

Abstract
Estrogen, in the clinical setting is used primarily for contraception and hormone replacement therapy. It has been well established that estrogen increases the risk of both arterial and venous thrombosis. While estrogen is known to induce a prothrombotic milieu through various effects on the hemostatic pathways, the exact molecular mechanism leading to those effects is not known. The most common clinical presentation of estrogen-related thrombosis is venous thromboembolism (VTE) of the deep veins of the legs or pulmonary vessels, usually within the first few months of use. Estrogen has also been associated with increased risk of “unusual site” thromboses, as well as arterial thrombosis. Women at high-risk of thrombosis need careful evaluation and counseling for contraception, pregnancy, menopausal hormonal therapy and other estrogen-related conditions or treatments in order to lower the risk of thromboses. We review the most recent evidence on management of high-estrogen states in women at high-risk of thrombosis, as well as emerging data on unique populations such as transgender women. More studies are needed to better understand the pathophysiology of hormone-related thrombosis, as well as more comprehensive techniques to stratify risks for thrombosis so as to enable tailoring of recommendations for each individual.
Introduction
Worldwide, hundreds of millions of women use exogenous estrogens either as contraceptives or as post-menopausal hormonal replacement. There is significant evidence that estrogen use in both these situations is associated with an increased risk of thrombosis, both arterial and venous. The increased risk is dependent both on the dose and well as the mode of delivery of the hormone, and studies identify alterations in numerous aspects of the hemostatic and fibrinolytic pathways that contribute to the generation of a prothrombotic milieu. This review summarizes the mechanisms identified, the various clinical situations predisposing to the increased risk of estrogen-associated thrombosis and the management thereof.
Discovery of Estrogens
In 1896, a young Austrian researcher excised the ovaries of adult rabbits, re-implanted pieces of the ovaries in different locations and noted that unlike oophorectomized rabbits, there was an absence of uterine atrophy. Based on this, he postulated that the ovaries must likely send some substance via the blood stream to the uterus.[1] By 1924, the estrus cycle and its effects on the uterus was well established. [2] In 1929, pure oestrone crystals called “theelin”, were purified by Doisy from the urine of pregnant women [3]. Doisy continued his work with his colleagues, and went on to describe “dihydrotheelin” in 1940 at St. Louis University, which was later renamed as oestradiol [4]. At the same time, another researcher, Adolf Butenandt purified oestrone, called “progynon”, and was awarded the Nobel prize in 1939 for the discovery of oestrogen and testosterone along with Leopold Ruzincka [5, 6]. Since then, the work on estrogens and the discoveries continue to evolve.
Physiology of Estrogen
The term estrogen is coined from the Greek word “Oistros” meaning “mad desire” and “andgennan” meaning “to produce”. It is a generic term for a family of hormones that affect the female reproductive system[7]. The female body naturally produces three forms of endogenous estrogen, which have many physiologic functions. 17β estradiol (E2) is a steroid hormone synthesized from cholesterol. E2 is the most potent form of estrogen produced by the ovary and has the highest affinity for the estrogen receptors. Estrone (E1) and Estriol (E3) are less potent metabolites of E2 in the periphery. Aromatization of the A rings of androgenic precursors (androstenedione or testosterone) also plays an important role in the production of E1, E2 and E3. [8, 9] E2 is the predominant form of estrogen in the pre-menopausal woman.
In post-menopausal women and men, extra gonadal tissues such as adipose tissue, osteoblasts, chondrocytes, vascular endothelium constitute a very important source of estrogen synthesis. Availability of C19 steroid precursors determines estrogen production in the extragonadal tissues. These include testosterone, androstenedione, dehydroepiandrosterone and dehydroepiandrosterone sulfate [10]. In post-menopausal women, the adrenal gland continues to make the precursor of E1 – androstenedione. Hence the E1 level remains unchanged, while E2 falls significantly. E3 is typically low in non-pregnant individuals, but high in pregnancy since it is the main estrogen produced by the placenta [8, 11, 12]. In a pre-menopausal female, circulating E2 levels range from 40–400pg/ml, whereas after menopause, the level drops to less than 20pg/mL [13].
A. Estrogen Receptors
Estrogen mediates its effects mainly via 2 nuclear receptors, namely, estrogen receptor α (ERα), ERβ, and a newly identified G-protein coupled receptor 30 (GPR30/GPER) [14]. Evolutionary studies show that estrogen receptors are ancient proteins expressed in all vertebrates and some invertebrates [15]. Estrogen receptors are expressed and produce specific effects in different organ systems of the body - the cardiovascular system, brain, bone, liver, adipose tissue, colon, skin, prostate, testes, epididymis and salivary gland [14]
ERα and ERβ are nuclear receptors, which share the same general structure with an amino (N) terminal domain (region A/B) which includes a transcription activation function domain (AF1), DNA binding domain (region C), a linker (region D), a ligand binding domain (region E) that includes the second activation function domains (AF2), and a carboxy terminal F domain which is unique to ERs [16, 17]. There exist at least 3 isoforms of ERα and 5 isoforms of ERβ [18]. A third nuclear receptor, ERγ has been described in only teleost fish, not in humans [19].
ERα and ERβ have different chromosomal loci, 6q25.1 and 14q22–24, respectively. Based on the cell type, ligand and promoter, ERα and ERβ have different transcriptional activities [20, 21]. While the DNA binding domains have 97% amino acid identity, there is only 16% similarity in the AF1 domains in the N terminal region between ERα and ERβ. Further, even though the ligand binding domain of ERα and ERβ are only 59% identical in amino acid sequences, the ligand binding pockets are very similar [22]. While ERα is primarily present in the ovarian thecal cells, uterus, testes, stroma of prostate, liver, kidney, heart, ERβ is predominantly seen in granulosa cell of ovary, epithelium of prostate, bladder, colon, and immune system. However, they are both co-expressed in many tissues such as mammary gland, epididymis, thyroid, adrenal, bone, adipose tissue and nervous system and form functional heterodimers [23, 24]. Over the last decade, several studies have identified the role for ERs in the plasma membrane and as well as the mitochondria [6]. Although discovered several years after ERα, ERβ is recognized to be dominant, and when co-expressed, has an antagonistic effect on the transcriptional activity of ERα [24].
Estrogen involves several distinct pathways to mediate its effects via estrogen receptors. These pathways can be ligand dependent and ligand independent. The ligand dependent pathway can be broadly classified as the direct pathway, an indirect/tethered pathway and a non-genomic rapid pathway. The direct pathway involves binding of ligand to the ER. Once a ligand binds to ERα and ERβ, the ligand bound receptors bind to the same DNA sequence known as the estrogen response element (ERE). [14]. The receptor activity can be affected by phosphorylation, co-activators and co-repressors [25–28]. The indirect or tethered pathway involves ligand activation, following which protein-protein interaction with other transcription factors occurs thereby affecting gene regulation by indirect DNA binding. The non-genomic rapid pathway, though not very clear, basically involves activation of a classical ER in the plasma membrane, followed by second messenger mediated activation of signaling cascades that affect nitric oxide levels or ion channels in the cytoplasm, leading to a physiological response that does not affect gene regulation [29]. The ligand-independent pathway involves activation of ER via other signaling pathways by mechanisms such as phosphorylation, resulting in dimerization of ER, DNA binding and gene regulation [29].
In tissues that do not have ERα and ERβ, estradiol mediates its effects via GPER [6]. GPER is a membrane protein, with 7 transmembrane regions and is known to activate several signal transduction pathways, resulting in phosphorylation of substrate proteins. ERα has been reported to be one of the downstream transcription factors, indicating possible interaction between genomic and non-genomic estrogen receptors [14, 30, 31]. GPER stimulates the release of heparin binding epidermal growth factor like growth factor (HB EGF) which is membrane bound, resulting in activation of the MAPK and PI3 kinase cascade, thereby resulting in the desired physiological response. Specifically GPERs have been identified in several tissues throughout the body – reproductive organs, brain, heart, vascular system, pancreas, skeletal muscle and kidney, with varying expression based on age, sex, tissue type and disease.[32, 33] Estrogen effects in cardiovascular, renal, reproductive, immune and central nervous systems have been linked to mediation via GPER [6, 34].
Estrogens have effects in almost every cell in the body. Estrogens increase proliferation and differentiation of osteoblasts, induce apoptosis of osteoclasts and shift the balance to new bone formation and maintenance. Thus, during menopause, bone mineral density decreases parallel to decline in estrogen levels. Studies have established that estrogen have an important role in myoblast differentiation and growth, indicating their role in skeletal muscle development and maintenance. Combined with the effects on the bone, estrogens overall reduce the risk of osteoporotic fractures. Estrogens further regulate adipocytes by genomic & non genomic signaling, thereby playing an important role in obesity and metabolic syndrome. In the skin, estrogens have been shown to prevent aging and promote wound healing. Estrogens regulate the heart and the vascular system, and have a protective effect on the cytoskeletal integrity of these cells. For a more detailed description of these effects, the reader is referred to Wend et al[35]. We summarize the chronology of the various discoveries leading to the understanding of the physiological mechanisms of estrogen and its receptors in the timeline in Figure 1.
Figure 1 -.
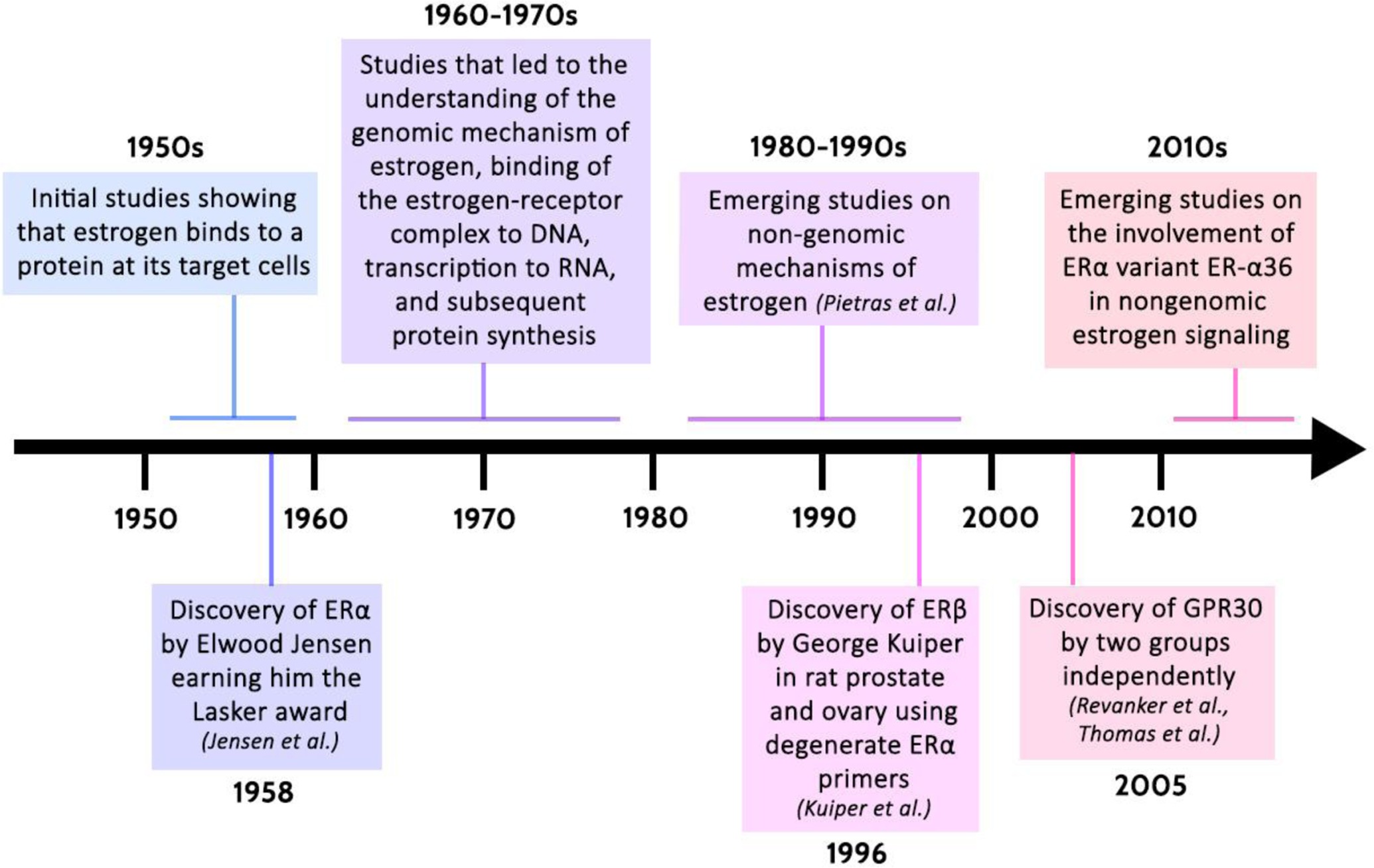
Timeline displaying the chronology of the various scientific discoveries that have shaped our understanding of the physiological mechanisms of estrogen. [31, 36–40]
ERα: Estrogen Receptor-alpha
ERβ: Estrogen Receptor-beta
B. Estrogens in Clinical Use
Estrogen, in the clinical setting is used primarily for contraception and hormone replacement therapy. Combined hormonal contraceptives (CHC) are the most commonly prescribed and preferred form of birth control [41, 42]. There are hundreds of CHCs available to patients, marketed by different pharmaceutical companies and at a quick glance are difficult to distinguish. However, CHCs are primarily classified based on the following (i) type and dose of estrogen, (ii) type of progesterone (iii) monophasic versus multiphasic pills (iv) active versus inactive pills [42].
Most CHCs contains one of the 4 available forms of estrogen - ethinyl estradiol (EE), 17β-estradiol (E2), estradiol valerate (E2V) or mestranol [42–44]. Mestranol, a 3-methyl ether of ethinyl estradiol (EE) was the first CHC to be approved in the 1960’s. A pro-drug that is demethylated to EE, mestranol is rarely used nowadays [44]. Natural E2 is the most potent naturally occurring estrogen in humans [8] and was introduced in the 1970s for oral contraception. Though a good contraceptive, a major limitation was relative inactivity when taken orally because of extensive hepatic first pass effect. Oral bioavailability of E2 is 5% versus the oral bioavailability of EE which is 38–48%. [45, 46]) Attempts to use E2 in CHCs in varying doses and combinations with progesterone also resulted in poor cycle control and bleeding [8, 45]. E2V is an esterified form of E2, which is rapidly hydrolyzed to estradiol after oral intake. E2V has been shown to have comparable or a slightly better impact on metabolic and hemostatic parameters. EE is the estrogen component used presently in majority of CHCs and has been heavily relied upon due to its good oral bioavailability in comparison to other estrogens. This is more active than the natural forms of estrogens.
Combined estrogen & progesterone: Combined oral contraceptive pills primarily act by inhibition of follicular development and inhibition of ovulation. Specifically, the progesterone component decreases gonadotropin releasing hormone pulse frequency by negative feedback to the hypothalamus, thereby decreasing FSH and LH secretion. This prevents development of the follicle and hence, the increase in estradiol levels, thereby preventing the mid cycle LH surge necessary for ovulation. [47, 48]
Enovid, the first combined estrogen and progesterone containing pills was approved in 1957 in the United states for menstrual disorders and in 1960 for contraception[44]. By 1970, as a dose relationship between estrogen and thrombosis was established, pharmaceutical companies started decreasing the dose of estrogen in the pill, and majority of the combined pills in the 1980s had less than 35 micrograms of estrogen, paired with a first or second generation progesterone. This was followed by the approval of third generation progesterones with norgestimate in 1989 and desogestrel in 1992[49]. With four generations of progesterones, there are multiple combination OCPs currently available in market.
Progesterones are classified into different generations as listed below:
Estrogen and Thrombosis
A. Mechanisms
Although estrogen is known to affect multiple hemostatic variables, the exact molecular mechanism of estrogen-induced thrombosis remains unclear. Given the delicate balance between hemostasis and thrombosis, it is possible that small changes induced by hormonal therapy may increase the overall risk of thrombosis [50]. In the paragraphs below, we summarize the animal and human studies that identify pathways/factors altered with estrogen treatment and that are likely to affect the risk for thrombosis.
1). Platelets and von Willebrand Factor (vWF):
The effect of estrogen on platelet number varies based on the dose of estrogen, duration of administration and time of measurement. Data on the effect of estrogen on platelet counts are conflicting. Treating mice with 10–100 micrograms/kg/day E2 over 10 days decreases platelet counts. Additionally, while 80 micrograms/kg/day E2 over 3 weeks does not alter platelet count, 200 micrograms/kg/day E2 over the same period of time tends to decrease platelet counts [51–54]. Estrogen also modulates platelet activation and aggregation, by either increasing or decreasing tail bleeding times in mouse models. Here again, these vary based on the dose of estrogen and duration of administration. In summary, as stated in a meta-analysis by Dupuis et al, the data in human studies examining the effect of hormonal therapy on platelet number, activation and aggregation is not clear at this time and is highly variable [51].
VWF, which plays an essential role in primary hemostasis as well as platelet adhesion and activation is regulated by different hormones including estrogen. Estrogen results in an elevation in vWF levels through direct stimulation of endothelial cells as well as endothelial cell replication [55]. Furthermore, pregnancy is known to be associated with an increase in vWF levels through increased production as well as prolongation of its half-life [56]. A study in post-menopausal women showed that there was a statistically significant increase in vWF after treatment with oral equine estrogen for 4 weeks [57]. However, other studies report no change in vWF level in post-menopausal women when checked at 12 months and 36 months [58].
2). Coagulation cascade:
Valera et al showed that in mice, treatment with estradiol with and without progesterone did not alter coagulation assays (Prothrombin time or PT, activated partial thromboplastin time or aPTT) and fibrinogen, [53, 54] which is contrary to the procoagulant profile noted in human studies [59]. Further, a study in rats shows that injection of 2 mg estradiol dipropinate (EDP) is associated with an increased incidence of thrombin-induced pulmonary thromboembolism. This study demonstrates that serum E2 level peaked on day 7 and dropped to baseline by day 21 when endogenous thrombin potential (ETP) was highest. Importantly, ETP was higher among rats injected with EDP when compared to those that were not [60]. Older studies in a rabbit model, where adult female rabbits were tube-fed either a contraceptive powder low in estrogen (0.8 pg/kg of estradiol), high in estrogen (2.0 pg/kg of estradiol), or a control powder for 7 weeks showed that estrogen per se did not affect thrombin times. However, when these rabbits were injected with thrombin, the thrombotic grades were significantly higher in rabbits treated with estrogen, and clearly greater with higher dose estrogens. [61]
Human studies clearly indicate that estrogen treatment results in a prothrombotic state. A randomized cross over study among women showed that combined oral contraceptives affect the coagulation cascade by increasing plasma levels of factors II, VII, VIII, X, and fibrinogen, and by decreasing factor V levels [62, 63]. Another study in post-menopausal women showed that estrogen increased factor VII levels, and overall resulted in a pro-thrombotic state [64]. These changes also vary based on the associated progesterones on the CHCs, with desogestrel containing CHCs showing more significant changes, when compared to levonorgenstrel containing CHC [62].
3). Anti-coagulant pathways:
Studies of the anticoagulant pathway show a decrease in plasma levels of naturally occurring inhibitors of hemostasis such as tissue factor pathway inhibitor and antithrombin with CHC use [63, 64]. Although CHCs also cause an increase in protein C antigen and activity, this is associated with a concomitant increase in the level of protein C inhibitors like α1 antitrypsin and α2 macroglobulin [63, 65]. Simultaneously, a pronounced decrease in levels of Protein S antigen (total and free) has been noted with CHC use, more significantly with use of 3rd generation progesterone containing CHCs[63, 65, 66] and in post-menopausal women [64].
4). Fibrinolysis:
CHCs also affect fibrinolysis by decreasing levels of plasminogen activator inhibitor (PAI −1) and increasing levels of tissue plasminogen activator (tPA). However, these changes are shown to be counteracted by higher levels of thrombin activatable fibrinolysis inhibitor in women taking CHCs [67]. Presently, it is unclear if this effect on fibrinolysis has clinical implications on the risk of VTE secondary to estrogen therapy [68]
5). Effect on hepatic synthesis of proteins involved in coagulation & fibrinolysis:
EE has been shown to impact synthesis of liver proteins when administered orally or parenterally [69, 70]. Studies have shown higher in vivo extraction of oral EE by the liver, relative to the brain or uterus[71]. A study by Moverare et al showed that 17β estradiol treatment in mice results in increased expression of bone marrow derived, not liver derived, factor V. [72]. However, another study demonstrates that ovariectomized mice treated with oral synthetic 17α estradiol had a significant downregulatory impact on hepatic transcript levels of both pro and anti coagulant genes, mediated via ER α. [73]
6). Inflammation:
Estrogens have a divergent effect on inflammation and immune modulation [74, 75] based on the dose and duration of treatment of the drug. Both in vitro and in vivo studies show that while low dose exposure to E2 (doses consistent with oral contraceptive pills) is associated with increased pro-inflammatory cytokine expression [74, 76, 77], high doses (pregnancy levels) tend to generate an anti-inflammatory response [74]. Similarly, a short duration of E2 treatment (2–6 hours) has an anti-inflammatory effect [78, 79], while chronic administration over 60 days is associated with a pro-inflammatory response [77]. In light of this data, it is interesting to note that the chronic low dose E2 therapy associated with a pro-inflammatory effect is similar to the dose of CHC used in females.
Some of the proposed mechanisms of how estrogens regulate inflammation include responses mediated via nuclear/membrane bound estrogen receptors, stimulation of toll like receptor ligand on dendritic cells and macrophages resulting in increased expression of pro-inflammatory cytokines, decreasing inhibitory PI3K signaling and Akt phosphorylation in macrophages, and effects on NF-κB transcriptional complexes. Estrogen induced epigenetic changes in hematopoietic stem cells influencing downstream pathways and responses in mature cells have also been proposed as a possible mechanism. Further studies are needed to increase our understanding of estrogen induced inflammatory changes and identify the molecular mechanisms involved [74, 80]
Taken together, all these changes tilt the ‘hemostatic balance’ to a prothrombotic state which is reflected in global tests of hemostasis. Thus, CHC users are noted to have significantly higher APC resistance, when compared to non CHC users [81]. We visually summarize the effects of estrogen on the different hemostatic pathways above in the diagram in Figure 2.
Figure 2 -.
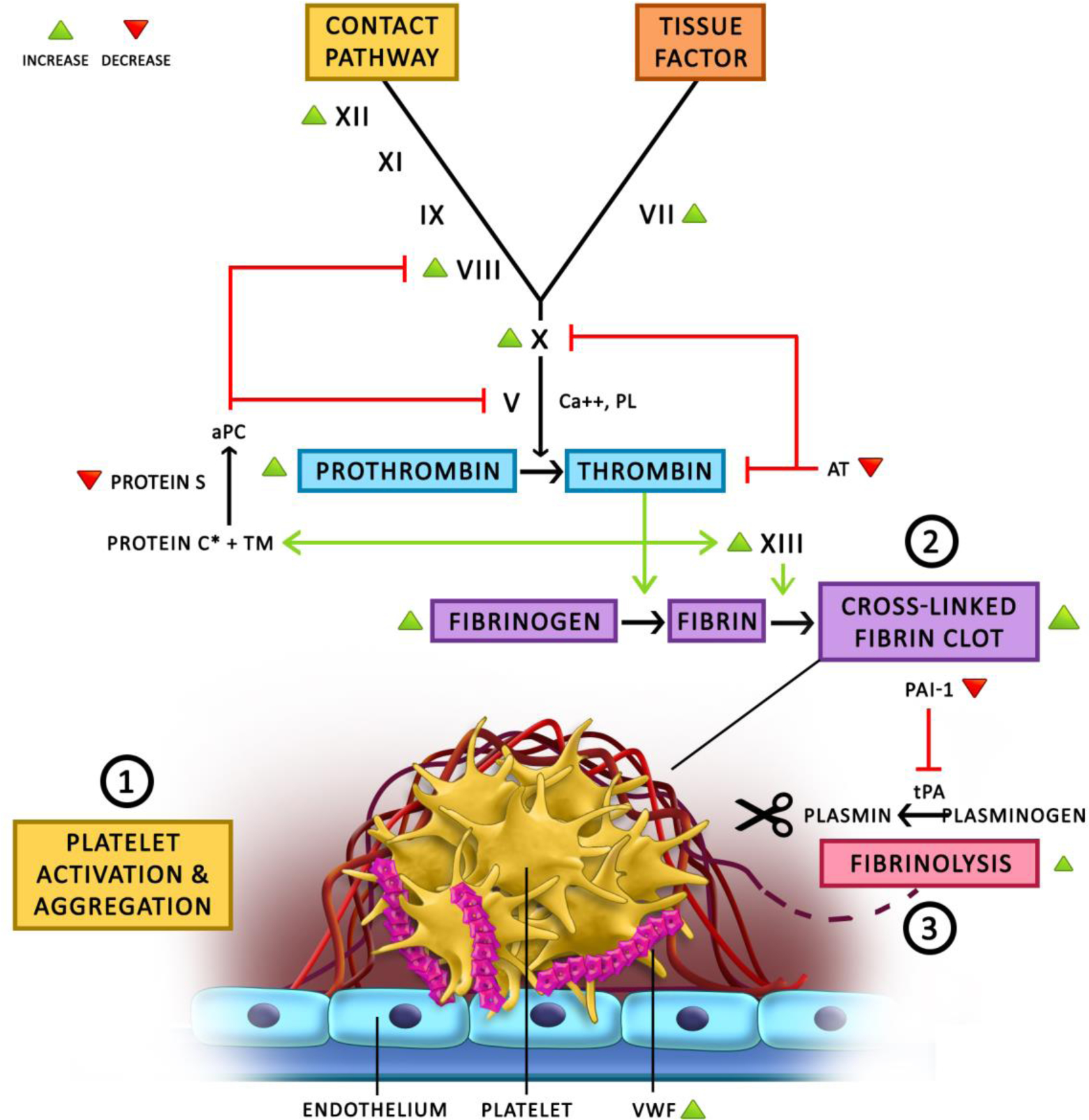
This diagram demonstrates our current understanding of the effects of estrogen on hemostasis & thrombosis. 1) The exact effect of estrogen on platelet activation and aggregation remains unclear, with conflicting reports in the literature showing altered platelet behavior. Estrogen is known to increase VWF levels which plays a central role in platelet adhesion and activation. 2) Estrogen leads to increased thrombin generation and fibrin clot formation by increasing the levels of variable coagulation proteins (green arrowheads) and decreasing the levels of anticoagulant proteins (red arrowheads). 3) Conversely, estrogen has also been shown to be associated with increased fibrinolysis due to decreased PAI-1 levels, which does not seem to balance out the increase in coagulation. Thus the net effect overall, has been shown to be prothrombotic.
aPC = activated Protein C
AT = anti-thrombin
PAI-1 = plasminogen activator inhibitor 1
PL = phospholipid
TM = thrombomodulin
tPA = tissue plasminogen activator
VWF = von Willebrand Factor
* In different studies, estrogen has been shown to increase Protein C antigen levels but also increase Protein C resistance
B. Estrogen Dose and Thrombosis
The EE dose in early formulations of CHC was as high as 150 micrograms (mcg). With evidence that higher doses results in increased risk of thrombosis, the dose has been gradually decreased over the years, and current pills have EE doses that range from 10 to 50 μg. [82] Large meta-analysis in recent years have established clearly that higher doses of estrogen are associated with increased risk of thrombosis (Table I)
Table 1 -.
ESTROGEN DOSES IN COMBINED ORAL CONTRACEPTIVES AND RELATIVE RISK OF VTE
| Meta-analysis | Studies includd | Non user | LNG/EE 20 | LNG/EE 30–40 | LNG/EE 50 | DSG/EE 20 | DSG/EE 30–40 | GSD/EE 20 | GSD/EE 30–40 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Oedingen 2018[41] | 11 case control studies 6 cohort studies | RR (95 % CI) | - | - | 1 | 1.46 (1.33–1.59) | 1.39 (1.16–1.67) | 1.18 (0.93–1.49) | 1.27 (1.15–1.41) | |
| De Bastos 2014 [83] | 9 cohort studies 17 case control studies | RR (95 % CI) | 1 | 2.2 (1.2–3.6) | 2.4 (1.8–3.2) | 5.2 (3.4–7.9) | 3.4 (2.5–4.6) | 3.7 (2.8–4.9) | 2.2 (1.4–3.2) | 3.7 (2.8–4.9) |
LNG - Levonorgestrel
DSG - Desogestrel
GSD - Gestodene
EE 20 - Ethinyl estrasdiol 20 micrograms
EE 30–40 - Ethinyl estradiol 30–40 micrograms
EE 50 - Ethinyl estradiol 50 micrograms
C. Combined Progesterone with Estrogen and Thrombosis
Interestingly, thrombosis has always been attributed to the estrogen component since the advent of CHCs. The role of progesterone in thrombosis was never looked into until a large observational study from WHO showed the increased risk of thrombosis with newer progesterones. [84] While The absolute risk of VTE in women who do not use OCPs is reported at 0.19 to 0.37 per 1,000 woman-years, that of of VTE with OCP use ranges from 0.55 to 1.41 per 1000 woman years, with higher risk among users of third and fourth generation progesterones.[83]
Over the years, there have been several studies that confirmed or denied the role of progesterone in thrombosis. Table 2 lists recent meta-analyses that compare the risk of VTE among CHCs with similar estrogen doses, but different progesterones and indicate that third and fourth generation progesterones have a higher risk of VTE.
Table 2 -.
Type of progesterone in CHC & VTE risk
| Meta-analysis | Studies included | EE 30–40 + LNG | EE 30–40 + DSG | EE 30–40 + GSD | EE 30–40 + Cyproteroneacetate | EE 30–40 + Drosperinone |
|---|---|---|---|---|---|---|
| RR (95%C) | RR (95%CI) | RR (95%CI) | RR (95%CI) | RR (95%CI) | ||
| Dragoman 2018 [85] | 17 case control studies 5 cohort studies (only 30 mcg) | 1 | 1.83 (1.55 – 2.13) | 1.67 (1.32 – 2.10) | 2.04 (1.55 – 2.49) | 1.58 (1.12 – 2.14) |
| Oedingen 2018 [41, 83] | 11 case control studies 6 cohort studies | 1 | 1.46 (1.33 – 1.59) | 1.27 (1.15 – 1.41) | 1.29 (1.12 – 1.49) | 1.40 (1.26 – 1.56) |
| De Bastos 2014[83] | 9 cohort studies 17 case control studies | 1 | 1.8 (1.4 – 2.2) | 1.5 (1.2 – 2.0) | 1.6 (1.1 – 2.2) | 1.6 (1.2 – 2.1) |
LNG - Levonorgestrel
DSG - Desogestrel
GSD - Gestodene
EE 30–40 - Ethinyl estradiol 30–40 micrograms
D. Route of Administration and Thrombosis
Estrogen, in CHC, is administered orally. Non-oral forms of estrogen used commonly include vaginal ring and combined injectable contraceptive [86]. The use of transdermal estrogen preparations began when investigators realized that estrogen is well absorbed through the skin. Transdermal form results in constant blood levels due to a slow and sustained release of estrogen from the patch [8]
A recent systematic review compared non-oral methods of contraception with CHC to determine risk of thrombosis. Analysis of 2 cohort studies and 4 case control studies to compare the risk of VTE among women taking CHCs versus transdermal patch show conflicting results. While 2 studies show significantly higher risk of VTE with transdermal patch, 4 studies fail to show a statistically significant difference in the risk of VTE [86]. The same systematic review further analyzed 3 studies (2 cohort studies and 1 case control study) comparing the risk of thrombosis in CHC users as compared to vaginal ring. While one study confirms a higher risk of VTE among CHC users (adjusted rate ratio 1.90; 95% CI 1.33–2.71), the other 2 studies fail to show a statistically significant difference in rates of thrombosis between the 2 groups. [86–88]
Finally, a meta-analysis of 15 observational studies showed that oral estrogen therapy was associated with a higher risk of first episode of VTE (RR, 1.63; 95% CI, 1.40–1.90) and deep venous thrombosis (DVT) (RR, 2.09; 95% CI, 1.35–3.23) when compared to transdermal estrogen therapy in post menopausal women. The level of evidence is low, since these were only observational studies and randomized control studies are not available [89]. In summary, the presently available data is insufficient to determine whether one route of administration of estrogen generates a higher thrombotic risk.
Clinical Presentation
A. Venous Thrombosis
The most common sites of thrombosis related to high estrogen states are the deep veins of the legs and the pulmonary veins. Consistently, approximately 80% of thromboses occurring in pregnancy are venous [90]. CHC use has also been associated with “unusual” sites of thrombosis. Multiple studies show an increase in the risk of cerebral venous sinus thrombosis, with an OR of 5.59 (95% CI 3.95 – 7.91) and 7.59 (95% CI 3.82—15.09) in two separate meta-analyses [91, 92]. However, the absolute risk remains very low, estimated around 2 per 100,000. There have also been many case reports of mesenteric venous thrombosis related to CHC use [93]. Using a nationwide database study, we found a significantly higher risk of retinal vein occlusion in women on estrogen-containing therapy as compared to women who do not use them [94]. Similarly, the absolute risk remains very low due to the rarity of these events.
B. Arterial Thrombosis
The effect of estrogen on the cardiovascular system is not entirely understood, and continues to be studied. There are several ways through which estrogen affects the risk for cardiovascular disease and myocardial infarction (MI). These include variable effects on the coagulation system, fibrinolytic pathway, platelet activation and aggregation, as well as its effects on cardiovascular risk factors such as cholesterol levels. In the past, there had been a long-held belief that estrogen decreased the risk for MI. The Heart and Estrogen-Progestin Replacement Study (HERS) in 1998 reported that after 2 years of menopausal hormonal therapy (MHT), women had a lower risk of MI and strokes [95]. However, a follow-up to the study (HERS II) published in 2002 concluded that there was no lasting decrease in MI or stroke from MHT after 6.8 years [96]. That same year, data from the Women’s Health Initiative randomized clinical trial showed an increased risk of MI and cardiovascular death in women on MHT (HR 1.29, 95% CI, 1.02–1.63) [97]. This lead to a change in recommendations and MHT was no longer recommended for prevention of heart attack or stroke.
In 2012, a study of more than a million women aged 15 to 49 by Lidegard et al showed an increased risk of stroke and myocardial infarction (MI) associated with use of CHC, estimated at a factor of 0.9 to 2.3 based on the estrogen dose and component. The absolute risk of stroke or MI, however, was very low at 21.4 and 10.1 per 100,000 respectively [98]. A 2015 meta-analysis of 24 studies by Roach et al showed a 1.6-fold increased risk of MI or ischemic stroke in women on CHCs, which appeared to increase with higher estrogen doses [99]. In summary, the most recent and compelling data suggests an increased risk of MI and stroke due to estrogen, however, the absolute risk remains low.
C. Timing of Thrombosis
In an international case-control study across 21 centers, the World Health Organization reported in 1995 that increased risk of VTE was apparent within 4 months of starting CHCs, was unaffected by the duration of current type of CHC use, and returned to baseline within 3 months of discontinuing CHCs [100]. Data from the population-based case-control Leiden Thrombophilia Study showed that the risk of developing VTE was greatest in the first 6 months of CHC use. The risk of developing VTE was 3-fold higher in the first 6 months (95% CI 0.6–14.8), and 2-fold higher in the first year (95% CI 0.6–6.1) compared to prolonged use. [101] In the Women’s Health Initiative (WHI) study, the risk of VTE due to MHT was highest in the first year of therapy, but persisted for the five years of follow-up [102]. We used a nation-wide data base and reported an increased risk of RVO in patients on CHCs that was highest within the first 30 days of initiation [94].
Management
A. Contraception and Thrombosis
i. Contraceptive Management in Women with High Risk of Thrombosis
Determining the thrombotic risk in women takes into consideration several factors, such as age, comorbidities, smoking history, family history, and history of thrombosis. Furthermore, while routine screening for hereditary thrombophilias remains controversial due to the rarity of these conditions, their presence can increase the risk of thrombosis.
The Medical Eligibility Criteria for contraceptive use by the World Health Organization lists history of VTE, surgery with prolonged immobilization, anti-phospholipid syndrome and presence of known hereditary thrombophilia as contraindications to CHC use (Category 4) [103]. Smoking in women of age 35 or older, uncontrolled hypertension, ischemic heart disease, migraine with aura, are also considered contraindications due to high risk of stroke or MI. Obesity, in otherwise healthy women, is listed as Category 2 (benefit of CHC outweighs risk) due to a low absolute risk of increased VTE.
In this review, we consider the following to be associated with a high risk of thrombosis:
Previous history of VTE
“Severe” thrombophilias: such as homozygous or double heterozygous FVL or prothrombin mutation, severe deficiencies in Protein C, S or antithrombin, anti-phospholipid syndrome. (See “Special Populations” section for more details on thrombophilias).
Strong family history of VTE without an identified cause or genetic defect
Other comorbidities including: uncontrolled hypertension, ischemic heart disease, stroke, migraine with aura, smoking in women of age 35 or older, or multiple cardiovascular comorbidities.
The selection of an optimal contraceptive method depends on several factors, including planning for pregnancy, menorrhagia, patient preference, and cost. In patients with high risk of thrombosis who are not fully anticoagulated, CHC and injectable depot medroxyprogesterone acetate (DMPA) should be avoided, and is generally contraindicated, as both have been shown to increase the risk of thrombosis[104, 105].
In the absence of contraindications, long-acting reversible contraception (LARC) is recommended first. This is due to their efficacy, which is comparable to that of sterilization, as well as their safety in patients at high-risk of thrombosis. Options include the copper IUD and progestin-only methods such as oral pills, levonorgestrel IUD, and the progestin subdermal implant, may induce amenorrhea, and have not been shown increase risk of VTE (contrary to etonogestrel implant package labeling) [98]. The injectable progestin DMPA, on the other hand, has been associated with increased risk of VTE[104, 105]. The levonorgestrel-releasing IUD is preferred over the progestin-only implant since IUD effects are primarily local (Table 3).
The levonorgestrel IUD prevents pregnancy by thinning the intrauterine lining, thickening the cervical mucous, and decreasing tubal motility. It is useful in women with heavy or irregular menses or on blood-thinners, as it can result in decreased menstrual flow and a higher likelihood of amenorrhea. In comparison with the other two LARCS, it has a more favorable bleeding profile.
The etonogestrel subdermal implant has a similar mechanism of contraception as well as suppressing ovulation, and is the most effective contraceptive option available for women. However, it can cause alteration in menstrual flow leading to irregular bleeding in ~20% of patients.
The copper IUD impairs sperm function and prevents fertilization by increasing copper ions, prostaglandins and macrophages in the uterine and tubal fluids. The most problematic side effect is an increase in painful, heavy menses after initiation.
In addition to the copper IUD, there are other non-hormonal contraceptive options that are less effective, such as spermicides and barrier methods including the diaphragm, cervical cap, and male and female condoms. CHC can be considered in special circumstances.
Table 3 -.
Summary of mechanisms, benefits, and risks of different LARC options.
| LARC Option | Mechanism | Benefits | Risks |
|---|---|---|---|
| Copper IUD |
|
|
|
| Levonorgestrel IUD |
|
|
|
| Progestin-only subdermal implant |
|
|
|
ii. Contraceptive Management in Women on Long-term Anticoagulation
While it is advised to avoid CHCs in patients with history of thrombosis not on anticoagulation, there has been question on their use in patients on therapeutic anticoagulation. An argument for their use is that these patients risk of thrombosis is lowered by anticoagulant therapy. Further, they lower the risk of heavy menstrual bleeding that can be associated with anticoagulation.
In a study by Martinelli et al, which analyzed data from the EINSTEIN DVT and Pulmonary Embolism (PE) anticoagulation trials found that women who were on anticoagulation and used CHCs were no more likely to suffer recurrent VTE than women who used progestin-only contraceptives or no hormonal contraceptives[106]. The study concluded that CHCs did not increase the risk of thrombosis in women who were on therapeutic anticoagulation.
We propose the following algorithm (Figure 3) as a guide for contraceptive management in women at risk of VTE. We include women with known hereditary thrombophilias in this algorithm, noting that routine screening for these conditions remains controversial and is not currently recommended.
Figure 3 -.
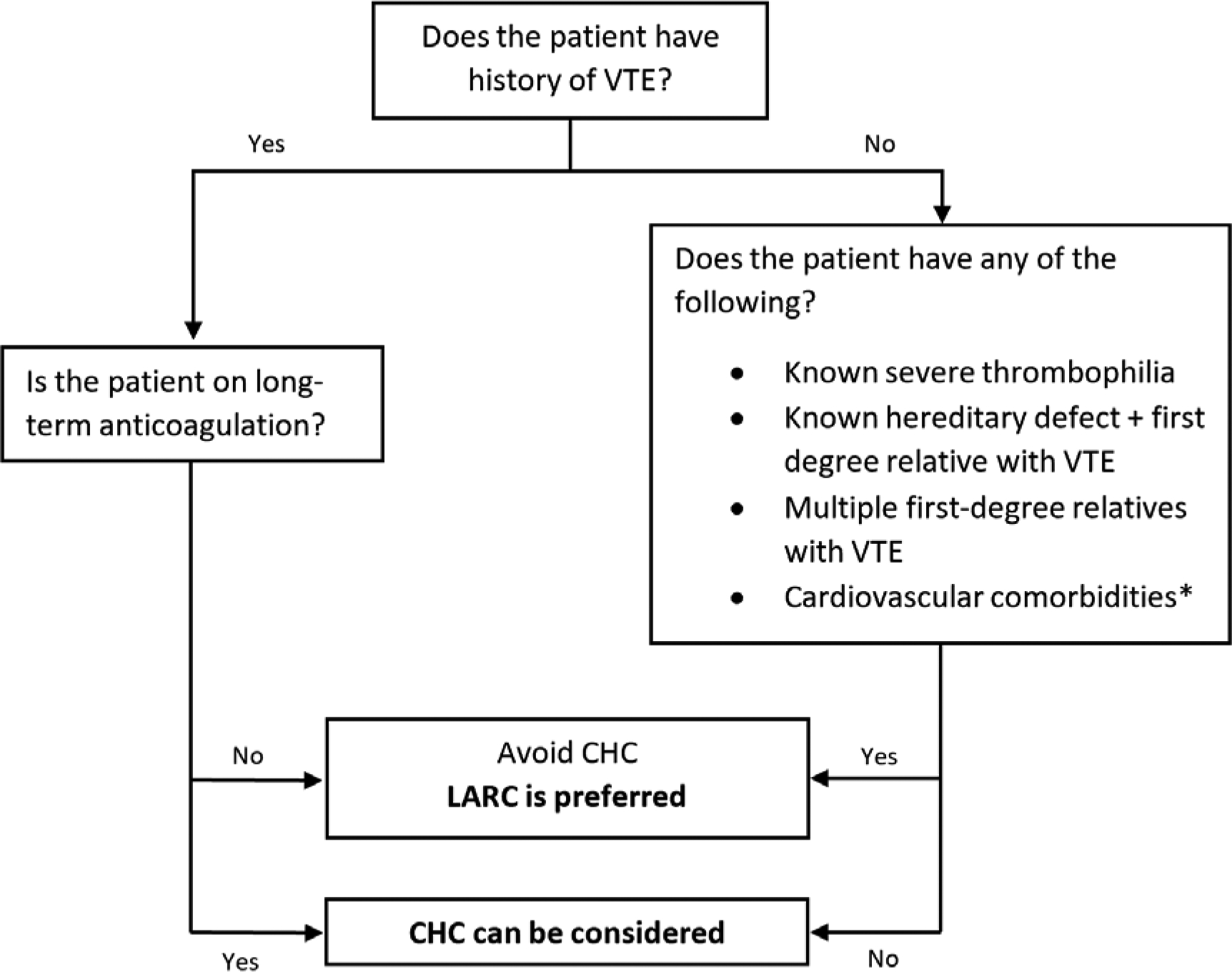
Proposed algorithm to guide contraceptive management for women at high risk of VTE based authors’ opinion.
*These include: uncontrolled hypertension, ischemic heart disease, stroke, migraine with aura, smoking in women of age 35 or older, or multiple cardiovascular comorbidities.
iii. Duration of Anticoagulation
In patients with thrombosis provoked by hormonal contraception, the risk is believed to return to baseline levels within one to three months of cessation of hormonal use [100, 107]. As such, therapeutic anticoagulation is not needed beyond the treatment of the acute event, unless there is an anticipated estrogen challenge such as pregnancy. The 2012 International Society of Thrombosis & Hemostasis (ISTH) guidelines do not require anticoagulant therapy beyond 3 months[108].
B. Menopausal Hormone Therapy and Thrombosis
The Women’s Health Initiative (WHI) trial conferred the most definitive clinical data regarding MHT. In the trial, VTE risk was increased with HR of 2.06 (95% CI, 1.6–2.7), which was similar for both DVT and PE [102]. A similar two-fold increase in VTE with CHC was noted in the Heart and Estrogen/Progestin Replacement Study (HERS) [95]. However, the absolute risk in both trials, particularly in healthy postmenopausal women, was extremely low.
The WHI trial showed a notably high risk of VTE in women with prior thromboembolic event, with HR of 3.9. As such, MHT is contraindicated in women with previous VTE or stroke. Furthermore, the trial showed a 6-fold increase in VTE (HR 6.7, 95% CI 3.1–14.5) in women with factor five leiden mutation (FVL) who were on CHC. Current recommendations suggest estrogens be avoided in women with FVL. Other genetic mutations did not affect the risk of VTE [102].
C. Pregnancy and Thrombosis
Pregnancy increases the risk of VTE due to hypercoagulable state, venous stasis, and endothelial injury. The risk is increased throughout all trimesters but highest during the post-partum period. Additional risk factors include a prior history of VTE, hospitalization for an acute illness or cesarean-section delivery, and the presence of an inherited thrombophilia (e.g. factor V Leiden mutation, prothrombin gene mutation, or antithrombin III, protein C, or protein S deficiencies).
The 2012 American College of Chest Physicians (ACCP), 2018 American College of Obstetricians and Gynecologists (ACOG), and the American Society of Hematology (ASH) offer guidelines on the selection criteria for antepartum pharmacologic thromboprophylaxis during pregnancy [90, 109, 110].
Patients who have had prior VTE related to high estrogen states (such as OCP use or previous pregnancy) are candidates for thromboprophylaxis during pregnancy given the increase in estrogen levels. The ASH 2018 guidelines strongly recommend both antepartum and postpartum thromboprophylaxis in these patients, although this is based on low-certainty evidence [109]. Thromboprophylaxis is also considered in patients with a history of idiopathic VTE, or in those with multiple VTEs regardless of the cause.
In contrast, women who have had a single VTE due to a transient risk factor such as trauma, immobility, or surgery, are considered to have a lower risk of recurrence. In these patients, clinical surveillance is preferred over pharmacologic thromboprophylaxis given the bleeding risks associated the pregnancy.
The ASH 2018 guidelines also offer recommendations regarding women with known hereditary thrombophilias, without personal history of VTE, and with or without family history of thrombosis[109]. We summarize those recommendations in our proposed algorithm in Figure 4 under the Special Populations, Inherited Thrombophilias section.
Figure 4 -.
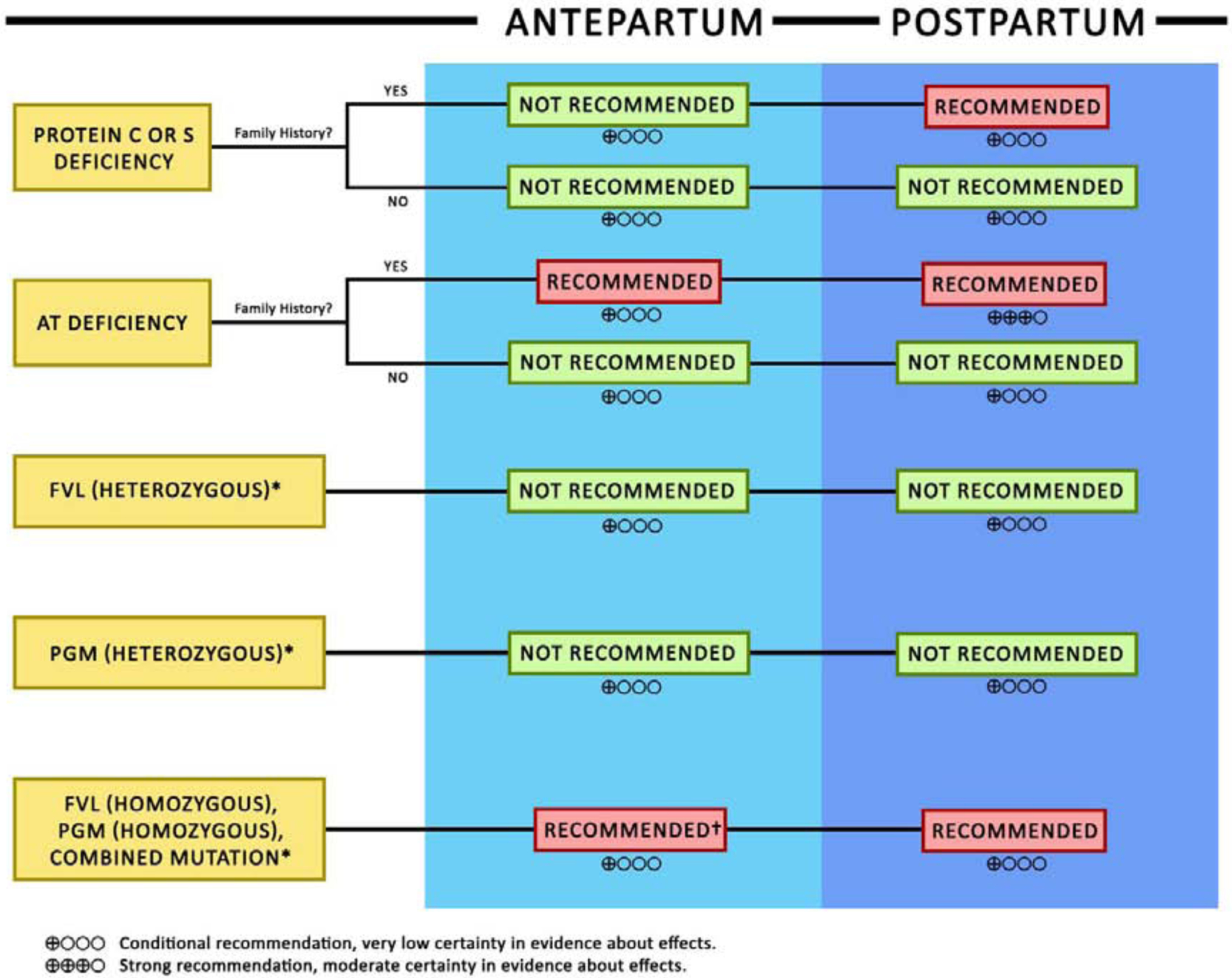
We summarize the 2018 American Society of Hematology recommendations on ante-partum and post-partum anticoagulation in pregnant women with hereditary thrombophilias without personal history of thrombosis in this algorithm.
AT: Anti-thrombin
FVL: Factor V Leiden
PGM: Prothrombin gene mutation
*Regardless of family history
†With the exception of homozygous PGM without family history, where antepartum anticoagulation was not recommended.
Special Populations
A. Antiphospholipid Antibodies
Women with anti-phospholipid syndrome (APS) on anticoagulation are continued on therapeutic anticoagulation with LMWH during pregnancy. There is a lack of robust data to guide the management of pregnancy or contraception in women with positive anti-phospholipid (aPL) antibodies without a history of thrombosis. A study by Lynch et al measured aPL antibody levels in 451 low-risk nulliparous women during early pregnancy, and found that 24.4% had positive aPL antibodies. The rate of fetal loss in these women was higher in those with aPL antibodies (15.8%) than in those without (6.5%), with a relative risk of 2.44 (95% CI, 1.29–4.62). An elevated IgG anti-cardiolipin antibody level at the first prenatal visit was the only aPL antibody significantly associated with fetal loss, with a relative risk of 3.5 (95% CI, 1.56–8.07). The risk of maternal complications was similar in both groups. The use of aspirin has been suggested in this group although the benefit is uncertain. Additionally, there is a paucity of data to provide strong recommendations on the use of CHCs in women who test positive for aPL antibodies without a history of thrombosis.
B. Inherited Thrombophilias
Women with inherited thrombophilias, such as FVL, Protein C or S deficiency, antithrombin deficiency, or prothrombin gene mutation, are at increased risk of VTE. This risk is further exacerbated by increased levels of estrogen due to medications or pregnancy. A systematic review and meta-analysis by Van Vlijmen et al, in 2017, showed that in CHC-users, mild or severe thrombophilia increased the risk of VTE almost 6-fold and 7-fold respectively. The absolute VTE risk was far higher in CHC-users with severe thrombophilia (4.3 – 4.6/100 pill-years) than in those with mild thrombophilia (0.5 – 2.0/100 pill-years). [111]
While there are guidelines on pregnancy management in women with hereditary thrombophilias (See Pregnancy section), there is lack of consensus guidelines on oral contraceptive management for different thrombophilias of varying degrees of severity. In 2016, the Center for Disease Control issued the U.S. Medical Eligibility Criteria for Contraceptive Use, in which CHC use with any hereditary thrombophilia was considered to be associated with unacceptable health risk (Category 4)[112]. Based on this, the ACOG 2019 guidelines considers CHC use with any hereditary thrombophilia to be contraindicated [113]. As such, CHC use is generally considered contraindicated in these patients, and particularly in those with “severe” thrombophilia and/or high-risk of thrombosis which includes:
homozygous or double heterozygous FVL mutation
homozygous or double heterozygous prothrombin mutation
severe deficiencies in Protein C, S or antithrombin.
same defect as a first-degree family member who developed VTE.
women with multiple first-degree relatives with VTE, but no evidence of inherited thrombophilia are at increased risk of VTE, and may have an unidentified defect.
In these patients, non-hormonal methods such as the copper IUD are the preferred method of contraception. Progestin-only pills are generally considered safe, however, injectable progesterone such as DMPA may have a small increase in VTE risk [104, 105].
In women with “mild” inherited thrombophilia, such as heterozygous FVL or prothrombin gene mutation, or mild deficiencies in Protein C, S, or anti-thrombin, and no personal or family history of VTE, CHC remains discouraged. However, they can be considered in special circumstances cases on an individual basis when they are highly needed.
In women with known hereditary defects without personal history of VTE, pregnancy management can be challenging and controversial due to limited evidence. The ASH 2018 guidelines offer recommendations which we summarize in our proposed algorithm in Figure 4 for consideration. Routine screening for hereditary thrombophilias before pregnancy is not recommended due to their rarity and the low certainty for evidence regarding their management.
See “Management” section for additional details on contraceptive and pregnancy management in patients with thrombophilia.
C. Myeloproliferative Neoplasms
Myeloproliferative neoplasms, such as essential thrombocythemia, polycythemia vera, and myelofibrosis have been shown to be associated with an increased risk of thrombosis [114]. However, data assessing the risk of VTE in patients with these conditions while taking estrogen-based therapy is lacking and insufficient to formulate guidelines. In one study of 305 female patients with essential thrombocythemia by Gangat et al, the use of MHT was not associated with an increase in venous or arterial thrombotic events. However, CHC use was associated with a threefold increase of VTE (HR), and a fivefold increased risk of splanchnic thrombosis (HR) [115] suggesting that alternative options for contraception need to be offered to these patients.
D. Transgender Women
Transgender patients who are assigned a male gender at birth and transition to female gender are treated with hormonal therapy aimed at suppressing endogenous sex hormone production and to maintain estrogen levels in the physiological range for the affirmed gender. In the US, most transwomen are treated with either oral or transdermal estradiol in association with spironolactone [116]. Oral estrogens seem to have similar effects on surrogate inflammatory endpoints in transgender women as they do in cisgender women [117], such as elevation in proinflammatory cytokines IL-1, IL-6, IL-8, and tumor necrosis factor-α [118]. Transdermal estrogen, on the other hand, has not been reported to increase proinflammatory cytokines and procoagulant factors [118–120].
A U.S. database study, which included the largest population of transgender persons on hormonal treatment to date, compared the incidence of VTE, stroke, and MI in 2,842 transwomen and matched cisgender men and women [121]. The researchers found that transwomen had a higher incidence of VTE than both reference populations, and that risk increased over time. The adjusted HR for VTE with estrogen use in transgender women was 3.2 (95% CI: 1.5–6.5) compared to cisgender men, and 2.5 (1.2–5.0) compared to ciswomen, which increased with longer duration of use. At 8 years, the hazard risks were 16.7 (CI, 6.4– 27.5) and 3.4 (CI, 1.1– 5.6) and 13.7 (CI, 4.1– 22.7), compared to cismen and ciswomen respectively. The increasing risk of VTE with duration of estrogen use is in contrast with menopausal hormonal therapy, where the risk is highest in the first year of use and decreases with time. [122]
The researchers of the same study also found an increased risk of ischemic stroke among transwomen using estrogens, where the hazard risks after 6 years of use were 9.9 (3.0– 33.1) compared to cismen and 4.1 (1.5– 11.4) compared to ciswomen. The incidence of myocardial infarction was similar in transwomen compared to the matched controls.
There currently is no role for thrombophilia screening in asymptomatic individuals transitioning genders. Data on the role of prophylactic aspirin or anticoagulation in patients with high risk of thrombosis does not exist, and is currently not recommended.
In transgender women with prior history of VTE, the decision to start hormonal therapy is challenging. The patient’s strong desire to embark on hormonal therapy should not be dismissed. The use of transdermal estrogen appears less thrombotic, and as such should be considered as first choice of therapy. Estrogen therapy in these patients should utilize the lowest possible dose of estrogen that is sufficient to achieve and maintain serum estradiol levels in the usual physiological female range.
Due to lack of data, there are currently no guidelines on anticoagulation management in transgender women, and as such management is extrapolated from data on cisgender women. Prospective data suggests that with therapeutic anticoagulation, estrogen therapy can be safely continued in cisgender women who develop VTE on estrogen [106]. As such, transgender women who develop VTE on estrogen may similarly have a low VTE recurrence rate if maintained on therapeutic anticoagulation. Thus, if estrogen therapy is continued, then therapeutic anticoagulation may need to be continued indefinitely while on estrogen. Women who develop thrombosis while on oral hormonal therapy are likely to have a reduction in risk of recurrence by changing to transdermal estrogen therapy. Discontinuing anticoagulation in this scenario is a complex decision, and data is lacking on the safety of discontinuation after switching to transdermal therapy. As such, discontinuing anticoagulation has not been recommended in the absence of contraindications [123].
Conclusion
CHCs are widely used and are safe and effective for regulation of fertility in the majority of women. Although thrombotic complications are the most important side effect, some clinical implications appear to be evident. The choice of a contraceptive method depends on a personal and family history of thrombosis, and those with a risk of thrombotic disease should be recommended alternatives to CHCs. Screening for prothrombotic mutations before initiating CHCs is not cost effective, and it is not clear that screening in women with a positive family history is better than offering alternative contraception. The presence of other risk factors for thrombosis such as dyslipidemia, smoking and obesity should be taken into consideration while counseling women on hormonal replacement therapy. Thus, all efforts should be made to ensure that women with contraindications against CHCs are not prescribed these drugs, and that women that use them are well informed about the symptoms of VTE.
In conclusion, a better understanding of the pathophysiology of hormone-related thrombosis is necessary to guide the development of safer alternatives. In the interim, we need to develop techniques to stratify risks for thrombosis so as to enable tailoring of recommendations for each individual considering hormonal therapy.
Highlights:
Estrogen affects the hemostatic pathway through various mechanisms
The overall effect of estrogen is prothrombotic
The dose and route of estrogen impacts its prothrombotic effect.
Evaluation of prothrombotic risk factors before contraception or pregnancy is needed
We present emerging data on unique populations such as transgender women.
Estrogen affects the hemostatic pathway through various mechanisms
The overall effect of estrogen is prothrombotic
The dose and route of estrogen impacts its prothrombotic effect.
Evaluation of prothrombotic risk factors before contraception or pregnancy is needed
We present emerging data on unique populations such as transgender women.
Acknowledgements
LN’s work is supported by National Heart, Lung and Blood Institute grants HL142647-01, HL121131-01, and U01 HL143402
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Watkins ES, The estrogen elixir: a history of hormone replacement therapy in America. 2007: JHU Press. [Google Scholar]
- 2.Allen E, et al. , The hormone of the ovarian follicle; its localization and action in test animals, and additional points bearing upon the internal secretion of the ovary. American Journal of Anatomy, 1924. 34(1): p. 133–181. [Google Scholar]
- 3.Doisy EA, Veler C, and Thayer S, Folliculin from urine of pregnant women. American Journal of Physiology, 1929. 90(2): p. 329–330. [Google Scholar]
- 4.Huffman MN, et al. , The isolation of α-dihydrotheelin from human pregnancy urine. Journal of Biological Chemistry, 1940. 134(2): p. 591–604. [Google Scholar]
- 5.Butenandt A, Über „Progynon “ein krystallisiertes weibliches Sexualhormon. Naturwissenschaften, 1929. 17(45): p. 879–879. [Google Scholar]
- 6.Simpson E and Santen RJ, Celebrating 75 years of oestradiol. J Mol Endocrinol, 2015. 55(3): p. T1–20. [DOI] [PubMed] [Google Scholar]
- 7.Canez M, Lee K, and Olive D, Progestogens and estrogens. Infertil Reprod Med Clin North Am, 1992. 3: p. 59–77. [Google Scholar]
- 8.Coelingh Bennink HJ, Are all estrogens the same? Maturitas, 2004. 47(4): p. 269–75. [DOI] [PubMed] [Google Scholar]
- 9.Birkhauser M, Chemistry, physiology, and pharmacology of sex steroids. J Cardiovasc Pharmacol, 1996. 28(Suppl 5): p. S1–S13.8891865 [Google Scholar]
- 10.Simpson ER, Sources of estrogen and their importance. J Steroid Biochem Mol Biol, 2003. 86(3–5): p. 225–30. [DOI] [PubMed] [Google Scholar]
- 11.Ruggiero RJ and Likis FE, Estrogen: physiology, pharmacology, and formulations for replacement therapy. J Midwifery Womens Health, 2002. 47(3): p. 130–8. [DOI] [PubMed] [Google Scholar]
- 12.Seifer DB and Speroff L, Clinical Gynecologic Endocrinology and Infertility: Self Assessment and Study Guide. 1999: Williams & Wilkins. [Google Scholar]
- 13.Society, N.A.M., Menopause Core Curriculum Study Guide, 2002. North American Menopause Society, Cleveland, OH, 2000. [Google Scholar]
- 14.Eyster KM, The estrogen receptors: an overview from different perspectives, in Estrogen Receptors. 2016, Springer; p. 1–10. [DOI] [PubMed] [Google Scholar]
- 15.Eick GN and Thornton JW, Evolution of steroid receptors from an estrogen-sensitive ancestral receptor. Mol Cell Endocrinol, 2011. 334(1–2): p. 31–8. [DOI] [PubMed] [Google Scholar]
- 16.Aranda A and Pascual A, Nuclear hormone receptors and gene expression. Physiol Rev, 2001. 81(3): p. 1269–304. [DOI] [PubMed] [Google Scholar]
- 17.Hewitt SC and Korach KS, Estrogen Receptors: New Directions in the New Millennium. Endocr Rev, 2018. 39(5): p. 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vrtacnik P, et al. , The many faces of estrogen signaling. Biochem Med (Zagreb), 2014. 24(3): p. 329–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawkins MB, et al. , Identification of a third distinct estrogen receptor and reclassification of estrogen receptors in teleosts. Proc Natl Acad Sci U S A, 2000. 97(20): p. 10751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menasce LP, et al. , Localization of the estrogen receptor locus (ESR) to chromosome 6q25.1 by FISH and a simple post-FISH banding technique. Genomics, 1993. 17(1): p. 263–5. [DOI] [PubMed] [Google Scholar]
- 21.Enmark E, et al. , Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab, 1997. 82(12): p. 4258–65. [DOI] [PubMed] [Google Scholar]
- 22.Jia M, Dahlman-Wright K, and Gustafsson JA, Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab, 2015. 29(4): p. 557–68. [DOI] [PubMed] [Google Scholar]
- 23.Paterni I, et al. , Estrogen receptors alpha (ERalpha) and beta (ERbeta): subtype-selective ligands and clinical potential. Steroids, 2014. 90: p. 13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthews J and Gustafsson JA, Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Interv, 2003. 3(5): p. 281–92. [DOI] [PubMed] [Google Scholar]
- 25.Carroll JS and Brown M, Estrogen receptor target gene: an evolving concept. Mol Endocrinol, 2006. 20(8): p. 1707–14. [DOI] [PubMed] [Google Scholar]
- 26.Lannigan DA, Estrogen receptor phosphorylation. Steroids, 2003. 68(1): p. 1–9. [DOI] [PubMed] [Google Scholar]
- 27.Shang Y, et al. , Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell, 2000. 103(6): p. 843–52. [DOI] [PubMed] [Google Scholar]
- 28.DiRenzo J, et al. , BRG-1 is recruited to estrogen-responsive promoters and cooperates with factors involved in histone acetylation. Mol Cell Biol, 2000. 20(20): p. 7541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heldring N, et al. , Estrogen receptors: how do they signal and what are their targets. Physiol Rev, 2007. 87(3): p. 905–31. [DOI] [PubMed] [Google Scholar]
- 30.Clark S, et al. , Estrogen receptor-mediated transcription involves the activation of multiple kinase pathways in neuroblastoma cells. J Steroid Biochem Mol Biol, 2014. 139: p. 45–53. [DOI] [PubMed] [Google Scholar]
- 31.Revankar CM, et al. , A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science, 2005. 307(5715): p. 1625–30. [DOI] [PubMed] [Google Scholar]
- 32.Olde B and Leeb-Lundberg LM, GPR30/GPER1: searching for a role in estrogen physiology. Trends Endocrinol Metab, 2009. 20(8): p. 409–16. [DOI] [PubMed] [Google Scholar]
- 33.Zimmerman MA, et al. , GPER-novel membrane oestrogen receptor. Clin Sci (Lond), 2016. 130(12): p. 1005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Filardo EJ and Thomas P, Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology, 2012. 153(7): p. 2953–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wend K, Wend P, and Krum SA, Tissue-Specific Effects of Loss of Estrogen during Menopause and Aging. Front Endocrinol (Lausanne), 2012. 3: p. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jensen EV, et al. , Estrogen action: a historic perspective on the implications of considering alternative approaches. Physiol Behav, 2010. 99(2): p. 151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pietras RJ, et al. , HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene, 1995. 10(12): p. 2435–46. [PubMed] [Google Scholar]
- 38.Pietras RJ and Szego CM, Partial purification and characterization of oestrogen receptors in subfractions of hepatocyte plasma membranes. Biochem J, 1980. 191(3): p. 743–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuiper GG, et al. , Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A, 1996. 93(12): p. 5925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas P, et al. , Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology, 2005. 146(2): p. 624–32. [DOI] [PubMed] [Google Scholar]
- 41.Oedingen C, Scholz S, and Razum O, Systematic review and meta-analysis of the association of combined oral contraceptives on the risk of venous thromboembolism: The role of the progestogen type and estrogen dose. Thromb Res, 2018. 165: p. 68–78. [DOI] [PubMed] [Google Scholar]
- 42.Powell A, Choosing the Right Oral Contraceptive Pill for Teens. Pediatr Clin North Am, 2017. 64(2): p. 343–358. [DOI] [PubMed] [Google Scholar]
- 43.Stanczyk FZ, Archer DF, and Bhavnani BR, Ethinyl estradiol and 17beta-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception, 2013. 87(6): p. 706–27. [DOI] [PubMed] [Google Scholar]
- 44.Christin-Maitre S, History of oral contraceptive drugs and their use worldwide. Best Pract Res Clin Endocrinol Metab, 2013. 27(1): p. 3–12. [DOI] [PubMed] [Google Scholar]
- 45.Fruzzetti F, et al. , [Oral contraceptive pill and thrombotic risk: epidemiological studies]. Minerva Ginecol, 2012. 64(6): p. 539–49. [PubMed] [Google Scholar]
- 46.Kuhl H, Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric, 2005. 8 Suppl 1: p. 3–63. [DOI] [PubMed] [Google Scholar]
- 47.Cooper DB and Mahdy H, Oral Contraceptive Pills, in StatPearls. 2020, StatPearls Publishing StatPearls Publishing LLC.: Treasure Island (FL). [Google Scholar]
- 48.Baird DT and Glasier AF, Hormonal contraception. N Engl J Med, 1993. 328(21): p. 1543–9. [DOI] [PubMed] [Google Scholar]
- 49.Lackie E and Fairchild A, The birth control pill, thromboembolic disease, science and the media: a historical review of the relationship. Contraception, 2016. 94(4): p. 295–302. [DOI] [PubMed] [Google Scholar]
- 50.Sandset PM, et al. , Mechanisms of thrombosis related to hormone therapy. Thromb Res, 2009. 123 Suppl 2: p. S70–3. [DOI] [PubMed] [Google Scholar]
- 51.Dupuis M, et al. , Effects of Estrogens on Platelets and Megakaryocytes. Int J Mol Sci, 2019. 20(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fox SW and Chambers TJ, The effect of oestrogen on megakaryocyte differentiation and platelet counts in vivo. Int J Cardiol, 2006. 109(3): p. 359–66. [DOI] [PubMed] [Google Scholar]
- 53.Valera MC, et al. , Chronic estradiol treatment reduces platelet responses and protects mice from thromboembolism through the hematopoietic estrogen receptor alpha. Blood, 2012. 120(8): p. 1703–12. [DOI] [PubMed] [Google Scholar]
- 54.Valera MC, et al. , Effect of chronic estradiol plus progesterone treatment on experimental arterial and venous thrombosis in mouse. PLoS One, 2017. 12(5): p. e0177043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harrison RL and McKee PA, Estrogen stimulates von Willebrand factor production by cultured endothelial cells. Blood, 1984. 63(3): p. 657–64. [PubMed] [Google Scholar]
- 56.Drury-Stewart DN, et al. , Complex changes in von Willebrand factor-associated parameters are acquired during uncomplicated pregnancy. PLoS One, 2014. 9(11): p. e112935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rabbani LE, et al. , Oral conjugated equine estrogen increases plasma von Willebrand factor in postmenopausal women. J Am Coll Cardiol, 2002. 40(11): p. 1991–9. [DOI] [PubMed] [Google Scholar]
- 58.Cushman M, et al. , Effect of postmenopausal hormones on inflammation-sensitive proteins: the Postmenopausal Estrogen/Progestin Interventions (PEPI) Study. Circulation, 1999. 100(7): p. 717–22. [DOI] [PubMed] [Google Scholar]
- 59.Zia A, et al. , Hypercoagulability in adolescent girls on oral contraceptives-global coagulation profile and estrogen receptor polymorphisms. Am J Hematol, 2015. 90(8): p. 725–31. [DOI] [PubMed] [Google Scholar]
- 60.Ohashi R, Sugimura M, and Kanayama N, Estrogen administration enhances thrombin generation in rats. Thromb Res, 2003. 112(5–6): p. 325–8. [DOI] [PubMed] [Google Scholar]
- 61.Wessler S, Estrogen-associated thromboembolism. Ann Epidemiol, 1992. 2(4): p. 439–43. [DOI] [PubMed] [Google Scholar]
- 62.Middeldorp S, et al. , Effects on coagulation of levonorgestrel- and desogestrel-containing low dose oral contraceptives: a cross-over study. Thromb Haemost, 2000. 84(1): p. 4–8. [PubMed] [Google Scholar]
- 63.Tans G, et al. , A randomized cross-over study on the effects of levonorgestreland desogestrel-containing oral contraceptives on the anticoagulant pathways. Thromb Haemost, 2000. 84(1): p. 15–21. [PubMed] [Google Scholar]
- 64.Cosman F, et al. , Short-term effects of estrogen, tamoxifen and raloxifene on hemostasis: a randomized-controlled study and review of the literature. Thromb Res, 2005. 116(1): p. 1–13. [DOI] [PubMed] [Google Scholar]
- 65.Kemmeren JM, et al. , Effect of second- and third-generation oral contraceptives on the protein C system in the absence or presence of the factor VLeiden mutation: a randomized trial. Blood, 2004. 103(3): p. 927–33. [DOI] [PubMed] [Google Scholar]
- 66.Koenen RR, et al. , Effect of oral contraceptives on the anticoagulant activity of protein S in plasma. Thromb Haemost, 2005. 93(5): p. 853–9. [DOI] [PubMed] [Google Scholar]
- 67.Meijers JC, et al. , Increased fibrinolytic activity during use of oral contraceptives is counteracted by an enhanced factor XI-independent down regulation of fibrinolysis: a randomized cross-over study of two low-dose oral contraceptives. Thromb Haemost, 2000. 84(1): p. 9–14. [PubMed] [Google Scholar]
- 68.Tchaikovski SN and Rosing J, Mechanisms of estrogen-induced venous thromboembolism. Thromb Res, 2010. 126(1): p. 5–11. [DOI] [PubMed] [Google Scholar]
- 69.Sitruk-Ware R and Nath A, Metabolic effects of contraceptive steroids. Rev Endocr Metab Disord, 2011. 12(2): p. 63–75. [DOI] [PubMed] [Google Scholar]
- 70.Maggi A and Della Torre S, Sex, metabolism and health. Mol Metab, 2018. 15: p. 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steingold KA, et al. , Enhanced hepatic extraction of estrogens used for replacement therapy. J Clin Endocrinol Metab, 1986. 62(4): p. 761–6. [DOI] [PubMed] [Google Scholar]
- 72.Moverare S, et al. , Estrogen increases coagulation factor V mRNA levels via both estrogen receptor-alpha and -beta in murine bone marrow/bone. Eur J Endocrinol, 2004. 151(2): p. 259–63. [DOI] [PubMed] [Google Scholar]
- 73.Cleuren AC, et al. , 17alpha-Ethinylestradiol rapidly alters transcript levels of murine coagulation genes via estrogen receptor alpha. J Thromb Haemost, 2010. 8(8): p. 1838–46. [DOI] [PubMed] [Google Scholar]
- 74.Straub RH, The complex role of estrogens in inflammation. Endocr Rev, 2007. 28(5): p. 521–74. [DOI] [PubMed] [Google Scholar]
- 75.Esmon C, Inflammation and thrombosis. Journal of Thrombosis and haemostasis, 2003. 1(7): p. 1343–1348. [DOI] [PubMed] [Google Scholar]
- 76.Calippe B, et al. , 17Beta-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor alpha signaling in macrophages in vivo. J Immunol, 2010. 185(2): p. 1169–76. [DOI] [PubMed] [Google Scholar]
- 77.Calippe B, et al. , Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: involvement of the phosphatidylinositol 3-kinase pathway. J Immunol, 2008. 180(12): p. 7980–8. [DOI] [PubMed] [Google Scholar]
- 78.Pelekanou V, et al. , Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERalpha36 and GPR30/GPER1. J Leukoc Biol, 2016. 99(2): p. 333–47. [DOI] [PubMed] [Google Scholar]
- 79.Vegeto E, et al. , Regulation of the lipopolysaccharide signal transduction pathway by 17beta-estradiol in macrophage cells. J Steroid Biochem Mol Biol, 2004. 91(1–2): p. 59–66. [DOI] [PubMed] [Google Scholar]
- 80.Kovats S, Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol, 2015. 294(2): p. 63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rosing J, et al. , Oral contraceptives and venous thrombosis: different sensitivities to activated protein C in women using second- and third-generation oral contraceptives. Br J Haematol, 1997. 97(1): p. 233–8. [DOI] [PubMed] [Google Scholar]
- 82.DeLoughery TG, Estrogen and thrombosis: controversies and common sense. Reviews in Endocrine and Metabolic Disorders, 2011. 12(2): p. 77–84. [DOI] [PubMed] [Google Scholar]
- 83.de Bastos M, et al. , Combined oral contraceptives: venous thrombosis. Cochrane Database Syst Rev, 2014(3): p. Cd010813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Effect of different progestagens in low oestrogen oral contraceptives on venous thromboembolic disease. World Health Organization Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception. Lancet, 1995. 346(8990): p. 1582–8. [PubMed] [Google Scholar]
- 85.Dragoman MV, et al. , A systematic review and meta-analysis of venous thrombosis risk among users of combined oral contraception. Int J Gynaecol Obstet, 2018. 141(3): p. 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tepper NK, et al. , Nonoral combined hormonal contraceptives and thromboembolism: a systematic review. Contraception, 2017. 95(2): p. 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dinger J, Mohner S, and Heinemann K, Cardiovascular risk associated with the use of an etonogestrel-containing vaginal ring. Obstet Gynecol, 2013. 122(4): p. 800–8. [DOI] [PubMed] [Google Scholar]
- 88.Lidegaard O, et al. , Venous thrombosis in users of non-oral hormonal contraception: follow-up study, Denmark 2001–10. Bmj, 2012. 344: p. e2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mohammed K, et al. , Oral vs Transdermal Estrogen Therapy and Vascular Events: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab, 2015. 100(11): p. 4012–20. [DOI] [PubMed] [Google Scholar]
- 90.ACOG Practice Bulletin No. 196: Thromboembolism in Pregnancy. Obstet Gynecol, 2018. 132(1): p. e1–e17. [DOI] [PubMed] [Google Scholar]
- 91.Dentali F, Crowther M, and Ageno W, Thrombophilic abnormalities, oral contraceptives, and risk of cerebral vein thrombosis: a meta-analysis. Blood, 2006. 107(7): p. 2766–73. [DOI] [PubMed] [Google Scholar]
- 92.Amoozegar F, et al. , Hormonal Contraceptives and Cerebral Venous Thrombosis Risk: A Systematic Review and Meta-Analysis. Front Neurol, 2015. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hassan HA, Oral contraceptive-induced mesenteric venous thrombosis with resultant intestinal ischemia. J Clin Gastroenterol, 1999. 29(1): p. 90–5. [DOI] [PubMed] [Google Scholar]
- 94.Citla Sridhar D, Gollamudi J, Cao S, Fu P, Nayak L, Incidence of retinal vein occlusion in women of reproductive age on estrogen. 2019, Haemophilia. [Google Scholar]
- 95.Hulley S, et al. , Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. Jama, 1998. 280(7): p. 605–13. [DOI] [PubMed] [Google Scholar]
- 96.Grady D, et al. , Cardiovascular disease outcomes during 6.8 years of hormone therapy: Heart and Estrogen/progestin Replacement Study follow-up (HERS II). Jama, 2002. 288(1): p. 49–57. [DOI] [PubMed] [Google Scholar]
- 97.Rossouw JE, et al. , Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. Jama, 2002. 288(3): p. 321–33. [DOI] [PubMed] [Google Scholar]
- 98.Lidegaard O, et al. , Thrombotic stroke and myocardial infarction with hormonal contraception. N Engl J Med, 2012. 366(24): p. 2257–66. [DOI] [PubMed] [Google Scholar]
- 99.Roach RE, et al. , Combined oral contraceptives: the risk of myocardial infarction and ischemic stroke. Cochrane Database Syst Rev, 2015(8): p. Cd011054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Venous thromboembolic disease and combined oral contraceptives: results of international multicentre case-control study. World Health Organization Collaborative Study of Cardiovascular Disease and Steroid Hormone Contraception. Lancet, 1995. 346(8990): p. 1575–82. [PubMed] [Google Scholar]
- 101.Bloemenkamp KW, et al. , Higher risk of venous thrombosis during early use of oral contraceptives in women with inherited clotting defects. Arch Intern Med, 2000. 160(1): p. 49–52. [DOI] [PubMed] [Google Scholar]
- 102.Cushman M, et al. , Estrogen plus progestin and risk of venous thrombosis. Jama, 2004. 292(13): p. 1573–80. [DOI] [PubMed] [Google Scholar]
- 103.Organization, W.H., Medical eligibility criteria for contraceptive use: Fifth edition. 2015. [PubMed]
- 104.Bergendal A, et al. , Association of venous thromboembolism with hormonal contraception and thrombophilic genotypes. Obstet Gynecol, 2014. 124(3): p. 600–9. [DOI] [PubMed] [Google Scholar]
- 105.Mantha S, et al. , Assessing the risk of venous thromboembolic events in women taking progestin-only contraception: a meta-analysis. Bmj, 2012. 345: p. e4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martinelli I, et al. , Recurrent venous thromboembolism and abnormal uterine bleeding with anticoagulant and hormone therapy use, in Blood. 2016. p. 1417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gomes MP and Deitcher SR, Risk of venous thromboembolic disease associated with hormonal contraceptives and hormone replacement therapy: a clinical review. Arch Intern Med, 2004. 164(18): p. 1965–76. [DOI] [PubMed] [Google Scholar]
- 108.Baglin T, et al. , Duration of anticoagulant therapy after a first episode of an unprovoked pulmonary embolus or deep vein thrombosis: guidance from the SSC of the ISTH. J Thromb Haemost, 2012. 10(4): p. 698–702. [DOI] [PubMed] [Google Scholar]
- 109.Bates SM, et al. , American Society of Hematology 2018 guidelines for management of venous thromboembolism: venous thromboembolism in the context of pregnancy, in Blood Adv. 2018. p. 3317–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bates SM, et al. , VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest, 2012. 141(2 Suppl): p. e691S–e736S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.van Vlijmen EF, et al. , Combined oral contraceptives, thrombophilia and the risk of venous thromboembolism: a systematic review and meta-analysis. J Thromb Haemost, 2016. 14(7): p. 1393–403. [DOI] [PubMed] [Google Scholar]
- 112.Curtis KM, et al. , U.S. Medical Eligibility Criteria for Contraceptive Use, 2016. MMWR Recomm Rep, 2016. 65(3): p. 1–103. [DOI] [PubMed] [Google Scholar]
- 113.ACOG Practice Bulletin No. 206: Use of Hormonal Contraception in Women With Coexisting Medical Conditions. Obstet Gynecol, 2019. 133(2): p. e128–e150. [DOI] [PubMed] [Google Scholar]
- 114.Hultcrantz M, et al. , Risk for Arterial and Venous Thrombosis in Patients With Myeloproliferative Neoplasms: A Population-Based Cohort Study. Ann Intern Med, 2018. 168(5): p. 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gangat N, et al. , Estrogen-based hormone therapy and thrombosis risk in women with essential thrombocythemia. Cancer, 2006. 106(11): p. 2406–11. [DOI] [PubMed] [Google Scholar]
- 116.Connors JM and Middeldorp S, Transgender patients and the role of the coagulation clinician. J Thromb Haemost, 2019. 17(11): p. 1790–1797. [DOI] [PubMed] [Google Scholar]
- 117.Goldstein Z, et al. , Managing the risk of venous thromboembolism in transgender adults undergoing hormone therapy, in J Blood Med. 2019. p. 209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wilson R, et al. , Effects of high dose oestrogen therapy on circulating inflammatory markers. Maturitas, 2009. 62(3): p. 281–6. [DOI] [PubMed] [Google Scholar]
- 119.Lowe GD, Hormone replacement therapy and cardiovascular disease: increased risks of venous thromboembolism and stroke, and no protection from coronary heart disease. J Intern Med, 2004. 256(5): p. 361–74. [DOI] [PubMed] [Google Scholar]
- 120.Vongpatanasin W, et al. , Differential effects of oral versus transdermal estrogen replacement therapy on C-reactive protein in postmenopausal women. J Am Coll Cardiol, 2003. 41(8): p. 1358–63. [DOI] [PubMed] [Google Scholar]
- 121.Getahun D, et al. , Cross-sex Hormones and Acute Cardiovascular Events in Transgender Persons: A Cohort Study. Ann Intern Med, 2018. 169(4): p. 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Canonico M, et al. , Hormone replacement therapy and risk of venous thromboembolism in postmenopausal women: systematic review and meta-analysis. Bmj, 2008. 336(7655): p. 1227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shatzel JJ, Connelly KJ, and DeLoughery TG, Thrombotic issues in transgender medicine: A review. Am J Hematol, 2017. 92(2): p. 204–208. [DOI] [PubMed] [Google Scholar]