

Abstract
Intracerebral microdialysis has proven useful for chemical monitoring in patients following traumatic brain injury. Recent studies in animals, however, have documented that insertion of microdialysis probes into brain tissues initiates a foreign-body response. Within a few days after probe insertion, the foreign body response impedes the use of microdialysis to monitor the K+ and glucose transients associated with spreading depolarization, a potential mechanism for secondary brain injury. Herein, we show that perfusing microdialysis probes with dexamethasone, a potent anti-inflammatory glucocorticoid, suppresses the foreign body response and facilitates the monitoring of spontaneous spreading depolarizations for at least 10 days following controlled cortical injury in the rat. In addition to spreading depolarizations, results of this study suggest that a progressive, apparently permanent, decline in pericontusional interstitial glucose may be an additional sequela of brain injury. This study establishes extended dexamethasone-enhanced microdialysis in the injured rodent cortex as a new paradigm for investigating trauma-induced metabolic crisis.
Keywords: Microdialysis, dexamethasone, biosensor, microfluidic, spreading depolarization, controlled cortical impact
INTRODUCTION
Traumatic brain injury (TBI) has emerged as a major health crisis and a leading cause of death in the younger populations of many countries, including the US.1, 2 Severe TBI (sTBI) often involves a primary and secondary injury. The primary injury is the acute mechanical trauma, which in severe cases might require surgical intervention. Secondary injury, which likely involves several mechanisms, results in the post-traumatic expansion of the brain lesion over the course of several days, and possibly weeks, into the vulnerable surrounding tissues, sometimes called the ischemic penumbra.3–9 Diagnosis of secondary injury in real time is challenging.
Real-time intracerebral chemical monitoring with clinical microdialysis has been used to detect episodes of post-TBI metabolic crisis, possibly associated with the phenomenon of spreading depolarization (SD).8–16 Due to the progressive nature of secondary injury, sTBI patients require extended monitoring times, up to 10 days in prior studies.17–19 However, during a recent study in the rat cortex, it became essentially impossible to monitor induced SDs with conventional microdialysis by 5 days after inserting the probes.20 Based on histochemical analyses, we attributed the difficulty to the foreign-body response of brain tissue initiated by insertion of the microdialysis probes.20–25 The foreign-body response includes vasoconstriction, activation of microglia, infiltration of macrophages, and activation of astrocytes, and a loss of nearby neurons and terminals.20–26 Immunostaining for the glial fibrillary acidic protein (GFAP) shows that astrocyte endfeet begin to engulf the probe track by 24 hrs after probe insertion. A fully formed glial barrier surrounds the probe tracks by 5 days after insertion.21, 22 Our working hypothesis is that the glial barrier impedes the detection of SDs, possibly by inhibiting diffusion of substances to the probe from the surrounding tissues, even though basal dialysate glucose levels, possibly derived from the glial barrier, remain relatively stable.
Dexamethasone (Dex) is a glucocorticoid with potent anti-inflammatory properties.27, 28 Recent studies in the rat striatum and cortex show that perfusing microdialysis probes with low-micromolar concentrations of Dex (also called Dex retrodialysis) effectively suppresses the foreign-body response to probe insertion.20–25, 29 Dex retrodialysis eliminates ischemia, glial activation, glial barrier formation, and neuronal losses in the tissues surrounding the probe track. Dex also facilitates reliable monitoring of induced SDs in the rat cortex at 1, 5, and 10 days after probe insertion. Interestingly, even though we stopped Dex perfusion 5 days after probe insertion, reliable SD monitoring at 10 days (5 days after Dex) was preserved.20
The objective of the present study was to investigate whether the Dex-enhanced microdialysis is translatable to the monitoring of spontaneous SDs in rats after controlled cortical impact (CCI), a widely studied rodent model of TBI.30–32 CCI and probe insertion were followed by 10 days (12 days in one case) of continuous perfusion with only brief interruptions to refill the perfusion syringe. Consistent with our previously developed protocol, the perfusion fluid contained 10-μM Dex on the first day, 2-μM Dex on days 2–5, and no Dex thereafter.20, 23, 25, 26 The outlet of the probe was connected in series to a K+ ion selective electrode (K+ ISE) and then to a rapid sampling microdialysis (rsMD) glucose analyzer.7, 33, 34 Although the probes were continuously perfused after insertion, the dialysate stream was not monitored “24/7”. Typically, we monitored the dialysate K+ and glucose concentrations for blocks of 10–15 hrs, with brief (typically <1 hr) interruptions to replace and calibrate the sensors as-needed (probe perfusion was not interrupted during calibration). We report herein recordings of K+ and glucose via Dex-enhanced microdialysis over the course of 10 days (12 days in one case) following CCI and probe insertion.
RESULTS AND DISCUSSION
Summary of observations
We recorded K+ and glucose in dialysates from 10 rats over the course of 10–12-days following CCI and microdialysis probe insertion. Recordings from n=8 rats exhibited 185 spontaneous SDs, hallmarked by a transient rise and fall in dialysate K+, typically over the course of 5–7 min. All SDs were spontaneous, i.e. none of them were induced by the pin-prick method of our previous work.20 Of the SDs observed while we were also monitoring glucose (n=126), some were accompanied by negative glucose transients (n=89), no obvious change in glucose (n=25), or transient increases in glucose (n=12). In one rat, SDs were observed as late as 12 days after CCI. Some rats (n=2) exhibited no SDs, which seems to be consistent with clinical reports that SDs are detected in some but not all TBI patients who undergo neuromonitoring.4, 8, 16 We have not observed spontaneous SDs in any CCI-naïve control rats (n=3) in this and prior study (n=31).20 Table 1 provides a summary of observations from each subject.
Table 1.
A summary of observations from each individual rat in this study.
| Probe location* | # of SDs observed | Glucose ND (hr) | SDs per hr observation | Notes | |
|---|---|---|---|---|---|
| Rat 1 | 1 mm | 58 | N/A | 3.149 | Rat broke the probe about 18 hours after implantation – until then, SDs nearly constant |
| Rat 2 | 1 mm | 12 | 81 | 0.067 | |
| Rat 3 | 1 mm | 11 | N/A | 0.070 | Large fluctuations in glucose concentrations over time |
| Rat 4 | 1 mm | 0 | 29 | 0 | |
| Rat 5 | 1 mm | 0 | 56 | 0 | |
| Rat 6 | 3 mm | 19 | 284 | 0.140 | SDs continued until day 12, stopping a few hours before glucose reached ND level |
| Rat 7 | 3 mm | 19 | 164 | 0.336 | |
| Rat 8 | 3 mm | 7 | 43 | 0.064 | Abnormal movements, presumably seizures, correlated with K+ fluctuations after glucose reached ND level |
| Rat 9 | 3 mm | 20 | 37 | 0.183 | |
| Rat 10 | 1 mm | 39 | 106 | 0.359 |
Probe location is the distance from the edge of the CCI lesion to the probe track: there was no systematic effect of probe location. The column titled Glucose ND (hr) gives the time, in hours from the CCI, when glucose declined to the non-detectable level.
We also observed a second post-CCI phenomenon consisting of a slow, progressive decline of dialysate glucose from basal to, eventually, concentrations below the detection limit of the rsMD glucose analyzer (<10 μM, see Methods). Once this occurred, glucose concentrations did not return to detectable levels. The onset of the glucose decline was highly variable between rats: in one case, it occurred on day 12 after CCI. In some cases, after glucose declined, the K+ level steadily increased, indicating a loss of K+ homeostasis. Occasional SDs were observed after glucose declined. We have not observed this glucose decline in any CCI-naïve control rats. Figures 1–5, below, report K+ in purple and glucose in red.
Figure 1.
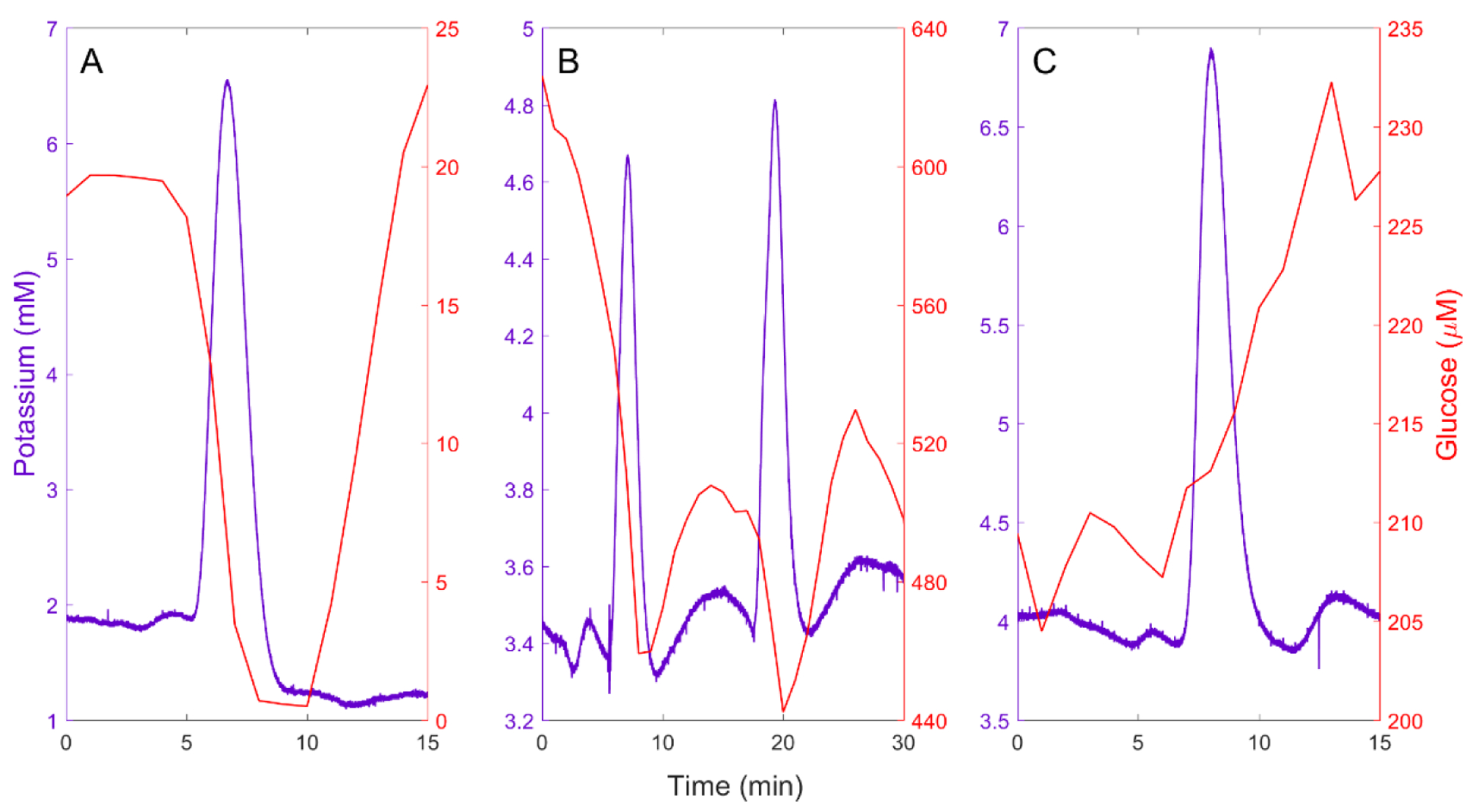
A) An isolated spontaneous SD accompanied by a negative glucose transient. B) A cluster of two closely spaced spontaneous SDs: the second SD triggers a negative glucose transient before the complete recovery of the prior negative glucose transient. C) A spontaneous SD accompanied by an increase in glucose, possibly indicating a change in blood flow.
Figure 5.

Spontaneous SDs before and after glucose reaches the ND level. A) Rat 9 exhibited SDs when K+ was relatively stable over time. B) Rat 10 had SDs as K+ exhibits a progressive rise. C) Rat 8 had numerous small-amplitude K+ fluctuations superimposed on a relatively high K+ background. The animal exhibited unusual movements during this time block. We assume the animal was having seizures. Arrows indicate spontaneous SDs.
Individual observations: spontaneous spreading depolarizations
Figure 1 contains representative examples of the spontaneous SDs observed in the injured rat cortex. Figure 1 clearly shows the SD profile consisting of a K+ transient lasting 5–7 minutes. Figure 1A shows a single, isolated SD accompanied by a negative glucose transient. The fall in glucose, which is coincident to the rise in K+, reflects the energy demand associated with tissue repolarization after the SD. Figure 1B shows two SDs about 10 min apart, which qualifies them as a SD cluster.35, 36 SD clusters are of special interest because, as in this example, the second SD might force a second negative glucose transient superimposed on the first one. Another noteworthy difference between Figures 1A and 1B is the magnitude of the glucose data: in Figure 1A, the glucose baseline is near 20 μM and is forced to “zero” (i.e. non-detectable) by the SD whereas in Figure 1B the glucose baseline is near 600 μM and only partially declines after the SDs. The origin of this wide variation in the glucose concentration is discussed further, below. Figure 1C shows a SD accompanied by a rise in glucose, possibly indicating a change of blood flow.
Figure 1 illustrates the potential value of microdialysis in the diagnosis of secondary injury. The K+ signal identifies SDs, as does electrocorticography (ECoG), a widely used neuromonitoring tool.5–7, 10, 20, 34, 37 However, ECoG and related recording techniques do not provide real time chemical information that might prove useful in identifying which SDs trigger glucose declines (Figures 1A and 1B) and which do not (Figure 1C). This is potentially important information because the glucose decline, a form of metabolic crisis that might deprive cells of the energy they need for basic survival, has been identified as a potential mechanism for secondary injury.12, 38–43
Figure 2 shows representative recordings of dialysate K+ and glucose over hours-long time blocks, from separate rats, at various times after probe insertion (spontaneous SDs are marked with arrows). The data in Figures 2A–E were obtained on day 1 of the recording session. Figures 2A and 2B show cases where multiple SDs and SD clusters were accompanied by negative glucose transients. Figure 2A also shows a likely seizure (asterisk). In the case of Figure 2C, no SDs were observed and the K+ and glucose concentrations were consistent with the normal basal range (370±40 μM, mean±SEM) from CCI-naïve animals.20 The data in Figures 2D and 2E exhibit SDs isolated in time and not part of a cluster. In Figure 2D the SDs are accompanied either by an increase or no change in the glucose level. In Figure 2E, the SDs are accompanied by negative glucose transients. The data in Figures 2F and 2G were collected 10–12 days after probe insertion. Figure 2F shows an isolated SD, confirming the potential of Dex-enhanced microdialysis to record SDs 12 days after injury: in this case, due to the low glucose baseline (explained below) it is unclear whether this SD caused a glucose transient. The data in Figure 2G are from a CCI-naïve, control rat: we have observed no spontaneous SDs in any CCI-naïve animals (this study, n=3: Varner et al., n=3120). In this case, K+ and glucose dropped below normal levels at hour 235.
Figure 2.
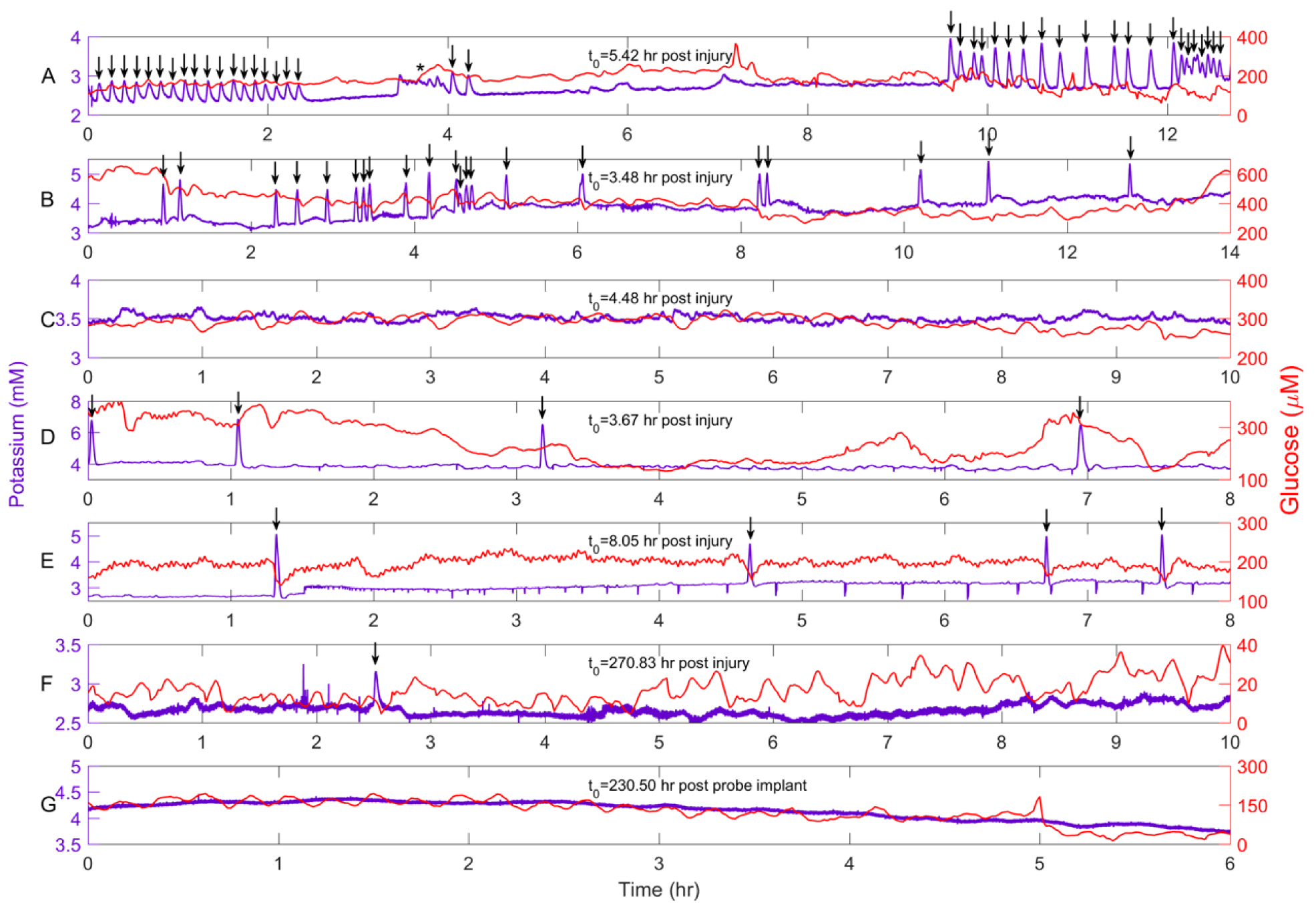
Representative recordings of dialysate K+ (purple) and glucose (red) from seven different rats. The data in Panels A-E were collected within the first 24 hr after CCI and probe insertion. A and B) Rats 1 and 10 exhibited numerous SDs and SD clusters, some of which were accompanied by negative glucose transients. C) Rat 4 exhibited no SDs. D and E) Rats 2 and 6 exhibited isolated SDs. F) Rat 6 exhibited a spontaneous SD 12 days after CCI and probe insertion. G) CCI-naïve control rats do not exhibit spontaneous SDs. Arrows mark spontaneous SDs.
Individual observations: the progressive glucose decline
All but two rats in the CCI group (one of which broke his probe, see Methods and Table 1) exhibited a progressive decline in dialysate glucose concentration (Figure 3). The glucose concentration declined from the normal basal range over a period of several hours, eventually falling below the detection limit of the rsMD glucose analyzer (<10 μM, see Methods). In Figure 3, the glucose level at the start of the time block is near 10% of the normal basal range (370±40 μM),20 so this recording shows the final stage of the decline to the nondetectable (ND) level (in the figures, “Glucose ND” means nondetectable glucose). Furthermore, in this case, the K+ trace is featureless, exhibiting no SDs and a relatively stable signal (see below for examples of the other K+ responses). With Dex-enhanced microdialysis, we have observed no such glucose declines in any CCI-naïve control animals (n total = 34).
Figure 3.
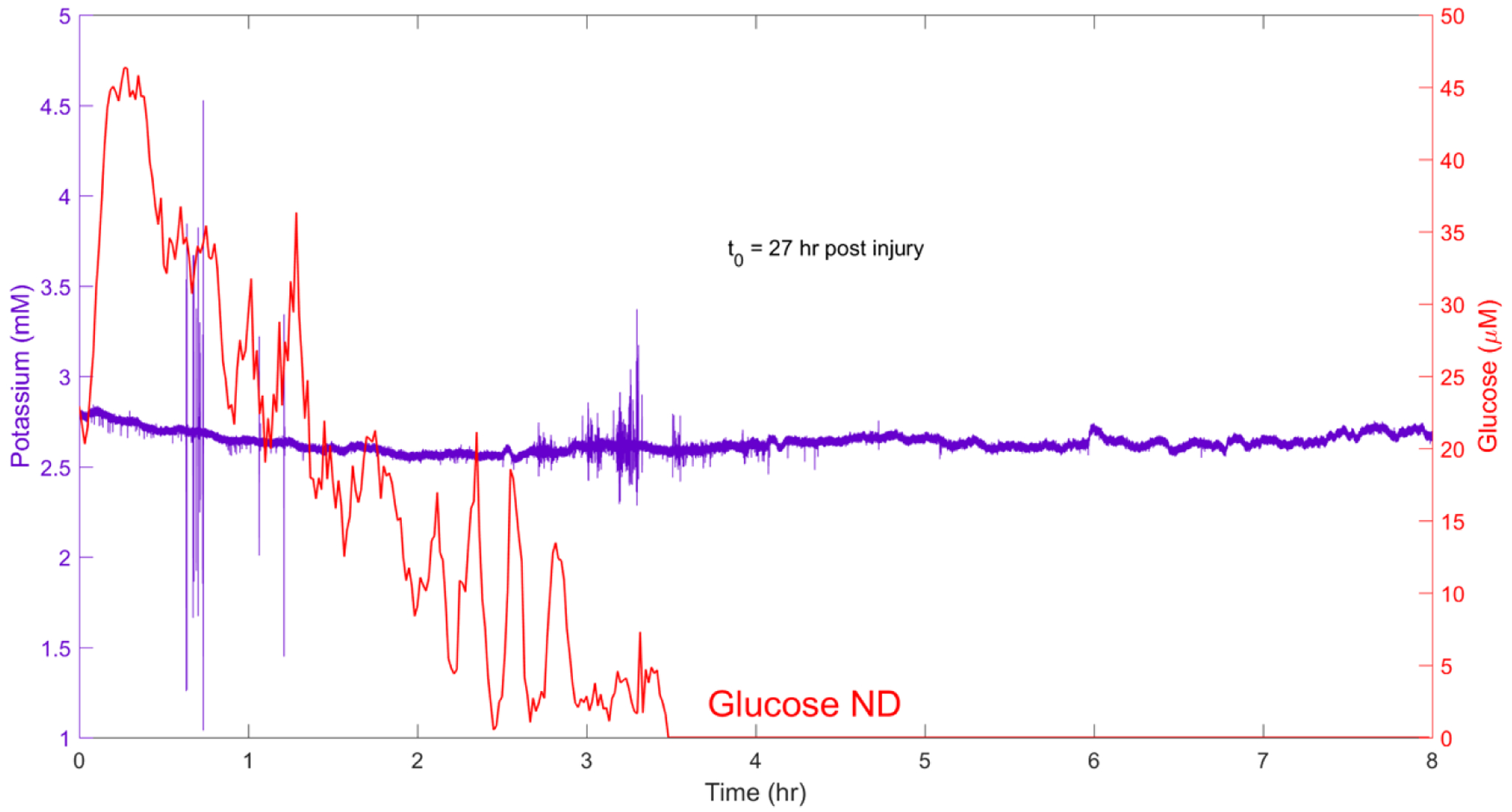
A representative example of the post-CCI progressive glucose decline from Rat 4. “Glucose ND” means non-detectable glucose below the detection limit of the rsMD glucose analyzer. In this case, SDs were not observed and the dialysate K+ concentration remained relatively stable.
Four of the eight rats exhibiting the progressive glucose decline also exhibited an accompanying rise in dialysate K+, indicating a loss of K+ homeostasis (Figure 4). In one case (Figure 4A), the K+ concentration rose suddenly when glucose finally reached the detection limit. In other cases (Figures 4B and 4C), K+ rose progressively as the glucose concentration declined. An isolated spontaneous SD that occurred while glucose was low, but still detectable, forced glucose transiently to the ND level (Figure 4C: see Figure 5A for another example). Typically, the glucose concentration hovered for several hours at a few percent of the normal basal range before eventually falling below the detection limit (Figures 4A and 4C). Two rats exhibited “huge” spikes of glucose, into the hundreds-of-micromolar range, just prior to glucose reaching the detection limit (e.g. Figure 4A): the origin of these rare spikes is unknown.
Figure 4.
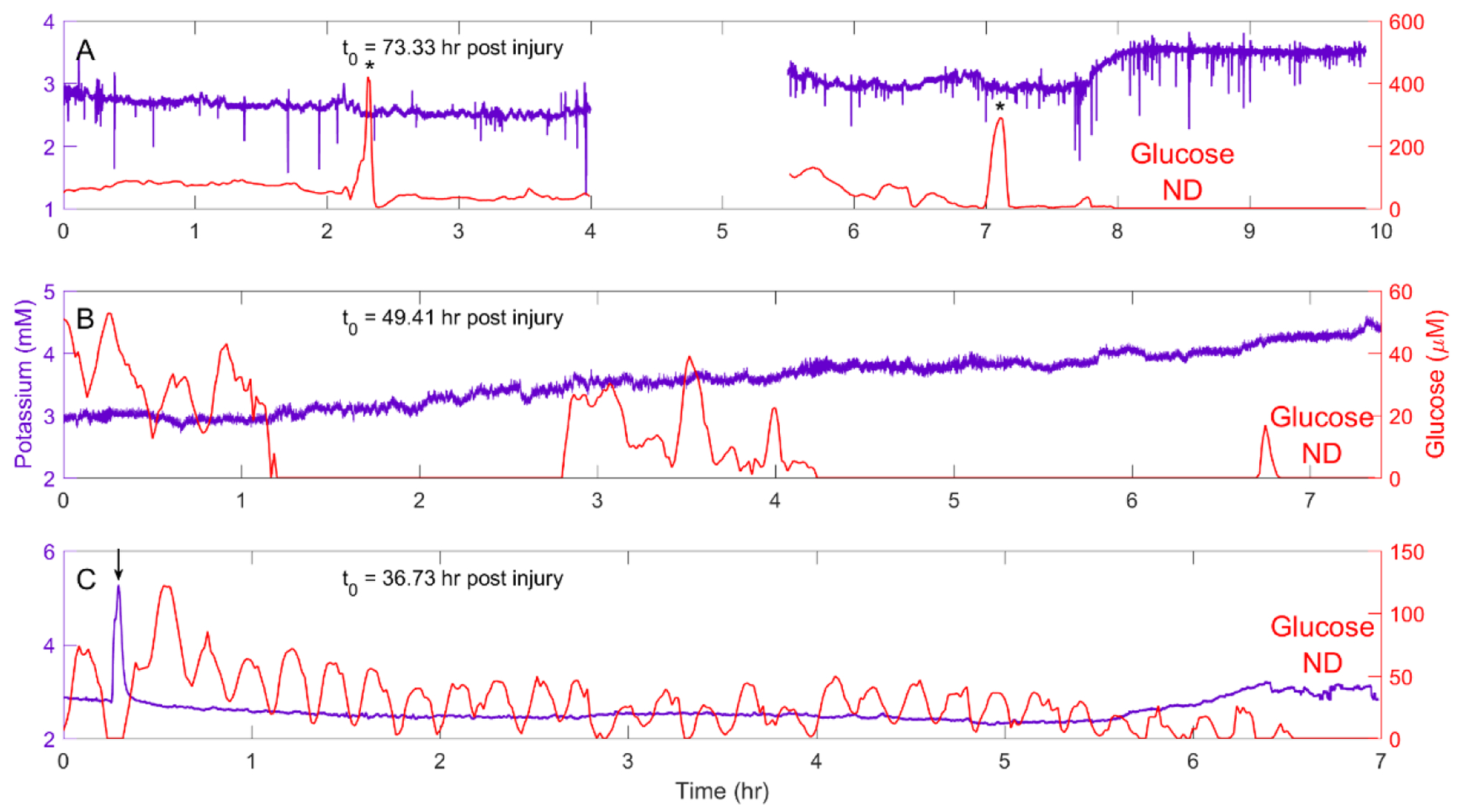
Examples of the progressive glucose decline accompanied by a loss of K+ homeostasis. A) Rat 2 exhibited rare but “huge” glucose spikes during the progressive glucose declines (asterisks). When glucose eventually fell below the detectable level (at 7.8 hr), K+ exhibited a sudden rise (the gap in the data occurred because it was necessary to refill the perfusion syringe.) B) Rat 5 exhibited a progressive glucose decline accompanied by a progressive rise of K+. C) Rat 8 exhibited a progressive rise in K+ just as glucose reaches the ND level: a spontaneous SD (arrow) forces glucose to fall from its already low level to ND.
Isolated SDs occurred during and after the progressive glucose decline (Figure 4C and Figures 5A and 5B). These SDs occurred regardless of whether the K+ concentration was relatively stable (Figure 5A) or exhibited a progressive rise (Figure 5B) once glucose reached the ND level. In one case, several days after glucose reached the ND level, K+ exhibited numerous small-amplitude fluctuations superimposed on an unusually high background (Figure 5C). These fluctuations lasted for several hours, during which the animal exhibit repeated abnormal movements of the head, trunk, and forelimbs. We assume the animal was having seizures.
Immunohistochemistry
At 10 or 12 days after CCI and probe insertion, the animals were re-anesthetized, the probes were carefully extracted, and the animals were perfused with saline and paraformaldehyde. Visual inspection of the intact brains after removal from the skull immediately showed a wide animal-to-animal variability in the severity of the injury. In some animals the injury was massive, with ablation of both cortical and subcortical tissues, while in others the brain surface was just dimpled (photographs are included in the Supplemental Information document). The three most severely injured rats (Rats X, Ym and Z) had to be excluded from further histochemical studies because it was impossible to prepare thin sections of their tissue. In these animals, the vasculature was sufficiently disrupted that tissue fixation failed. The results below are representative of the remaining 7 rats.
The brain tissues from 7 of the 10 rats were sliced (30-μm sections) in the plane perpendicular to the microdialysis probe, and processed for immunohistochemistry. Figure 6 shows a representative DIC image of a CCI lesion with an adjacent probe track: a separate panel shows the probe track at higher magnification. The CCI lesion appears as a hole surrounded by a darkened, presumably necrotic border. Figure 7A shows fluorescence images of two probe tracks. The markers in this image are for blood vessels (nanobeads), macrophages (ED-1), astrocytes (GFAP), and neurons (NeuN). The left and right columns show probe tracks placed 1 and 3 mm, respectively, from the CCI lesion: as mentioned in the Methods section, these placements had no apparent systematic impact on the outcome of any of the measurements reported herein. Essentially, these probe tracks are similar in appearance to those of Dex-perfused probes in CCI-naïve animals reported in our previous studies.20, 22, 23, 26, 44 Quantitative analysis of blood vessel and GFAP images of probe tracks from CCI and CCI-naïve animals found no statistically significant difference (Figure 7B). Specifically, the images show intact blood vessels, minimal glial activation, and no evidence of a glial barrier. These findings confirm that Dex retrodialysis is an effective means of mitigating the foreign-body response to the insertion of microdialysis probes into the injured rat cortex. We emphasize again that these images show tissues 10 days after CCI and probe insertion but 5 days after halting Dex retrodialysis.
Figure 6.

DIC image of a thin section of brain tissue passing through the CCI lesion and the track of a microdialysis probe: the image on the right shows the probe track at higher magnification.
Figure 7.
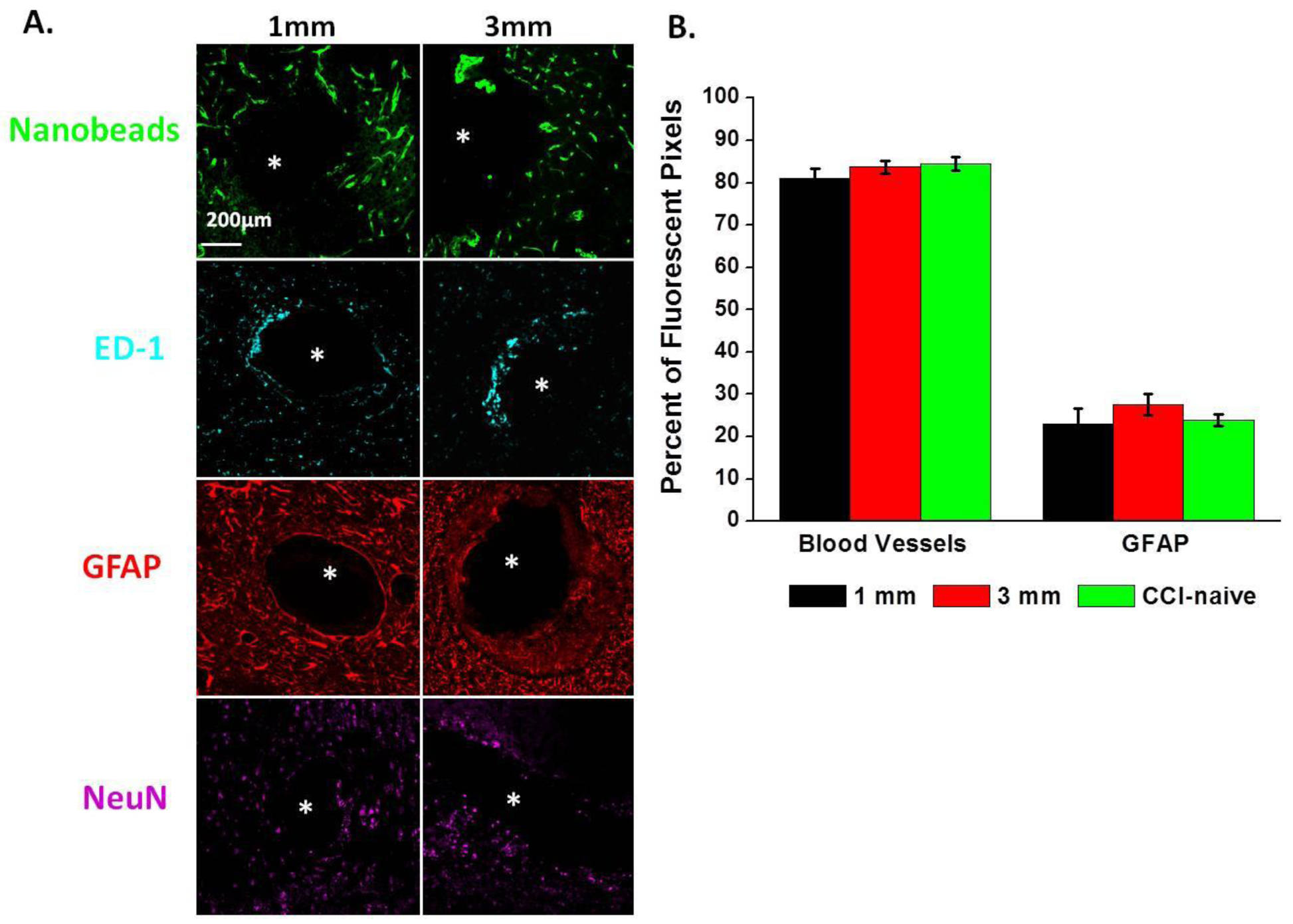
A. Images of two probe tracks from the CCI group (Rat X and Y) showing markers for blood vessels (nanobeads), macrophages (ED-1), astrocytes (GFAP), and neurons (NeuN). The left and right columns show probe tracks located 1 and 3 mm, respectively, from the CCI lesion. B. Quantitative analysis of blood vessel and GFAP images of probe tracks from CCI and CCI-naïve control animals: results are reported as percent of fluorescent pixels (mean ± SEM, n=x rats (y images per rat). No significant quantitative differences between CCI and controls, t-test).
Figure 8 shows images of the border of the CCI lesion (see Figure 6). These images show that the border is a zone of ischemic tissue that lacks signs of blood flow (nanobeads), contains activated macrophages and glia, with few if any surviving neurons. The images do not show any clear signs that Dex delivered via the microdialysis probe had any mitigating effect on the CCI lesion, such as a local reduction in ED-1 or GFAP or increase in blood flow or NeuN. Thus, Dex appears to have acted only to mitigate the foreign-body response of the brain tissue to the probe insertion, which is consistent with our previous observation of Dex diffusing no more than about 100 μm from microdialysis probes.26 This is important in the context of future efforts to translate Dex-enhanced microdialysis to the clinical setting, where the goal will be to monitor, rather than treat, the injured brain.
Figure 8.

Fluorescence microscopy of the boundary region of the CCI lesion. The markers show (A) macrophages (ED-1), (B) astrocytes (GFAP), (C) neurons (NeuN), (D) blood vessels (nanobeads), and (E) nuclei (DAPI): a white dotted line has been added to (D) to indicate the location of the tissue boarder. These images show that the boarder of the CCI lesion is an ischemic zone with activated glia and a loss of neurons.
Dialysate analysis by MALDI-MS
We wished to confirm that the progressive decline in dialysate glucose level (Figures 3–5) is not caused by an unexpected loss in permeability of the microdialysis membrane, due perhaps to increased inflammation in the injured tissue. To this end, we collected dialysate samples for offline analysis by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS). Samples were collected from control rats (no CCI) on days 1 and 10 after probe insertion and from CCI-injured rats before and after the progressive glucose decline: Figure 9 reports representative mass spectra from each of the four experimental conditions. The spectra all show that the dialysate contains brain-derived species in the m/z range of 300–1500, whereas the molecular weight of glucose is 180 Da. These spectra confirm that the microdialysis membranes remain permeable to a range of species in both control and CCI-treated rats. Consistent with the histology of the probe tracks, these data further verify the anti-inflammatory efficacy of Dex-retrodialysis in both control and CCI-treated cortex.
Figure 9.
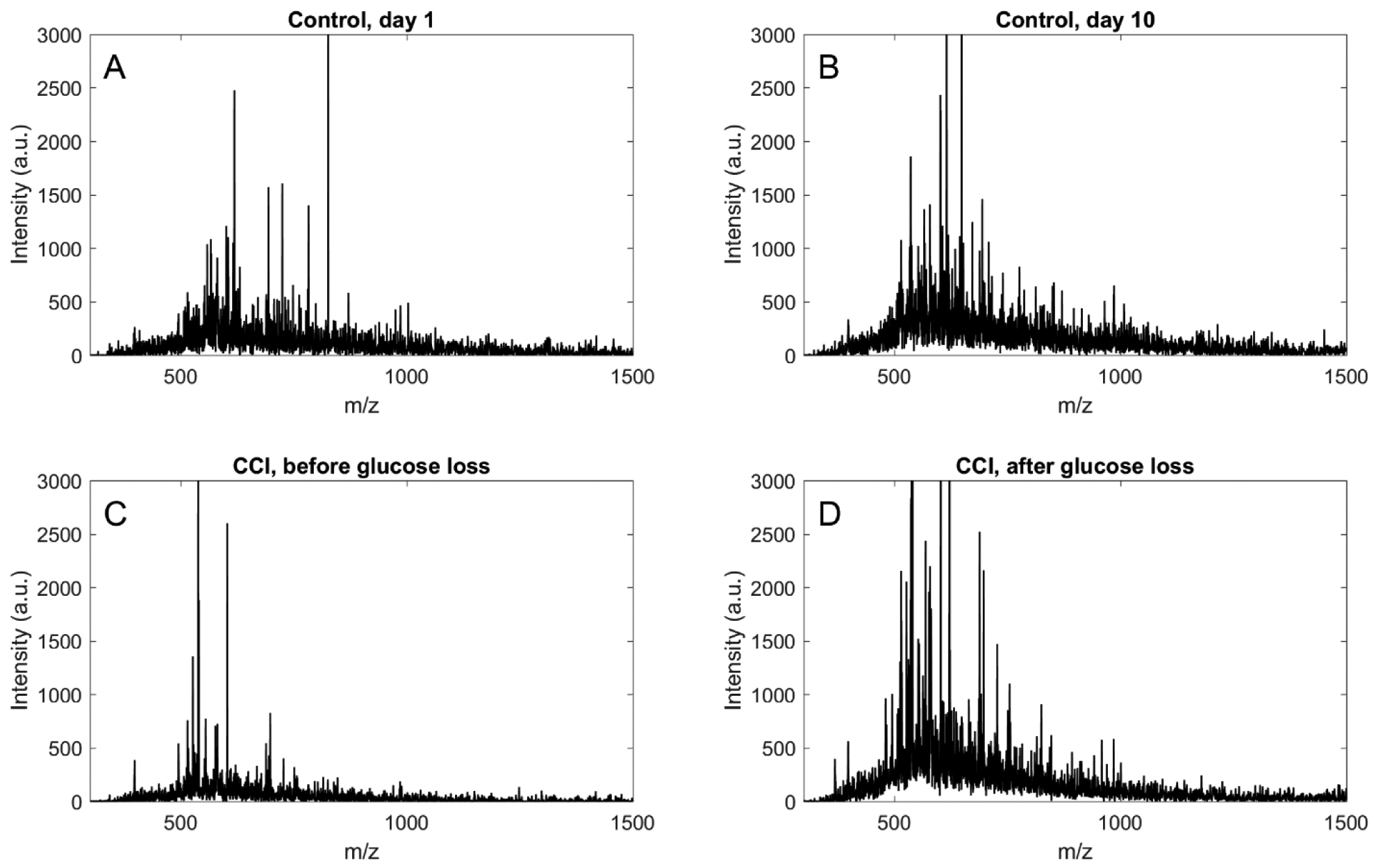
MALDI-MS spectra of dialysate samples collected from control rats (no CCI) on days 1 (A) and 10 (B) after probe insertion and from CCI rats before (C) and after (D) the progressive decline in dialysate glucose levels.
CONCLUSIONS
The findings of this study confirm the successful translation of Dex-enhanced microdialysis to the CCI rodent model of TBI. Specifically, Dex retrodialysis effectively suppressed the foreign-body response of brain tissue to probe insertion adjacent to a CCI lesion, thereby facilitating the pericontusional monitoring of K+ and glucose during the course of 10–12 days post-CCI. Perfusion of the probe was continuous over this time interval. K+ and glucose levels were monitored for 10–15 hr time blocks but not “24/7”.
We found a high degree of animal-to-animal variability in the outcome of chemical monitoring after CCI. Individual animals varied widely in the number and frequency of SDs and the onset and duration of progressive glucose decline. We tentatively attribute this to the varied extent of the injury induced by CCI. Whereas the CCI per se was tightly controlled, the resultant injury was not. In some animals, the injury was confined to the cortex but in others it penetrated into subcortical structures, with variability in the overall volume of ablated tissue. We positioned probes at 1 and 3 mm from the edge of the impact site (Table 1) in anticipation of observing some sign that secondary injury spreads over time into the penumbra. But, this did not happen. Two of the animals with probes at 3 mm exhibited the latest onset of progressive glucose decline, but two others exhibited onsets similar to those in animals with probes at the 1 mm location (Table 1). Given the variability in the extent of injury, it is likely that our study was too small to reliably detect differences between the 1 and 3 mm location.
Despite the animal-to-animal variability, this study yielded several consistent observations. Most rats (8 of 10) exhibited spontaneous SDs, either isolated or in clusters. It is not altogether surprising that no SDs were observed in two subjects. First, clinical neuromonitoring detects SDs in only about 60% of TBI patients, suggesting that SDs may not be a universal sequela of TBI.45 Of course, the incidence of SDs likely affected by the severity of TBI, which was variable in this study. Second, it is important to acknowledge that the microdialysis probe only monitors at a single tissue location, so it is possible that SDs occurred at remote locations. Numerous spontaneous SDs were accompanied by negative glucose transients, which have become a subject of intense interest as a potential mechanism of SD-associated secondary injury.14, 18, 46, 47,48 However, some SDs appeared to have no effect on glucose or were accompanied by an increase in glucose possibly caused by a change in blood flow.12, 41 Discriminating between SDs that do and do not induce metabolic crisis could be of clinical importance. A second consistent observation, but apparently independent of SDs, was the progressive decline in dialysate glucose output, which occurred in all CCI rats except two, one of which prematurely broke its probe (Table 1). The decline started at various times after the CCI, 12 days after CCI in one case, and typically lasted for several hours until glucose concentrations eventually became too low to detect. Once glucose reached the nondetectable level, it did not return to detectable levels at a later time. Spontaneous SDs occurred before, during, and after the glucose decline. In one animal, the glucose decline occurred in the absence of SDs. In approximately half the cases, the decline in glucose was accompanied by a rise in K+, suggesting a failure of K+ homeostasis. Although additional data would be desirable to confirm this, SDs and the progressive declines in glucose appeared to be independent sequalae of CCI.
Vespa et al. also observed dramatic declines in glucose by microdialysis in TBI patients.19, 49 In contrast to our observations, in which glucose fell to non-detectable levels in 8 of 10 rats, Vespa et al. reported this in 6 of their 30 patients.19, 49 Moreover, Vespa et al. reported that the decline was associated with terminal herniation, whereas our CCI protocol induced no terminal events. Additionally, it is important to mention that Vespa et al. intentionally avoided pericontusional probe locations, whereas we placed the probes adjacent to the CCI lesions. These findings, and their contrast with those of Vespa, might suggest that a progressive glucose decline may be a common but non-terminal event in ischemic penumbral zones and might signal a local metabolic crisis involved in secondary injury and expansion of the CCI lesion.
We wish to be duly cautious in interpreting the progressive glucose decline. We must consider the possibility that it is related to the permeability of the microdialysis membrane. Whereas data from control animals shows that microdialysis probes continue to recover glucose from the brain for up to 10 days, it is possible that CCI triggers a more intense inflammatory response to probe insertion, resulting a loss of glucose recovery. Several observations speak against this possibility. First, even in animals exhibiting the glucose decline, examination of the probe tracks did not show the presence of a glial barrier, absence of blood flow, or profound losses of neurons: these observations indicate the anti-inflammatory efficacy of Dex in the presence of CCI. Second, even after the progressive decline in glucose we continued to detect K+, including increases in K+ levels, SDs, and seizures: this confirms that the probes remain permeable to K+. Third, MALDI-MS did not indicate systematic differences in composition of the dialysate under four conditions (without CCI on days 1 and 8, with CCI before and after the glucose decline of glucose). The MALDI data indicate that the probes remain permeable to a range of brain-derived species, including species with higher molecular weight than glucose. These observations support the conclusion that the progressive decline in glucose is not attributable to a loss in permeability of the microdialysis membrane.
The loss of detectable glucose in the dialysate comes as something of a surprise, considering that our histological studies indicate blood flow and NeuN-positive neurons at the probe track. It is important to mention that the absence of detectable glucose in the dialysate, while indicating a glucose aberration, does not necessarily imply an absence of glucose in the tissue. Dialysate levels of any substance, including glucose, reflect the balance between its delivery and removal from the interstitial space. Thus, a low dialysate glucose concentration indicates that glucose consumption (nonzero) in the tissue near the probe exceeds delivery via the vasculature, leaving insufficient glucose to be recovered by the microdialysis probe. Explaining whether the apparent imbalance is due to increased consumption (e.g. glycolysis) or decreased delivery (e.g. ischemia) is not possible with the data we have in hand: this will be the subject of a future report.
This study adds to a mounting body of evidence that Dex retrodialysis facilitates extended intracranial microdialysis in the rat brain over the course of at least 10, and possibly 12, days with a single probe insertion. The use of Dex in this manner represents a technical advance, given our previous difficulty with monitoring evoked dopamine release and pin-prick-induced SDs 5 days after probe insertion without Dex.20, 23, 25, 26 We attribute these difficulties to the foreign-body response, rather than to probe failure per se. Dex retrodialysis offers the advantage of a single probe insertion as an alternative to extended microdialysis based on removal and replacement of multiple probes.50 In the clinical setting, for example, removing and replacing multiple probes would not be medically practical.
In this work, Dex retrodialysis was combined with rapid measurements by the K+ ISE and the rsMD glucose analyzer.6, 7, 10, 20, 33, 51 This has facilitated real time monitoring of spontaneous SDs at 10 and, in one case 12, days after CCI and probe insertion. Although clinical 10-day microdialysis post injury has been reported before, those prior studies used 1-hr sampling times, which are much longer than SDs.5–7, 9–11, 33, 38, 51, 52 Thus, herein we have documented a microdialysis protocol that uniquely combines extended monitoring with high temporal resolution. This study introduces extended Dex-enhanced microdialysis in the CCI rodent model of TBI as a new paradigm for investigating metabolic crisis in the injured brain.
METHODS
Reagents, solutions, and materials
All solutions were made with ultrapure water (Nanopure; Barnstead, Dubuque, IA). Artificial cerebrospinal fluid (aCSF) contained 142 mM NaCl, 1.2 mM CaCl2, 2.7 mM KCl, 1.0 mM MgCl2, and 2.0 mM NaH2PO4 (all from Sigma-Aldrich) adjusted to a pH of 7.4. Dexamethasone sodium phosphate (APP Fresenius Kabi USA, LLC, Lake Zurich IL) was prepared in aCSF at 10 μM and 2 μM. Microdialysis perfusion fluids were filtered with 0.2 μm pore size Nalgene sterile filters (Fisher, Pittsburgh, PA; PES). Glucose oxidase (100–200 units/mg) and horseradish peroxidase (≥250 units/mg) were obtained from Sigma-Aldrich. Ferrocene solution was prepared with 1.5 mM ferrocenecarboxylic acid, 1 mM EDTA, 150 mM sodium chloride and 100 mM sodium citrate and was filtered through 0.1 and 0.02 μm pore size filters.
Microdialysis
In-house concentric style microdialysis probes were built using hollow fiber membranes (13 kDa MWCO, Specta/Por RC, Spectrum, Ranco Dominguez, CA), 280 μm in outer diameter and 4 mm in length. Fused silica capillaries (75 μm ID, 150 μm OD, Polymicro Technologies, Phoenix, AZ) were used for the inlet and outlet lines. Probes were soaked in 70% ethanol prior to use, and then flushed with aCSF with DEX for several hours prior to implantation into the rat brain. The probe inlet was connected to a 1 mL gastight syringe driven by a microliter syringe pump (Harvard Apparatus, Holliston, MA) operating at 1.67 μL/min. The probe outlet (1.6 m long) was connected first to the K+ ISE and then to the rsMD glucose analyzer: both systems have been described in full detail in previous papers. Data were collected with LabChart (AD Instruments) and analyzed with published algorithms in MATLAB (Mathworks Inc). The K+ and glucose analyzers were calibrated at least daily. The glucose oxidase beds in the rsMD analyzer were changed at least daily, meaning that during the course of this study we used over one hundred glucose oxidase beds. Each one was calibrated with 25, 50, 125, 500, and 1000 μM glucose standard solutions. During this work, glucose was defined as non-detectable (see Results and Discussion) when the rsMD response was less than 3X the baseline noise of the detector: with this criterion, we could often quantify glucose concentrations down to the single-digit micromolar range (see righthand axes of Figures 1–5). When dialysate glucose levels reached the rsMD detection limit (see Results and Discussion) the glucose sensor was calibrated to confirm its function. In addition, monitoring was continued with a new enzyme bed.
Surgical procedures and experimental protocol
All experiments involving animals were approved by the University of Pittsburgh’s Institutional Animal Care and Use Committee. Male Sprague-Dawley rats, 250–275 g (Charles River, Raleigh, NC) were anesthetized with isoflurane (5% initially, 2.5% for maintenance and 2:1 N2O:O2) and placed in a stereotaxic frame. While anesthetized, the rats were wrapped in a homeothermic blanket. A CCI device (Pittsburgh Precision Instruments, Inc. Pittsburgh, PA, USA) was used to create a model of mild to moderate TBI. A craniotomy was performed on the right hemisphere posterior to bregma, and the exposed dura was impacted at an average velocity of 3.9 ± 0.3 m/s, a 100 ms dwell time, to a depth of 2.2 mm. The removed bone was not replaced. The microdialysis probe was inserted 1 mm or 3 mm anterior to the craniotomy at a 50° angle so that the entire dialysis membrane was placed into the cortex. We observed no systematic effect of the 1- or 3-mm probe location, so data from the two locations were pooled. The probe was secured with bone screws and dental cement, and the incision was closed with sterile sutures. Following surgery, the rats were housed in a Raturn Microdialysis Bowl (MD-1404, BASI, West Lafayette, IN) with ad libitum access to food and water. The probes were continuously perfused with 10-μM DEX for the first 24 hr, followed by 2-μM DEX for days 2–5, and aCSF (no Dex) for the remainder of the experiment. Microdialysis monitoring was performed in a total of 10 rats. In 8 rats, microdialysis perfusion continued for 10 days. In one rat, at the end of 10 days, both the K+ and glucose levels were stable and at normal baseline levels, so monitoring was extended to 12 days. The first rat in this study broke the connections to the microdialysis probe 18 hrs after surgery: up to that point, the experiment had been progressing normally, so the data are included in this report (Rat #1, Table 1). Subsequent rats were fitted with an Elizabethan collar (MF-53721, BASi, West Lafayette, IN). Three CCI-naïve, control rats underwent the same 10-day Dex-enhanced microdialysis protocol as just described: we ran 31 CCI-naïve control rats in our prior study.20
Immunohistochemistry and fluorescence microscopy
The tissue processing and imaging procedures are fully described in our previous papers.22, 23, 26 Briefly, rats were anesthetized with isoflurane (2.5% by volume O2) and transcardially perfused with 200 mL 0.01 M of pH 4 phosphate-buffered saline (PBS), 160 mL of 4% paraformaldehyde, and 50 mL of PBS containing 100-nm fluorescent beads (Molecular Probes, 1:1000 dilution in PBS). The brain and probe were removed and further fixed in 4% paraformaldehyde for 24 h at 4 °C. The tissue containing the probe track was cut to 35-μm thin horizontal sections (3 slices per antibody per animal). Free floating sections were rinsed in PBS (3 × 10 min), blocked (3% goat serum in PBS containing 0.1% Triton X-100) for 1 h at room temperature, and immunostained for 24 hours at 4 °C. Primary antibodies were anti-glial fibrillary acidic protein (GFAP; 1:100, DAKO Agilent Technologies), NeuN (1:100, Chemicon, Temecula, CA), and ED-1 (1:100, AbD Serotec, Raleigh, NC). Sections were washed with PBS (10 min) and incubated in 3% goat serum, 0.1% Triton X-100, and 1:500 goat anti-rabbit Alexa 568 (Invitrogen, Carlsbad CA) for 2 h at room temperature. Sections were rinsed with PBS (10 min) and cover-slipped with Fluoromount-G (Southern Biotech, Birmingham AL). Sections were imaged with a FluoView 1000 (Olympus, Inc., Tokyo, Japan) at 20× magnification.
MALDI-MS
Samples of dialysate were collected offline and frozen at −20C until use. For MALDI analysis, samples were mixed with matrix in a 5:1 matrix to sample ratio. The matrix solution was a 10 mg/mL solution of sinapinic acid (Sigma-Aldrich) in 50:50 water and acetonitrile with 1% difluoroacetic acid. Samples were plated with the dry drop method and run on a Bruker Daltonics UltrafleXtreme MALDI TOF-TOF instrument equipped with a Nd:YAG laser (Bruker, Billerica, MA). Samples were run in reflectron positive ion mode with 10 consecutive laser shots.
Acknowledgement
This work was funded by the National Institutes of Health (R01NS102725, R21NS109875) and the University of Pittsburgh Center for Biological Imaging (1S10RR028478-01).
Abbreviations
- TBI
traumatic brain injury
- sTBI
severe traumatic brain injury
- SD
spreading depolarization
- GFAP
glial fibrillary acidic protein
- Dex
dexamethasone
- CCI
controlled cortical impact
- ISE
ion selective electrode
- rsMD
rapid sampling microdialysis
- ECoG
electrocorticography
- ED-1
anti-CD68 antibody
- NeuN
neuron-specific nuclear protein
- MALDI
matrix-assisted laser desorption/ionization
- MS
mass spectrometry
- aCSF
artificial cerebrospinal fluid
- PBS
phostphate-buffered saline
- EDTA
ethylene diamine tetra-acetic acid
Footnotes
The authors declare no competing interests.
References
- 1.Bruns J Jr.; Hauser WA, The epidemiology of traumatic brain injury: a review. Epilepsia 2003, 44 (s10), 2–10. [DOI] [PubMed] [Google Scholar]
- 2.Faul M; Xu L; Wald MM; Coronado V, Traumatic brain injury in the United States Atlanta, GA: national Center for injury Prevention and Control, Centers for disease Control and Prevention 2010. [Google Scholar]
- 3.Dohmen C; Sakowitz OW; Fabricius M; Bosche B; Reithmeier T; Ernestus RI; Brinker G; Dreier JP; Woitzik J; Strong AJ, Spreading depolarizations occur in human ischemic stroke with high incidence. Annals of neurology 2008, 63 (6), 720–728. [DOI] [PubMed] [Google Scholar]
- 4.Dreier JP; Fabricius M; Ayata C; Sakowitz OW; Shuttleworth CW; Dohmen C; Graf R; Vajkoczy P; Helbok R; Suzuki M, Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: review and recommendations of the COSBID research group. Journal of Cerebral Blood Flow & Metabolism 2016, 0271678X16654496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feuerstein D; Manning A; Hashemi P; Bhatia R; Fabricius M; Tolias C; Pahl C; Ervine M; Strong AJ; Boutelle MG, Dynamic metabolic response to multiple spreading depolarizations in patients with acute brain injury: an online microdialysis study. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2010, 30 (7), 1343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkin M; Hopwood S; Jones DA; Hashemi P; Landolt H; Fabricius M; Lauritzen M; Boutelle MG; Strong AJ, Dynamic changes in brain glucose and lactate in pericontusional areas of the human cerebral cortex, monitored with rapid sampling on-line microdialysis: relationship with depolarisation-like events. Journal of Cerebral Blood Flow & Metabolism 2005, 25 (3), 402–413. [DOI] [PubMed] [Google Scholar]
- 7.Rogers ML; Feuerstein D; Leong CL; Takagaki M; Niu X; Graf R; Boutelle MG, Continuous online microdialysis using microfluidic sensors: dynamic neurometabolic changes during spreading depolarization. ACS chemical neuroscience 2013, 4 (5), 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartings JA; Watanabe T; Bullock MR; Okonkwo DO; Fabricius M; Woitzik J; Dreier JP; Puccio A; Shutter LA; Pahl C, Spreading depolarizations have prolonged direct current shifts and are associated with poor outcome in brain trauma. Brain 2011, awr048. [DOI] [PubMed] [Google Scholar]
- 9.Strong AJ; Boutelle MG; Vespa PM; Bullock MR; Bhatia R; Hashemi P, Treatment of critical care patients with substantial acute ischemic or traumatic brain injury. Crit. Care Med 2005, 33 (9), 2147–9; author reply 2149. [DOI] [PubMed] [Google Scholar]
- 10.Bhatia R; Hashemi P; Razzaq A; Parkin MC; Hopwood SE; Boutelle MG; Strong AJ, Application of rapid-sampling, online microdialysis to the monitoring of brain metabolism during aneurysm surgery. Neurosurgery 2006, 58 (4), ONS-313-ONS-321. [DOI] [PubMed] [Google Scholar]
- 11.Córcoles EP; Boutelle MG, Biosensors and invasive monitoring in clinical applications. Springer: 2013. [Google Scholar]
- 12.de Lima Oliveira M; Kairalla AC; Fonoff ET; Martinez RCR; Teixeira MJ; Bor-Seng-Shu E, Cerebral microdialysis in traumatic brain injury and subarachnoid hemorrhage: state of the art. Neurocritical care 2014, 21 (1), 152–162. [DOI] [PubMed] [Google Scholar]
- 13.Griesdale DE; Tremblay M-H; McEwen J; Chittock DR, Glucose control and mortality in patients with severe traumatic brain injury. Neurocritical care 2009, 11 (3), 311–316. [DOI] [PubMed] [Google Scholar]
- 14.Guiou M; Sheth S; Nemoto M; Walker M; Pouratian N; Ba A; Toga AW, Cortical spreading depression produces long-term disruption of activity-related changes in cerebral blood volume and neurovascular coupling. Journal of biomedical optics 2005, 10 (1), 011004–0110047. [DOI] [PubMed] [Google Scholar]
- 15.Haddad SH; Arabi YM, Critical care management of severe traumatic brain injury in adults. Scandinavian journal of trauma, resuscitation and emergency medicine 2012, 20 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartings JA; Bullock MR; Okonkwo DO; Murray LS; Murray GD; Fabricius M; Maas AI; Woitzik J; Sakowitz O; Mathern B, Spreading depolarisations and outcome after traumatic brain injury: a prospective observational study. The Lancet Neurology 2011, 10 (12), 1058–1064. [DOI] [PubMed] [Google Scholar]
- 17.Hartings JA; Rolli ML; Lu X-CM; Tortella FC, Delayed Secondary Phase of Peri-Infarct Depolarizations after Focal Cerebral Ischemia: Relation to Infarct Growth and Neuroprotection. The Journal of Neuroscience 2003, 23 (37), 11602–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seule M; Keller E; Unterberg A; Sakowitz O, The hemodynamic response of spreading depolarization observed by near infrared spectroscopy after aneurysmal subarachnoid hemorrhage. Neurocritical care 2015, 23 (1), 108–112. [DOI] [PubMed] [Google Scholar]
- 19.Vespa P; Bergsneider M; Hattori N; Wu H-M; Huang S-C; Martin NA; Glenn TC; McArthur DL; Hovda DA, Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. Journal of Cerebral Blood Flow & Metabolism 2005, 25 (6), 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varner EL; Leong CL; Jaquins-Gerstl A; Nesbitt KM; Boutelle MG; Michael AC, Enhancing Continuous Online Microdialysis Using Dexamethasone: Measurement of Dynamic Neurometabolic Changes during Spreading Depolarization. ACS chemical neuroscience 2017, 8 (8), 1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaquins-Gerstl A; Michael AC, Comparison of the brain penetration injury associated with microdialysis and voltammetry. J Neurosci Methods 2009, 183 (2), 127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaquins-Gerstl A; Shu Z; Zhang J; Liu Y; Weber SG; Michael AC, Effect of dexamethasone on gliosis, ischemia, and dopamine extraction during microdialysis sampling in brain tissue. Anal Chem 2011, 83 (20), 7662–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nesbitt KM; Jaquins-Gerstl A; Skoda EM; Wipf P; Michael AC, Pharmacological Mitigation of Tissue Damage during Brain Microdialysis. Analytical Chemistry 2013, 85 (17), 8173–8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nesbitt KM; Varner EL; Jaquins-Gerstl A; Michael AC, Microdialysis in the Rat Striatum: Effects of 24 h Dexamethasone Retrodialysis on Evoked Dopamine Release and Penetration Injury. ACS Chemical Neuroscience 2015, 6 (1), 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varner EL; Jaquins-Gerstl A; Michael AC, Enhanced Intracranial Microdialysis by Reduction of Traumatic Penetration Injury at the Probe Track. ACS chemical neuroscience 2016, 7 (6), 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nesbitt KM; Varner EL; Jaquins-Gerstl A; Michael AC, Microdialysis in the rat striatum: effects of 24 h dexamethasone retrodialysis on evoked dopamine release and penetration injury. ACS chemical neuroscience 2014, 6 (1), 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shain W; Spataro L; Dilgen J; Haverstick K; Retterer S; Isaacson M; Saltsman M; Turner JN, Controlling cellular reactive responses around neural prosthetic devices using peripheral and local intervention strategies. IEEE Transaction on Neural Systems and Rehabilitation Engineering 2003, 11, 186–188. [DOI] [PubMed] [Google Scholar]
- 28.Kozai TDY; Jaquins-Gerstl AS; Vazquez AL; Michael AC; Cui XT, Dexamethasone retrodialysis attenuates microglial response to implanted probes in vivo. Biomaterials 2016, 87, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaquins-Gerstl A; Varner EL; Nesbitt KM; Michael AC, ADVANCING INTRACRANIAL MICRODIALYSIS SAMPLING AND MEASUREMENTS. Compendium Of In Vivo Monitoring In Real-time Molecular Neuroscience-Volume 2: Microdialysis And Sensing Of Neural Tissues 2017, 375. [Google Scholar]
- 30.Wagner AK; Sokoloski JE; Ren D; Chen X; Khan AS; Zafonte RD; Michael AC; Dixon CE, Controlled cortical impact injury affects dopaminergic transmission in the rat striatum. J. Neurochem 2005, 95 (2), 457–65. [DOI] [PubMed] [Google Scholar]
- 31.Dixon CE; Lyeth BG; Povlishock JT; Findling RL; Hamm RJ; Marmarou A; Young HF; Hayes RL, A fluid percussion model of experimental brain injury in the rat. J. Neurosurg 1987, 67 (1), 110–9. [DOI] [PubMed] [Google Scholar]
- 32.Xiong Y; Mahmood A; Chopp M, Animal models of traumatic brain injury. Nature Reviews Neuroscience 2013, 14 (2), 128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papadimitriou KI; Wang C; Rogers ML; Gowers SA; Leong CL; Boutelle MG; Drakakis EM, High-Performance Bioinstrumentation for Real-Time Neuroelectrochemical Traumatic Brain Injury Monitoring. Frontiers in human neuroscience 2016, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogers ML; Leong CL; Gowers SA; Samper IC; Jewell SL; Khan A; McCarthy L; Pahl C; Tolias CM; Walsh DC, Simultaneous monitoring of potassium, glucose and lactate during spreading depolarization in the injured human brain–Proof of principle of a novel real-time neurochemical analysis system, continuous online microdialysis. Journal of Cerebral Blood Flow & Metabolism 2017, 37 (5), 1883–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pietrobon D; Moskowitz MA, Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nature reviews. Neuroscience 2014, 15 (6), 379–93. [DOI] [PubMed] [Google Scholar]
- 36.Bosche B; Graf R; Ernestus RI; Dohmen C; Reithmeier T; Brinker G; Strong AJ; Dreier JP; Woitzik J, Recurrent spreading depolarizations after subarachnoid hemorrhage decreases oxygen availability in human cerebral cortex. Ann Neurol 2010, 67 (5), 607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roitbak T; Sykova E, Diffusion barriers evoked in the rat cortex by reactive astrogliosis. Glia 1999, 28 (1), 40–8. [DOI] [PubMed] [Google Scholar]
- 38.Strong AJ; Fabricius M; Boutelle MG; Hibbins SJ; Hopwood SE; Jones R; Parkin MC; Lauritzen M, Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke 2002, 33 (12), 2738–2743. [DOI] [PubMed] [Google Scholar]
- 39.Somjen GG, Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiological reviews 2001, 81 (3), 1065–1096. [DOI] [PubMed] [Google Scholar]
- 40.Bramlett HM; Dietrich WD, Long-term consequences of traumatic brain injury: current status of potential mechanisms of injury and neurological outcomes. Journal of neurotrauma 2015, 32 (23), 1834–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lauritzen M; Dreier JP; Fabricius M; Hartings JA; Graf R; Strong AJ, Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. Journal of Cerebral Blood Flow & Metabolism 2011, 31 (1), 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinzman JM; Andaluz N; Shutter LA; Okonkwo DO; Pahl C; Strong AJ; Dreier JP; Hartings JA, Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain 2014, 137 (11), 2960–2972. [DOI] [PubMed] [Google Scholar]
- 43.Hashemi P; Bhatia R; Nakamura H; Dreier JP; Graf R; Strong AJ; Boutelle MG, Persisting depletion of brain glucose following cortical spreading depression, despite apparent hyperaemia: evidence for risk of an adverse effect of Leao’s spreading depression. J. Cereb. Blood Flow Metab 2009, 29 (1), 166–75. [DOI] [PubMed] [Google Scholar]
- 44.Jaquins-Gerstl A; Michael AC, A review of the effects of FSCV and microdialysis measurements on dopamine release in the surrounding tissue. Analyst 2015, 140 (11), 3696–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dreier JP; Bhatia R; Major S; Drenckhahn C; Lehmann TN; Sarrafzadeh A; Willumsen L; Hartings JA; Sakowitz OW; Seemann JH; Thieme A; Lauritzen M; Strong AJ, Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 2006, 129 (12), 3224–3237. [DOI] [PubMed] [Google Scholar]
- 46.Ayata C; Lauritzen M, Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiological reviews 2015, 95 (3), 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hall CN; Reynell C; Gesslein B; Hamilton NB; Mishra A; Sutherland BA; O’Farrell FM; Buchan AM; Lauritzen M; Attwell D, Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508 (7494), 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balanca B; Meiller A; Bezin L; Dreier JP; Marinesco S; Lieutaud T, Altered hypermetabolic response to cortical spreading depolarizations after traumatic brain injury in rats. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2017, 37 (5), 1670–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vespa PM; McArthur D; O’Phelan K; Glenn T; Etchepare M; Kelly D; Bergsneider M; Martin NA; Hovda DA, Persistently Low Extracellular Glucose Correlates With Poor Outcome 6 Months After Human Traumatic Brain Injury Despite a Lack of Increased Lactate: A Microdialysis Study. Journal of Cerebral Blood Flow & Metabolism 2003, 23 (7), 865–877. [DOI] [PubMed] [Google Scholar]
- 50.Di Chiara G; Tanda G; Carboni E, Estimation of in-vivo neurotransmitter release by brain microdialysis: the issue of validity. Behav Pharmacol 1996, 7 (7), 640–657. [PubMed] [Google Scholar]
- 51.Hopwood SE; Parkin MC; Bezzina EL; Boutelle MG; Strong AJ, Transient changes in cortical glucose and lactate levels associated with peri-infarct depolarisations, studied with rapid-sampling microdialysis. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2005, 25 (3), 391–401. [DOI] [PubMed] [Google Scholar]
- 52.Fabricius M; Fuhr S; Bhatia R; Boutelle M; Hashemi P; Strong AJ; Lauritzen M, Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain 2006, 129 (3), 778–790. [DOI] [PubMed] [Google Scholar]