

Abstract
Background
Congenital hemolytic anemia constitutes a heterogeneous group of rare genetic disorders of red blood cells. Diagnosis is based on clinical data, family history and phenotypic testing, genetic analyses being usually performed as a late step. In this study, we explored 40 patients with congenital hemolytic anemia by whole exome sequencing: 20 patients with hereditary spherocytosis and 20 patients with unexplained hemolysis.
Results
A probable genetic cause of disease was identified in 82.5% of the patients (33/40): 100% of those with suspected hereditary spherocytosis (20/20) and 65% of those with unexplained hemolysis (13/20). We found that several patients carried genetic variations in more than one gene (3/20 in the hereditary spherocytosis group, 6/13 fully elucidated patients in the unexplained hemolysis group), giving a more accurate picture of the genetic complexity of congenital hemolytic anemia. In addition, whole exome sequencing allowed us to identify genetic variants in non-congenital hemolytic anemia genes that explained part of the phenotype in 3 patients.
Conclusion
The rapid development of next generation sequencing has rendered the genetic study of these diseases much easier and cheaper. Whole exome sequencing in congenital hemolytic anemia could provide a more precise and quicker diagnosis, improve patients’ healthcare and probably has to be democratized notably for complex cases.
Keywords: Hemolysis, Red blood cell, Membrane, NGS, Anemia, Congenital, Mutation
Background
Congenital hemolytic anemia (CHA) is a group of rare genetic disorders characterized by increased destruction of red blood cells (RBC) [1]. They result from corpuscular causes such as hemoglobin disorders, membrane diseases, RBC enzyme deficiencies or congenital dyserythropoietic anemia (CDA), or from extra-corpuscular causes such as atypical hemolytic and uremic syndromes (aHUS) due to complement pathways dysfunctions.
Membrane disorders [2–5] include hereditary spherocytosis (HS, the most frequent cause in European population with nearly 1/2000 subjects affected, MIM 182900, 182,870, 270,970, 612,653, 612,690) [6]; hereditary elliptocytosis (HE, from 1/5000–1/10,000 in European population to 1/100 in West African populations, MIM 130600, 109,270, 611,804) [7]; hereditary pyropoïkilocytosis (HPP, MIM 266140), which is a rare and severe subtype of HE, and hereditary stomatocytosis, with a dehydrated form (incidence of 1/50,000 individuals, MIM 616689) and an overhydrated form (incidence lower than 1/100,000, MIM 185000) [8]. Transmission is often autosomal dominant (AD) but de novo mutations or autosomal recessive (AR) forms (as is the case in HPP) can occur.
Defaults in enzymatic pathways of RBC metabolism are numerous [9] the most frequent being Glucose-6 Phosphate Dehydrogenase (G6PD, MIM 300908) and Pyruvate Kinase deficiencies (MIM 266200) [10]. Most of them have AR inheritance but some are X-Linked (XL) (as is the case for G6PD deficiency).
CDA (MIMs 224,120, 615,631, 224,100, 105,600, 613,673) is characterized by ineffective central erythropoiesis with progressive secondary iron overload and has AR, AD or XL transmission [11, 12]. Peripheral hemolysis can occur and misdiagnosis is often due to phenotypic overlap with other types of CHA [13, 14].
Clinical features of CHA vary from severe neonatal or even prenatal anemia with high morbidity and transfusion dependence to well-compensated hemolysis without anemia. In addition, clinical manifestations may be increased by associated comorbidities (such as hemochromatosis or hemoglobin variant) or during acute diseases, inflammatory conditions, or pregnancy.
Mutations causing CHA have been identified in more than 100 different genes, some of which are very large. Next generation sequencing (NGS) allows massive parallel sequencing of numerous genes and therefore appears as a very suitable approach for genetic dissection of CHA. Different NGS strategies have been developed over the last few years for CHA (Additional file 1, Supplementary Table S1). They included targeted gene panels (comprising 28 to 76 genes) with a success rate for diagnosis varying from 38 to 90%, depending on proband numbers (ranging from 2 to 62) and subtypes of anemia [15–23], and whole-exome sequencing (WES) in a few studies (limited to 1 to 7 probands, except two studies focusing on 38 Chinese cases of HS [24] and 24 cases of autosomal recessive HS [25]), with success rates between 29 and 100% [14, 24–32]. We used WES to explore 40 CHA patients: 20 with suspected HS and 20 with unexplained hemolysis (UH) despite available phenotype exploration. We first analyzed genomic data using a predefined list of 71 CHA genes (Additional file 1, Supplementary Table 2) and extended the analysis to WES when necessary. This approach allowed us to find a probable molecular cause in 82.5% of patients.
Methods (see supplementary material for more details)
Biochemical and hematologic tests
Standard laboratory tests included cell blood count, blood smear examination, hemolysis markers, RBC density (measured by the phthalate density-distribution method, as described by Bartolucci et al in 2012 [33], with density curves profiles), hemoglobin study (beginning with isoelectric focusing and HPLC. In case of abnormalities, acid agar gel electrophoresis, Itano test, capillary electrophoresis and reverse phase HPLC could be performed, RBC enzymes activity (G6PD, Pyruvate Kinase, Hexokinase routinely measured) and EMA test (performed following the methods described by King et al in 2000 [34] and Girodon et al in 2008 [35]). When possible, osmolar gradient ektacytometry (Technicon®) and membrane protein electrophoresis (MPE) were performed.
DNA extraction
Peripheral blood EDTA sample was obtained from patients after written informed consent for genetic analysis. Genomic DNA was extracted using the Flexigene extraction kit, following manufacturer’s recommendations (Qiagen; Hilden, Germany).
Sequencing and data analysis
Library preparation was performed from 100 ng of enzymatically fragmented genomic DNA using the Kapa library hyperprep kit, following manufacturer’s recommendations (KAPABIOSYSTEMS, Wilmington, Massachusetts). Enrichment for exonic sequences was performed using the Medexome kit, following manufacturer’s recommendations (NimbleGen, Madison, Wisconsin).
NGS sequencing
Captured libraries were sequenced on a Nextseq500 instrument (Illumina, San Diego, California, USA) with high-output FlowCell and reagents, in order to obtain 150 bp paired-end reads.
Bioinformatic analyses
Demultiplexing and .fastq files generation were performed using Bcl2fastq software (v.1.8.4). Trimming of low-quality bases (Phred score < 10) and adapter-contaminated ends was performed with Trimmomatic (v.0.6.1) (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Alignment and variant calling were performed in duplicates, using both an in-house pipeline following the BWA/GATK best practices and the Seqnext software (JSI, Ettenheim, Germany). For our in-house bioinformatic pipeline, high-quality reads were mapped on the Human reference genome (GRCh37, hg19) using BWA-MEM (v.0.7.1). SAMtools (http://www.htslib.org/) and Picard (v.1.106, https://broadinstitute.github.io/picard/) were run to remove the duplicate reads. Mean depth of coverage was >171x for all samples with > 97.6% covered at least 30X. Targeted regions with depth under 30X were selected with the Depth Of Coverage GATK tool and manually checked using bam files loaded on two reads viewers, i.e. Alamut Visual (Interactive Biosoftware, Rouen, France) and IGV (Broad institute, Cambridge, MA 02142, USA). For our first-line list of genes, all targeted regions were above 30X, excepted GPX1 and PGD first exons, which were sequenced by Sanger. Exon 1 of PIEZO1 was usually covered ~15X but was validated after a manual check of high-quality reads on bam viewers.
Variant calling was performed using the Genome Analysis Toolkit (v.3.8) with the Haplotype Caller tool in GVCF mode. VQSR filters were applied according to GATK guidelines (when QD < 2, FS > 60 for snps and > 200 for indels, ReadPosRankSum <− 8 for snps and > − 20 for indels, when MQ < 40, MQRankSum <− 12.5 and SOR < 3).
Variant annotation
The quality-filtered variant calling files (VCF) obtained were gene- and region-based annotated on GRCh37, hg19 reference genome using Annovar [36] and dbSNP141.
Functional annotation was made using in silico prediction tools and amino acid conservation scores (SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Taster (http://www.mutationtaster.org/), align GVGD (http://agvgd.iarc.fr/) and CADD (https://cadd.gs.washington.edu/)). MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html) and Human Splicing Finder3.0 (http://www.umd.be/HSF/) were used for splicing effect predictions. Protein domain functions were determined by using the SMART software (http://smart.embl-heidelberg.de/) and are available in Table S3. Functional classification following ACMG criteria [37] was performed using InterVar [38] and Ingenuity Variant Analysis (IVA) software (Qiagen).
Variants filtering and selection
According to ACMG guidelines, we excluded all variants tagged as Benign.
According to Annovar gene- or region-based annotations, we excluded variants disrupting microRNA binding sites, promoter, intergenic, ncRNA as well as intronic variants > 10 nucleotides of a splice site (−splicing_threshold 10) and kept only those present on an alternative transcript. Variants in regions with segmental duplications were excluded (genomicSuperDups).
All the possible inheritance patterns were tested according to disease knowledge. Variants with population frequency (Minor Allele Frequency, MAF) in gnomAD, ExAC, 1000 g and ESP6500 higher than 5% for AR disorders and higher than 1% for AD disorders were filtered out, except variants known to have modulating effects on phenotype, such as HFE variants (H63D or C282Y) involved in iron overload and the SPTA1 alpha-Lely polymorphism (c.6531-12C > T) involved in membrane disorders. This latter variant is known to be pathogenic when associated in trans with a deleterious variant of SPTA1 [39].
The first-line analysis was focused on 71 known CHA genes as a target gene panel (Additionnal file 1, Supplementary Table S2). An extended WES analysis was performed using IVA filters “biological context” and “phenotype driven”. In addition, we performed a complementary analysis of 2138 genes encoding erythrocyte proteins identified by proteomic studies [40–42].
New variants
All new variants classified as VUS, likely pathogenic or pathogenic have been registered in the Clinvar database (https://www-ncbi-nlm-nih-gov.gate2.inist.fr/clinvar/ see Additional file 1, Supplementary Table S4).
Sanger sequencing
Sanger sequencing was used to confirm each potentially deleterious variation found by NGS. Primers sequences and PCR conditions are available upon request.
Patients
Forty patients suffering from non-immune hemolysis with negative direct antiglobulin tests were submitted to WES sequencing. Clinical and biological data are available in Supplementary Tables S5 and S6 (normal values in Additional file 1 and Supplementary Table S7). Blood smear results and genetic results are available in Tables S8 and S11. In silico analysis of genetic variants is available in Additional file 1, supplementary Table S3.
Twenty patients had a HS phenotype, among which four were apparently sporadic cases (P1, P3, P13, P14). The 20 other patients had UH, with either discordant or non-conclusive phenotypical tests. For several patients, EMA test results were discordant with ektacytometry. In the other cases, EMA test and ektacytometry were either normal or atypical. Three of these patients had UH associated to a sickle cell trait (A/S). Several patients had no familial history. Despite of this fact they were considered as potential cases of CHA because phenotypic exploration of common acquired causes of hemolytic anemia such as complete antiglobulin tests (Coombs test) and paroxysmal nocturnal hemoglobinuria test remained negative.
Results
Genetic results are available in Tables 1 and 2 and in Additional file 1, supplementary Tables S3 and S8 to S11.Clinico-biological data are available in Additional file 1, supplementary Tables S5 to S7.
Table 1.
Genetic results of HS patient
| Patient ID | Gene name | transcript | Nucleotide change | AA change | zygosity | References rsNumber |
gnomAD allele frequency | Polyphen-2 | Mutation taster | MaxEnt Scan | CADD score | ACMG class |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | ANK1 | NM_020476 | c.5152C > T | p.Gln1718* | het | No | 0 | NA | NA | 37 | LP | |
| HBA1 | NM_0005558 | c.389 T > C | p.L130P | het | Darbellay et al 1995 | 0 | PD | DC | 23.4 | LP | ||
| P2 | ANK1 | NM_020476 | c.1702-2A > C | het | No ref | 0 | NA | NA | −100% | 34 | LP | |
| HFE | NM_000410 | c.187C > G | p.H63D | het |
Kaczorowska-Hac et al 2016 rs1799945 |
10.83% | B | P | 12.8 | VUS | ||
| HFE | NM_000410 | c.845G > A | p.C282Y | het |
Kaczorowska-Hac et al 2016 rs1800562 |
3.37% | PD | DC | 25.2 | P | ||
| P3 | SLC4A1 | NM_000342 | c.1458C > G | p.Y486* | het | No | 0 | NA | NA | 35 | LP | |
| P4 | SLC4A1 | NM_000342 | c.486-2A > G | het | no | 0 | NA | NA | −100% | 25.3 | P | |
| P5 | SPTB | NM_001024858 | c.1331_1338del | p.Leu444Profs*3 | het | Dhermy et al 1998 | 0 | NA | DC | P | ||
| P6 | ANK1 | NM_020476 | c.5497C > T | p.R1833* | het | Hayette et al 1998 | 0 | 47 | P | |||
| P7 | SLC4A1 | NM_000342 | c.1322 T > G | p.L441R | het | No | 0 | PD | DC | No effect | 26.8 | VUS |
| P8 | ANK1 | NM_020476 | c.1801-17G > A | het |
Duru et al rs786205243 |
0 | Creation of a cryptic acceptor | 8.2 | LP | |||
| P9 | ANK1 | NM_020476 | c.4462C > T | p.R1488* | het |
Ozcan et al 2003 rs777701149 |
0 | 36 | P | |||
| P10 | ANK1 | NM_020476 | c.1A > G | p.? | het | no | 0 | NA | NA | NA | 14.3 | P |
| P11 | SPTB | NM_001024858 | c.2863C > T | p.R955* | het | no | 0 | NA | NA | NA | 37 | P |
| SPTA1 | NM_003126 | c.6421C > T | p.R2141W | het |
Niss et al 2016 rs41273519 |
0.2% | PD | DC | 27.4 | LP | ||
| P12 | SPTB | NM_001024858 | c.4973 + 5G > A | het | no | 0 | NA | NA | −100% | 16.48 | VUS | |
| P13 | ANK1 | NM_020476 | c.534delC | p.H178Qfs*75 | het | no | 0 | NA | NA | LP | ||
| P14 | SPTB | NM_001024858 | c.5623C > T | p.Q1875* | het | no | 0 | NA | NA | 48 | LP | |
| P15 | SLC4A1 | NM_000342 | c.1462G > A | p.V488M | het |
Alloisio et al 1997 rs28931584 |
0.00041% | PD | DC | No effect | 24.5 | P |
| P16 | ANK1 | NM_020476 | c.712-2A > G | het | no | 0 | NA | NA | −100% | 34 | LP | |
| P17 | SLC4A1 | NM_000342 | c.2423G > A | p.R808H | het |
Bogardus et al 2012 rs866727908 |
0 | PD | DC | No effect | 33 | LP |
| PIEZO1 | c.2578G > A | p.V860M | het | rs532390680 | 0.0028% | PossD | DC | No effect | VUS | |||
| P18 | SLC4A1 | NM_000342 | c.2279G > A | p.R760Q | het | Jarolim et al 1995 | 0 | PD | DC | No effect | 29.6 | LP |
| P19 | SPTB | NM_001024858 | c.3436dup | p.L1146Pfs*36 | het | no | 0 | NA | NA | LP | ||
| SPTB | NM_001024858 | c.6101G > A | p.S2034N | het | no | 0.00041% | B | DC | No effect | 22.9 | VUS | |
| P20 | SPTB | NM_001024858 | c.3916C > T | p.R1306* | het |
No rs150471537 |
0 | NA | NA | 38 | LP |
Table 1 legend: Variants description and classification according to ACMG guidelines as benign likely benign (LB) variant of uncertain significance (VUS) likely pathogenic (LP) or pathogenic (P). In silico study of missense variations was assessed thanks to Polyphen-2, Mutation taster and CADD score algorithm. HGMD professional and pubmed web interface were used to check for variants description in litterature. Abbreviations: het: heterozygous state; hom: homozygous state; hem: hemizygous state; F: female; M: male; HS: hereditary spherocytosis; gnomAD: genome agregation database https://gnomad.broadinstitute.org; ND: not done; NA: not applicable; DC: disease causing; P: polymorphism; PD: probably damaging; PossD: possibly damaging; B: benign.
Table 2.
Genetic results of UH patients
| Patient ID | Gene name | Transcript | Nucleotide change | AA change | zygosity | references | gnomAD allele frequency | Polyphen-2 | Mutation taster | MaxEnt Scan | CADD score | ACMG class |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P21 | SPTA1 | NM_003126 | c.6600 + 5G > T | het | no | 0 | NA | NA | −62.5% | 18.74 | VUS | |
| SPTA1 | NM_003126 | c.6531-12C > T | het |
Alpha-Lely polymorphism rs28525570 |
25% | |||||||
| P22 | SPTA1 | NM_003126 | C.2898G > A | p.(=) | het | no | 0 | NA | NA | −29.3% | 14.2 | VUS |
| SPTA1 | NM_003126 | c.6531-12C > T | het |
Alpha-Lely polymorphism rs28525570 |
25% | |||||||
| P23 | ALAS2 | NM_000032 | c.-258C > G | het |
Bekri et al 2003 rs140772352 |
0.54% | NA | NA | NA | VUS | ||
| P24 | TRPV4 | NM_021625 | c.1913C > T | p.P638L | hom |
No rs35058636 |
0.03% (no homozygotes) | B | DC | No effect | VUS | |
| ADAR | NM_001111 | c.1586C > T | p.P529L | het | no | 0 | PD | DC | No effect | VUS | ||
| P25 | SEC23B | NM_001172745 | c.40C > T | p.R14W | het |
Russo et al 2011 rs121918222 |
0.022% | PossD | DC | No effect | P | |
| SEC23B | NM_001172745 | c.325G > A | p.E109K | het |
Russo et al 2011 Rs121918221 |
0.023% | PD | DC | No effect | P | ||
| P26 | HAMP | NM_021175 | c.49_54del | p.L17_L18del | het | no | 0 | NA | NA | No effect | VUS | |
| HFE | NM_000410 | c.845G > A | p.C282Y | het | rs1800562 | 3.37% | PD | P | No effect | P | ||
| CD46 | NM_172359 | c.402 T > G | p.I134M | het | no | 0 | PossD | P | No effect | VUS | ||
| P27 | CFH | NM_00186 | c.2850G > T | p.Q950H | het |
Mohlin et al 2015 rs149474608 |
0.39% | B | P | No effect | LB | |
| P28 | SEC23B | NM_001172745 | c.1276G > A | p.V426I | het |
Schwartz et al 2009 rs41309927 |
4.3% | B | P | No effect | VUS | |
| CDAN1 | NM_138477 | c.256C > T | p.P86S | het |
No rs543791953 |
0.052% | B | P | No effect | VUS | ||
| P29 | SPTA1 | NM_003126 | c.1688G > A | p.R563Q | het |
No rs202243588 |
0.11% | PD | DC | Possible new acceptor site | 25.1 | VUS |
| HBB | NM_000518 | c.20A > T | p.E7V | het |
Yes coding for HbS rs334 |
0.44% | B | P | 13.8 | P | ||
| SPTA1 | NM_003126 | c.6531-12C > T | het |
Alpha-Lely polymorphism rs28525570 |
||||||||
| P30 | PIEZO1 | NM_001142864 | c.1126C > G | p.P376A | het | no | 0 | B | P | Possible new acceptor site | VUS | |
| P31 | KCNN4 | NM_002250 | c.1055G > A | p.R352H | het |
Rappetti Mauss et al 2015 rs774455945 |
0 | PossD | DC | No effect | P | |
| PIEZO1 | NM_001142864 | c.3629C > T | p.A1210V | het |
No rs761971227 |
0.006% | B | DC | No effect | LP | ||
| PIEZO1 | NM_001142864 | c.3629C > T | p.A1210V | absence | ||||||||
| P32 | SPTB | NM_001024858 | c.[6706C > A;6737C > T] | p.[L2236M;A2246V] | Het in cis | no | 0 | B/PD | DC/P | No effect | VUS | |
| HBB | NM_000518 | c.20A > T | p.E7V | het |
Yes coding for HbS rs334 |
0.44% | B | P | 13.8 | P | ||
| P33 | G6PD | NM_000402 | c.538G > A | p.V180I | het | no | 0 | PossD | DC | No effect | VUS | |
| SPTB | NM_001024858 | c.6271C > A | p.P2091T | het |
No rs372733273 |
0.0065% | B | DC | no effect | VUS | ||
| P34 | HFE | NM_000410 | c.187C > G | p.H63D | hom |
Kaczoeowska-Hac et al 2016 rs1799945 |
10.83% | B | P | No effect | LB | |
| ABCG8 | NM_022437 | c.-27G > A | het | no | 0 | NA | NA | NA | VUS | |||
| ADAMTS13 | NM_139025 | c.119C > G | p.A40G | het |
No rs782213090 |
0.00041% | B | P | VUS | |||
| ADAMTS13 | NM_139025 | c.4007G > A | p.R1336Q | het |
No No rs |
0.0012% | PD | P | VUS | |||
| P35 | SH2B3 | NM_005475 | c.1A > G | p.0? | het | no | 0 | NA | NA | No effect | LP | |
| SCN9A | NM_002977 | c.2938G > T | p.A980S | het | no | 0 | PossD | DC | No effect | VUS | ||
| P36 | SPTA1 | NM_003126 | c.6672A > C | p.E2224D | hom |
No Rs142775522 |
1.5% no homozygotes | PD | DC | No effect | 22.3 | VUS |
| SLC4A1 | NM_000342 | c.1199_1225del | p.A400_A408del | het |
Wilder et al 2009 rs769664228 |
0.0047% | PD | DC | LP | |||
| PIEZO1 | NM_001142864 | c.1369C > T | p.R457C | het | Russo et al 2018 | 0 | PD | DC | No effect | LP | ||
| G6PD | NM_000402 | c.292G > A | p.V98M | het |
Vulliamy et al 1988 rs1050828 |
1.15% | LP | |||||
| HBB | NM_000518 | c.20A > T | p.E7V | het |
Yes coding for HbS rs334 |
0.44% | B | P | 13.8 | P | ||
| P37 | SPTA1 | NM_003126 | c.3291G > A | p.W1097* | het | no | 0 | NA | NA | NA | 42 | LP |
| SPTA1 | NM_003126 | c.6531-12C > T | het |
Alpha-Lely polymorphism rs28525570 |
||||||||
| HFE | NM_000410 | c.187C > G | p.H63D | hom |
Kaczoeowska-Hac et al 2016 rs1799945 |
10.83% | B | P | No effect | LB | ||
| P38 | CFH | NM_00186 | c.157C > T | p.R53C | het |
Servais et al 2012 rs757785149 |
0.0014% | PD | DC | No effect | LP | |
| PIEZO1 | NM_001142864 | c.4246G > A | p.G1416R | het |
No rs771605269 |
0.00033% | PD | DC | New cryptic acceptor site | VUS | ||
| P39 | SPTA1 | NM_003126 | c.779 T > C | p.L260P | het |
Marchesi et al 1987 Rs121918634 |
0.017% (Afr) | PD | DC | No effect | LP | |
| SPTA1 | NM_003126 | c.6531-12C > T | het |
Alpha-Lely polymorphism rs28525570 |
||||||||
| P40 | ATP11C | NM_173694 | c.2434C > T | p.P812S | hem | no | 0.00055% no hemizygous | PD | DC | No effect | VUS | |
| ANK1 | NM_020476 | c.4558G > C | p.E1520Q | het | no | 0.0021% | B | DC | No effect | 26 | VUS |
Table 2 Legend: Variants description and classification according to ACMG guidelines as benign likely benign (LB), variant of uncertain significance. (VUS), likely pathogenic (LP) or pathogenic (P). In silico study of missense variations was assessed thanks to Polyphen-2, Mutation taster andCADD score algorithm. HGMD professional and pubmed web interface were used to check for variants description in litterature. Abbreviations: het: heterozygous state; hom: homozygous state; hem: hemizygous state; F: female; M: male; HS: hereditary spherocytosis; gnomAD: genome agregation database https://gnomad.broadinstitute.org; ND: not done; NA: not applicable; DC: disease causing; P: polymorphism; PD: probably damaging; PossD: possibly damaging; B: benign.
We defined two groups: one with clear phenotype (HS group) and one with unclear phenotype (UH group). The different types of variants identified are summarized in Fig. 1.
Fig. 1.

Distribution of mutations types
HS group
We identified the probable genetic cause of hemolysis in all HS patients using target genes panel analysis (71 CHA genes) on WES data. Nineteen patients were heterozygous carriers of one mutation in ANK1 (8 patients), SLC4A1 (6 patients), or SPTB (5 patients). All patients had different mutations, among which 13 were novel and 8 had already been reported. In one family with typical HS (P11 and affected relatives), affected members harbored 2 heterozygous probably damaging variations, one in SPTB (stop-gained) and the other in SPTA1 (already-reported missense mutation, previously found in a case of HPP which also harbored the alpha-Lely polymorphism in trans). Such an association has rarely been reported in the literature [15, 16, 43, 44]. In 3 families (P4, P10 and P11), genetic tests could be extended to other affected family members and showed cosegregation of mutations and disease (Fig. 2). Two HS patients also had additional mutations in other CHA genes. P1 carried a heterozygous mutation of alpha-globin gene, called Tunis-Bizerte hemoglobin, responsible for an alpha- thalassemia trait [45]. P17 carried a variant of uncertain significance (VUS) in PIEZO1 (p.V860M). In total, 3 out of 20 HS patients (P1, P11 and P17) had variations in two different CHA genes. Patient P2, who had significant iron overload also harbored 2 variants in HFE (H63D and C282Y) which had been previously found during iron overload exploration.
Fig. 2.
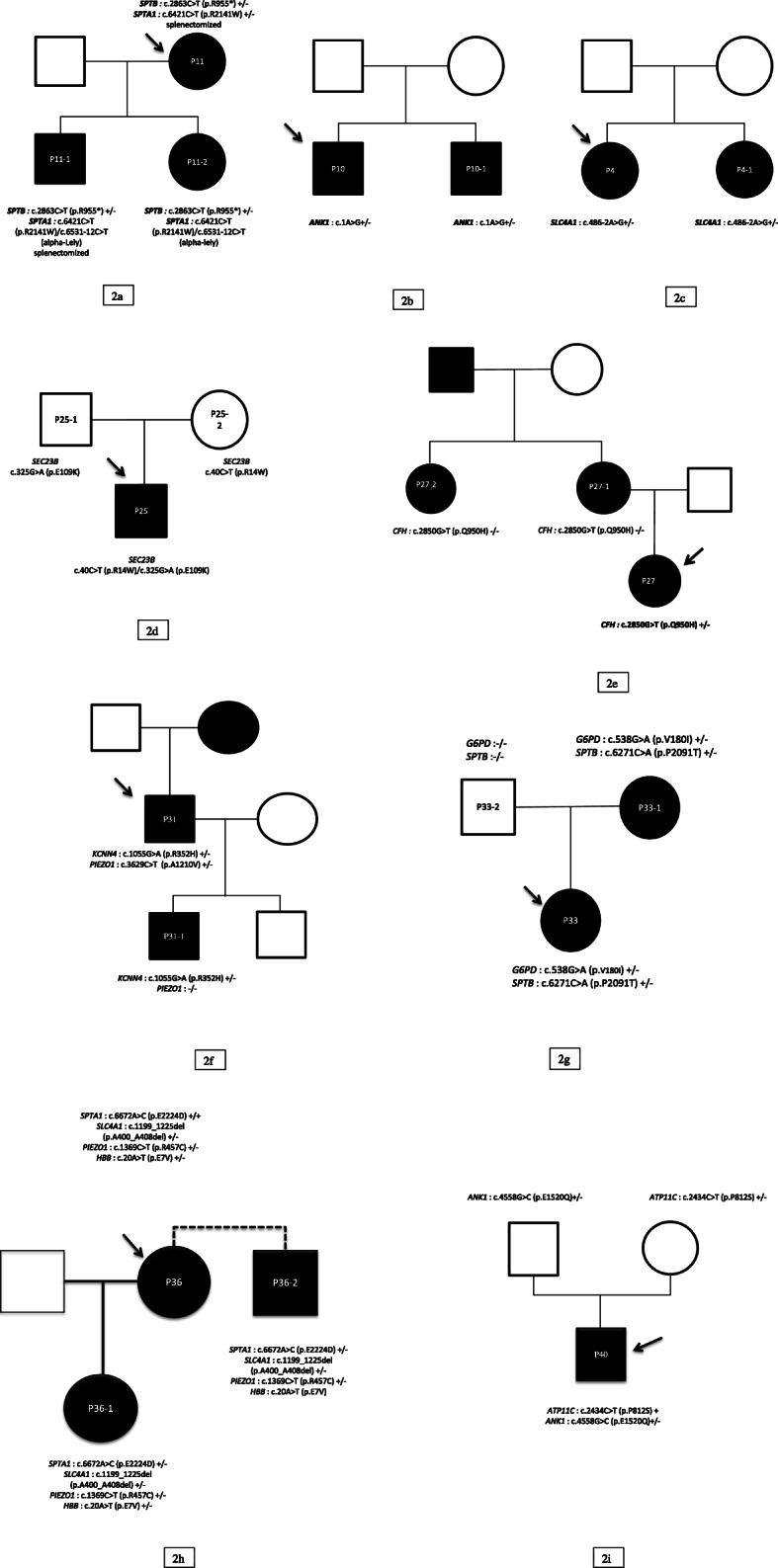
Pedigrees of 3 HS families and 6 UH families. Legend: Black squares: affected males; black circles: affected females; open squares: unaffected males; open circles: unaffected females; arrow: the proband. 2a = P11 family pedigree (HS); 2b = P10 family pedigree (HS); 2c = P4 family pedigree (HS); 2d = P25 family pedigree (CDA2); 2e = P27 family pedigree (UH); 2f: P31 family pedigree (GARDOS); 2 g: P33 family pedigree (UH); 2 h: P36 family pedigree (multiple association); 2i: P40 family pedigree (ATP11C hemolysis)
UH GROUP
Twenty patients were classified as UH because of discordant or non-contributive clinical features and/or biological tests. Familial segregation study could be performed in 6 families (P25, P27, P31, P33, P36, P40, Fig. 2).
Twelve UH patients have been fully characterized (P21, P22, P25, P29, P30, P31, P32, P36, P37, P38, P39, and P40) by target gene panel analysis of 71 CHA genes on the WES data. One UH patient (P35) could be fully characterized thanks to WES analysis. Seven other UH patients have not been fully characterized (P23, P24, P26, P27, P28, P33 and P34) despite extended WES analysis. Interestingly, in those patients WES identified two new possible target genes.
Among the 12 fully characterized UH patients, nine harbored mutations in genes encoding membrane proteins. SPTA1 was the most frequently mutated gene (in 6 probands: P21, P22, P29, P36, P37, and P39). Variations in SPTA1 result in HE, HPP or HS depending on variants type and phase. In P37 and P39, genotypes and phenotypic data (clinical features, blood smear) were suggestive of HPP. We could not clearly decipher between HE or HPP for P21, 22 and P29. Other membrane genes with mutations were PIEZO1 (P30 and P36, DHSt), KCNN4 (P31, GARDOS MIM 616689), SPTB (P32, elliptocytosis) and SLC4A1 (P36, SEA ovalocytosis, MIM 166900). Among them, some had original presentation. P30 had myelodysplasia and important hemolysis without any family history. She carried a constitutional (present in blood and hair bulb DNA) heterozygous PIEZO1 variant of uncertain significance (VUS) (p.P376A) and had atypical ektacytometry results. P36 is one of the 3 A/S symptomatic patients explored in our study (P29, P32 and P36). This woman originating from Comoros islands experienced spleen infarct after a long-distance flight and showed the association of a HbS trait, SEA ovalocytosis (SLC4A1: p.Ala400_Ala408del), a G6PD MATERA A- p.V98M variant at heterozygous state [46], a homozygous SPTA1 variation (p.E2224D) and a PIEZO1 mutation (p.R457C) already involved in DHSt [16]. Her daughter (P36–1) also presented with hemolytic anemia and harbored the same mutations (the SPTA1 variant being heterozygous). Her blood smear showed anisopoikilocytosis and some stomatocytes. Another relative (P36–2) was explored and carried the same mutations as P36–1. He had a hemolytic anemia and a retinopathy typical of sickle cell disease.
Three fully characterized UH patients had mutations in genes not encoding membrane proteins: SEC23B in P25 with CDA type 2; CFH in P38 with aHUS; ATP11C in P40. P25 harbored 2 already described mutations in SEC23B gene at compound heterozygous state (Fig. 2) allowing correction of diagnosis towards CDA type 2 and not HS as initially suspected.
P38 carried a heterozygous mutation in CFH (p.R53C), which had previously been found in patients with preeclampsia-related SHUa [47], in complement-related glomerulopathies [48] and in familial forms of AMD (age-related macular degeneration, MIM 610698) [49]. She also carried a heterozygous PIEZO1 VUS (p.G1416R) but had normal ektacytometry and blood smear. In this case one single mutation in CFH probably explains the entire phenotype (AMD, preeclampsia, hemolysis and altered renal function). P40 carried a likely pathogenic hemizygous variation in ATP11C (p.P812S). This X chromosome variation is recorded in gnomAD in only one heterozygous female (no hemizygous males or homozygous females recorded). He also carried one VUS in ANK1. His blood smear was normal and ektacytometry showed atypical profile with only dehydration and no change in osmotic resistance. Functional testing of flippase activity is in progress.
Among the 12 fully characterized UH patients, 6 had mutations in several CHA genes:
P29HBB S mutation and SPTA1
P31, KCNN4 and PIEZO1
P32, HBB S mutation and SPTB
P36,HBB S mutation, SPTA1, SLC4A1, PIEZO1 and G6PD
P38,CFH and PIEZO1
P40, ATP11C and ANK1,
One additional UH patient was characterized thanks to WES analysis
P35 was found to harbor a heterozygous deleterious variation in SH2B3: c.1A > G (initiation codon loss). This result allowed to reconsider diagnosis towards a probable myeloproliferative condition. SH2B3 somatic mutations have been reported in myeloproliferative neoplasms such as primary myelofibrosis [50]. He also carried a heterozygous VUS in the SCN9A gene c.2938G > T (p.A980S) which likely explained the severity of painful crises reported in this patient. SCN9A is involved in neurogenic painful syndromes [51].
Seven UH patients remained unsolved (P23, P27 and P28) or partially solved (P24, P26, P33 and P34) despite WES extended analysis. P23 carried a heterozygous ALAS2 promoter variation (c.-258C > G), which had previously been reported as a cause of X-linked sideroblastic anemia (MIM 300751) [52]. This variation is present in gnomAD at an allelic frequency of 0.54%, with 39 hemizygous males, which suggests that it is most probably a rare benign polymorphism. No other relevant genetic variation was found. P27 was a female child with major hemolytic anemia at birth and neonatal splenomegaly with thrombocytopenia. Her mother, maternal aunt and maternal grand-father had the same phenotype. She carried a heterozygous variation in CFH (p.Q950H) [53], which was absent in the mother and the maternal aunt and is therefore not responsible for CHA in this family. No other relevant genetic variation was found. P28 was carrier of two heterozygous variants in two genes involved in CDA: one in SEC23B (p.V426I) and one in CDAN1 (p.P86S). No case of digenic inheritance has yet been reported in CDA. No other relevant genetic variation was found. The other patients had a part of their phenotype explained by WES. In P24, candidate gene variations could be identified thanks to WES. A heterozygous missense variation was found in the gene encoding the RNA-specific Adenosine Deaminase (ADAR) (p.P529L). No ADAR mutation has been associated to date with congenital hemolysis in humans but several studies showed a crucial role for ADAR in mouse erythropoiesis. This patient also carried an apparently homozygous variation of TRPV4 (p.P638L), present in the gnomAD database at an allelic frequency of 0.03% without any homozygotes recorded (over > 138,000 subjects tested). TRPV4 mutations have been found in skeletal dysplasia, arthropathies and in a familial form of osteonecrosis [54]. The TRPV4 mutation could explain osteonecrosis but not hemolysis. P26 carried HAMP and HFE variations probably explaining iron overload and CD46 variation for susceptibility to hemolysis. HAMP gene is out of our genes panel analysis. P33 harbored G6PD and SPTB variations, which combination cannot explain the severe phenotype. P34 had UH and iron overload, and variations in 3 genes: a homozygous p.H63D variation in HFE, which may contribute to iron overload even though its implication is not totally clear [55, 56], a heterozygous ABCG8 VUS and 2 heterozygous ADAMTS13 VUS. ABCG8 mutations are found in AR sitosterolemia and AD xanthelasma [57]. A single heterozygous ABCG8 variation is not sufficient to explain hemolysis. This patient had a normal platelet count and ADAMTS13 enzyme activity testing showed a 39% decrease (suggestive of a heterozygous loss-of-function). Normal ADAMTS13 activity in P34 does not support a diagnosis of AR Thrombotic thrombocytopenic purpura (MIM 274150), and therefore cannot explain hemolysis.
In summary, among n UH patients, 12 patients could be fully characterized thanks to targeted analysis on CHA genes. Eight benefited from extended WES analysis leading to full characterization in one additionnal patient and 7 UH patients not fully characterized but two new potential target genes were identified by WES.
Discussion
In this study, WES sequencing allowed the identification of a genetic cause of CHA in 33 out of 40 patients (82.5%), including 20 HS patients (100%) and 13 UH patients (65%). All HS patients harbored mutations in already-known HS genes: ANK1, SLC4A1, SPTB, SPTA1. All identified variants (21, among which 13 novel) were classified as likely pathogenic or pathogenic loss-of-function mutations. A recent exomic study of 38 Chinese patients with suspected HS found mutations of ANK1 or SPTB in all patients [24]. Gallagher et al in 2019 [25] reported on a group of 24 recessive HS patients explored by WES and whole genome sequencing (WGS), all having mutations found in SPTA1. Our results, together with those previously published, suggest that there are probably no other major HS genes. In mouse, beta-adducin deficiency has been shown to cause spherocytosis [58], but no mutations of adducins have yet been identified in human HS.
In UH patients, NGS analysis allowed diagnosis in 13 patients (P21, P25, P29, P31, P22, P30, P32, P35, P36, P37, P38, P39, and P40). Rate of positivity is lower in UH patients than in HS cases, probably because of the lack of a clear phenotype.
One important finding of our study is the frequent association of several genetic variants in different genes in a same patient, emphasizing the genetic complexity of CHA. Twenty two percents (9/40) of patients harbored several variations in different CHA genes. Among HS patients, only 3 (15%) had variations in other genes, whereas in the UH group, 9/20 (45%) had an association of variants in different CHA genes. The frequent occurrence of multiple genetic variants in single UH patients suggests that this association could contribute to the complexity of the phenotype. Phenotypical tests alone are not able to detect all those variants associations, some of which can have therapeutic consequences (contra-indication of splenectomy in DHSt for example). Such associations of several deleterious variants in genes causing RBC diseases have rarely been reported [59–61] and could be explained in part by the patients’origins. For example, the P36 family that carried variations in 5 CHA genes originated from Comoros Islands, a region of endemic malaria. Plasmodium has been shown to exert a selective pressure on various RBC proteins [62].
Our study also emphasizes the usefulness of exomic approach in the field of CHA, both for discovery of candidate genes (such as TRPV4 or ADAR) and for diagnosis reorientation. Thanks to exomic capture, we reorientated initial diagnosis for P35 (probable myeloproliferative syndrome) and explained a part of the phenotype in P24 (osteonecrosis and TRPV4), P26 (HAMP and iron overload) and P35 (painful crisis and SCN9A). The cases of P27 remained unsolved despite large familial investigations. Exomic capture allowed us to find a possible cause for P24’s osteonecrosis since this patient harbored a homozygous TRPV4 variation. TRPV4 encodes a non-selective calcium-permeant cation channel expressed in different cell types [63] but not in erythrocytes and it has never been associated with hemolysis. Mutations have already been involved in skeletal dysplasia, hereditary neuropathies, arthropathies [64] and recently in a familial case of osteonecrosis [65]. All variants reported were heterozygous and only one paper described a familial case of complex phenotype with severe intellectual disability and neuropathy associated with compound heterozygosity for TRPV4 mutations [66]. Our patient does not present any neurological symptoms. In this patient, we also found an ADAR variation that could be a good candidate for hemolysis. No mutations in ADAR have yet been associated with hemolysis in humans. Previous studies in mouse showed that ADAR is implicated in erythropoiesis [67, 68]. ADAR heterozygous mutations are been found in dyschromatosis symmetrica hereditaria [69] and homozygous or compound heterozygous variations in Aicardi-Goutières syndrome [70].
During the last few years, several NGS studies of CHA have been published [14–24, 26–32, 71] and have shown their usefullness (Additional file 1, Supplementary Table S1). Two recent studies using targeted approaches, focused on 21 index cases and 62 families respectively [16, 17]. They obtained a positive rate (elucidation of the genetic cause of anemia) of 62 and 65% of cases respectively. In our study, this rate was 82.5%. In the two former studies and ours, the positive rate appears to be considerably higher than the WES success rate obtained in other genetic diseases, such as cardiomyopathies or intellectual deficiencies: 62 to 82.5% for CHA versus 30% on average for cardiomyopathies or intellectual deficiencies [72, 73]. The two recent studies of Wang R. et al 2018 and Gallagher et al 2019 reporting WES findings in relatively large CHA samples [24, 25] were only dedicated to HS patient. Our results suggest that WES is highly useful in CHA patients, whether they have HS or UH. Recommendations proposed by King et al in 2015 [13], and compiled by Kim et al in 2017 [74] concerning the indications of genetic testing for CHA diagnosis need to be updated, as they only recommended genetic testing in a limited number of cases and as a second diagnostic step. A recent article of Rets et al in 2019 suggests that targeted NGS becomes the diagnosis standard tool in CHA molecular testing and that WES and WGS could represents the future [75]. Our results show that molecular diagnosis with WES could easily be democratized and be of great help to understand patients’ phenotype, adapting therapeutic approach (such as splenectomy) and to allow genetic counseling. Indeed, exomic approach appeared as particularly useful in UH cases and for complex phenotypes.
Conclusion
This work emphasizes the usefulness of WES in CHA in order to reach a right diagnosis for each patient. This allows clinical geneticists to provide a personally fitted genetic counseling and specialized clinicians to adjust treatment in some cases. When first-line phenotypic analyses have not been successful in elucidating the disease cause, genetic tests using NGS and especially WES sequencing appears as very helpful to uncover the intricate genetic defects causing CHA and associated manifestations. The recent developments of exomic or genomic sequencing technologies make them the most suitable and cheapest approaches for the genetic diagnosis of these disorders, keeping in mind the absolute necessity of pluridisciplinar teams able to deal with incidental or unexpected findings if needed.
Democratization of NGS should lead to update the current recommendations and diagnosis strategy concerning the place of genetic testing in CHA on Fig. 3. The frequent discovery of several mutations in different genes in a same patient also suggests to reconsider the genetic bases of CHA: apparently monogenic diseases may in fact be oligogenic, with satellite variations acting as modulators of the phenotype or resulting in new clinical entities.
Fig. 3.

Diagnosis strategy proposition for CHA
Deeper mechanistic studies are warranted to better understand the relationship between genotype and phenotype, especially in patients with atypical or poorly described variants association and to explain VUS. However, some interactions may prove difficult to investigate with currently existing tools. The development of next generation phenotypic tools, such as RBC metabolomics and proteomics, may represent new steps in the exploration of RBC genetic disorders and key tools for variant interpretation.
Supplementary information
Acknowledgements
We thank Ms. Cécile Aubrun for her help in patients’ assessment.
Disclosures of conflicts of interest
none
Abbreviations
- AMD
Age-Related Macular Degeneration
- AR
Autosomic recessive
- AD
Autosomic dominant
- CHA
Congenital hemolytic anemia
- DHSt
Dehydrated hereditary stomatocytosis
- GFF
Glomerular filtration flow
- HS
Hereditary spherocytosis
- HPP
Hereditary pyropoïkilocytosis
- HE
Hereditary elliptocytosis
- MDS
Myelodysplastic syndrome
- RBC
Red blood cell
- MPE
Membrane protein electrophoresis
- MPN
Myeloproliferative neoplasms
- NGS
Next generation sequencing
- TPI
Triose phosphate isomerase
- UH
Unexplained hemolysis
- aHUS
Atypical hemolytic and uremic syndrome
- VUS
Variant of uncertain significance
- WES
Whole exome sequencing
- WGS
Whole genome sequencing
- XLD
X-linked disorder
Authors’ contributions
L.M.H. analyzed and validated genetic data and wrote the paper; A.A. developed and performed bioinformatic analyses and reviewed the paper; B.B. performed EMA tests, examinations of blood smears and reviewed the paper; M.S. was in charge of enzymatic activities measurement; C.G. and V.O. performed genetic tests; O.W.B. performed EMA tests and reviewed the paper; S.P. was responsible for sickle cell disease genetic exploration; V.P. performed ektacytometry analysis and reviewed the paper; K.G. made membrane proteins electrophoresis; M.B. made enzymatic activities measurement and designed panel genes list; C.G. Z.M., M.D., A.P., N.C.C. and M.M. provided patients for study; P.B. provided patients for study and technical support for research; S.M. supervised enzymatic activities measurement and hemoglobin studies; B.F. supervised the entire study, validated genetic data and wrote the paper; F.G. supervised the entire study, provided patients and reviewed the paper. The author(s) read and approved the final manuscript.
Funding
No funding.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
All patients cited in the text have filled consent form for genetic analysis and consent to participate to this study.
Consent for publication
Consent for publication has been obtained.
Competing interests
No competing of interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13023-020-01425-5.
References
- 1.Gallagher PG. Diagnosis and management of rare congenital nonimmune hemolytic disease. ASH Educ Program Book. 2015;2015(1):392–399. doi: 10.1182/asheducation-2015.1.392. [DOI] [PubMed] [Google Scholar]
- 2.Gallagher PG. Hereditary elliptocytosis: spectrin and protein 4.1R. Semin Hematol. 2004;41(2):142–164. doi: 10.1053/j.seminhematol.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Mohandas N, Gallagher PG. Red cell membrane: past, present, and future. Blood. 2008;112(10):3939–3948. doi: 10.1182/blood-2008-07-161166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallagher PG. Abnormalities of the erythrocyte membrane. Pediatr Clin N Am. 2013;60(6):1349–1362. doi: 10.1016/j.pcl.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andolfo I, Russo R, Gambale A, Iolascon A. New insights on hereditary erythrocyte membrane defects. Haematologica. 2016;101(11):1284–1294. doi: 10.3324/haematol.2016.142463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372(9647):1411–1426. doi: 10.1016/S0140-6736(08)61588-3. [DOI] [PubMed] [Google Scholar]
- 7.Glele-Kakai C, Garbarz M, Lecomte MC, Leborgne S, Galand C, Bournier O, et al. Epidemiological studies of spectrin mutations related to hereditary elliptocytosis and spectrin polymorphisms in Benin. Br J Haematol. 1996;95(1):57–66. doi: 10.1046/j.1365-2141.1996.d01-1869.x. [DOI] [PubMed] [Google Scholar]
- 8.Picard V, Guitton C, Thuret I, Rose C, Bendelac L, Ghazal K, et al. Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos-channelopathy: a retrospective series of 126 patients. Haematologica. 2019;104(8):1554–1564. doi: 10.3324/haematol.2018.205328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prchal JT, Gregg XT. Red cell enzymes. ASH Educ Program Book. 2005;2005(1):19–23. doi: 10.1182/asheducation-2005.1.19. [DOI] [PubMed] [Google Scholar]
- 10.Bianchi P, Fermo E, Glader B, Kanno H, Agarwal A, Barcellini W, et al. Addressing the diagnostic gaps in pyruvate kinase deficiency: consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am J Hematol. 2019;94(1):149–161. doi: 10.1002/ajh.25325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iolascon A, Esposito MR, Russo R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. Haematologica. 2012;97(12):1786–1794. doi: 10.3324/haematol.2012.072207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gambale A, Iolascon A, Andolfo I, Russo R. Diagnosis and management of congenital dyserythropoietic anemias. Expert Rev Hematol. 2016;9(3):283–296. doi: 10.1586/17474086.2016.1131608. [DOI] [PubMed] [Google Scholar]
- 13.King M-J, Garçon L, Hoyer JD, Iolascon A, Picard V, Stewart G, et al. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015;37(3):304–325. doi: 10.1111/ijlh.12335. [DOI] [PubMed] [Google Scholar]
- 14.Hamada M, Doisaki S, Okuno Y, Muramatsu H, Hama A, Kawashima N, et al. Whole-exome analysis to detect congenital hemolytic anemia mimicking congenital dyserythropoietic anemia. Int J Hematol. 2018;108(3):306–311. doi: 10.1007/s12185-018-2482-7. [DOI] [PubMed] [Google Scholar]
- 15.Niss O, Chonat S, Dagaonkar N, Almansoori MO, Kerr K, Rogers ZR, et al. Genotype-phenotype correlations in hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood Cells Mol Dis. 2016;61:4–9. doi: 10.1016/j.bcmd.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russo R, Andolfo I, Manna F, Gambale A, Marra R, Rosato BE, et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am J Hematol. 2018;93(5):672–682. doi: 10.1002/ajh.25058. [DOI] [PubMed] [Google Scholar]
- 17.Averbuch NS, Steinberg-Shemer O, Dgany O, Krasnov T, Noy-Lotan S, Yacobovich J, et al. Targeted next generation sequencing for the diagnosis of patients with rare congenital anemias. Eur J Haematol. 2018;101(3):297–304. doi: 10.1111/ejh.13097. [DOI] [PubMed] [Google Scholar]
- 18.Barreto RDO, Arrizabalaga B, la Hoz ABD, García-Orad Á, Tejada MI, Garcia-Ruiz JC, et al. Detection of new pathogenic mutations in patients with congenital haemolytic anaemia using next-generation sequencing. Int J Lab Hematol. 2016;38(6):629–638. doi: 10.1111/ijlh.12551. [DOI] [PubMed] [Google Scholar]
- 19.Agarwal AM, Nussenzveig RH, Reading NS, Patel JL, Sangle N, Salama ME, et al. Clinical utility of next-generation sequencing in the diagnosis of hereditary haemolytic anaemias. Br J Haematol. 2016;174(5):806–814. doi: 10.1111/bjh.14131. [DOI] [PubMed] [Google Scholar]
- 20.Roy NBA, Wilson EA, Henderson S, Wray K, Babbs C, Okoli S, et al. A novel 33-gene targeted resequencing panel provides accurate, clinical-grade diagnosis and improves patient management for rare inherited anaemias. Br J Haematol. 2016;175(2):318–330. doi: 10.1111/bjh.14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kedar PS, Gupta V, Dongerdiye R, Chiddarwar A, Warang P, Madkaikar MR. Molecular diagnosis of unexplained haemolytic anaemia using targeted next-generation sequencing panel revealed (p.Ala337Thr) novel mutation in GPI gene in two Indian patients. J Clin Pathol. 2019;72(1):81–85. doi: 10.1136/jclinpath-2018-205420. [DOI] [PubMed] [Google Scholar]
- 22.Moreno Carralero M-I, Horta Herrera S, Morado Arias M, Ricard Andrés M-P, Lemes Castellano A, Abio Calvete M, et al. Clinical and genetic features of congenital dyserythropoietic anemia (CDA) Eur J Haematol. 2018;101(3):368–378. doi: 10.1111/ejh.13112. [DOI] [PubMed] [Google Scholar]
- 23.Aydin Koker S, Karapinar T, Oymak Y, Bianchi P, Fermo E, Gozmen S, et al. Identification of a novel mutation in the SEC23B gene associated with congenital dyserythropoietic anemia type II through the use of next-generation sequencing panel in an undiagnosed case of nonimmune hereditary hemolytic anemia. J Pediatr Hematol Oncol. 2018;40(7):e421–e423. doi: 10.1097/MPH.0000000000001207. [DOI] [PubMed] [Google Scholar]
- 24.Wang R, Yang S, Xu M, Huang J, Liu H, Gu W, et al. Exome sequencing confirms molecular diagnoses in 38 Chinese families with hereditary spherocytosis. Sci China Life Sci. 1 2018;61(8):947–53. [DOI] [PubMed]
- 25.Gallagher PG, Maksimova Y, Lezon-Geyda K, Newburger PE, Medeiros D, Hanson RD, et al. Aberrant splicing contributes to severe α-spectrin–linked congenital hemolytic anemia. J Clin Invest. 2019;129(7):2878–2887. doi: 10.1172/JCI127195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han JH, Kim S, Jang H, Kim SW, Lee MG, Koh H, et al. Identification of a novel p.Q1772X ANK1 mutation in a Korean family with hereditary spherocytosis. PLOS ONE. 2015;10(6):e0131251. [DOI] [PMC free article] [PubMed]
- 27.Lacy JN, Ulirsch JC, Grace RF, Towne MC, Hale J, Mohandas N, et al. Exome sequencing results in successful diagnosis and treatment of a severe congenital anemia. Mol Case Stud. 2016;2(4):a000885. doi: 10.1101/mcs.a000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khurana M, Edwards D, Rescorla F, Miller C, He Y, Sierra Potchanant E, et al. Whole-exome sequencing enables correct diagnosis and surgical management of rare inherited childhood anemia. Mol Case Stud. 2018;4(5):a003152. doi: 10.1101/mcs.a003152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim M, Park J, Lee J, Jang W, Chae H, Choi H, et al. Hemolytic anemia with null PKLR mutations identified using whole exome sequencing and cured by hematopoietic stem cell transplantation combined with splenectomy. Bone Marrow Transplant. 2016;51(12):1605–1608. doi: 10.1038/bmt.2016.218. [DOI] [PubMed] [Google Scholar]
- 30.Errichiello E, Vetro A, Mina T, Wischmeijer A, Berrino E, Carella M, et al. Whole exome sequencing in the differential diagnosis of diamond-Blackfan anemia: clinical and molecular study of three patients with novel RPL5 and mosaic RPS19 mutations. Blood Cells Mol Dis. 2017;64:38–44. doi: 10.1016/j.bcmd.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin P-C, Chiou S-S, Lin C-Y, Wang S-C, Huang H-Y, Chang Y-S, et al. Whole-exome sequencing for the genetic diagnosis of congenital red blood cell membrane disorders in Taiwan. Clin Chim Acta. 2018;487:311–317. doi: 10.1016/j.cca.2018.10.020. [DOI] [PubMed] [Google Scholar]
- 32.Ittiwut C, Natesirinilkul R, Tongprasert F, Sathitsamitphong L, Choedamphai C, Fanhchaksai K, et al. Novel mutations in SPTA1 and SPTB identified by whole exome sequencing in eight Thai families with hereditary pyropoikilocytosis presenting with severe fetal and neonatal anaemia. Br J Haematol. 2019;185(3):578–582. doi: 10.1111/bjh.15559. [DOI] [PubMed] [Google Scholar]
- 33.Bartolucci P, Brugnara C, Teixeira-Pinto A, Pissard S, Moradkhani K, Jouault H, et al. Erythrocyte density in sickle cell syndromes is associated with specific clinical manifestations and hemolysis. Blood. 2012;120(15):3136–3141. doi: 10.1182/blood-2012-04-424184. [DOI] [PubMed] [Google Scholar]
- 34.King M-J, Telfer P, MacKinnon H, Langabeer L, McMahon C, Darbyshire P, et al. Using the eosin-5-maleimide binding test in the differential diagnosis of hereditary spherocytosis and hereditary pyropoikilocytosis. Cytometry B Clin Cytom. 2008;74B(4):244–250. doi: 10.1002/cyto.b.20413. [DOI] [PubMed] [Google Scholar]
- 35.Girodon F, Garçon L, Bergoin E, Largier M, Delaunay J, Fénéant-Thibault M, et al. Usefulness of the eosin-5′-maleimide cytometric method as a first-line screening test for the diagnosis of hereditary spherocytosis: comparison with ektacytometry and protein electrophoresis. Br J Haematol. 2008;140(4):468–470. doi: 10.1111/j.1365-2141.2007.06944.x. [DOI] [PubMed] [Google Scholar]
- 36.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.On behalf of the ACMG Laboratory Quality Assurance Committee. Richards S, Aziz N, Bale S, Bick D, Das S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100(2):267–280. doi: 10.1016/j.ajhg.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delaunay J, Nouyrigat V, Proust A, Schischmanoff P-O, Cynober T, Yvart J, et al. Different impacts of alleles αLEPRA and αLELY as assessed versus a novel, virtually null allele of the SPTA1 gene in trans. Br J Haematol. 2004;127(1):118–122. doi: 10.1111/j.1365-2141.2004.05160.x. [DOI] [PubMed] [Google Scholar]
- 40.Barasa B, Slijper M. Challenges for red blood cell biomarker discovery through proteomics. Biochim Biophys Acta. 2014;1844(5):1003–1010. doi: 10.1016/j.bbapap.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Gautier E-F, Leduc M, Cochet S, Bailly K, Lacombe C, Mohandas N, et al. Absolute proteome quantification of highly purified populations of circulating reticulocytes and mature erythrocytes. Blood Adv. 2018;2(20):2646–2657. doi: 10.1182/bloodadvances.2018023515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hegedűs T, Chaubey PM, Várady G, Szabó E, Sarankó H, Hofstetter L, et al. Inconsistencies in the red blood cell membrane proteome analysis: generation of a database for research and diagnostic applications. Database (Oxford). 2015;2015:bav056.. [DOI] [PMC free article] [PubMed]
- 43.Dhermy D, Galand C, Bournier O, King M-J, Cynober T, Roberts I, et al. Coinheritance of α- and β-Spectrin gene mutations in a case of hereditary elliptocytosis. Blood. 1998;92(11):4481–4482. [PubMed] [Google Scholar]
- 44.Christensen RD, Nussenzveig RH, Reading NS, Agarwal AM, Prchal JT, Yaish HM. Variations in both α-Spectrin (SPTA1) and β-Spectrin (SPTB) in a neonate with prolonged jaundice in a family where nine individuals had hereditary elliptocytosis. Neonatology. 2014;105(1):1–4. doi: 10.1159/000354884. [DOI] [PubMed] [Google Scholar]
- 45.Darbellay R, Mach Pascual S, Rose K, Graf J, Beris P. Haemoglobin Tunis-Bizerte: a new a globin 129 Leu–> pro unstable variant with thalassaemic phenotype. Br J Haematol. 1995;90(1):71–76. doi: 10.1111/j.1365-2141.1995.tb03382.x. [DOI] [PubMed] [Google Scholar]
- 46.Vulliamy TJ, D’Urso M, Battistuzzi G, Estrada M, Foulkes NS, Martini G, et al. Diverse point mutations in the human glucose-6-phosphate dehydrogenase gene cause enzyme deficiency and mild or severe hemolytic anemia. Proc Natl Acad Sci. 1988;85(14):5171–5175. doi: 10.1073/pnas.85.14.5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fakhouri F, Roumenina L, Provot F, Sallée M, Caillard S, Couzi L, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol JASN. 2010;21(5):859–867. doi: 10.1681/ASN.2009070706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Servais A, Noël L-H, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey M-A, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82(4):454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 49.Yu Y, Triebwasser MP, Wong EKS, Schramm EC, Thomas B, Reynolds R, et al. Whole-exome sequencing identifies rare, functional CFH variants in families with macular degeneration. Hum Mol Genet. 2014;23(19):5283–5293. doi: 10.1093/hmg/ddu226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oh ST, Simonds EF, Jones C, Hale MB, Goltsev Y, Gibbs KD, et al. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood. 2010;116(6):988–992. doi: 10.1182/blood-2010-02-270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 52.Bekri S. A promoter mutation in the erythroid-specific 5-aminolevulinate synthase (ALAS2) gene causes X-linked sideroblastic anemia. Blood. 2003;102(2):698–704. doi: 10.1182/blood-2002-06-1623. [DOI] [PubMed] [Google Scholar]
- 53.Mohlin FC, Nilsson SC, Levart TK, Golubovic E, Rusai K, Müller-Sacherer T, et al. Functional characterization of two novel non-synonymous alterations in CD46 and a Q950H change in factor H found in atypical hemolytic uremic syndrome patients. Mol Immunol. 2015;65(2):367–376. doi: 10.1016/j.molimm.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 54.Mah W, Sonkusare SK, Wang T, Azeddine B, Pupavac M, Carrot-Zhang J, et al. Gain-of-function mutation in TRPV4 identified in patients with osteonecrosis of the femoral head. J Med Genet. 2016;53(10):705–709. doi: 10.1136/jmedgenet-2016-103829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelley M, Joshi N, Xie Y, Borgaonkar M. Iron overload is rare in patients homozygous for the H63D mutation. Can J Gastroenterol Hepatol. 2014;28(4):198–202. doi: 10.1155/2014/468521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barbara K-H, Marcin L, Jedrzej A, et al. The impact of H63D HFE gene carriage on hemoglobin and iron status in children. Ann Hematol. 2016;95(12):2043–2048. doi: 10.1007/s00277-016-2792-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bardawil T, Rebeiz A, Chaabouni M, et al. Mutations in the ABCG8 gene are associated with sitosterolaemia in the homozygous form and xanthelasmas in the heterozygous form. Eur J Dermatol. 2017;27(5):519–523. doi: 10.1684/ejd.2017.3087. [DOI] [PubMed] [Google Scholar]
- 58.Gilligan DM, Lozovatsky L, Gwynn B, Brugnara C, Mohandas N, Peters LL. Targeted disruption of the beta-adducin gene (Add2) causes red blood cell spherocytosis in mice. Proc Natl Acad Sci U S A. 1999;96(19):10717–10722. doi: 10.1073/pnas.96.19.10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nivaggioni V, Léonnet C, Ebbo M, Grados A, Schleinitz N, Picard V, et al. Multiple splenic infarctions in a malagasy patient whith both southeast asian ovalocytosis and a sickle cell trait. Hématologie. 2016;7–8;(4):284–287.
- 60.Favale F, Gardembas M, Pajot O, Saada V, Fénéant-Thibault M, Delaunay J, et al. Southeast Asian ovalocytosis and a sickle cell trait in a young patient with sudden retinal stroke: a fortuitous association? Hemoglobin. 2009;33(6):475–479. doi: 10.3109/03630260903344440. [DOI] [PubMed] [Google Scholar]
- 61.Ravindranath Y, Goyette G, Johnson R. Southeast Asian ovalocytosis in an African-American family. Blood. 1994;84:2823–2824. [PubMed] [Google Scholar]
- 62.Ma S, Cahalan S, LaMonte G, Grubaugh ND, Zeng W, Murthy SE, et al. Common PIEZO1 allele in African populations causes RBC dehydration and attenuates Plasmodium infection. Cell. 2018;173(2):443–455. doi: 10.1016/j.cell.2018.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White JPM, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I. TRPV4: molecular conductor of a diverse orchestra. Physiol Rev. 2016;96(3):911–973. doi: 10.1152/physrev.00016.2015. [DOI] [PubMed] [Google Scholar]
- 64.Nilius B, Voets T. The puzzle of TRPV4 channelopathies. EMBO Rep. 2013;14(2):152–163. doi: 10.1038/embor.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mah W, Sonkusare SK, Wang T, Azeddine B, Pupavac M, Carrot-Zhang J, et al. Gain-of-function mutation in TRPV4 identified in patients with osteonecrosis of the femoral head. J Med Genet. 2016;53:705–709. doi: 10.1136/jmedgenet-2016-103829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thibodeau ML, Peters CH, Townsend KN, Shen Y, Hendson G, Adam S, et al. Compound heterozygous TRPV4 mutations in two siblings with a complex phenotype including severe intellectual disability and neuropathy. Am J Med Genet A. 2017;173(11):3087–3092. doi: 10.1002/ajmg.a.38400. [DOI] [PubMed] [Google Scholar]
- 67.Wang Q, Khillan J, Gadue P, Nishikura K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science. 2000;290(5497):1765–1768. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- 68.Liddicoat BJ, Hartner JC, Piskol R, Ramaswami G, Chalk AM, Kingsley PD, et al. Adenosine-to-inosine RNA editing by ADAR1 is essential for normal murine erythropoiesis. Exp Hematol. 2016;44(10):947–963. doi: 10.1016/j.exphem.2016.06.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ansai O, Shigehara Y, Ito A, Abe R, Shimomura Y. A novel splice site mutation in the ADAR gene leading to exon skipping and dyschromatosis symmetrica hereditaria in a Japanese patient. Clin Exp Dermatol. 2016;41(8):933–934. doi: 10.1111/ced.12981. [DOI] [PubMed] [Google Scholar]
- 70.Rice GI, Kitabayashi N, Barth M, Briggs TA, Burton ACE, Carpanelli ML, et al. Genetic, phenotypic, and interferon biomarker status in ADAR1-related neurological disease. Neuropediatrics. 2017;48(3):166–184. doi: 10.1055/s-0037-1601449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He Y, Jia S, Dewan RK, Liao N. Novel mutations in patients with hereditary red blood cell membrane disorders using next-generation sequencing. Gene. 2017;627:556–562. doi: 10.1016/j.gene.2017.07.009. [DOI] [PubMed] [Google Scholar]
- 72.Seidelmann SB, Smith E, Subrahmanyan L, Dykas D, Ziki MDA, Azari B, et al. The application of whole exome sequencing in the clinical diagnosis and management of inherited cardiovascular diseases in adults. Circ Cardiovasc Genet. 2017;10(1):e001573. doi: 10.1161/CIRCGENETICS.116.001573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nambot S, Thevenon J, Kuentz P, Duffourd Y, Tisserant E, Bruel AL, et al. Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet Med Off J Am Coll Med Genet. 2018;20(6):645–654. doi: 10.1038/gim.2017.162. [DOI] [PubMed] [Google Scholar]
- 74.Kim Y, Park J, Kim M. Diagnostic approaches for inherited hemolytic anemia in the genetic era. Blood Res. 2017;52(2):84. doi: 10.5045/br.2017.52.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rets A, Clayton AL, Christensen RD, Agarwal AM. Molecular diagnostic update in hereditary hemolytic anemia and neonatal hyperbilirubinemia. Int J Lab Hematol. 2019;41(Suppl. 1):95–101. doi: 10.1111/ijlh.13014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.