevalent in patients with sleep disorders, type 2 diabetes, chronic kidney disease, and resistant hypertension. The dosing of anti-hypertensive drugs according to circadian rhythms positively impacts the control of hypertension and sleep-time hypertension and the adverse effects associated with these drugs.76
The morning symptoms of RA are attributed to an abnormal circadian increase in inflammation during the night, along with inadequate cortisol secretion. As a result, joint pain and stiffness are most pronounced in the morning. The upregulation of inflammatory reactions and cytokine secretion during the night is better controlled using night-time-release formulations of steroids. Coordinating the timing of glucocorticoid therapy to coincide with the increase in serum interleukin (IL)-6 levels at night reduces morning stiffness and pain compared with morning administration.78 Bedtime methotrexate administration results in a greater improvement in RA symptoms compared to standard dosing methods.79 Similarly, trials of nonsteroidal anti-inflammatory drugs (NSAIDs) have shown that chronotherapy can improve outcomes and reduce adverse effects in RA patients.78 Inflammatory pain is linked to circadian rhythms, and diurnal patterns have been described for peripheral neuropathic pain conditions. This knowledge of circadian patterns enables the development of pain-control strategies, while minimizing adverse effects.80
Symptoms of bipolar disorders are partially associated with dysfunction of the circadian system. Antidepressants and mood stabilizers affect the circadian clock. Circadian rhythms and the manifestation of mood disorders are associated with polymorphisms in molecular clock genes.81 The rate of urine clearance of water-soluble drugs and their metabolites is associated with renal function, renal blood flow, glomerular filtration rate, the ability of the kidney to reabsorb or secrete drugs, urine flow, and urine pH, all of which have circadian behavior.82 Mesial temporal lobe epilepsy is characterized by complex partial seizures originating from the hippocampus and is a common and refractory type of epilepsy. These types of spontaneous seizures show a dependency on the endogenous clock system for seizure threshold. Temporal dysfunction of the circadian clock in the hippocampus, combined with multiple uncoupled oscillators, leads to the periodic occurrence of seizures, suggesting that chronotherapy may provide beneficial effects in these patients.83
Variability in anti-cancer therapy was described in association with cancer treatment schedules.84, 85, 86 The clock gene machinery regulates multiple cellular functions, including DNA damage recognition and repair, as well as cell proliferation.87 Chronotherapy-based regimens were proposed for improving drug effectiveness and tolerability of anticancer drugs. Dysfunction of the circadian clock is involved in tumorigenesis and impacts host-tumor and tumor-drug interactions.88 Abnormal expression levels of core clock genes in colon cancer tissue were demonstrated.87 The Circadian locomotor output cycles kaput (Clock) gene is a regulatory gene of circadian rhythm and is associated with the development of breast tumors.89
Current challenges of chronotherapy mainly rely on its personalization. A strong impact of sex, genetic background, and chronotype were described to affect the circadian timing system, drug chrono-pharmacokinetics/pharmacodynamics, and optimal timing.85,90,91 The inter-patient variability may explain the results of studies that assumed the same rhythm for the whole patient population, mistakenly concluding to an absence of circadian effect in drug efficacy and toxicities. The use of wearables to non-invasively record circadian biomarkers in individual patients and their integration into dedicated platforms was proposed to be mandatory for the future development of personalized chronotherapies.92, 93, 94, 95, 96
Taken together, these examples support the notion that drug efficacy can be improved by re-scheduling their time of administration.
Introducing Variability in Chronic Drug Administration to Overcome Compensatory Effects and Improve Long-Term Efficacy
The concept of alterations in compensatory mechanisms that are associated with disease progression and with the loss drug efficacy is thought to underlie many chronic disorders. For many disorders, both adaptation and compensation maintain homeostasis, based on a dynamic equilibrium involving non-linear interactions between the genome, epigenome, and the environment. The progress of the pathology of such disorders is a non-linear course, involving adaptation, compensation, and decompensation, eventually leading to system failure. Regulatory processes at the levels of gene expression, the cell cycle, and tissue repair are dynamic and time-dependent. Diseases involve both spatial and temporal dynamics of non-linear pathophysiological mechanisms, which affect homeostasis and may lead to system failure.97,98 The control of time-dependent adaptive and compensatory responses can modify the disease course and may also improve the response to chronic medication administration. However, this is likely to require a personalized approach.
The prominent effects of chronobiology on drug efficacy and toxicity may be associated with an alteration of the compensatory mechanisms that are activated by chronic drug administration, some of which are deleterious. Regimens are currently being developed in an attempt to overcome these compensatory effects. In addition, identifying simultaneous aberrant molecular pathways and pathway-based molecular signatures has been proposed to assist in the study of compensatory mechanisms and in stratifying patients and generating a personalized treatment regimen to improve response rates and reduce side effects.97
Marked irregularities are observed in the effects of chronic drug intake. Dosing regimens are usually designed according to the pharmacokinetics of the drug. However, this does not take into consideration the large inter- and intra-individual variability, the potentially altered volume of distribution in patients with obesity, congestive heart failure, and edema, or changes in renal or non-renal drug excretion.
A drug holiday has been proposed as a potential way for overcoming partial or complete loss of response and for reducing the side effects of chronic medication administration. Tyrosine kinase inhibitors of the EGFR are used to treat patients with non-small-cell lung cancer. Despite an initial response, patients with an activating EGFR mutation invariably relapse. Retreatment following a drug holiday is effective in patients who initially benefited from treatment, but whose disease subsequently progressed.99 A favorable safety profile and the feasibility of drug holidays have been shown for trabectedin in patients with metastatic sarcomas and in patients with metastatic renal-cell carcinoma.100,101 The loss of response to levodopa in patients with Parkinson’s disease is attributed to disease progression and an altered receptor sensitivity. Drug holidays have been used for re-sensitizing dopamine receptors in the striatum, but they are associated with neuroleptic malignant-like syndrome. Intravenous administration of amantadine during the drug holiday prevents this effect. Following drug holidays, when levodopa administration is resumed at the same dose, improvements are seen in both motor condition and side effects.102 A dosing regimen consisting of cyclic withdrawal of interferon β is not inferior to the full regimen in relapsing, remitting multiple sclerosis patients and it did not affect long-term outcome.103 RA patients with long-standing, low disease activity have been shown to be eligible for tapering and/or ceasing anti-rheumatic medications.104 A disease-modifying anti-rheumatic drug-tapering algorithm, based on a multi-biomarker disease activity score and anti-citrullinated protein antibody levels, enabled risk stratification for successful tapering in these patients.105 In patients with early RA who had achieved sustained, low disease activity, reduced-frequency regimens of certolizumab pegol with methotrexate maintain the low disease activity and are not inferior to the standard full dosing.106 A randomized controlled trial in RA patients who achieved sustained remission demonstrated that more than half of the patients maintained remission after tapering or stopping conventional and biological treatments.107 In contrast, small trials in patients with multiple sclerosis have failed to show similar results following drug holidays.108,109
Methods are being developed to collect data from patients regarding their real-life drug scheduling. Classical pharmacological methods have been reconsidered to enable the use of a stochastic context for a patient’s drug intake irregularity. A repeated one-point method (ROPM) is a model that assumes equal maintenance doses at equal dosing intervals. However, in real-world practice, many patients do not consume their medications at equal intervals. A modified ROPM has been developed for unequal dosing intervals, in an attempt to maintain the appropriate the drug concentration, by using different dose sizes to maintain trough concentrations at steady-state (Cssmin) and peak concentrations at steady-state (Cssmax) at equal dose sizes, where the peak does not exceed a certain maximum concentration or fall below a certain minimum concentration.110 A different stochastic model has been developed based on an irregular drug intake schedule for drugs that require multiple oral doses and is based on patients with poor medication adherence. The model assumes that an irregular dosing schedule follows a Poisson distribution, and it has been used to analyze alterations in drug concentrations and the cumulative probability distribution. This model is based on four variables, including continuous time or discrete time, with each using deterministic or random doses. The mean rate of intake, elimination rate, absorption rate, and mean dose are calculated using the model. Analyzing the effect of poor adherence on drug efficacy showed that a random dosing schedule is associated with inconsistent drug concentration behaviors.111 While this model was designed to improve dosing under conditions of poor adherence, it also supports the option for the prevention of drug level plateaus, which are associated with unwanted compensatory effects and drug toxicity. Similarly, a model has been developed for HIV treatment, using a combination of lopinavir/ritonavir for once daily and twice daily regimens. Missing doses and deviations from nominal times have been analyzed using this model. The probabilistic model of measuring the impact of noncompliance has been used to optimize dosing under situations of noncompliance.112 While these methods are aimed at reaching a certain drug level, the fact that many patients take the drugs using unequal interval regimens may, however, assist them in achieving an improved response rate and lower toxicity.
Introducing irregularity into therapeutic regimens may have a beneficial impact on drug efficacy. Amlodipine administered alone or together with a beta-blocker, reduces myocardial ischemia when patients frequently forget to take medication or dose irregularly, including drug-free intervals.113 While this is attributed to the relatively prolonged half-life of the drug, the irregularity may have directly affected the results. Secondary loss of response to anti-TNF therapy in patients with inflammatory bowel disease is attributed to inadequate drug exposure and sub-therapeutic drug levels. It has been suggested that the “one size fits all” concept does not apply to all patients and that attempts to improve efficacy solely by improving drug levels are insufficient.114 In a prospective trial in these patients, a loss of clinical response occurred in 36% of those receiving a standard anti-TNF dosing regimen, but in only 13% of patients who were dosed based on a de-escalation dashboard, leading to alternating increases or decreases in the dosage. These data showed that simple alterations in dosage can improve the efficacy of anti-TNF drugs, as compared with the fixed dosing regimen.115 In contrast, a retrospective, small observational trial of patients treated with maintenance infliximab showed that those who continued an uninterrupted maintenance dosing regimen had a lower incidence of hospitalization and surgery than did those who received an irregular or interrupted regimen.116 These discrepancies may be attributed to the lack of personalization of the regimens used.
Intra- and inter-patient variability in the response to drugs and in the heterogeneity of disease and the diversity of parameters associated with different compensatory mechanisms are all contributing to drug resistance. Many of these systems are dynamic and change over time even in the same patient.40,117
A dynamic platform, based on signatures of patient-tailored variability, is being developed to ensure the long-term, sustainable beneficial effect of drugs, with minimal toxicity.4,40,58,63,118 The platform is being developed in steps. In the first step, the effect of introducing variability into therapeutic regimens of patients who partially or completely lost their response to chronic medications is evaluated using algorithms that introduce pseudo-random number generators, which introduce variability in times of administration and dosages within an approved range. In the second step, a closed-loop algorithm is implemented wherein inputs are based on clinically meaningful endpoints used for generating therapeutic regimens. In the third step, host and disease-related patterns of variability are quantified in a personalized way and are implemented into a true-random number generator. Inputs from quantifiable variability parameters that are related with the disease, host response, and mechanism of action of the drug are introduced.
Ongoing clinical trials implementing these personalized variability-based algorithms are evaluating the effects of these regimens in patients with inflammatory bowel disease who lost their response to anti-TNFs, and in patients with epilepsy who lost response to anti-epileptics (ClinicalTrials.gov: NCT03843697 and NCT03747705). These studies will shed light on their potential effect in patients with chronic diseases. These studies are aimed at implementing dynamic personalized-based variability signatures that are based on the disease for which the algorithms are being developed in an individualized way. The results of these studies will enable the overcoming of compensatory mechanisms that prohibit the full response to chronic medication administration, while reducing toxicity.
Conflicts of Interest
Y.I. is the founder of Oberon Sciences and a consultant for Teva, ENZO, Protalix, Betalin Therapeutics, Immuron, SciM, Natural Shield, Oberon Sciences, Tiziana Pharma, Plantylight, and Exalenz Bioscience.
Acknowledgments
This work was supported by the Roaman-Epstein Research Foundation.
References
- 1.Gurevich E.V., Gurevich V.V. Beyond traditional pharmacology: new tools and approaches. Br. J. Pharmacol. 2015;172:3229–3241. doi: 10.1111/bph.13066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lopez J.S., Banerji U. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017;14:57–66. doi: 10.1038/nrclinonc.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottenberg J.E., Brocq O., Perdriger A., Lassoued S., Berthelot J.M., Wendling D., Euller-Ziegler L., Soubrier M., Richez C., Fautrel B. Non-TNF-targeted biologic vs a second anti-TNF drug to treat rheumatoid arthritis in patients with insufficient response to a first anti-TNF drug: a randomized clinical trial. JAMA. 2016;316:1172–1180. doi: 10.1001/jama.2016.13512. [DOI] [PubMed] [Google Scholar]
- 4.Khoury T., Ilan Y. Introducing patterns of variability for overcoming compensatory adaptation of the immune system to immunomodulatory agents: a novel method for improving clinical response to anti-TNF therapies. Front. Immunol. 2019;10:2726. doi: 10.3389/fimmu.2019.02726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fafá B.P., Louzada-Junior P., Titton D.C., Zandonade E., Ranza R., Laurindo I., Peçanha P., Ranzolin A., Hayata A.L., Duarte A., BIOBADABRASIL Drug survival and causes of discontinuation of the first anti-TNF in ankylosing spondylitis compared with rheumatoid arthritis: analysis from BIOBADABRASIL. Clin. Rheumatol. 2015;34:921–927. doi: 10.1007/s10067-015-2929-7. [DOI] [PubMed] [Google Scholar]
- 6.Souto A., Maneiro J.R., Gómez-Reino J.J. Rate of discontinuation and drug survival of biologic therapies in rheumatoid arthritis: a systematic review and meta-analysis of drug registries and health care databases. Rheumatology (Oxford) 2016;55:523–534. doi: 10.1093/rheumatology/kev374. [DOI] [PubMed] [Google Scholar]
- 7.Biggioggero M., Favalli E.G. Ten-year drug survival of anti-TNF agents in the treatment of inflammatory arthritides. Drug Dev. Res. 2014;75(Suppl 1):S38–S41. doi: 10.1002/ddr.21192. [DOI] [PubMed] [Google Scholar]
- 8.Kalden J.R., Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat. Rev. Rheumatol. 2017;13:707–718. doi: 10.1038/nrrheum.2017.187. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Horin S., Kopylov U., Chowers Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun. Rev. 2014;13:24–30. doi: 10.1016/j.autrev.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Gisbert J.P., Panés J. Loss of response and requirement of infliximab dose intensification in Crohn’s disease: a review. Am. J. Gastroenterol. 2009;104:760–767. doi: 10.1038/ajg.2008.88. [DOI] [PubMed] [Google Scholar]
- 11.Billioud V., Sandborn W.J., Peyrin-Biroulet L. Loss of response and need for adalimumab dose intensification in Crohn’s disease: a systematic review. Am. J. Gastroenterol. 2011;106:674–684. doi: 10.1038/ajg.2011.60. [DOI] [PubMed] [Google Scholar]
- 12.Ma C., Huang V., Fedorak D.K., Kroeker K.I., Dieleman L.A., Halloran B.P., Fedorak R.N. Crohn’s disease outpatients treated with adalimumab have an earlier secondary loss of response and requirement for dose escalation compared to infliximab: a real life cohort study. J. Crohn’s Colitis. 2014;8:1454–1463. doi: 10.1016/j.crohns.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Barrios V., Escobar C. Aliskiren in the management of hypertension. Am. J. Cardiovasc. Drugs. 2010;10:349–358. doi: 10.2165/11584980-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 14.Packer M., McMurray J.J.V. Importance of endogenous compensatory vasoactive peptides in broadening the effects of inhibitors of the renin-angiotensin system for the treatment of heart failure. Lancet. 2017;389:1831–1840. doi: 10.1016/S0140-6736(16)30969-2. [DOI] [PubMed] [Google Scholar]
- 15.van der Klaauw A.A. Neuropeptides in obesity and metabolic disease. Clin. Chem. 2018;64:173–182. doi: 10.1373/clinchem.2017.281568. [DOI] [PubMed] [Google Scholar]
- 16.Wolf Greenstein A., Majumdar N., Yang P., Subbaiah P.V., Kineman R.D., Cordoba-Chacon J. Hepatocyte-specific, PPARγ-regulated mechanisms to promote steatosis in adult mice. J. Endocrinol. 2017;232:107–121. doi: 10.1530/JOE-16-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker M.C., Kullmann D.M. In: Tonic GABAA receptor-mediated signaling in epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies. 4th edition. Noebels J.L., Avoli M., Rogawski M.A., Olsen R.W., Delgado-Escueta A.V., editors. National Center for Biotechnology Information [US]; 2012. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z., Wang X., Rong R., Xu Y., Zhang B., Wang Z. Impaired hippocampal functional connectivity in patients with drug resistant, generalized tonic-clonic seizures. Neuroreport. 2019;30:700–706. doi: 10.1097/WNR.0000000000001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simioni A.C., Dagher A., Fellows L.K. Compensatory striatal-cerebellar connectivity in mild-moderate Parkinson’s disease. Neuroimage Clin. 2015;10:54–62. doi: 10.1016/j.nicl.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang H.C., Mallidi S., Liu J., Chiang C.T., Mai Z., Goldschmidt R., Ebrahim-Zadeh N., Rizvi I., Hasan T. Photodynamic therapy synergizes with irinotecan to overcome compensatory mechanisms and improve treatment outcomes in pancreatic cancer. Cancer Res. 2016;76:1066–1077. doi: 10.1158/0008-5472.CAN-15-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rueff J., Rodrigues A.S. Cancer drug resistance: a brief overview from a genetic viewpoint. Methods Mol. Biol. 2016;1395:1–18. doi: 10.1007/978-1-4939-3347-1_1. [DOI] [PubMed] [Google Scholar]
- 22.Mancini M., Yarden Y. Mutational and network level mechanisms underlying resistance to anti-cancer kinase inhibitors. Semin. Cell Dev. Biol. 2016;50:164–176. doi: 10.1016/j.semcdb.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 23.Rosenzweig S.A. Acquired resistance to drugs targeting tyrosine kinases. Adv. Cancer Res. 2018;138:71–98. doi: 10.1016/bs.acr.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray B.W., Miller N. Durability of kinase-directed therapies—a network perspective on response and resistance. Mol. Cancer Ther. 2015;14:1975–1984. doi: 10.1158/1535-7163.MCT-15-0088. [DOI] [PubMed] [Google Scholar]
- 25.Goltsov A., Faratian D., Langdon S.P., Bown J., Goryanin I., Harrison D.J. Compensatory effects in the PI3K/PTEN/AKT signaling network following receptor tyrosine kinase inhibition. Cell. Signal. 2011;23:407–416. doi: 10.1016/j.cellsig.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 26.Pedini F., De Luca G., Felicetti F., Puglisi R., Boe A., Arasi M.B., Fratini F., Mattia G., Spada M., Caporali S. Joint action of miR-126 and MAPK/PI3K inhibitors against metastatic melanoma. Mol. Oncol. 2019;13:1836–1854. doi: 10.1002/1878-0261.12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cruickshanks N., Zhang Y., Hine S., Gibert M., Yuan F., Oxford M., Grello C., Pahuski M., Dube C., Guessous F. Discovery and therapeutic exploitation of mechanisms of resistance to MET inhibitors in glioblastoma. Clin. Cancer Res. 2019;25:663–673. doi: 10.1158/1078-0432.CCR-18-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romaniello D., Mazzeo L., Mancini M., Marrocco I., Noronha A., Kreitman M., Srivastava S., Ghosh S., Lindzen M., Salame T.M. A combination of approved antibodies overcomes resistance of lung cancer to osimertinib by blocking bypass pathways. Clin. Cancer Res. 2018;24:5610–5621. doi: 10.1158/1078-0432.CCR-18-0450. [DOI] [PubMed] [Google Scholar]
- 29.Lucido C.T., Miskimins W.K., Vermeer P.D. Propranolol promotes glucose dependence and synergizes with dichloroacetate for anti-cancer activity in HNSCC. Cancers (Basel) 2018;10:476. doi: 10.3390/cancers10120476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinto M.P., Sotomayor P., Carrasco-Avino G., Corvalan A.H., Owen G.I. Escaping antiangiogenic therapy: strategies employed by cancer cells. Int. J. Mol. Sci. 2016;17:1489. doi: 10.3390/ijms17091489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gacche R.N., Assaraf Y.G. Redundant angiogenic signaling and tumor drug resistance. Drug Resist. Updat. 2018;36:47–76. doi: 10.1016/j.drup.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Sharan S., Woo S. Systems pharmacology approaches for optimization of antiangiogenic therapies: challenges and opportunities. Front. Pharmacol. 2015;6:33. doi: 10.3389/fphar.2015.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adamska A., Elaskalani O., Emmanouilidi A., Kim M., Abdol Razak N.B., Metharom P., Falasca M. Molecular and cellular mechanisms of chemoresistance in pancreatic cancer. Adv. Biol. Regul. 2018;68:77–87. doi: 10.1016/j.jbior.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Wang X., Huang Y., Christie A., Bowden M., Lee G.S., Kantoff P.W., Sweeney C.J. Cabozantinib inhibits abiraterone’s upregulation of IGFIR phosphorylation and enhances its anti-prostate cancer activity. Clin. Cancer Res. 2015;21:5578–5587. doi: 10.1158/1078-0432.CCR-15-0824. [DOI] [PubMed] [Google Scholar]
- 35.González-Guerrero C., Ocaña-Salceda C., Berzal S., Carrasco S., Fernández-Fernández B., Cannata-Ortiz P., Egido J., Ortiz A., Ramos A.M. Calcineurin inhibitors recruit protein kinases JAK2 and JNK, TLR signaling and the UPR to activate NF-κB-mediated inflammatory responses in kidney tubular cells. Toxicol. Appl. Pharmacol. 2013;272:825–841. doi: 10.1016/j.taap.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 36.Chen J., Huang X.F., Shao R., Chen C., Deng C. Molecular mechanisms of antipsychotic drug-induced diabetes. Front. Neurosci. 2017;11:643. doi: 10.3389/fnins.2017.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenbaum L., Lerer B. Pharmacogenetics of antipsychotic-induced movement disorders as a resource for better understanding Parkinson’s disease modifier genes. Front. Neurol. 2015;6:27. doi: 10.3389/fneur.2015.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ilan Y. Why targeting the microbiome is not so successful: can randomness overcome the adaptation that occurs following gut manipulation? Clin. Exp. Gastroenterol. 2019;12:209–217. doi: 10.2147/CEG.S203823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ilan Y. Randomness in microtubule dynamics: an error that requires correction or an inherent plasticity required for normal cellular function? Cell Biol. Int. 2019;43:739–748. doi: 10.1002/cbin.11157. [DOI] [PubMed] [Google Scholar]
- 40.Ilan Y. Generating randomness: making the most out of disordering a false order into a real one. J. Transl. Med. 2019;17:49. doi: 10.1186/s12967-019-1798-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yasuda S.U., Zhang L., Huang S.M. The role of ethnicity in variability in response to drugs: focus on clinical pharmacology studies. Clin. Pharmacol. Ther. 2008;84:417–423. doi: 10.1038/clpt.2008.141. [DOI] [PubMed] [Google Scholar]
- 42.Urquhart B.L., Tirona R.G., Kim R.B. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs. J. Clin. Pharmacol. 2007;47:566–578. doi: 10.1177/0091270007299930. [DOI] [PubMed] [Google Scholar]
- 43.Theken K.N. Variability in analgesic response to non-steroidal anti-inflammatory drugs. Prostaglandins Other Lipid Mediat. 2018;139:63–70. doi: 10.1016/j.prostaglandins.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 44.Bruno A., Tacconelli S., Patrignani P. Variability in the response to non-steroidal anti-inflammatory drugs: mechanisms and perspectives. Basic Clin. Pharmacol. Toxicol. 2014;114:56–63. doi: 10.1111/bcpt.12117. [DOI] [PubMed] [Google Scholar]
- 45.Lee S.J., Park M.K., Shin D.S., Chun M.H. Variability of the drug response to nonsteroidal anti-inflammatory drugs according to cyclooxygenase-2 genetic polymorphism. Drug Des. Devel. Ther. 2017;11:2727–2736. doi: 10.2147/DDDT.S143807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bauer M., Tournier N., Langer O. Imaging P-glycoprotein function at the blood-brain barrier as a determinant of the variability in response to central nervous system drugs. Clin. Pharmacol. Ther. 2019;105:1061–1064. doi: 10.1002/cpt.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petrykiv S.I., de Zeeuw D., Persson F., Rossing P., Gansevoort R.T., Laverman G.D., Heerspink H.J.L. Variability in response to albuminuria-lowering drugs: true or random? Br. J. Clin. Pharmacol. 2017;83:1197–1204. doi: 10.1111/bcp.13217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El Desoky E.S., Derendorf H., Klotz U. Variability in response to cardiovascular drugs. Curr. Clin. Pharmacol. 2006;1:35–46. doi: 10.2174/157488406775268273. [DOI] [PubMed] [Google Scholar]
- 49.Sarkar A.X., Sobie E.A. Quantification of repolarization reserve to understand interpatient variability in the response to proarrhythmic drugs: a computational analysis. Heart Rhythm. 2011;8:1749–1755. doi: 10.1016/j.hrthm.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Materson B.J. Variability in response to antihypertensive drugs. Am. J. Med. 2007;120(4, Suppl 1):S10–S20. doi: 10.1016/j.amjmed.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Garbacz G., Wedemeyer R.S., Nagel S., Giessmann T., Mönnikes H., Wilson C.G., Siegmund W., Weitschies W. Irregular absorption profiles observed from diclofenac extended release tablets can be predicted using a dissolution test apparatus that mimics in vivo physical stresses. Eur. J. Pharm. Biopharm. 2008;70:421–428. doi: 10.1016/j.ejpb.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 52.Contin M., Alberghini L., Candela C., Benini G., Riva R. Intrapatient variation in antiepileptic drug plasma concentration after generic substitution vs stable brand-name drug regimens. Epilepsy Res. 2016;122:79–83. doi: 10.1016/j.eplepsyres.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 53.van Bezooijen J.S., Schreurs M.W.J., Koch B.C.P., Velthuis H.T., van Doorn M.B.A., Prens E.P., van Gelder T. Intrapatient variability in the pharmacokinetics of etanercept maintenance treatment. Ther. Drug Monit. 2017;39:333–338. doi: 10.1097/FTD.0000000000000384. [DOI] [PubMed] [Google Scholar]
- 54.Whalen H.R., Glen J.A., Harkins V., Stevens K.K., Jardine A.G., Geddes C.C., Clancy M.J. High intrapatient tacrolimus variability is associated with worse outcomes in renal transplantation using a low-dose tacrolimus immunosuppressive regime. Transplantation. 2017;101:430–436. doi: 10.1097/TP.0000000000001129. [DOI] [PubMed] [Google Scholar]
- 55.Shuker N., Bouamar R., Hesselink D.A., van Gelder T., Caliskan K., Manintveld O.C., Balk A.H., Constantinescu A.A. Intrapatient variability in tacrolimus exposure does not predict the development of cardiac allograft vasculopathy after heart transplant. Exp. Clin. Transplant. 2018;16:326–332. doi: 10.6002/ect.2016.0366. [DOI] [PubMed] [Google Scholar]
- 56.Hande K., Messenger M., Wagner J., Krozely M., Kaul S. Inter- and intrapatient variability in etoposide kinetics with oral and intravenous drug administration. Clin. Cancer Res. 1999;5:2742–2747. [PubMed] [Google Scholar]
- 57.Finn E.H., Misteli T. Molecular basis and biological function of variability in spatial genome organization. Science. 2019;365 doi: 10.1126/science.aaw9498. eaaw9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ilan Y. Advanced tailored randomness: a novel approach for improving the efficacy of biological systems. J. Comput. Biol. 2020;27:20–29. doi: 10.1089/cmb.2019.0231. [DOI] [PubMed] [Google Scholar]
- 59.Elgart V., Lin J.R., Loscalzo J. Determinants of drug-target interactions at the single cell level. PLoS Comput. Biol. 2018;14:e1006601. doi: 10.1371/journal.pcbi.1006601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ilan-Ber T., Ilan Y. The role of microtubules in the immune system and as potential targets for gut-based immunotherapy. Mol. Immunol. 2019;111:73–82. doi: 10.1016/j.molimm.2019.04.014. [DOI] [PubMed] [Google Scholar]
- 61.Pharris M.C., Patel N.M., Kinzer-Ursem T.L. Competitive tuning among Ca2+/calmodulin-dependent proteins: analysis of in silico model robustness and parameter variability. Cell. Mol. Bioeng. 2018;11:353–365. doi: 10.1007/s12195-018-0549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hagai T., Chen X., Miragaia R.J., Rostom R., Gomes T., Kunowska N., Henriksson J., Park J.E., Proserpio V., Donati G. Gene expression variability across cells and species shapes innate immunity. Nature. 2018;563:197–202. doi: 10.1038/s41586-018-0657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ilan Y. β-Glycosphingolipids as mediators of both inflammation and immune tolerance: a manifestation of randomness in biological systems. Front. Immunol. 2019;10:1143. doi: 10.3389/fimmu.2019.01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maharaj S., Lu K.D., Radom-Aizik S., Zaldivar F., Haddad F., Shin H.W., Leu S.Y., Nussbaum E., Randhawa I., Cooper D.M. Inter- and intra-subject variability of nitric oxide levels in leukocyte subpopulations. Nitric Oxide. 2018;72:41–45. doi: 10.1016/j.niox.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jenkins B.N., Hunter J.F., Cross M.P., Acevedo A.M., Pressman S.D. When is affect variability bad for health? The association between affect variability and immune response to the influenza vaccination. J. Psychosom. Res. 2018;104:41–47. doi: 10.1016/j.jpsychores.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mermet J., Yeung J., Naef F. Systems chronobiology: global analysis of gene regulation in a 24-hour periodic world. Cold Spring Harb. Perspect. Biol. 2017;9:9. doi: 10.1101/cshperspect.a028720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chowdhury D., Wang C., Lu A.P., Zhu H.L. Understanding quantitative circadian regulations are crucial towards advancing chronotherapy. Cells. 2019;8:883. doi: 10.3390/cells8080883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wallach T., Kramer A. Chemical chronobiology: toward drugs manipulating time. FEBS Lett. 2015;589:1530–1538. doi: 10.1016/j.febslet.2015.04.059. [DOI] [PubMed] [Google Scholar]
- 69.Farrow S.N., Solari R., Willson T.M. The importance of chronobiology to drug discovery. Expert Opin. Drug Discov. 2012;7:535–541. doi: 10.1517/17460441.2012.689283. [DOI] [PubMed] [Google Scholar]
- 70.Lunn R.M., Blask D.E., Coogan A.N., Figueiro M.G., Gorman M.R., Hall J.E., Hansen J., Nelson R.J., Panda S., Smolensky M.H. Health consequences of electric lighting practices in the modern world: a report on the National Toxicology Program’s workshop on shift work at night, artificial light at night, and circadian disruption. Sci. Total Environ. 2017;607-608:1073–1084. doi: 10.1016/j.scitotenv.2017.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ulhôa M.A., Marqueze E.C., Burgos L.G., Moreno C.R. Shift work and endocrine disorders. Int. J. Endocrinol. 2015;2015:826249. doi: 10.1155/2015/826249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Friedman E.A., Banerji M.A. Chronobiology impacts response to antihypertensive drug regimen in type 2 diabetes. Diabetes Care. 2011;34:1438–1439. doi: 10.2337/dc11-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaur G., Phillips C., Wong K., Saini B. Timing is important in medication administration: a timely review of chronotherapy research. Int. J. Clin. Pharm. 2013;35:344–358. doi: 10.1007/s11096-013-9749-0. [DOI] [PubMed] [Google Scholar]
- 74.Izquierdo-Palomares J.M., Fernandez-Tabera J.M., Plana M.N., Añino Alba A., Gómez Álvarez P., Fernandez-Esteban I., Saiz L.C., Martin-Carrillo P., Pinar López Ó. Chronotherapy versus conventional statins therapy for the treatment of hyperlipidaemia. Cochrane Database Syst. Rev. 2016;11:CD009462. doi: 10.1002/14651858.CD009462.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buurma M., van Diemen J.J.K., Thijs A., Numans M.E., Bonten T.N. Circadian rhythm of cardiovascular disease: the potential of chronotherapy with aspirin. Front. Cardiovasc. Med. 2019;6:84. doi: 10.3389/fcvm.2019.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hermida R.C., Ayala D.E., Smolensky M.H., Fernández J.R., Mojón A., Portaluppi F. Chronotherapy with conventional blood pressure medications improves management of hypertension and reduces cardiovascular and stroke risks. Hypertens. Res. 2016;39:277–292. doi: 10.1038/hr.2015.142. [DOI] [PubMed] [Google Scholar]
- 77.Mochel J.P., Danhof M. Chronobiology and pharmacologic modulation of the renin-angiotensin-aldosterone system in dogs: what have we learned? Rev. Physiol. Biochem. Pharmacol. 2015;169:43–69. doi: 10.1007/112_2015_27. [DOI] [PubMed] [Google Scholar]
- 78.Buttgereit F., Smolen J.S., Coogan A.N., Cajochen C. Clocking in: chronobiology in rheumatoid arthritis. Nat. Rev. Rheumatol. 2015;11:349–356. doi: 10.1038/nrrheum.2015.31. [DOI] [PubMed] [Google Scholar]
- 79.Cutolo M. Glucocorticoids and chronotherapy in rheumatoid arthritis. RMD Open. 2016;2:e000203. doi: 10.1136/rmdopen-2015-000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gilron I. Impact of chronobiology on neuropathic pain treatment. Pain Manag. 2016;6:241–247. doi: 10.2217/pmt-2015-0007. [DOI] [PubMed] [Google Scholar]
- 81.Dallaspezia S., Benedetti F. Chronobiology of bipolar disorder: therapeutic implication. Curr. Psychiatry Rep. 2015;17:606. doi: 10.1007/s11920-015-0606-9. [DOI] [PubMed] [Google Scholar]
- 82.De Lavallaz L., Musso C.G. Chronobiology in nephrology: the influence of circadian rhythms on renal handling of drugs and renal disease treatment. Int. Urol. Nephrol. 2018;50:2221–2228. doi: 10.1007/s11255-018-2001-z. [DOI] [PubMed] [Google Scholar]
- 83.Leite Góes Gitai D., de Andrade T.G., Dos Santos Y.D.R., Attaluri S., Shetty A.K. Chronobiology of limbic seizures: potential mechanisms and prospects of chronotherapy for mesial temporal lobe epilepsy. Neurosci. Biobehav. Rev. 2019;98:122–134. doi: 10.1016/j.neubiorev.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dallmann R., Okyar A., Lévi F. Dosing-time makes the poison: circadian regulation and pharmacotherapy. Trends Mol. Med. 2016;22:430–445. doi: 10.1016/j.molmed.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 85.Ballesta A., Innominato P.F., Dallmann R., Rand D.A., Lévi F.A. Systems chronotherapeutics. Pharmacol. Rev. 2017;69:161–199. doi: 10.1124/pr.116.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shuboni-Mulligan D.D., Breton G., Smart D., Gilbert M., Armstrong T.S. Radiation chronotherapy-clinical impact of treatment time-of-day: a systematic review. J. Neurooncol. 2019;145:415–427. doi: 10.1007/s11060-019-03332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mazzoccoli G., Panza A., Valvano M.R., Palumbo O., Carella M., Pazienza V., Biscaglia G., Tavano F., Di Sebastiano P., Andriulli A., Piepoli A. Clock gene expression levels and relationship with clinical and pathological features in colorectal cancer patients. Chronobiol. Int. 2011;28:841–851. doi: 10.3109/07420528.2011.615182. [DOI] [PubMed] [Google Scholar]
- 88.Mazzoccoli G., Vinciguerra M., Papa G., Piepoli A. Circadian clock circuitry in colorectal cancer. World J. Gastroenterol. 2014;20:4197–4207. doi: 10.3748/wjg.v20.i15.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li X., Wang S., Yang S., Ying J., Yu H., Yang C., Liu Y., Wang Y., Cheng S., Xiao J. Circadian locomotor output cycles kaput affects the proliferation and migration of breast cancer cells by regulating the expression of E-cadherin via IQ motif containing GTPase activating protein 1. Oncol. Lett. 2018;15:7097–7103. doi: 10.3892/ol.2018.8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bailey M., Silver R. Sex differences in circadian timing systems: implications for disease. Front. Neuroendocrinol. 2014;35:111–139. doi: 10.1016/j.yfrne.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jones S.E., Lane J.M., Wood A.R., van Hees V.T., Tyrrell J., Beaumont R.N., Jeffries A.R., Dashti H.S., Hillsdon M., Ruth K.S. Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nat. Commun. 2019;10:343. doi: 10.1038/s41467-018-08259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grant A.D., Wilsterman K., Smarr B.L., Kriegsfeld L.J. Evidence for a coupled oscillator model of endocrine ultradian rhythms. J. Biol. Rhythms. 2018;33:475–496. doi: 10.1177/0748730418791423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hannay K.M., Booth V., Forger D.B. Macroscopic models for human circadian rhythms. J. Biol. Rhythms. 2019;34:658–671. doi: 10.1177/0748730419878298. [DOI] [PubMed] [Google Scholar]
- 94.Komarzynski S., Bolborea M., Huang Q., Finkenstädt B., Lévi F. Predictability of individual circadian phase during daily routine for medical applications of circadian clocks. JCI Insight. 2019;4:e130423. doi: 10.1172/jci.insight.130423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Skeldon A.C., Dijk D.J., Derks G. Mathematical models for sleep-wake dynamics: comparison of the two-process model and a mutual inhibition neuronal model. PLoS ONE. 2014;9:e103877. doi: 10.1371/journal.pone.0103877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smarr B.L. Digital sleep logs reveal potential impacts of modern temporal structure on class performance in different chronotypes. J. Biol. Rhythms. 2015;30:61–67. doi: 10.1177/0748730414565665. [DOI] [PubMed] [Google Scholar]
- 97.Hampel H., Vergallo A., Aguilar L.F., Benda N., Broich K., Cuello A.C., Cummings J., Dubois B., Federoff H.J., Fiandaca M., Alzheimer Precision Medicine Initiative (APMI) Precision pharmacology for Alzheimer’s disease. Pharmacol. Res. 2018;130:331–365. doi: 10.1016/j.phrs.2018.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Noble D. Evolution viewed from physics, physiology and medicine. Interface Focus. 2017;7:20160159. doi: 10.1098/rsfs.2016.0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Becker A., Crombag L., Heideman D.A., Thunnissen F.B., van Wijk A.W., Postmus P.E., Smit E.F. Retreatment with erlotinib: regain of TKI sensitivity following a drug holiday for patients with NSCLC who initially responded to EGFR-TKI treatment. Eur. J. Cancer. 2011;47:2603–2606. doi: 10.1016/j.ejca.2011.06.046. [DOI] [PubMed] [Google Scholar]
- 100.Pierantoni F., Maruzzo M., Brunello A., Chiusole B., Pusole G., Bezzon E., Basso U., Zagonel V. Trabectedin drug holiday and rechallenge in soft tissue sarcomas: report of 4 cases and literature review. Front. Oncol. 2019;9:553. doi: 10.3389/fonc.2019.00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mittal K., Derosa L., Albiges L., Wood L., Elson P., Gilligan T., Garcia J., Dreicer R., Escudier B., Rini B. Drug holiday in metastatic renal-cell carcinoma patients treated with vascular endothelial growth factor receptor inhibitors. Clin. Genitourin. Cancer. 2018;16:e663–e667. doi: 10.1016/j.clgc.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 102.Koziorowski D., Friedman A. Levodopa “drug holiday” with amantadine infusions as a treatment of complications in Parkinson’s disease. Mov. Disord. 2007;22:1033–1036. doi: 10.1002/mds.21448. [DOI] [PubMed] [Google Scholar]
- 103.Romano S., Ferraldeschi M., Bagnato F., Mechelli R., Morena E., Caldano M., Buscarinu M.C., Fornasiero A., Frontoni M., Nociti V. Drug holiday of interferon beta 1b in multiple sclerosis: a pilot, randomized, single blind study of non-inferiority. Front. Neurol. 2019;10:695. doi: 10.3389/fneur.2019.00695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Markusse I.M., Akdemir G., Huizinga T.W., Allaart C.F. Drug-free holiday in patients with rheumatoid arthritis: a qualitative study to explore patients’ opinion. Clin. Rheumatol. 2014;33:1155–1159. doi: 10.1007/s10067-014-2500-y. [DOI] [PubMed] [Google Scholar]
- 105.Hagen M., Englbrecht M., Haschka J., Reiser M., Kleyer A., Hueber A., Manger B., Figueiredo C., Cobra J.F., Tony H.P. Cost-effective tapering algorithm in patients with rheumatoid arthritis: combination of multibiomarker disease activity score and autoantibody status. J. Rheumatol. 2019;46:460–466. doi: 10.3899/jrheum.180028. [DOI] [PubMed] [Google Scholar]
- 106.Weinblatt M.E., Bingham C.O., 3rd, Burmester G.R., Bykerk V.P., Furst D.E., Mariette X., van der Heijde D., van Vollenhoven R., VanLunen B., Ecoffet C. A phase III study evaluating continuation, tapering, and withdrawal of certolizumab pegol after one year of therapy in patients with early rheumatoid arthritis. Arthritis Rheumatol. 2017;69:1937–1948. doi: 10.1002/art.40196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Haschka J., Englbrecht M., Hueber A.J., Manger B., Kleyer A., Reiser M., Finzel S., Tony H.P., Kleinert S., Feuchtenberger M. Relapse rates in patients with rheumatoid arthritis in stable remission tapering or stopping antirheumatic therapy: interim results from the prospective randomised controlled RETRO study. Ann. Rheum. Dis. 2016;75:45–51. doi: 10.1136/annrheumdis-2014-206439. [DOI] [PubMed] [Google Scholar]
- 108.Killestein J., Vennegoor A., Strijbis E.M., Seewann A., van Oosten B.W., Uitdehaag B.M., Polman C.H. Natalizumab drug holiday in multiple sclerosis: poorly tolerated. Ann. Neurol. 2010;68:392–395. doi: 10.1002/ana.22074. [DOI] [PubMed] [Google Scholar]
- 109.Kerbrat A., Le Page E., Leray E., Anani T., Coustans M., Desormeaux C., Guiziou C., Kassiotis P., Lallement F., Laplaud D. Natalizumab and drug holiday in clinical practice: an observational study in very active relapsing remitting multiple sclerosis patients. J. Neurol. Sci. 2011;308:98–102. doi: 10.1016/j.jns.2011.05.043. [DOI] [PubMed] [Google Scholar]
- 110.Thompson G.A., Ritschel W.A. The repeated one-point method for predicting dose sizes with irregular dosing intervals. Int. J. Clin. Pharmacol. Ther. Toxicol. 1986;24:337–343. [PubMed] [Google Scholar]
- 111.Fermín L.J., Lévy-Véhel J. Variability and singularity arising from poor compliance in a pharmacokinetic model II: the multi-oral case. J. Math. Biol. 2017;74:809–841. doi: 10.1007/s00285-016-1041-1. [DOI] [PubMed] [Google Scholar]
- 112.Li J., Nekka F. A probabilistic approach for the evaluation of pharmacological effect induced by patient irregular drug intake. J. Pharmacokinet. Pharmacodyn. 2009;36:221–238. doi: 10.1007/s10928-009-9119-7. [DOI] [PubMed] [Google Scholar]
- 113.Deanfield J.E., Detry J.M., Sellier P., Lichtlen P.R., Thaulow E., Bultas J., Brennan C., Young S.T., Beckerman B., CAPE II Trial Investigators Medical treatment of myocardial ischemia in coronary artery disease: effect of drug regime and irregular dosing in the CAPE II trial. J. Am. Coll. Cardiol. 2002;40:917–925. doi: 10.1016/s0735-1097(02)02050-8. [DOI] [PubMed] [Google Scholar]
- 114.Strik A.S., Berends S.E., Löwenberg M. Therapeutic drug monitoring-based dosing of TNF inhibitors in inflammatory bowel disease: the way forward? Expert Rev. Clin. Pharmacol. 2019;12:885–891. doi: 10.1080/17512433.2019.1642745. [DOI] [PubMed] [Google Scholar]
- 115.Strik A.B., Mould S., Mathôt D., Ponsioen R., van den Brande C., Jansen J., Hoekman J., Brandse D., Löwenberg J., D’Haens M. DOP56 Dashboard driven vs. conventional dosing of infliximab in inflammatory bowel disease patients: the PRECISION trial. J. Crohn’s Colitis. 2019;13(Suppl 1):S063. [Google Scholar]
- 116.Stein D.J., Ananthakrishnan A.N., Issa M., Williams J.B., Beaulieu D.B., Zadvornova Y., Ward A., Johnson K., Knox J.F., Skaros S., Binion D.G. Impact of prior irregular infliximab dosing on performance of long-term infliximab maintenance therapy in Crohn’s disease. Inflamm. Bowel Dis. 2010;16:1173–1179. doi: 10.1002/ibd.21164. [DOI] [PubMed] [Google Scholar]
- 117.El-Haj M., Kanovitch D., Ilan Y. Personalized inherent randomness of the immune system is manifested by an individualized response to immune triggers and immunomodulatory therapies: a novel platform for designing personalized immunotherapies. Immunol. Res. 2019;67:337–347. doi: 10.1007/s12026-019-09101-y. [DOI] [PubMed] [Google Scholar]
- 118.Kenig A., Ilan Y. A personalized signature and chronotherapy-based platform for improving the efficacy of sepsis treatment. Front. Physiol. 2019;10:1542. doi: 10.3389/fphys.2019.01542. [DOI] [PMC free article] [PubMed] [Google Scholar]