

Abstract
In this issue of Neuron, Santello and Nevian (2015) report HCN channel plasticity and increased temporal summation in layer 5 ACC neurons following nerve injury. They are able to restore HCN channel function and reduce behavioral hypersensitivity with selective serotonin receptor targeting.
Pain is both a sensory and emotional experience where noxious stimuli trigger intense aversion (Basbaum et al., 2009; King et al., 2009). While acute pain is essential to avoid potentially damaging stimuli, for many patients chronic pain induces unwanted and debilitating distress from which it is impossible to escape (Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education, 2011). We lack effective medication against chronic pain, in part because the molecular mechanisms underlying the emotional component of the disease remain poorly understood. In this issue of Neuron, Santello and Nevian (2015) provide evidence that hyperpolarization-activated cyclic nucleotide-gated (HCN) channel dysfunction contributes to anterior cingulate cortex (ACC) hyperactivity during neuropathic chronic pain. Using the difficult feat of dendritic patch recordings in layer 5 pyramidal neurons of mice, they show how the activity-dependent loss of function of HCN channels increases temporal summation and contributes to nerve injury-induced behavioral hypersensitivity.
Pain perception emerges from distributed neural pathways that coordinately shape its distinct dimensions. Incoming nociceptive signals branch at early stages of processing to act on many parallel, yet interconnected, circuits (Figure 1A). The somatosensory cortical branch (S1 and S2) encodes the sensory-discriminative component of pain, i.e., the body location, duration, intensity, and nature of noxious stimuli. Other cortical and limbic regions (anterior cingulate cortex, amygdala, and striatum) give rise to the affective-motivational component, i.e., pain evokes unpleasant feelings and motivation to escape. The multiplicity of ascending pain signals ensures that the brain as-signs the appropriate affective-motivational context to the concomitantly received sensory signal. Descending signals then modulate nociception at the spinal cord level.
Figure 1. A Speculative Model of ACC-Enhanced Dendritic Integration and Rescue during Chronic Pain.
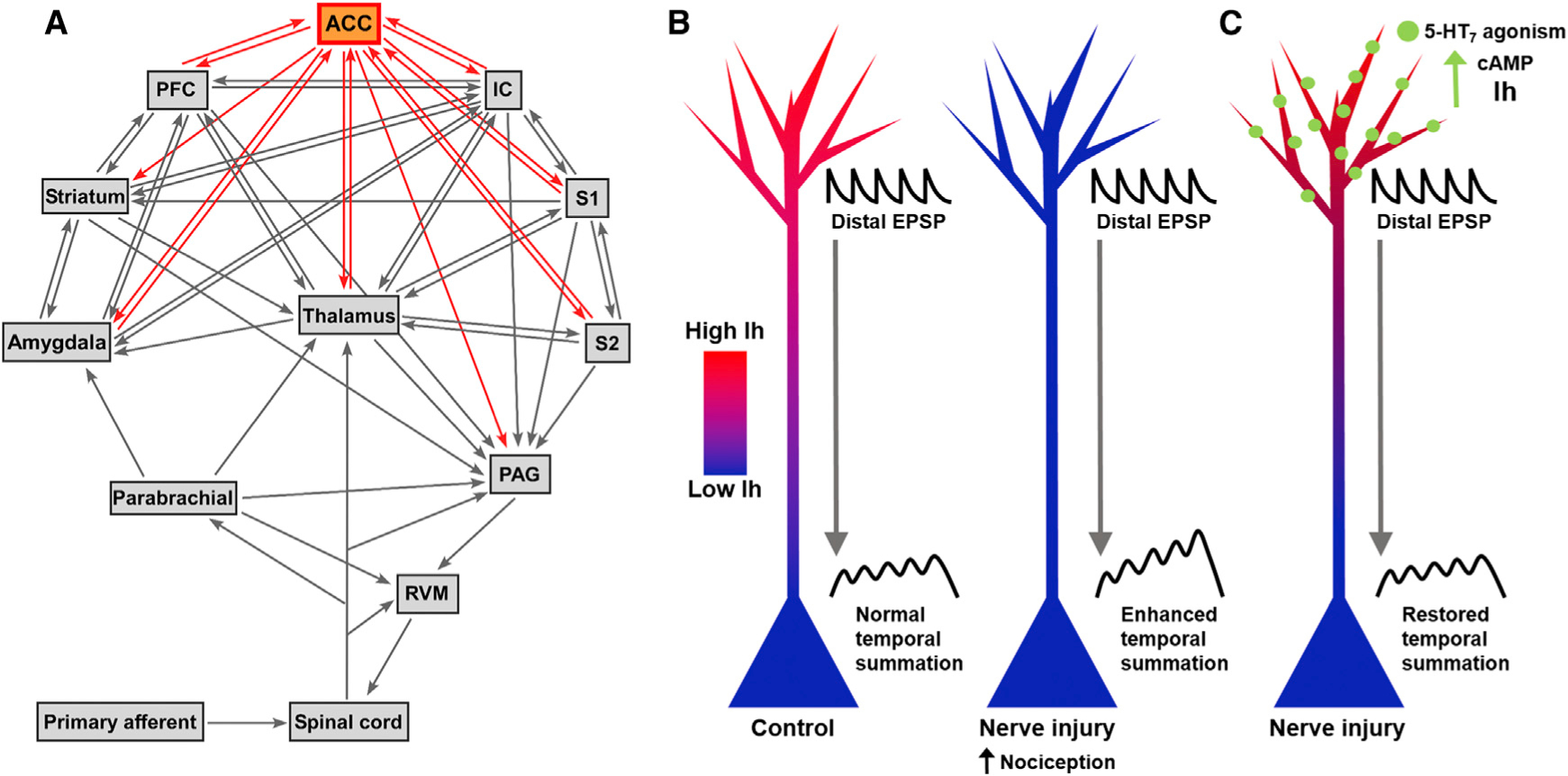
(A) Pain neural pathways. ACC, anterior cingulate cortex; IC, insular cortex; S1, primary somatosensory cortex; S2, secondary somatosensory cortex; PFC, prefrontal cortex; PAG, periaqueductal gray; RVM, rostral ventromedial medulla. (B) Nerve injury reduces Ih, leading to enhanced temporal summation at the soma and increased nociception. (C) 5-HT7 agonism at distal dendrites increases cAMP levels which restores Ih and normal temporal summation, reducing injury-induced hypersensitivity.
Recent research and therapeutic efforts to relieve pain have focused on primary somatosensory neurons and spinal circuits to limit transmission of pain information. Unfortunately, these approaches have not led to major progress in pain management and patients’ well-being (Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education, 2011). Additionally, long after tissue damage heals, many patients still chronically suffer from pain, which could suggest supraspinal maintenance of pain states. Chronic pain patients are at increased risk of additional emotional dysfunction, including anxiety and depression, accentuating the importance of understanding affective pain-processing regions whose abnormalities may contribute to the development of co-morbid mood disorders. To successfully treat pain, we must better understand the cellular and molecular mechanisms that subserve its development and maintenance, including in the non-sensory regions that shape pain perception.
One of these critically important structures for pain experience is the ACC. Many human fMRI studies show increased ACC activity in direct relation-ship to the emotional dimension of pain (Price, 2000). ACC lesions in rodents reduce noxious stimulus-evoked negative emotional associations in conditioned place aversion tests but do not change sensitivity to noxious stimuli (Johansen et al., 2001). Interestingly, increasing ACC excitation both generates place aversion and increases sensory sensitivity (Johansen and Fields, 2004). This asymmetry suggests that while the ACC’s primary role in pain may not be to process the sensory component of pain, it can heighten both sensation and emotion. Targeting ACC abnormalities may there-fore be a key mechanism to generate pain relief.
The ACC also undergoes substantial plastic changes in response to bodily injury, the most prominent of which is synaptic strengthening including long-term potentiation (LTP). ACC neuron hyperex-citability and both pre-and postsynaptic LTP have been observed in multiple pain models. Peripheral nerve injury increases presynaptic glutamate release and postsynaptic AMPA receptor function in the ACC, both of which intensify nociception (Xu et al., 2008; Koga et al., 2015). During inflammatory pain, NMDA NR2B receptors are upregulated in the ACC, and their blockade reduces behavioral sensitization (Wu et al., 2005a). A rodent model of phantom limb pain results in long-lasting membrane depolarization in ACC neurons, which can be significantly reduced by an NMDA receptor antagonist (Wu et al., 2005b). These data illustrate how the brain molecularly encodes a “memory” of painful stimuli in the ACC, which probably influences the transition of pain from acute to chronic.
Beyond pain-induced changes in synaptic strength in the ACC, Santello and Nevian (2015) investigate HCN channel plasticity. HCN channels are voltage-gated and cAMP-sensitive ion channels that activate with membrane hyperpolarization and cAMP binding and deactivate with depolarization. When activated at hy-perpolarized membrane voltages, HCN channels conduct an inward non-selective Na+ and K+ current (Ih) that depolarizes the membrane but ultimately limits excitation via an increase in membrane conductance (shunting). Ih corrects varying temporal summation resulting from inputs received at different distances from the soma (Magee, 2000). As an excitatory postsynaptic potential (EPSP) decrements across the membrane over space and time, its amplitude decreases and its waveform broadens due to passive cable properties of dendrites. There is thus a relatively larger decay time when EPSPs are generated distally from the soma compared to when they are generated more proximally, which subjects distally generated EPSPs to longer temporal integration windows. Without correction, this imbalance in temporal summation distorts the readout of total synaptic activity used to generate action potentials. An increasing Ih gradient from the soma to-ward the distal dendrites, as seen in hippocampal CA1 and neocortical layer 5 pyramidal neurons, corrects these problems by normalizing dendritic temporal summation relative to site of generation. Experimental evidence for normalization comes from both pharmacological blockade and genetic deletion of HCN channels, which increases temporal summation of distal EPSPs (Magee, 2000; Nolan et al., 2004). During the depolarization of an EPSP, HCN channels deactivate, which results in a virtual outward hyper-polarizing current and promotes voltage decay. The more Ih that was initially present, the greater the virtual outward current and the steeper the EPSP decay, which mitigates its ability to temporally sum with other EPSPs as they propagate to the soma.
Santello and Nevian (2015) are the first to show HCN channel plasticity in layer 5 ACC neurons of mice in a chronic pain model. They found that peripheral nerve injury-induced chronic pain decreases the number of HCN channels present, which consequently decreases Ih. This Ih reduction leads to increased temporal summation of inputs onto the neuron, which increases its spiking output (Figure 1B). Although the phenomenon of HCN channel reduction leading to enhanced dendritic integration has been previously documented, Santello and Nevian (2015) extend previous artificial HCN channel expression manipulations to a behaviorally relevant model in which similar effects are endogenously produced. Their results also link Ih mechanisms most thoroughly documented in hippocampal CA1 neurons to other brain regions, which until now has been mostly the product of speculative extrapolation. Santello and Nevian (2015)’s work adds a new layer of complexity to our knowledge of pain-induced CNS plasticity. Reduced Ih may facilitate NMDA receptor-dependent LTP by increasing the peak voltage of postsynaptic potential bouts and the net amount of time the dendritic membrane is depolarized, thus increasing the probability that NMDA receptors will be released from Mg2+ block. Also, since these enhanced integrated potentials propagate along the dendritic membrane between their initiation sites and the soma, this raises the possibility that synapses from non-pain-related inputs may become potentiated over time. This phenomenon could explain the emergence of co-morbid mood disorders due to the enhanced inclusion of these “pain” ACC neurons in non-pain circuits.
Santello and Nevian (2015) further show that Ih hypofunction in ACC layer 5 neurons can be selectively rescued. By targeting the 5-HT7 serotonin receptor expressed by these neurons, they were able to increase intracellular cAMP levels and restore Ih (Figure 1C). Such restoration reversed the enhanced dendritic integration and reduced nociception. However, the diversity of Ih function in this brain region should signal caution for viewing HCN channels as drug targets. In a recent issue of Neuron, Koga et al. (2015) showed that presynaptic HCN channels enhance LTP at ACC layer 2/3 synapses. Infusing an HCN channel blocker reversed this potentiation and reduced mechanical hypersensitivity. Furthermore, deleting HCN channels in primary afferent nociceptors reduces hy-peralgesia (Emery et al., 2011). Based on these results, pharmacological blockade of HCN channels seems like a valid analgesic strategy. But Santello and Nevian (2015)’s work shows that blocking HCN channels will lead to pain-facilitating activity in ACC layer 5 neurons. Instead of direct pharmacological targeting of HCN channels to solve one problem but create another, a better strategy might be to indirectly target HCN activity with cell-type-and spatially specific modulation. Targeting the 5-HT7 receptor achieves this specificity and will hopefully be the first of many exploitations of other membrane receptors to evoke downstream effects on Ih to relieve pain.
The identification of nerve injury-induced HCN channel plasticity in the ACC raises even more questions than it answers. First, given the ACC’s function in the emotional component of pain, does Ih in the ACC also modulate pain affect in a conditioned place aversion paradigm? Next, how does reduced Ih impact ACC network activity, given that enhanced distal temporal summation and Ih significantly contribute to spike patterns and local field potential oscillations (Hutcheon et al., 1996)? Finally, can other conditions, such as chronic stress or chronic itch, evoke a similar Ih reduction in the ACC? Can similar selective targeting methods be used to alleviate these conditions? As research on pain-induced plasticity receives increasing attention, accumulating evidence for long-lasting plastic modifications in the CNS provides support for the concept that pain is not only a symptom of tissue damage, but can also be a pathology in and of itself.
REFERENCES
- Basbaum AI, Bautista DM, Scherrer G, and Julius D (2009). Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery EC, Young GT, Berrocoso EM, Chen L, and McNaughton PA (2011). Science 333, 1462–1466. [DOI] [PubMed] [Google Scholar]
- Hutcheon B, Miura RM, and Puil E (1996). J. Neurophysiol 76, 683–697. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine (US) Committee on Advancing Pain Research, Care, and Education (2011). Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. (National Academies Press; ). [PubMed] [Google Scholar]
- Johansen JP, and Fields HL (2004). Nat. Neurosci 7, 398–403. [DOI] [PubMed] [Google Scholar]
- Johansen JP, Fields HL, and Manning BH (2001). Proc. Natl. Acad. Sci. USA 98, 8077–8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Van-derah TW, Dussor G, Lai J, Fields HL, and Porreca F (2009). Nat. Neurosci 12, 1364–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K, Descalzi G, Chen T, Ko HG, Lu J, Li S, Son J, Kim T, Kwak C, Huganir RL, et al. (2015). Neuron 85, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC (2000). Nat. Rev. Neurosci 1, 181–190. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, and Morozov A (2004). Cell 119, 719–732. [DOI] [PubMed] [Google Scholar]
- Price DD (2000). Science 288, 1769–1772. [DOI] [PubMed] [Google Scholar]
- Santello M, and Nevian T (2015). Neuron 86, this issue, 233–246. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Toyoda H, Zhao MG, Lee YS, Tang J, Ko SW, Jia YH, Shum FW, Zerbinatti CV, Bu G, et al. (2005a). J. Neurosci 25, 11107–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, Pang ZP, Zhuo M, and Xu ZC (2005b). Mol. Pain 1, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, and Zhuo M (2008). J. Neurosci 28, 7445–7453. [DOI] [PMC free article] [PubMed] [Google Scholar]