

Abstract
The transforming growth factor beta (TGFβ) superfamily of secreted signaling molecules and their cognate receptors regulate cell fate and behaviors relevant to many developmental and disease processes. Disruption of TGFβ signaling during embryonic development can, for example, affect morphogenesis and differentiation through complex pathways that may be SMAD (Small Mothers Against Decapentaplegic) -dependent or SMAD-independent. In the present study, the SMAD Binding Element (SBE)-beta lactamase (bla) HEK 293T cell line, which responds to the activation of the SMAD2/3/4 complex, was used in a quantitative high-throughput screening (qHTS) assay to identify potential TGFβ disruptors in the Tox21 10K compound library. From the primary screening, we identified several kinase inhibitors, organometallic compounds, and dithiocarbamates (DTCs) that inhibited TGFβ1-induced SMAD signaling of reporter gene activation independent of cytotoxicity. Counter-screen of SBE antagonists on human embryonic neural stem cells demonstrated cytotoxicity, providing additional evidence to support evaluation of these compounds for developmental toxicity. We profiled the inhibitory patterns of putative SBE antagonists toward other developmental signaling pathways, including wingless-related integration site (WNT), retinoic acid α receptor (RAR) and sonic hedgehog (SHH). The profiling results from SBE-bla assay identify chemicals that disrupt TGFβ/SMAD signaling as part of an integrated qHTS approach for prioritizing putative developmental toxicants.
Keywords: TGFβ/SMAD, developmental and neurotoxicity, quantitative high throughput screening (qHTS)
Graphical Abstract
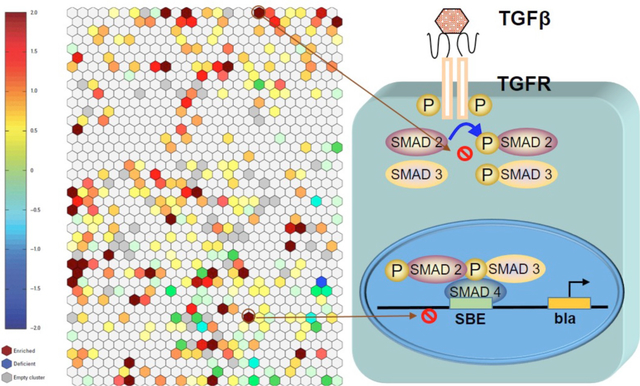
1. Introduction
The transforming growth factor β (TGFβ) superfamily of secreted signaling molecules and their cognate receptor systems are critical regulators of embryogenesis and tissue homeostasis.1, 2 Various ligands including TGFβs, nodal, activins, bone morphogenic proteins (BMPs), and growth/differentiation factor (GDF), regulate cell fate and behavior that underlie differential growth and patterning via complex biological networks.3 Despite the complexity of combinatorial ligand-receptor interaction in the TGFβ superfamily, the downstream regulation of target gene expression has been shown to be mediated through SMAD (Small Mothers Against Decapentaplegic).4 Canonical SMAD-dependent signaling is transduced by two distinct SMAD complexes, SMAD2/3/4 or SMAD1/5/8, in response to different ligands.5, 6
Transcriptional activity regulated by the SMAD2/3/4 complex plays an important role in neurogenesis, hematopoiesis, angiogenesis, osteogenesis and epithelial-mesenchymal transitions.7–10 Evidence exists to implicate SMAD-dependent TGFβ signaling pathway during developmental toxicity. For example, a recent study on gestational exposure to the carbamate pesticide carbofuran in rat revealed increased mRNA level of TGFβ, SMAD2 and 3 in hippocampus, and that adverse consequences of these compounds on neurogenic stem cells could be ameliorated with a pharmacological inhibitor (SB431542) and Smad-3 siRNA.11 In contrast, genomic analysis of zebrafish embryos exposed to the fungicides thiram and disulfiram implicated inhibition of the TGFβ pathway, including an essential transcription factor for zebrafish craniofacial development, SRY-box-containing gene 9a (sox9a), which resulted in craniofacial abnormalities.12 These findings, together with a recognition of the critical role for SMAD-dependent TGFβ signaling in normal embryogenesis10 and tissue homeostasis13, motivated the broad screening of chemical compound libraries for potential disrupters of this pathway, as few environmental and industrial chemicals have been tested for such effects. This study focused on the antagonist modes because inhibition of TGFβ/SMAD signaling leads to failure in mesoderm formation and during gastrulation in the mouse development.14
To identify potential TGFβ/SMAD inhibitors, the current study used a cell-based reporter gene assay to determine whether a chemical has the potential to disrupt TGFβ1 signaling via the SMAD 2/3/4 complex.15 TGFβ1 binding to its cognate receptor results in the phosphorylation of SMAD2 and SMAD3, which then dissociate from the receptor and bind to SMAD4.16 Nuclear translocation of the SMAD2/3/4 complex results in a trans-activation of the beta-lactamase (bla) reporter via the SMAD Binding Element (SBE). Inhibition of SBE-bla activty17, independent of significant cytotoxic effect, indicates suppression of SMAD2/3/4-dependent transcriptional activity. Here, we performed a quantitative high-throughput screen (qHTS) against the Tox21 10K compound library (7872 unique chemicals defined by CASRNs) using the SBE-bla HEK 293T cell line to identify potential TGFβ/SMAD pathway inhibitors (dataset available in PubChem AID: 1347032). Since we have in-house assays of other developmental related pathways (WNT, RAR and SHH), the selected compounds’ effects in SBE-bla, WNT-, RAR-, and SHH-luc assays were evaluated and compared. WNT signaling is critical for neurodevelopmental processes, such as central nervous system (CNS) patterning and differentiation.18, 19 Retinoic acid (RA) is an essential morphogen in vertebrate development20, 21 through binding with homeobox genes regulated by nuclear retinoic acid receptors (RARs) for patterning the neuroaxis.21–23 SHH plays a crucial role in early patterning and organization of CNS as well.24–26 Therefore, perturbations in WNT, RAR, or SHH signaling may disrupt neurodevelopmental patterning and result in disorders related to the structure and function of the CNS. The experimental design and workflow are outlined in Figure 1. We identified several known TGFβ/SMAD pathway inhibitors [e.g. flavopiridol hydrochloride (CASRN: 131740-09-5) and dasatinib (CASRN: 302962-49-8)], and several compounds including pharmaGSID_48519 (CASRN: 686756-87-6) and ziram (CASRN: 137-30-4) that were not previously known to interact with the TGFβ/SMAD pathway and found that they inhibited SMAD3 phosphorylation. As such, a SBE-bla reporter gene assay coupled to SMAD3 phosphorylation conveys a robust and efficient means to identify potential disruptors of TGFβ/SMAD signaling for mechanistic evaluation in other assays relevant to developmental signaling (eg, WNT, RAR, SHH).
Figure 1.

A flowchart of identification of potential TGFβ/SMAD inhibitors. The primary screening of Tox21 10K library in SBE-bla assay generates a dataset with 5.6% active SBE-bla antagonists. From the 68 potent antagonists, 65 inhibitory compounds were confirmed in the confirmation screening of SBE-bla assay. These 65 compounds were further tested in a group of follow up studies including pSMAD3 assay, cell viability in NSCs, WNT-, RAR-, SHH-luc assays. Selected compounds were further tested in the neurite outgrowth assay.
2. Materials and Methods
2.1. Cell lines and culture conditions:
CellSensor® (SBE-bla HEK 293T) cell line, containing a beta-lactamase reporter gene under control of the SBE stably integrated into HEK 293T cells, was purchased from Life Technologies Corporation (Carlsbad, CA). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% dialyzed fetal bovine serum (FBS), 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 25 mM HEPES, 100 U/mL penicillin and 100 μg/mL streptomycin, and 5 μg/mL blasticidin (Life Technologies Corporation). HepG2 cells, purchased from American Type Culture Collection (ATCC, Manassas, VA), were cultured in Eagle’s Minimum Essential Medium (EMEM, ATCC) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA), and 100 U/mL penicillin and 100 μg/mL streptomycin. Neural Stem Cells (NSC) derived from human embryonic stem cell (hESC-H9) were obtained from Life Technologies (Carlsbad, CA) and cultured in StemPro NSC serum free medium from Thermo Fisher (Grand Island, NY). The cells were subcultured by release with StemPro Accutase and seeded on Matrigel-coated flasks.
Human cortical glutamatergic neurons were ready-to-use iPSC-derived neurons purchased from BrainXell (Madison, WI) and were cultured in DMEM/F12 and neurobasal medium supplemented with B27 supplement, N2 supplement, GlutaMAX, Geltrex (Thermo Fisher, Grand Island, NY) and BDNF, GDNF and TGFβ1 (Peprotech, Rocky Hill, NJ). TCF/LEF-luc HEK293 cells were cultured in DMEM supplemented with 10% FBS, and 100 U/mL penicillin and 100 μg/mL streptomycin (Life Technologies Corporation, Carlsbad, CA). C3RL4 cells stably transfected with RARE-luc were cultured in Eagle’s Basal Medium (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 2 μg/mL puromycin and 100 U/mL penicillin and 100 μg/mL streptomycin. Gli1-luc NIH/3T3 cells were cultured in DMEM medium supplemented with 10% bovine calf serum, BCS (ATCC, Manassas, VA), 2 mM L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin, and 2 μg/mL puromycin (Life Technologies Corporation, Carlsbad, CA). All the cells were maintained at 37°C under a humidified atmosphere and 5% CO2.
2.2. SBE-bla assay and qHTS:
SBE-bla HEK 293T cells (passage 6) were seeded at 4000 cells/well/5 μL culture medium without blasticidin into 1536-well tissue culture treated black-wall/clear bottom tissue culture plates (Greiner Bio-One North America, NC) by using a Multidrop Combi (Thermo Fisher, Waltham, MA). Assay plates were incubated at 37°C overnight for cell attachment. The compound treatment of the Tox21 10K compound library was executed by a Wako Pintool station (Wako Automation, San Diego, CA) which transfer 23 nL of test compounds dissolved in DMSO into 5 μL culture medium, followed by addition of 1 μl of 0.25 ng/mL human TGFβ1 using BioRaptr FRD (Beckman Coulter, Brea, CA) to each well. The final compound concentrations in the wells were in the range of 0.98 nM to 77 μM, with assays run in 15-point concentration response in triplicates. After compound treatment, the assay plates were incubated for 5 h at 37°C. For SBE-bla readout, 1 μL of LiveBLAzer™ FRET B/G CCF4-AM substrate (Invitrogen, Carlsbad, CA) was added to each well and the fluorescence intensity was measured after 2 h using an Envision plate reader (Perkin Elmer, Waltham, MA) with filter setting at Ex 320 nm, and Em 460 nm and 530 nm. SBE-bla activity was measured as ratio of reading at 460 nm/530 nm. As for SBE-bla assay data normalization, the readings of wells adding TGFβ1 only were used as 0% inhibition, and vehicle control DMSO wells were used as 100% inhibition since no TGFβ1 activity should be observed.
After measurement of SBE-bla activity, cell viability was measured by adding 5 μL CellTiter-Glo reagent (Promega, Madison, WI) to each well, and luminescence signal was measured through ViewLux plate reader (Perkin Elmer, Waltham, MA) after 30 min incubation at room temperature. Cell viability was measured by relative luminescence units. The positive control in the cell viability assay was tetra-N-octylammonium bromide (Sigma-Aldrich, St. Louis, MO). The readings of vehicle control DMSO wells were used as 0% activity, and positive control tetra-N-octylammonium bromide wells were used as −100% activity for cell viability data normalization.
2.3. Measurement of phosphor-SMAD3 (pSMAD3) level by homogeneous time resolved fluorescence (HTRF) technology:
HepG2 cells (passage 6) were seeded at 5000 cells/well/5 μL of culture medium into 1536-well PDL-coated tissue culture treated white plates using a BioRaptr FRD. The assay plates were incubated overnight at 37°C for cell attachment. The next day, 23 nL of test compound or positive control, a TGFβ1 receptor (ALK5) inhibitor, SB525334 (CASRN: 356559-20-1, Sigma-Aldrich), was added into assay plates, followed by addition of 1 μl of 1.5 ng/mL human TGFβ1 (R&D Systems, Minneapolis, MN) to stimulate SMAD3 phosphorylation. The final compound concentrations in the wells were in the range of 75 nM to 77 μM, with assays run in 11-point concentration response in triplicates. After the assay, plates were incubated at 37°C for 1 h and the supernatant from each well was removed with a light spin evacuation step using Bluewasher (GC biotech, Waddinxveen, The Netherlands). Then, 4 μL of 1X lysis buffer with blocking reagent (Cisbio, Bedford, MA) was added to each well. The assay plates were centrifuged at 1000 rpm for 20 s and incubated for 30 min at room temperature. Antibody mixture to detect pSMAD3 (Ser 423/425) was added after 30 min cell lysis, followed by 4 h incubation at room temperature. Fluorescence intensity was measured with filter setting at Ex 320 nm and Em 620 nm/ 665nm. The relative pSMAD3 level was expressed as the ratio of readings at 665nm/620nm. The readings of vehicle control DMSO wells were used as 0% activity, and positive control SB525334 (CASRN: 356559-20-1, Sigma-Aldrich) wells were used as −100% activity for pSMAD3 activity normalization.
2.4. Cell viability assays in human embryonic neural stem cells:
Human NSCs (passage 5) were seeded at 2000 cells/ well/5 μL culture medium supplemented with 1% Vitronectin (Thermo Fisher, Grand Island, NY)27 into 1536-well white, solid bottom plates using a Multidrop Combi. The cells were incubated at 37°C for 16 h for attachment. Then, 23 nL of compound or positive control, tetra-N-octylammonium bromide, was transferred to the assay plates to reach 11 final concentrations from 0.8 nM to 92 μM. The cells were treated for 5 h or 24 h. Cell viability was measured with CellTiter-Glo luminescent cell viability assay.28 After compound treatment, 5 μL CellTiter-Glo reagent was added to each well and luminescence signal was measured through ViewLux plate reader after 30 min incubation at room temperature. The readings of DMSO was used as a vehicle control (0% activity) and tetra-N-octylammonium bromide was the positive control (−100% activity) for data normalization.
2.5. Neurite outgrowth assay in iPSC-derived neurons:
iPSC-derived GFP-expressing cortical glutamatergic neurons (immediately usage after receiving from vendor) were thawed and directly seeded at the density of 800 cells per well in 5 μL culture medium into low-base poly-D-lysine-coated 1536-well black-wall/clear-bottom assay plates (Aurora Microplates, Whitefish, MT) using BioRaptr FRD. The cells were incubated at 37°C in a humidified environment of 5% CO2 in air for 42 h, followed by addition of 23 nL compounds to reach final concentrations from 90 nM to 92 μM. The treated cells were cultured for 24 h.
GFP-expressing neurons allows imaging analysis of neurite outgrowth. Confocal 20×water lens was used to capture 4-field images per well through Operetta CLS (Perkin Elmer, Shelton, CT). EGFP fluorescence intensities (460–490 nm excitation, 500–550 nm emission) were analyzed by Harmony 4.6 High-Content Imaging Analysis System. The neurite length was quantified based on the default image analysis function, maximum neurite length. Number of objects which indicated cell body number was used to normalize the number of cells in each well. The reduction of maximum neurite length comparing to untreated group was inhibition of neurite outgrowth. Normalized percentage of inhibition was according to the difference between maximum neurite length of vehicle control DMSO and positive control, rotenone (final concentration 11.5 μM).
2.6. WNT-, RAR- and SHH-luc reporter gene assays:
2.6.1. WNT-luc assay:
TCF/LEF-luc HEK293 cells (passage 9) were seeded at 2000 cells/well/4 μl culture medium into 1536-well tissue culture treated white-solid plates using a BioRaptr FRD. The assay plates were cultured overnight at 37°C and 5% CO2. Then 23 nL of compounds were transferred to the assay plates using Wako Pintool station, followed by the addition of 1 μl of 200 ng/mL recombinant human WNT-3a (R & D Systems, Minneapolis, MN) using a BioRaptr FRD. The cells were treated for 24 h. The cell viability assay was multiplexed with reporter-gene readout by addition of 1 μl of CellTiter-Fluor reagent into each well of the assay plates using a BioRaptr FRD and the assay plates were incubated for 1 h at 37°C before measuring luciferase reporter activity. Fluorescence intensity was measured using a ViewLux plate reader. For the luciferase reporter assay, 4 μl ONE-Glo reagent (Promega) was added to each well and luminescence signal was measured using ViewLux plate reader after 30 min incubation at room temperature. Data were expressed as relative fluorescence units (cell viability assay) and relative luminescence units (luciferase reporter assay).
2.6.2. RAR-luc assay:
The use of C3RL4 cells (passage 8) stably transfected with the retinoic acid response element (RARE) in qHTS screening was previously reported.29 The cells were seeded at 1000 cells/well/4 μl culture medium in 1536-well white-solid plates using a Multidrop Combi. After assay plates were incubated at 37°C and 5% CO2 overnight, 23 nL of compounds were transferred to the assay plates by Wako Pintool station, followed by addition of 1 μL if 1.0 μM retinol using a BioRaptr FRD. The assay plates were incubated for 6 h. To multiplex the cell viability assay with reporter-gene readout, 1 μl CellTiter-Fluor reagent was added into each well of the assay plates using a BioRaptr FRD and the assay plates were incubated for 1 h at 37°C before measuring luciferase reporter activity. Fluorescence intensity was measured using a ViewLux plate reader. For luciferase reporter assay, 4 μl of ONE-Glo reagent was added to each well, and luminescence signal was measured through a ViewLux plate reader after 30 min incubation at room temperature. Data were expressed as relative fluorescence units (cell viability assay) and relative luminescence units (luciferase reporter assay).
2.6.3. SHH-luc assay:
3T3 Gli1-luc cells (passage 7) were seeded at density of 2000 cells per well in 4 μL of assay medium into a 1536-well tissue culture treated white plates using a Multidrop Combi. The assay plates were incubated for 4 h at 37°C for cell attachment, and then followed by the transfer of 23 nL test compounds by a Wako Pintool station. To screen for SHH signaling inhibitors, 1 μl of conditioned medium was added using a Bioraptr, FRD and the assay plates were then incubated for 24 h. To multiplex the cell viability assay with reporter-gene readout, 1 μl of CellTiter-Fluor reagent was added into each well of the assay plates using BioRaptr FRD after 23 h of compound treatment. The assay plates were further incubated for additional 1 h at 37°C. Fluorescence intensity was measured using a ViewLux plate reader (Perkin Elmer, Waltham, MA). Then 4 μl of Amplite Luciferase reporter gene assay reagent (AAT Bioquest, Sunnyvale, CA) was added to each well of the assay plates. After 30 min incubation at room temperature, the luminescence intensity was measured using a ViewLux plate reader. Data were expressed as relative fluorescence units (cell viability assay) and relative luminescence units (luciferase reporter assay).
2.7. Data analysis:
qHTS data analysis.
Data normalization and concentration-response curve fitting for the data from the qHTS screening and follow up studies were performed as previously described.30 Briefly, raw plate reads for each titration point were first normalized relative to the positive control compound and 32 DMSO-only wells (0.25 ng/μl human TGFβ−1 = 0%, DMSO = −100%) as follows: % Activity = ((Vcompound − VDMSO)/(Vpos − VDMSO)) ×100, where Vcompound denotes the compound well values, Vpos denotes the median value of the positive control wells, and VDMSO denotes the median values of the DMSO-only wells (final concentrations ranging from 0.38 to 0.45%), and then corrected by applying a NCATS in-house pattern correction algorithm using compound-free control plates (i.e., DMSO-only plates) at the beginning and end of the compound plate stack.31 Concentration-response titration points for each compound were fitted to a four-parameter Hill equation yielding concentrations of half-maximal inhibitory activity (IC50) and maximal response (efficacy) values.32
Compounds were designated as Class 1–4 according to the type of concentration-response curve observed.33, 34 Each compound was assigned an activity outcome, inactive, active or inconclusive, based on the type of concentration response curve and reproducibility (3 independent runs) as described previously33 and summarized in Table S3. Each replicate curve is first assigned an activity class based on its curve class as follows: inactive (class 4), active antagonist (class −1.1, −2.1; class 5 due to super potency (IC50 < lowest test concentration), antagonist (class −1.2, −2.2), inconclusive antagonist (all other cases). Each activity class is then assigned a score: active antagonist (−3), antagonist (−2), inconclusive antagonist (−1), inactive (0). The pair-wise activity score differences for all replicate curves of each sample are then averaged and the % of inactive calls for the sample calculated to determine the reproducibility call: active match (average score difference <1.1, %inactive call <25 %), inactive match (average score difference <1.1, %inactive call >50 %), mismatch (average score difference >2.5), inconclusive (all other cases). Compounds with active or inactive match calls are considered reproducible.
2.8. Chemical structure cluster analysis.
The Tox21 10K compound collection was first grouped into 1014 clusters based on structural similarity (9242-bit fingerprints, Leadscope®, Columbus, OH, USA) using the self-organizing map (SOM) algorithm.35, 36 Each cluster was evaluated for its enrichment of active antagonists and significance of enrichment was determined by the Fisher’s exact test (p<0.01).
2.9. Compound management.
The Tox21 10K compound collection was dissolved in DMSO and stored at −80°C. Three sets of the Tox21 10K compound collection was made as replicates. Each copy of a compound was located in the different plate location. The primary screening against three sets of the compounds were run independently. When screening started, the plates were kept in the room temperature for 4 months after thawing. Quality control testing assess the compound stability and reported in https://tripod.nih.gov/tox21/samples.
3. Results
3.1. SBE-bla assay optimization:
The SBE-bla assay was used to measure the activation of SMAD2/3/4 signaling. As shown in Figure 2A, all three native TGFR ligands (TGFβ1, TGFβ2, and TGFβ3) triggered activity in the SBE-bla assay with effects that were both concentration-dependent and similar potency (EC50 = 0.11 ng/mL). Accordingly, we chose antagonism of TGFβ1-induced SBE-bla activity to indicate potential inhibition of TGFβ signaling. To identify chemicals and drugs that inhibit TGFβ/SMAD signaling, we screened the Tox21 10K compound library in an SBE-bla antagonist mode against stimulation by 0.25 ng/mL (EC85) TGFβ1 multiplexed with a cell viability assay (CellTiter-Glo). Compounds that were reproducibly active in both the ratio of 460/530 nm and 460 nm readouts and not apparently cytotoxic (meaning the IC50 value is significantly more potent (at least 3-fold; p< 0.05) in the SBE-bla assay than the IC50 value in the viability assay) in the primary screen were selected for confirmation and follow-up studies.
Figure 2.

Effects of agonist and antagonist of SBE-bla assay in primary screening. SBE-bla activity of natural ligand TGFβ1 (A), reported TGFβ/SMAD inhibitors (B), and potential TGFβ/SMAD inhibitors (C), were plotted in concentration-response curves. Data were expressed as mean ± SD from three independent experiments.
3.2. qHTS performance and reproducibility:
The primary screening tested 7872 unique compounds at 15 concentrations ranging from 0.98 nM to 77 μM in three independent runs (n=3). To evaluate qHTS performance, a set of parameters were calculated as described previously.37 For SBE-bla assay screening, the signal to background (S/B) ratio was 2.89 ± 0.22, the coefficient of variation (CV) of DMSO vehicle control was 4.66 ± 1.16, and the average Z′ factor from 408 plates of DMSO vehicle control and TGFβ1 across all the plates was 0.76 ± 0.03. The Z’ was calculated based on the standard deviation (SD) and mean of negative (vehicle control) and positive control (TGFβ) wells in each plate. This indicated excellent performance for a large-scale screen. Cell viability assayed simultaneously yielded an S/B ratio of 166.80 ± 4.64 with CV of 7.18 ± 0.68 and Z’ factor of 0.79 ± 0.03, again consistent with excellent results. The qHTS reproducibility parameters in Table 1 are described in the data analysis section. The activity for each compound from three independent runs was defined as an active, inconclusive, or inactive based on curve classes. It showed that the three independent runs reproduced well; no mismatch was found.
Table 1.
qHTS reproducibility parameters. See Methods Section 2.7 for description of detailed criteria for assigning compounds to each reproducibility category.
| Assay names | Reproducibility | |||
|---|---|---|---|---|
| Active match | Inactive match | Inconclusive | Mismatch | |
| qHTS SBE-bla assay | 0.57% | 98.82% | 0.61% | 0.00% |
| qHTS viability assay | 2.75% | 94.53% | 2.72% | 0.00% |
During the primary screening of the Tox21 10K compound library, each compound was tested with SBE-bla assay multiplexed with cell viability assay in HEK293T cells. Data reproducibility was measured by the percentage of active match, inactive, mismatch and inconclusive compounds. As every compound in the Tox21 10K collection was tested three or more times with various activity outcomes, we assessed the reproducibility of these activity calls. “Active match” or “inactive match” means that most of the calls were active or inactive. “Mismatch” means that most of the calls were contradictory, e.g., half of the calls were active and half inactive. For cases where the discrepancies among the calls were not large enough to meet the “mismatch” criteria, these were grouped into the “inconclusive” category.
3.3. Identification of SBE-bla antagonists:
The primary screen identified 68 compounds that potently inhibited TGFβ1-induced SBE-bla activity with efficacy > 50% and IC50 < 10 μM. Re-testing in SBE-bla HEK 293T cells using in-house compound stock solutions from the existing Tox21 10K library confirmed 65 of 68 (95%) actives which are listed in Table S1. Note that these compounds are mainly anti-cancer drugs and pesticides. Concentration response curves of several known TGFβ/SMAD pathway inhibitors are shown in Figure 2B, such as the cyclin-dependent kinase inhibitor flavopiridol hydrochloride (IC50 at 0.02 μM) and the SRC/ABL inhibitor dasatinib (IC50 at 0.26 μM). We identified several potential novel TGFβ/SMAD actives such as ziram and PharmaGSID_48519, with IC50 values of 0.52 μM and 0.57 μM, respectively (Figure 2C).
The Tox21 10K compounds were grouped into clusters based on structural similarity using a self-organizing map (SOM). A heatmap was generated to show the enrichment of SBE-bla antagonists in each structural class (Figure 3A). Among the structural clusters containing active SBE-bla antagonists in the Tox21 10K compound collection, a cluster of dithiocarbamates (DTCs, cluster k35.18) is the most significantly enriched (p<10−20) with 10 selected active SBE-bla antagonists reported in Figure 3B. We sub-categorized the DTCs based on the type of metal ion, R- group and the number of sulfides in the molecule. As shown in Figure 3B, dialkyl dithiocarbamate metal that complexes with different metal ions (M), and tetraalkyl thiuram disulfides/polysulfides that have different R-groups both exhibited different IC50 and efficacy values in the SBE-bla assay. Among the dialkyl dithiocarbamate metal complexes, the ones with heavy metals and higher valent metals were more potent in blocking TGFβ1-induced SBE-bla activity. For example, ferbam, ziram, copper dimethyl dithiocarbamates and sodium dimethyl dithiocarbamate share the same organic component, but different metal ions, i.e., Fe3+, Zn2+, Cu2+ and Na+, respectively. The rank order of potency (based on IC50 value) is Fe3+ (0.05 μM) >Zn2+ (0.66 μM) > Cu2+ (1.62 μM) > Na+ (9.33 μM).
Figure 3.
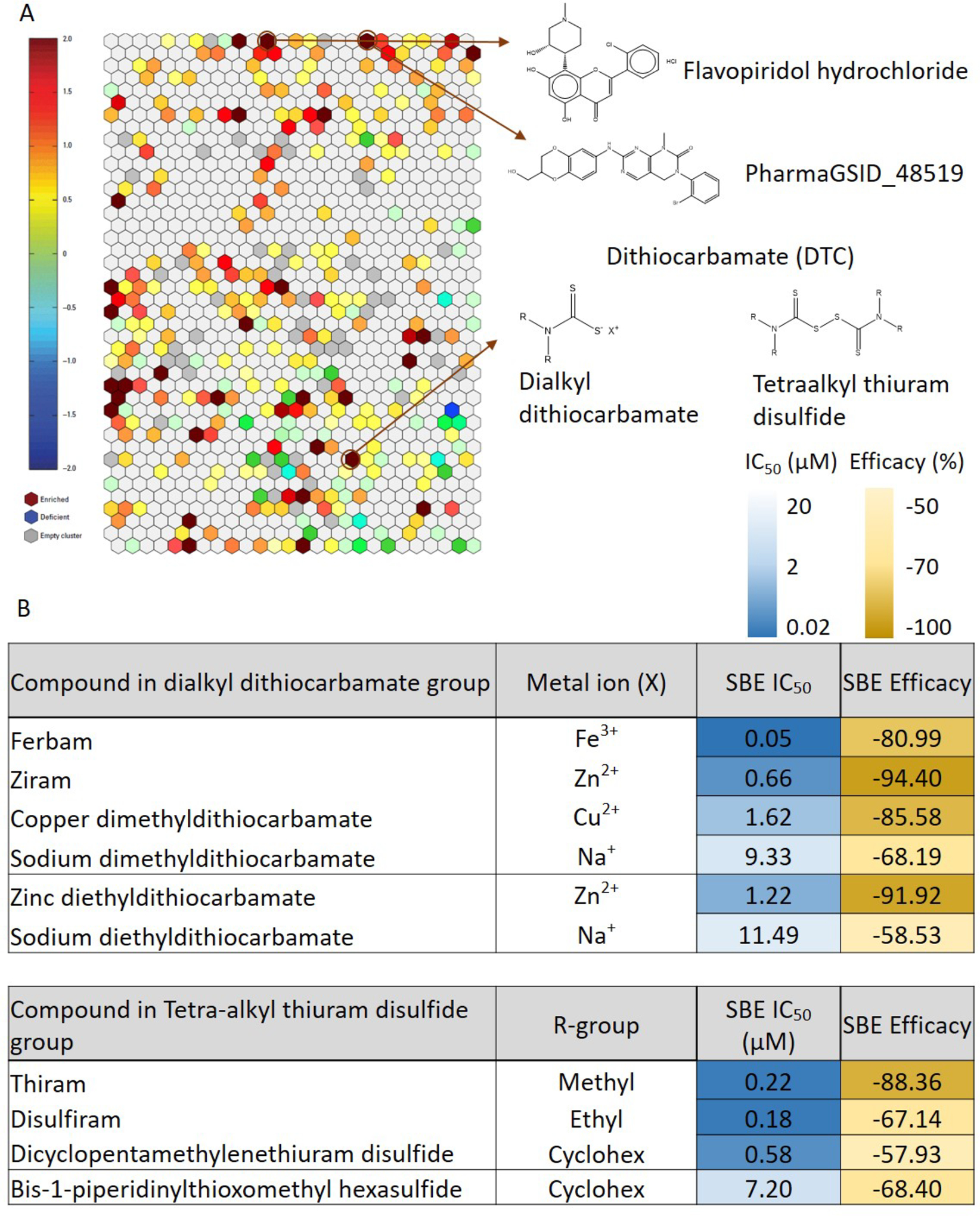
(A) Compound structural clustering of SBE-bla antagonist activity. Each cluster was screened in qHTS SBE-bla assay. Clusters were colored according to the enrichment of active compounds which is calculated by -log p-value. Some clusters were highlighted to represent the compounds of interest in follow up study. (B) Differential inhibitory effects of DTCs in SBE-bla activity. Ten SBE-bla antagonists in cluster k35.18 were sub-categorized in to (1) dialkyl dithiocarbamate complexed with metal ion and (2) tetraalkyl thiuram disulfide/polysulfide have different R-groups. Their IC50 and efficacy values in SBE-bla assay were colored based on potencies and efficacies.
Tetraalkyl thiuram disulfide was the most potent inhibitor in DTCs. Thiram, disulfiram and dicyclopentamethylenethiuram blocked more than 50% of TGFβ1-induced SBE-bla activity at <1 μM. The size of the R group seemed to have an effect on compound potency, such that compounds with the larger polysulfide group were less potent in inhibiting SBE-bla activity. For example, bis-1-piperidinylthioxomethyl hexasulfide (IC50 = 7.2 μM) was 12-fold less potent than its disulfide counterpart, dicyclopentamethylenethiuram (IC50 = 0.58 μM). To further investigate their structure-activity relationship, the SBE-bla antagonists in the DTC cluster were selected for follow-up targeted studies.
3.4. Inhibition of SMAD3 phosphorylation:
To investigate the effect of potential SBE-bla antagonists on inhibition of downstream SMAD3 phosphorylation, intracellular pSMAD3 (Ser 423/425) level was measured using HTRF technology. HepG2 cells were co-treated with 0.15 ng/mL TGFβ1 and test compound for 1 h. As shown in Figure 4A, pSMAD3 level was decreased upon treatment of dasatinib38 or cantharidin39, which are known inhibitors in TGFβ1-induced SMAD3 phosphorylation. The novel SBE antagonists identified here reduced TGFβ1-induced pSMAD3 level with potency similar to dasatinib. For example, at a concentration of 15 μM, pharmaGSID _48519 eliminated TGFβ1-elevated pSMAD3 levels; and ziram inhibited SMAD3 phosphorylation with an IC50 at 22 μM. Interestingly, a group of organometallics decreased the pSMAD3 level (Figure 4B). The most potent example is phenylarsine oxide, with an IC50 value of 5 μM and complete abolishment of TGFβ’s effect around 10 μM. None of the compounds that reduced pSMAD3 levels had cytotoxicity at the measure time point.
Figure 4.

Inhibitory effect of SBE-bla antagonists on SMAD3 phosphorylation in HepG2 cells. HepG2 cells were lysed after 1 h co-treatment of TGFβ1 and test compound. The phosphorylated SMAD3 level was measured by using HTRF technology. Inhibition of TGFβ1-induced SMAD3 phosphorylation were concentration dependent upon the treatment of known TGFβ/SMAD inhibitors (A) and metal-organics (B). Data are expressed as mean ± SD from three independent experiments.
3.5. Cytotoxic effects of potential SBE antagonists on human embryonic H9-derived neural stem cells (NSCs):
The primary and confirmatory screens identified 65 SBE antagonists independent of cytotoxic effect in HEK293T cells. A counter screen was performed on NSC cell viability. IC50 values of viability in HEK293T cells (SBE viability) and NSCs were listed in Table S1. Among the 65 SBE-bla antagonists, 36 did not exhibit cytotoxicity in SBE-bla HEK 293T cells after 5 h compound treatment. However, 16 out of 35 showed cytotoxicity in NSCs after 5 h treatment, and 33 of them showed cytotoxicity after 24 h treatment (Table S1). Compared to iPSC-derived neurons, NSCs appeared to be more sensitive to cytotoxic effects due to their active proliferation state. For example, several kinase inhibitors (dasatinib, flavopiridol hydrochloride, pp242, PharmaGSID_48519) invoked NSC cytotoxicity at 24 h, but not at 5 h. Additionally, nine organometallics (dicyanoaurate, phenylmercuric acetate/lactate, phenylarsine oxide, triphenyltin fluoride, dibutyltin and zinc pyrithione) had cytotoxic effects in NSCs but were 10-fold less cytotoxic in SBE-bla HEK 293T cells. Among these organometallics, phenylarsine and phenylmercuric and dicyanoaurate inhibited TGFβ1-elevated SMAD3 phosphorylation (Figure 4). The cytotoxicity in NSCs motivated follow-up studies related to developmental or neurotoxicity.
3.6. Inhibitory effects of SBE-antagonists in WNT, SHH and RAR pathways:
Regarding NSC cytotoxicity of SBE antagonists in NSCs, their potential toxicity was evaluated through profiling developmental-related pathways using three different luciferase reporter gene assays.
For the DTCs, we found similar inhibition patterns across WNT, RAR and SHH pathways (blue box, Figure S1). DTCs are organosulfur compounds represented by a general structure [(R1R2)N-(C=S)-S]n-M, where R can be substituted by an alkyl group, and M usually by a metal ion. Since differences in the R-group and metal ion affected SBE-bla activity (Figure 3B), we sub-grouped these DTCs by either metal ion (Table 2) or R group (Table 3). As shown in Table 2, dimethyldithiocarbmates complexed with higher oxidization state metal ions demonstrated more potent and efficacious inhibition of TGFβ, WNT, RAR, and SHH pathways. Among them, ferbam (Fe3+) was the most potent in inhibiting all pathways with IC50 values of 0.05 μM, 0.57 μM, 0.83 μM and 0.05 μM in SBE-bla, WNT-luc, RAR-luc, and SHH-luc assays, respectively. We also compared outcomes in developmental pathways based on the size of R group (Table 3). Sodium dimethyldithiocarbamate inhibited all developmental pathways at 10 μM while sodium dibutyldithiocarbamate was inactive. The rank order of general inhibition effects in WNT, RAR and SHH pathways were in accordance with their potency in inhibiting SBE-bla activity (Table 3). In addition, most of the anti-cancer drugs inhibited SBE and RAR pathways, but not SHH or WNT pathways (purple box, Figure S1). These compounds inhibited SBE and RAR reporter gene activities at concentrations that did not compromise cell viability, as evidenced by the comparison of the IC50 values of reporter gene assay and viability assay resulting in a ratio greater than 3.
Table 2.
Outcomes of dimethyldithiocarbamates complexed with different metals in SBE-bla, WNT-, RAR-, SHH-luc assays.
| Sample Name | CASRN | Structure | SBE Antagonist | WNT Antagonist | RAR Antagonist | SHH Antagonist | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | Efficacy (%) | IC50 (μM) | Efficacy (%) | IC50 (μM) | Efficacy (%) | IC50 (μM) | Efficacy (%) | |||
| Ferbam | 14484-64-1 | 0.05 | −80.99 | 0.57 | −66.42 | 0.83 | −93.29 | 0.05 | −87.88 | |
| Ziram | 137-30-4 |  |
0.52 | −93.60 | 0.85 | −81.79 | 1.39 | −106.31 | 0.42 | −92.50 |
| Sodium dimethyldithiocarbamate | 128-04-1 | 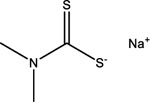 |
9.14 | −68.19 | Inactive | Inactive | 17.37 | −93.31 | 6.73 | −87.30 |
Potency (IC50) and efficacy (activity %) in inhibiting TGFβ WNT, RAR, and SHH pathways by ferbam, ziram, and sodium dimethyldithiocarbamates were compared in the table.
Table 3.
Outcomes of Na-DTCs with different size of alkyl group in SBE-bla, WNT-, RAR-, SHH-luc assays.
| Sample Name | CASRN | Structure | SBE Antagonist | WNT Antagonist | RAR Antagonist | SHH Antagonist | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | Efficacy (%) | IC50 (μM) | Efficacy (%) | IC50 (μM) | Efficacy (%) | IC50 (μM) | Efficacy (%) | |||
| Sodium dimethyldithiocarbamate | 128-04-1 | 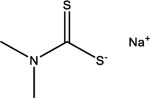 |
9.14 | −68.19 | Inactive | Inactive | 17.37 | −93.31 | 6.73 | −87.30 |
| Sodium diethyldithiocarbamate | 148-18-5 | 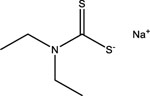 |
11.49 | −58.53 | 3.69 | −74.76 | 15.31 | −113.16 | 8.92 | −96.63 |
| Sodium dibutylcarbamodithioate | 136-30-1 | 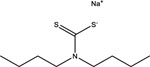 |
Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
Potency (IC50) and efficacy (activity %) in inhibiting TGFβ WNT, RAR, and SHH pathways by sodium dimethyl-, diethyl-, and dibutyl- dithiocarbamates were compared in the table.
3.7. Inhibition of neurite outgrowth by selected SBE antagonists:
To further explore potential neurotoxicity, selected SBE antagonists were tested in a neurite outgrowth assay. iPSC derived neurons were treated by 6 DTCs and 13 anti-cancer drugs for 24 h post-neurite extension. High-content imaging of neurite length and viable cell body relative to the control (DMSO) group are shown in Figure 5. Ziram and ferbam inhibited neurite outgrowth at 0.1 μM and were 18-fold more potent than sodium dimethyldithiocarbamate (Figure 5B); all three of these compounds share a similar structure in their organic moiety but differ in metal ion. A general pattern among organically-similar DTCs was decreasing IC50 values (increase of potency) for neurite inhibition with increasing valency in the metal ions. Differential inhibition of neurite outgrowth was also observed for DTCs having different R groups. For example, disulfiram and thiram had IC50 values of 0.18 and 0.1 μM, respectively, versus that for dicyclopentamethylenethiuram disulfide at 0.82 μM. The R group in the case of dicyclopentamethylenethiuram disulfide is dicyclohex versus thiram (R=methyl) and disulfiram (R=ethyl) that were 10-fold more potent. This pattern followed the structure-related activity of DTCs observed in the SBE-bla assay (Figure 3B), indicating that potency toward neurite outgrowth was dependent both on metal ion status and R group.
Figure 5.

Inhibitory effects of DTCs and anti-cancer drugs on neurite outgrowth. Neurite length is measured by maximum length method. The number of cells are counted as object number according to the shape of cell body. Neurons treated with DTCs and anti-cancer drugs for 24 h showed inhibition of neurite outgrowth. (A) Representative images of ferbam and thiram induced-neurite outgrowth inhibition. (B) Differential inhibitory effects of DTCs in neurite outgrowth and object number. Dialkyl dithiocarbamates with different metal ions, and tetra-alkyl thiuram disulfide with different R-group were compared separately. (C) Representative images of anti-cancer drugs-induced neurite outgrowth inhibition. (D) Differential inhibitory effects of anti-cancer drugs in neurite outgrowth assay and object number. Data from (B) and (D) are expressed as mean ± SD from three experiments.
Among anti-cancer drugs, flavopiridol hydrochloride, canertinib, 5-Azacitidine, idarubicin hydrochloride, vinblastine sulfate, bortezomib, pharmaGSID_48505, and nemorubicin inhibited neurite outgrowth (Figure 5D). Surprisingly, dasatinib, pharmaGSID_48519 and pp242 did not inhibit neurite outgrowth in iPSC-derived neurons despite their activity on the SBE-bla assay.
4. Discussion
Normal biological development requires specific signaling pathways that regionalize and pattern the growth of tissue. Many pathways including TGFβ, WNT, BMP, hedgehog, RAR and Notch-delta were identified as essential signaling in development and they were conserved from model organisms, laboratory animals to humans.40, 41 The current study used the SBE-bla assay as a first-tier screen to determine whether a chemical has the potential to inhibit TGFβ1-induced SMAD2/3/4 signaling. TGFβ signaling plays an important role in early (before organogenesis and cytodifferentiation) and later development (tissue growth and renewal).42, 43 For example, disruption of TGFβ signaling leads to craniofacial defects44 or embryolethality.45 TGFβ is also essential for the survival of embryonic dopaminergic neurons and survival of embryonic dopaminergic neurons.46, 47 Our results showed the following main findings: (1) several kinase inhibitors, organometallic compounds, and DTCs were found across the Tox21 library to inhibit TGFβ1-induced SMAD signaling independent of cytotoxicity; (2) counter-screen of SBE antagonists demonstrated differential cytotoxicity on human NSCs providing evidence for potential developmental toxicity; (3) SBE antagonist profiles showed potential effects on other key developmental signaling pathways, including WNT, RAR, and SHH; and (4) some but not all compounds with inhibitory effects on the SBE assay also impaired neurite outgrowth. These results show that the SBE-bla assay is an effective qHTS approach for identifying chemicals that disrupt TGFβ/SMAD signaling in a manner relevant to predicting a potential developmental hazard.
The qHTS SBE-bla platform identified both known and previously unreported inhibitors of SMAD-dependent TGFβ1 signaling. For example, a failed drug, pharmaGSID_48519, which was initially designed as a kinase inhibitor, inhibited SMAD3 phosphorylation and consequently directly blocked TGFβ1 signaling through its cognate receptor (ALK5). Flavopiridol hydrochloride (a pan-CDK/GSK3 inhibitor), pp242 (a mTOR inhibitor) and dasatinib (a BCR-ABL inhibitor) inhibited TGFβ1-induced SBE-bla activity at a similar concentration as reported in Mv1Lu cells16, normal fibroblast48, panc-1 and colo-357 cells, respectively.49 Dasatinib and flavopiridol hydrochloride’s inhibition on TGFβ1’s effect has been reported as a treatment or for prevention of pathophysiologic conditions caused by excessive TGFβ activity, such as cancer38, pulmonary fibrosis50 or atherosclerosis.51 In the current toxicology study, dasatinib, pp242 and pharmaGSID_48519 were cytotoxic in NSCs after culturing for 24 h but didn’t inhibit neurite outgrowth. These results point to a potential effect on developmental toxicity, but not via a mechanism involving impact on neurite outgrowth.
Some organometallic environmental chemicals were active in the SBE-bla and pSMAD3 assays with broad toxicity observed in NSCs as well. The mechanism of organometallic chemicals’ inhibition in TGFβ signaling remains unknown. It has been reported that phenylarsine oxide inhibited internalization of TGFR and limited the access of activated receptor to intracellular substrates, which may explain the inhibition in TGFβ signaling.52 Organometallics has been used as anticancer agents because of their inhibition in protein kinases.53 Therefore, they may inhibit SMAD phosphorylation which is required for SBE-bla activity. Transition metals are generally toxic due to oxidative stress.54-42 Heavy metals such as mercury, lead, and arsenic lead to an alteration of neurogenesis, resulting in motor and cognitive function deficits.55, 56 Perhaps adverse effects on TGFβ1 signaling may contribute to neuroinflammation57 and neurological disorders.58
DTCs are used as fungicides, accelerators in the rubber industry, animal repellants, and biocides in many household products. Previous studies have suggested that DTCs were neurotoxic at different stages of life and affected multiple organs.59 In our study, the rank order of DTC-induced inhibition in WNT-, RAR and SHH-luc assays were associated with a reduction in SBE-bla activity. Thiram, disulfiram, ziram, and ferbam were the most potent actives in the SBE-bla assay, and showed a similar trend inhibiting SHH, WNT and RAR pathways. These results are consistent with existing evidence about their broad developmental toxicity, including embryo-fetal toxicity (e.g., thiram and disulfiram induced-skeletal malformations in rats, human, hamster and rabbits, and cleft palate in mice);60–62 neurodevelopmental defects (e.g., ferbam-induced hydrocephalus in rat development and thiram-induced anophthalmia and microphthalmia);63 and peripheral neuropathy effects (e.g., ziram induced-spinal cord histopathology in rat, and thiram-induced degeneration of ventral horn and lower lumbar region of spinal cord).64
With respect to redox status65, the pattern observed was that less stable DTCs show a stronger inhibition of TGFβ/SMAD signaling. This may be explained by metal ionization state and/or increasing R group size. For the former, DTCs complexed with transition metals, such as Zn2+ (ziram), and Fe3+ (ferbam), were more effective in the inhibition of SBE-bla activity than the Na+ salt. Under physiological conditions, most DTCs reoxidize to thiuram disulfide. Higher valent heavy metals, such as Zn2+ or Fe3+, are relatively unstable and can be easily more readily oxidized to thiuram disulfide. Thiuram disulfides can then react with other molecules to form thiol groups suppressing a ‘cysteine knot’ in the SMAD2/3/4 complex, resulting in dysregulation of TGFβ downstream targets. As to the R-group pattern, inhibition of SBE-bla activity by the DTC series declined with longer alkyl chains. Steric hindrance from large alkyl groups against reoxidization may decrease their ability to generate isothiocyanates, which are believed to be responsible for the antifungal activity and general inhibition of developmental pathways. Subsequent studies showed that the mutagenicity of DTCs also followed this pattern.66 Rank order potency of DTCs on WNT-, RAR-, and SHH-luc assays in the present study also followed this pattern. This can help explain their potential to cause developmental effects through the impact of complexed metals or alkyl groups. We speculate that the ability to form thiuram disulfides may be critical to the differential neuropathic effects of DTCs.
Current prenatal toxicity testing guidelines monitor the potential action of toxicants at many levels of normal development and assess disruption of overall processes of developmental, reproductive, and teratogenic effects of pharmaceuticals, food additives, and pesticides.67–69 Faster, inexpensive high throughput in vitro assays, such as the SBE-bla assay, could be used as a quantitative high-throughput (qHTS) screen to identify chemicals for developmental toxicity assessment. For example, 15 SBE-bla antagonists in the Tox21 SBE-bla assay have been reported to downregulate TGFβ protein levels as determined by ELISA in a ToxCast Bioseek study (noted with * in Table S1).70 The concordance of downregulation of TGFβ expression and inhibition of TGFβ/SMAD signaling showed associations in these in vitro endpoints, which suggests TGFβ perturbation by chemicals such as ziram, thiram, disulfiram may be predictable through cheminformatics analysis. We recognize the identification of chemical-induced TGFβ1 blockage effects on reporter gene assay will not necessarily predict specific developmental or neurotoxicity. Since TGFβ subtypes are indistinguishable in most biological assays with cell lines cultured in vitro, it is expected the SBE-bla assay responds to TGFβ’s −1,− 2, −3 non-differentially. Therefore, it is important to conduct follow-up tests that measure structurally similar chemicals in functional assays to maximize the probability of detecting neurotoxicity. Further experiments can be done in more biologically complex functional assays involving neurobehavioral, neurochemical, and neuropathologic measurement. For developmental toxicants in general, the cell viability results in hESC-derived NSC offers a potential counter-screen. Because some but not all SBE inhibitors impaired neurite outgrowth in human neurogenic cells derived from H9 embryonic stem cells, the modeling the impact of TGFβ/SMAD, WNT, RAR, and SHH pathways provides a pragmatic means to address developmental toxicity (and potential neurotoxicity) at a systems level.
Supplementary Material
Acknowledgements
This work was supported in part by the Intramural Research Program of the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH) and the interagency agreement IAG #NTR 12003 from the National Institute of Environmental Health Sciences/Division of the National Toxicology Program to the NCATS, NIH. The authors thank Xiuli Huang for helping to optimize SMAD3 phosphorylation assay.
Footnotes
Publisher's Disclaimer: Disclaimer: The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency, the National Institute of Environmental Health Sciences, the National Center for Advancing Translational Sciences, National Institutes of Health, or the United States government. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Supporting Information
Table S1: 65 compounds in 4 reporter gene assays related with development (SBE-bla, WNT-, RAR-, SHH-luc) and pSMAD assay, and cell viability assay on SBE-HEK 293T and NSCs; Table S2: chemicals in enriched clusters from primary screening of SBE-bla assay; Table S3: reference to assign curve classes and activity category as described in method section 2.7. Figure S1: inhibitory effects on SBE, WNT, RAR, and SHH pathways and cytotoxicity of 65 confirmed SBE antagonists.
References
- 1.Wu MY; Hill CS, Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell 2009, 16 (3), 329–43. [DOI] [PubMed] [Google Scholar]
- 2.Linask KK; D’Angelo M; Gehris AL; Greene RM, Transforming growth factor-beta receptor profiles of human and murine embryonic palate mesenchymal cells. Exp Cell Res 1991, 192 (1), 1–9. [DOI] [PubMed] [Google Scholar]
- 3.Massagué J, TGFβ signalling in context. Nature reviews Molecular cell biology 2012, 13 (10), 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng X-H; Derynck R, Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol 2005, 21, 659–693. [DOI] [PubMed] [Google Scholar]
- 5.Lucarelli P; Schilling M; Kreutz C; Vlasov A; Boehm ME; Iwamoto N; Steiert B; Lattermann S; Wasch M; Stepath M; Matter MS; Heikenwalder M; Hoffmann K; Deharde D; Damm G; Seehofer D; Muciek M; Gretz N; Lehmann WD; Timmer J; Klingmuller U, Resolving the Combinatorial Complexity of Smad Protein Complex Formation and Its Link to Gene Expression. Cell Syst 2018, 6 (1), 75–89 e11. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y; Massague J, Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113 (6), 685–700. [DOI] [PubMed] [Google Scholar]
- 7.Araujo AP; Diniz LP; Eller CM; de Matos BG; Martinez R; Gomes FC, Effects of Transforming Growth Factor Beta 1 in Cerebellar Development: Role in Synapse Formation. Front Cell Neurosci 2016, 10, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Praag H; Schinder AF; Christie BR; Toni N; Palmer TD; Gage FH, Functional neurogenesis in the adult hippocampus. Nature 2002, 415 (6875), 1030–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millan FA; Denhez F; Kondaiah P; Akhurst RJ, Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development 1991, 111 (1), 131–43. [DOI] [PubMed] [Google Scholar]
- 10.Gehris AL; Greene RM, Regulation of murine embryonic epithelial cell differentiation by transforming growth factors beta. Differentiation 1992, 49 (3), 167–73. [DOI] [PubMed] [Google Scholar]
- 11.Seth B; Yadav A; Agarwal S; Tiwari SK; Chaturvedi RK, Inhibition of the transforming growth factor-β/SMAD cascade mitigates the anti-neurogenic effects of the carbamate pesticide carbofuran. Journal of Biological Chemistry 2017, jbc. M117. 798074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Boxtel AL; Pieterse B; Cenijn P; Kamstra JH; Brouwer A; van Wieringen W; de Boer J; Legler J, Dithiocarbamates induce craniofacial abnormalities and downregulate sox9a during zebrafish development. Toxicol Sci 2010, 117 (1), 209–17. [DOI] [PubMed] [Google Scholar]
- 13.Mercado-Pimentel ME; Runyan RB, Multiple transforming growth factor-beta isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs 2007, 185 (1–3), 146–56. [DOI] [PubMed] [Google Scholar]
- 14.Dunn NR; Vincent SD; Oxburgh L; Robertson EJ; Bikoff EK, Combinatorial activities of Smad2 and Smad3 regulate mesoderm formation and patterning in the mouse embryo. Development 2004, 131 (8), 1717–1728. [DOI] [PubMed] [Google Scholar]
- 15.Wilkinson P Functional Adenosine Receptor 2B Drives Profibrotic and ProinflammatorySignaling in Renal Fibroblasts Through Modulation of TGFβ Receptor Signaling. University of the Sciences in Philadelphia, 2017. [Google Scholar]
- 16.Wang G; Matsuura I; He D; Liu F, Transforming growth factor-{beta}-inducible phosphorylation of Smad3. J Biol Chem 2009, 284 (15), 9663–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chin J; Adams AD; Bouffard A; Green A; Lacson RG; Smith T; Fischer PA; Menke JG; Sparrow CP; Mitnaul LJ, Miniaturization of cell-based beta-lactamase-dependent FRET assays to ultra-high throughput formats to identify agonists of human liver X receptors. Assay Drug Dev Technol 2003, 1 (6), 777–87. [DOI] [PubMed] [Google Scholar]
- 18.Rosso SB; Inestrosa NC, WNT signaling in neuronal maturation and synaptogenesis. Frontiers in cellular neuroscience 2013, 7, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bielen H; Houart C, The Wnt cries many: Wnt regulation of neurogenesis through tissue patterning, proliferation, and asymmetric cell division. Developmental neurobiology 2014, 74 (8), 772–780. [DOI] [PubMed] [Google Scholar]
- 20.Niederreither K; Dollé P, Retinoic acid in development: towards an integrated view. Nature Reviews Genetics 2008, 9 (7), 541. [DOI] [PubMed] [Google Scholar]
- 21.Duester G, Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134 (6), 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das BC; Thapa P; Karki R; Das S; Mahapatra S; Liu T-C; Torregroza I; Wallace DP; Kambhampati S; Van Veldhuizen P, Retinoic acid signaling pathways in development and diseases. Bioorganic & medicinal chemistry 2014, 22 (2), 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zieger E; Schubert M, New insights into the roles of retinoic acid signaling in nervous system development and the establishment of neurotransmitter systems In International review of cell and molecular biology, Elsevier: 2017; Vol. 330, pp 1–84. [DOI] [PubMed] [Google Scholar]
- 24.Choy SW; Cheng SH, Hedgehog signaling In Vitamins & Hormones, Elsevier: 2012; Vol. 88, pp 1–23. [DOI] [PubMed] [Google Scholar]
- 25.Álvarez-Buylla A; Ihrie RA In Sonic hedgehog signaling in the postnatal brain, Seminars in cell & developmental biology, Elsevier: 2014; pp 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirschen GW; Xiong Q, Primary cilia as a novel horizon between neuron and environment. Neural regeneration research 2017, 12 (8), 1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai S; Li R; Long Y; Titus S; Zhao J; Huang R; Xia M; Zheng W, One-Step Seeding of Neural Stem Cells with Vitronectin-Supplemented Medium for High-Throughput Screening Assays. J Biomol Screen 2016, 21 (10), 1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia M; Huang R; Shi Q; Boyd WA; Zhao J; Sun N; Rice JR; Dunlap PE; Hackstadt AJ; Bridge MF, Comprehensive Analyses and Prioritization of Tox21 10K Chemicals Affecting Mitochondrial Function by in-Depth Mechanistic Studies. Environmental health perspectives 2018, 126 (7), 077010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y; Sakamuru S; Huang R; Reese DH; Xia M, Identification of compounds that modulate retinol signaling using a cell-based qHTS assay. Toxicology in Vitro 2016, 32, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang R, A Quantitative High-Throughput Screening Data Analysis Pipeline for Activity Profiling. Methods Mol Biol 2016, 1473, 111–22. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y; Huang R, Correction of Microplate Data from High Throughput Screening In High-Throughput Screening Assays in Toxicology, 1 ed.; Zhu H; Xia M, Eds. Humana Press: 2016; Vol. 1473. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y; Jadhav A; Southal N; Huang R; Nguyen DT, A grid algorithm for high throughput fitting of dose-response curve data. Curr Chem Genomics 2010, 4, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang R, A Quantitative High-Throughput Screening Data Analysis Pipeline for Activity Profiling In High-Throughput Screening Assays in Toxicology, 1 ed.; Zhu H; Xia M, Eds. Humana Press: 2016; Vol. 1473. [Google Scholar]
- 34.Inglese J; Auld DS; Jadhav A; Johnson RL; Simeonov A; Yasgar A; Zheng W; Austin CP, Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A 2006, 103 (31), 11473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohonen T, Self-organizing neural projections. Neural Netw 2006, 19 (6–7), 723–33. [DOI] [PubMed] [Google Scholar]
- 36.Attene-Ramos MS; Miller N; Huang R; Michael S; Itkin M; Kavlock RJ; Austin CP; Shinn P; Simeonov A; Tice RR, The Tox21 robotic platform for the assessment of environmental chemicals–from vision to reality. Drug discovery today 2013, 18 (15–16), 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J-H; Chung TD; Oldenburg KR, A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of biomolecular screening 1999, 4 (2), 67–73. [DOI] [PubMed] [Google Scholar]
- 38.Bartscht T; Rosien B; Rades D; Kaufmann R; Biersack H; Lehnert H; Gieseler F; Ungefroren H, Dasatinib blocks transcriptional and promigratory responses to transforming growth factor-beta in pancreatic adenocarcinoma cells through inhibition of Smad signalling: implications for in vivo mode of action. Mol Cancer 2015, 14, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li K; Shi C; Huang J; Tang K, Cisplatin plus norcantharidin alter the expression of TGF-β1/Smads signaling pathway in hepatocellular carcinoma. Bratislavske lekarske listy 2017, 118 (2), 85–88. [DOI] [PubMed] [Google Scholar]
- 40.Grandjean P; Landrigan PJ, Developmental neurotoxicity of industrial chemicals. The Lancet 2006, 368 (9553), 2167–2178. [DOI] [PubMed] [Google Scholar]
- 41.National Research Council (U.S.). Committee on Developmental Toxicology.; National Research Council (U.S.). Commission on Life Sciences., Scientific frontiers in developmental toxicology and risk assessment. National Academy Press: Washington, DC, 2000; p xviii, 327 p. [PubMed] [Google Scholar]
- 42.McCartney-Francis NL; Mizel DE; Frazier-Jessen M; Kulkarni AB; McCarthy JB; Wahl SM, Lacrimal gland inflammation is responsible for ocular pathology in TGF-beta 1 null mice. The American journal of pathology 1997, 151 (5), 1281. [PMC free article] [PubMed] [Google Scholar]
- 43.Sanford LP; Ormsby I; Gittenberger-de Groot AC; Sariola H; Friedman R; Boivin GP; Cardell EL; Doetschman T, TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124 (13), 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greene RM; Pisano MM, Perspectives on growth factors and orofacial development. Curr Pharm Des 2004, 10 (22), 2701–17. [DOI] [PubMed] [Google Scholar]
- 45.Dickson MC; Martin JS; Cousins FM; Kulkarni AB; Karlsson S; Akhurst RJ, Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1995, 121 (6), 1845–54. [DOI] [PubMed] [Google Scholar]
- 46.Roussa E; und Halback O. v. B.; Krieglstein K, TGF-β in Dopamine Neuron Development, Maintenance and Neuroprotection In Development and Engineering of Dopamine Neurons, Springer: 2009; pp 81–90. [DOI] [PubMed] [Google Scholar]
- 47.Hegarty SV; Sullivan AM; O’Keeffe GW, Roles for the TGFβ superfamily in the development and survival of midbrain dopaminergic neurons. Molecular neurobiology 2014, 50 (2), 559–573. [DOI] [PubMed] [Google Scholar]
- 48.Chang W; Wei K; Rosen G, Suppression Of Lung Fibrosis By Dual MTOR Inhibitors. In B28. SCARRING IN THE LUNG: PRINCIPLES AND PERSPECTIVES, American Thoracic Society: 2011; pp A2718–A2718. [Google Scholar]
- 49.Huang R; Xia M; Cho MH; Sakamuru S; Shinn P; Houck KA; Dix DJ; Judson RS; Witt KL; Kavlock RJ; Tice RR; Austin CP, Chemical genomics profiling of environmental chemical modulation of human nuclear receptors. Environ Health Perspect 2011, 119 (8), 1142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanemaru R; Takahashi F; Kato M; Mitsuishi Y; Tajima K; Ihara H; Hidayat M; Wirawan A; Koinuma Y; Hayakawa D, Dasatinib Suppresses TGFβ-Mediated Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells and Inhibits Pulmonary Fibrosis. Lung 2018, 1–11. [DOI] [PubMed] [Google Scholar]
- 51.Rostam MA; Shajimoon A; Kamato D; Mitra P; Piva TJ; Getachew R; Cao Y; Zheng W; Osman N; Little PJ, Flavopiridol Inhibits TGF-β-Stimulated Biglycan Synthesis by Blocking Linker Region Phosphorylation and Nuclear Translocation of Smad2. Journal of Pharmacology and Experimental Therapeutics 2018, 365 (1), 156–164. [DOI] [PubMed] [Google Scholar]
- 52.Zwaagstra JC; El-Alfy M; O’Connor-McCourt MD, Transforming Growth Factor (TGF)-β1 Internalization MODULATION BY LIGAND INTERACTION WITH TGF-β RECEPTORS TYPES I AND II AND A MECHANISM THAT IS DISTINCT FROM CLATHRIN-MEDIATED ENDOCYTOSIS. Journal of Biological Chemistry 2001, 276 (29), 27237–27245. [DOI] [PubMed] [Google Scholar]
- 53.Zaki M; Hairat S; Aazam ES, Scope of organometallic compounds based on transition metal-arene systems as anticancer agents: starting from the classical paradigm to targeting multiple strategies. RSC advances 2019, 9 (6), 3239–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hogarth G, Transition metal dithiocarbamates: 1978–2003. Progress in inorganic chemistry 2005, 53, 71–561. [Google Scholar]
- 55.Jaako-Movits K; Zharkovsky T; Romantchik O; Jurgenson M; Merisalu E; Heidmets LT; Zharkovsky A, Developmental lead exposure impairs contextual fear conditioning and reduces adult hippocampal neurogenesis in the rat brain. Int J Dev Neurosci 2005, 23 (7), 627–35. [DOI] [PubMed] [Google Scholar]
- 56.Sigel A; Sigel H; Sigel RKO, Neurodegenerative diseases and metal ions. Wiley: Chichester, West Sussex, England ; Hoboken, NJ, 2006; p xxiii, 463 p. [Google Scholar]
- 57.Shinoda Y; Ehara S; Tatsumi S; Yoshida E; Takahashi T; Eto K; Kaji T; Fujiwara Y, Methylmercury-induced neural degeneration in rat dorsal root ganglion is associated with the accumulation of microglia/macrophages and the proliferation of Schwann cells. The Journal of toxicological sciences 2019, 44 (3), 191–199. [DOI] [PubMed] [Google Scholar]
- 58.Salehi F; Normandin L; Krewski D; Kennedy G; Philippe S; Zayed J, Neuropathology, tremor and electromyogram in rats exposed to manganese phosphate/sulfate mixture. Journal of Applied Toxicology: An International Journal 2006, 26 (5), 419–426. [DOI] [PubMed] [Google Scholar]
- 59.Kanchi S; Singh P; Bisetty K, Dithiocarbamates as hazardous remediation agent: A critical review on progress in environmental chemistry for inorganic species studies of 20th century. Arabian Journal of Chemistry 2014, 7 (1), 11–25. [Google Scholar]
- 60.Miller DB, Neurotoxicity of the pesticidal carbamates. Neurobehav Toxicol Teratol 1982, 4 (6), 779–87. [PubMed] [Google Scholar]
- 61.Lee CC; Peters PJ, Neurotoxicity and behavioral effects of thiram in rats. Environ Health Perspect 1976, 17, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frisoni GB; MONDA VD, Disulfiram neuropathy: a review (1971–1988) and report of a case. Alcohol and Alcoholism 1989, 24 (5), 429–437. [PubMed] [Google Scholar]
- 63.Hodge HC; Maynard EA; Downs WL; Coye RD Jr.; Steadman LT, Chronic oral toxicity of ferric dimethyldithiocarbamate (ferbam) and zinc dimethyldithiocarbamate (ziram). J Pharmacol Exp Ther 1956, 118 (2), 174–81. [PubMed] [Google Scholar]
- 64.Mulkey M The grouping of a series of dithiocarbamate pesticides based on a common mechanism of toxicity US Environmental Protection Agency, Office of Pesticide Programs, Office of Prevention, Pesticides and Toxic Substances, Washington, DC; SAP Report: 2001. [Google Scholar]
- 65.Mathieu C; Duval R; Xu X; Rodrigues-Lima F; Dupret J-M, Effects of pesticide chemicals on the activity of metabolic enzymes: focus on thiocarbamates. Expert opinion on drug metabolism & toxicology 2015, 11 (1), 81–94. [DOI] [PubMed] [Google Scholar]
- 66.Hedenstedt A; Rannug U; Ramel C; Wachtmeister CA, Mutagenicity and metabolism studies on 12 thiuram and dithiocarbamate compounds used as accelerators in the Swedish rubber industry. Mutation Research/Genetic Toxicology 1979, 68 (4), 313–325. [DOI] [PubMed] [Google Scholar]
- 67.Guideline, I. H. T., Detection of toxicity to reproduction for medicinal products & toxicity to male fertility S5 (R2). Citeseer: 2005. [Google Scholar]
- 68.Cooper RL, Current developments in reproductive toxicity testing of pesticides. Reproductive Toxicology 2009, 28 (2), 180–187. [DOI] [PubMed] [Google Scholar]
- 69.Kimmel CA; Makris SL, Recent developments in regulatory requirements for developmental toxicology. Toxicology letters 2001, 120 (1–3), 73–82. [DOI] [PubMed] [Google Scholar]
- 70.Kleinstreuer NC; Yang J; Berg EL; Knudsen TB; Richard AM; Martin MT; Reif DM; Judson RS; Polokoff M; Dix DJ; Kavlock RJ; Houck KA, Phenotypic screening of the ToxCast chemical library to classify toxic and therapeutic mechanisms. Nat Biotechnol 2014, 32 (6), 583–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.