

Abstract
The virus that causes COVID-19, SARS-CoV-2, has a large RNA genome that encodes numerous proteins that might be targets for antiviral drugs. Some of these proteins, such as the RNA-dependent RNA polymerase, helicase and main protease, are well conserved between SARS-CoV-2 and the original SARS virus, but several others are not. This study examines one of the proteins encoded by SARS-CoV-2 that is most different, a macrodomain of nonstructural protein 3 (nsp3). Although 26% of the amino acids in this SARS-CoV-2 macrodomain differ from those observed in other coronaviruses, biochemical and structural data reveal that the protein retains the ability to bind ADP-ribose, which is an important characteristic of beta coronaviruses and a potential therapeutic target.
Keywords: COV1D-19, Antiviral Drug target, Severe Acute Respiratory Syndrome, Coronavirus
Graphical Abstract
INRODUCTION
The development of antivirals targeting Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), the causative agent of the present COVID-19 pandemic,1 will most likely focus on viral proteins and enzymes needed for replication.2 Similar to other coronaviruses, SARS-CoV-2 has a large positive sense (+)RNA genome greater than 30,000 nucleotides long with several open reading frames. Most of the proteins that form the viral replicase are encoded by the “rep lab” reading frame, which codes for a 7,096 amino acid-long polyprotein that is ultimately processed into at least 15 functional peptides, five of which are only produced by a translational frameshift event occurring after nsp10 (Fig. 1). Parts of the SARS-CoV-2 rep lab polyprotein are very similar to the rep lab protein of the coronavirus that caused the SARS epidemic in 2003 (which will be referred to here as SARS-CoV), suggesting the that drugs targeting the SARS-CoV nsp5-14 might be effective against SARS-CoV-2. However, some portions of the SARS rep lab polyproteins are quite different.
FIGURE 1. Sequence divergence between potential drug targets in SARS-CoV and SARS-CoV-2.

The SARS-CoV-2 rep lab peptide sequence was aligned with each of the PDB files listed, which describe an atomic structure of a homologous region of the SARS-CoV rep lab polyprotein. Nsps are shown in sequence as black arrows (note: there is no “nsp11”, and the translational frameshift occurs after nsp10). The percent of amino acids that differ in each protein is plotted. Nsp1 is an interferon antagonist.28 The nsp3 Mac1 domain is studied here. Mac2 and Mac3 are of tandem macrodomains that bind G-quadruplex structures,4 and nsp 3 dC is the C-terminus of the Mac3.29 Nsp3pro is a papain-like protease,30 and nsp3 RBD is another possible RNA binding domain.31 Nsp5 is the main viral protease.32 Nsp7 and nsp8 are polymerase cofactors.33 Nsp9 is an RNA binding protein.34 Nsp10 is a zinc-binding cofactor for Nsp14 and Nsp16.35 Nsp12 is an RNA polymerase.33 Nsp13 is a helicase.36 Nsp14 is a 3’-5’ exonuclease and a 7-methyltransferase.35 Nsp15 is an RNA endonuclease.37 Nsp16 is an RNA cap 2’-O-methyltransferase.38
In contrast to the well-conserved SARS-CoV nsp5 protease, nsp12 polymerase and nsp13 helicase enzymes, significantly more differences exist between the nsp3 proteins encoded by SARS-CoV and SARS-CoV-2. The most variation occurs in a domain of nsp3 domain suspected to bind ADP-ribose, which will be referred to here as the Mac1 domain,3 to differentiate it from the two downstream macrodomains (Mac2 and Mac3), which do not bind ADP-ribose.4 The Mac1 domain of SARS-CoV also catalyzes the hydrolysis of ADP-ribose 1 phosphate, albeit at a slow rate.5 Some viral macrodomains also remove ADP-ribose from proteins,6 and this de-ADP-ribosylation activity correlates with virulence and ability to evade the innate immune response.6-11 Jean-Michel Claverie recently suggested that the putative ability of Mac1 to remove ADP-ribose from proteins might be related to the cytokine storm syndrome seen in severe cases of COVID-19.12
Compounds blocking ADP-ribose binding could be used to test this important hypothesis. However, the many sequence differences preclude the use of the SARS-CoV Mac1 domain structures as scaffolds to design compounds that might target this nsp3 region in SARS-CoV-2. ADP-ribose binding must also be confirmed, especially in light of the observation that the same nsp3 domain from gamma coronaviruses does not bind ADP-ribose in vitro.13 The ability of the SARS-CoV-2 Mac1 domain to bind ADP-ribose was therefore examined here using a recombinant purified protein and isothermal titration calorimetry (ITC). We also determined the structure of the SARS-CoV-2 Mac1 domain to examine the biochemical context of ADP-ribose binding and to provide data for rational inhibitor design or in silico screening.
MATERIAL & METHODS
Gene Synthesis—
To facilitate the comparison between SARS-CoV and SARS-CoV-2, a protein expression vector was generated that is similar to that used by Eglott et al.14 To this end, a codon optimized open reading frame was synthesized by GenScript (Piscataway, NJ) that encodes the Mac1 domain with an N-terminal TEV-cleavage site flanked by NheI and BamH1 restriction sites. This open reading frame was cloned into pET21b to yield plasmid pET21-COVID-Mac1. The pET11-COVID-Mac1 plasmid was used to transform BL21(DE3) cells.
Protein Purification—
Colonies of BL21(DE3) cells harboring the pET21-COVID-Mac1 plasmid were used to inoculate 3 ml of lysogeny broth containing 100 mg/ml ampicillin. The starter culture was incubated at 37 °C with shaking at 225 rpm. After the cells grew to an OD600 of 1.0, they were transferred to 1 liter of fresh medium containing ampicillin. After the cells reached an OD600 of 1.0 again, protein production was induced with 1 mM isopropyl-β-D-thiogalactoside. After growing 16 h at 23 °C, cells were harvested by centrifugation at 4,000 rpm and 4 °C. The resulting cell pellet was suspended in 25 ml of IMAC buffer (20 mM Tris pH 8, 0.5 M NaCl), sonicated on ice for five 1-min bursts with 2-min rests between, and clarified by centrifugation at 10,000 g for 30 min. The supernatant was loaded onto a 5-ml Ni-NTA column, and the fractions were eluted with a step gradient from 5 to 500 mM imidazole. Fractions containing the Mac1 domain protein (5 ml total) were loaded on a 250 ml Sephacryl S300 gel filtration column and eluted with 10 mM MOPS and 150 mM NaCl. Purified protein concentrations were determined by measuring absorbance at 260 nm using a molar extinction coefficient of 10,555 M−1 cm−1, which was calculated using the ProtParam tool (https://web.expasy.org/protparam/).
Isothermal Titration Calorimetry (ITC)—
Binding of ADP-ribose to the SARS-CoV-2 Mac1 domain was measured using a Nano ITC (TA Instruments). Before starting the measurement, samples of both ligand and protein were diluted in 10 mM MOPS and 150 mM NaCl (pH 7) and were degassed at 400 mmHg for 30 min Measurements were taken at 20 °C by injecting 2.0-μl aliquots of 500 μM ADP-ribose (Sigma) to 50 μM protein (175 μl initial volume) with stirring at 250 rpm. Using NanoAnalyze Software (v. 3.11.0), data were fitted by nonlinear regression to an independent binding model. Briefly, after baseline correction, background heats from ligand-to-buffer titrations were subtracted, and the corrected heats from the binding reaction were used to identify best fit parameters for the stoichiometry of the binding (n), free energy of binding (ΔG), apparent enthalpy of binding (ΔH), and entropy change (ΔS). Dissociation constants (Kd) were calculated from the ΔG.
Crystallization and Structure Determination—
In preparation for crystallization experiments, purified SARS-CoV-2 Mac1 domain protein was cleaved with tobacco etch virus (TEV) protease to remove the N-terminal His6-tag and passed back through the Ni-NTA column. The flow-through fractions were desalted into 10 mM HEPES (pH 7.2) using a 2x5 ml HiTrap desalting column (GE Life Sciences) and concentrated to 10 mg/mL in a centrifugal concentrator. This preparation of the protein was mixed at a 1:1 μL ratio with Morpheus HT screen reagents (Molecular Dimensions) in a 96-well SwissSci MRC UV-transmissible sitting drop plate. Large, diffraction-quality crystals grew directly from a number of the screen conditions. The crystal ultimately used for structure determination grew from condition D9: 0.12 M alcohols [0.02 M each 1,6-hexanediol, 1-butanol, 1,2-propanediol, 2-propanol, 1,4-butanediol, and 1,3-propanediol]; 0.1 M buffer system 3, pH 8.5 [0.05 M each TRIS and bicine]; and 30% precipitant mix 1 [20% poly(ethylene glycol) (PEG) 500 monomethylether, 10% PEG 20,000]. Large, thick plates grew within 1 week at 22 °C. Given the high concentration of PEG 500 MME, the crystal did not require additional cryo-protection and was flash-cooled by looping it directly from the sitting drop and plunging it into liquid nitrogen.
Diffraction data were collected on Life Sciences Collaborative Access Team (LS-CAT) beamline 21-ID-F at the Advanced Photon Source of Argonne National Laboratory. The wavelength at this station is fixed at 0.9787 Å; the detector is a MarMosaic M300 charge-coupled device. The data were collected with an oscillation width of 0.5° per image for a total oscillation of 180°. The data were indexed and integrated with DIALS 15, 16 as implemented in version 7.2 of the CCP4 software suite.17, 18 Data scaling and reduction were performed using AIMLESS.19-21 Data collection statistics are provided in Table 1.
Table 1.
Crystallographic data collection and model refinement statistics for the SARS-CoV-2 Mac1 domain.
| Data collection | |
|---|---|
| Resolution range (Å) (last shell)a | 43.32 – 0.95 (0.97 – 0.95) |
| Space Group | P 21 21 21 |
| a, b, c (Å) | 43.3, 54.4, 67.6 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Rmerge a | 0.063 (0.348) |
| Rmeas a | 0.075 (0.475) |
| Rpim a | 0.039 (0.320) |
| CC1/2 a | 99.5 (89.1) |
| No. of unique reflections a | 99442 (4199) |
| Completeness (%) a | 98.4 (85.0) |
| Multiplicity a | 6.4 (2.6) |
| ⟨I/σ(I)⟩ a | 13.1 (1.5) |
| Model Refinement | |
| Reflections used in refinement | 99335 (2606)b |
| Reflections used for Rfree | 5046 (125) |
| Rcryst (Rfree) | 0.119 (0.137) |
| Wilson B-factor (Å2) | 7.4 |
| Average B factor (Å2) | 13.9 |
| Protein atoms | 10.2 |
| Solvent | 28.2 |
| Root-mean-square (RMS) deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.16 |
| Coordinate error (Å) | 0.06 |
| Ramachandran statistics | |
| Favored/allowed/outliers (%) | 98.2/1.8/0.0 |
| Rotamer outliers (%) | 1.2 |
| Clashscore | 1.70 |
Values in parentheses apply to the high-resolution shell indicated in the resolution row
The limits of the high-resolution bin for refinement were 0.96 – 0.95 Å
The structure was determined by molecular replacement in PHASER22 using the model of the SARS-CoV Mac1 domain as the search model (PDB ID 2FAV14). The model underwent iterative rounds of (re)building in COOT23 and refinement in PHENIX.refine.24, 25 The very high resolution of the data justified a full anisotropic treatment of the protein and solvent temperature factors. Model refinement and validation statistics are provided in Table 1. The coordinates were deposited in the Protein Data Bank with accession code 6WEY.
RESULTS AND DISCUSSION
Variability in the nsp3 Mac1 domain—
The structures of most of the soluble portions of the SARS-CoV nsp proteins were examined at atomic resolution to help understand coronavirus replication and facilitate antiviral drug discovery. The amino acid sequences of each of these proteins were compared with the homologous regions of the rep lab protein encoded by SARS-CoV-2 (GenPept Accession YP_009724389). The most similar proteins were the RNA helicases (nsp13), which are identical in all but one of their 603 amino acids, namely a conservative Val to Ile substitution near their C-termini. The RNA-dependent RNA polymerases (nsp12) are also well conserved, sharing all but 34 of 955 amino acids. The primary protease that cleaves the polyprotein (nsp5) is also similar in SARS-CoV and SARS-CoV-2, with only 13 amino acids that differ among 306 (4.2% different) (Fig. 1).
At the other end of the spectrum are the nsp3 proteins, which are notably more different in the two SARS viruses. Nsp3 is a large multidomain membrane-bound protein,26 and its clearest role in viral replication is cleaving the rep polyprotein. Greater than 17% of the amino acids in the nsp3 protease domain differ between SARS-CoV and SARS-CoV-2. Other parts of nsp3 are even more variable, such as the macrodomains that lie N-terminal to the nsp3 protease domain. Macrodomains consist of four helices that surround a mixed beta sheet. A ligand-binding pocket that typically binds ADP-ribose or related compounds lies between the helices and the sheet.27 SARS-CoV has three macrodomains in tandem, but only the first binds ADP-ribose. The amino acid sequences of this Mac1 domain differ by 26% between SARS-CoV and SARS-CoV-2 (Fig. 1).
Six of the 47 variant residues in the 180-amino acid long SARS Mac1 domain are clustered in a 21 amino acid region near its N-terminus, which is particularly variable in the three Coronaviridae genera (Fig. 2) and sometimes not included in other macrodomains. As shown by the conservation plot above the alignment (Fig. 2), there are several highly variable regions throughout the protein, which might affect protein function. Mac1 domains from coronaviruses that cause the common cold (alpha coronaviruses)13 and the beta coronaviruses, such as SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV)39, all bind ADP-ribose. However, reports with gamma coronaviruses suggest that ADP-ribose binding might not be conserved. Sequence differences exist between infectious bronchitis virus (IBV) strain M41, which binds ADP-ribose, and IBV strain Beaudette, which does not.13
FIGURE 2. Variation in the Mac1 domains of coronaviruses.

Mac1 structures were aligned using the “MatchMaker” function of UCSF Chimera (v. 1.14).40 Amino acids are colored by class. Beta sheets are noted in green boxes, and alpha helices are indicated in yellow boxes. Arrows mark key residues F360, F336, D226, and N244 (see text.)
Expression and purification of the SARS-CoV-2 Mac1 domain—
An E. coli expression vector for the Mac1 domain was generated to express and N-terminally His-tagged protein similar to a SARS-CoV protein studied by Egloff et al.14 Upon induction, a one-liter culture of BL21(DE3) cells harboring the vector expresses 50-100 mg of the Mac1 domain protein that can be purified in one step to apparent homogeneity using immobilized metal affinity chromatography (Fig. 3A). The protein was polished further with gel filtration chromatography and concentrated before analysis and crystallization.
FIGURE 3. The SARS-CoV-2 Mac1 domain binds ADP ribose.

(A) 15% SDS PAGE showing 10 μL samples of a soluble crude lysate of induced BL21(DE3) cells harboring the plasmid p21-COVID-Mac1 (lane C), proteins that do not bind a Ni-NTA column (F), and fractions eluted from a Ni-nitrilotriacetic acid column during an imidazole step gradient from 0 mM (lanes 1-3), 5 mM (lanes 4-6), 40 mM (lanes 7-9), and 500 mM (lanes 10-12). Protein markers (lane M) are 116, 66.2, 45, 35, and 25 kDa. (B) Example ITC experiment in which the purified SARS-CoV-2 nsp3 macrodomain was titrated with ADP ribose. (C) ITC experiments such as those shown in panel B were repeated thrice with each of the compounds listed. Means are plotted, and error bars are standard deviations. Average (±SD) dissociation constants were 10 ± 4 μM for ADP-ribose, 8 ± 9 μM for ADP, 3 ± 3 μM for ATP, 6± 4 μM for ADP-glucose, 2 ± 1 μM for cAMP, and 2 ± 1 μM for adenosine. (D) Comparison of the thermodynamics of ADP-ribose binding by Mac1 domains from SARS-CoV-2 (data from panel C), SARS-CoV, MERS-CoV, and an alpha coronavirus. aData from Egloff et al.34 bData from Cho et al.39 cData from Piotrowski et al.13
Nucleotide binding by the SARS-CoV-2 Mac1 domain—
Repeated ITC experiments revealed that the purified recombinant protein bound ADP-ribose (Fig. 3C) with a dissociation constant of 10 ± 4 μM (uncertainty is the standard deviation of Kds from independent titrations). To examine binding specificity, similar titrations were repeated with related nucleotides. The SARS-CoV-2 protein bound ADP, cAMP, ATP and ADP-glucose (Fig. 3D). All nucleotides lacking the ribose moiety bound with similar high affinities, but none bound with an enthalpy change similar to that observed with ADP-ribose, suggesting specific contacts form between ADP-ribose the SARS-CoV-2 protein. Based on findings from the SARS-CoV-2 structures below, these contacts likely occur with the conserved D226 and N244 (positions 30 and 43 in the numbering above the alignment in Fig. 2). None of the other nucleotides bind with an entropic penalty as observed with ADP-ribose, suggesting that the ribose moiety becomes structured when bound to the macrodomain.
The energetics of ADP-ribose binding to the SARS-CoV protein are similar to those noted for the same protein from SARS-CoV14 and MERS-CoV.39 Enthalpy and entropy of binding were also very similar for all three proteins (Fig. 3D). In contrast to findings from Mac1 protein from an alpha coronavirus,13 enthalpy appears to drive ADP-ribose binding to the Mac1 domains of the three beta coronaviruses. Variation in the ADP-ribose binding cleft might account for these differences. For example, nsp3 amino acid 360, which is near the adenine base, is a Phe in SARS-CoV-2, an Asn in both SARS-CoV and MERS-CoV, and an aliphatic amino acid in the alpha and gamma coronaviruses (Fig. 2)
Structure of the SARS-CoV-2 Mac1 domain—
The SARS-CoV-2 Mac1 domain (nsp3 residues 207 to 277) crystallized in space group P212121 with 1 molecule per asymmetric unit. These crystals had a solvent content of 43% and diffracted extremely well. The final resolution limit of the data was set at 0.95 Å (Table 1). The quality of the electron density maps is correspondingly excellent (Fig. 4A). The section of the structure depicted in this image is located on the surface of the protein, and the B-factors of these residues are close to the average B-factor of the protein (7.2 vs. 10.2 Å2), indicating that this sample accurately represents the overall quality of the maps. The final model contains the entire sequence from V207 to S377 of nsp3, an N-terminal glycine residue that was left from the TEV-protease cleavage, and 374 solvent molecules. The Rcryst and Rfree values of the final model were 0.119 and 0.137, respectively (Table 1)
FIGURE 4. The SARS-CoV-2 Mac1 domain structure.

(A) The electron density is shown for a representative portion of the structure (residues 359-361) on the surface of the protein. The 2mFo-DFc map is contoured at 1.5σ and is shown as a magenta mesh. The mFo-DFc (difference) maps are shown at + and − 3.0σ as green and red mesh, respectively. (B) Ribbon diagram of the SARS-CoV-2 Mac1 domain structure colored according to the sequence conservation plot in Fig. 2 as a gradient from red (low conservation; <10%) to blue (highly conserved; 100%) through magenta. As observed in the sequence alignment, the N- and C-termini are particularly poorly conserved. (C) Overlay of the structure of the SARS-CoV-2 Mac1 domain bound to ADP-ribose determined by Michalska. et al. of the CGSID (PDB ID 6W02) with the ultrahigh-resolution structure of the unliganded protein determined here. ADP-ribose is presented as a ball-and-stick model with the carbon atoms colored gold. The backbone trace of the unliganded structure is colored cyan, and that of the ADP-ribose-bound model is colored gray. There are three loops with significantly different conformations in the two structures. In the unliganded structure, the β2-α2 loop is colored bright red, the β4-α4 loop is colored purple, and the β5-α5 loop is colored bright green. The same regions of the ADP-ribose-bound structure are colored pale red, pale purple, and pale green, respectively. The transparent blue and yellow spheres represent water molecules bound to the unliganded (transparent blue) and ADP-ribose-bound (transparent yellow) forms of the protein. Interestingly, several of the water molecules interacting with ADP-ribose in 6W02 can also be found in the unliganded structure of the protein. The inset shows a close-up of the boxed region colored according to the same scheme. The β2-α2 and β5-α5 loops, which contact ADP-ribose, are presented as a ball-and-stick model. Note that the β2-α2 loop rotates ~180° to allow it to make a hydrogen-bonding interaction with the 1’-hydroxyl of the ribose moiety. Additionally, the phenylalanine residue in the β5-α5 loop (F336) would clash with the β-phosphate and ribose of ADP-ribose if the β5-α5 loop did not adopt a different conformation.
As expected, the tertiary structure ranges from approximately identical to very similar to those of other coronavirus macrodomains, including SARS CoV (2FAV14; 74.7% sequence identity) with a root mean square deviation (RMSD) value for 162 of 172 Cα atoms of 0.6 Å, MERS-CoV (5DUS39; 42.2% identical) with a 1.2 Å RMSD for 161 of 172 Cα atoms, human alpha coronavirus 229E (3EWR41; 32.5% identical) with a 1.5 Å RMSD for 154 of 172 Cα atoms, feline coronavirus (FCoV; 3JZT42 26.8% identical) with a 1.5 Å RMSD for 153 of 172 Cα atoms, and the gamma CoV IBV; 3EWP41 26.7% identical) with a 2.1 Å RMSD for 150 of 172 Cα atoms. The regions of high sequence conservation are not clustered in any particular region(s) of the molecule, as is clear when the ribbon is colored as a gradient from red (poorly conserved) to blue (highly conserved) (Fig. 4B). This finding is consistent with the fact that the protein atoms involved in hydrogen bonding interactions with the ligands in these structures are more often part of the main chain; relatively few interactions of side chains with ligands are observed.
At the time of writing, we discovered that Michalska et al. of the Center for Structural Genomics of Infectious Diseases (CSGID) deposited coordinates for a very similar construct of the SARS-CoV-2 Mac1 domain including the region from E206 to E275 of the nsp3 protein plus an additional 4 residues at the N-terminus (6VSX; unpublished). Their crystals also allowed binding of ADP-ribose (6W02) and AMP (6W6Y), whereas ours seemed to be packed too tightly to permit ligands to access the binding site (data not shown). We compared our ultrahigh-resolution model of the unliganded protein to the ADP-ribose-bound form. The RMSD values for the fitting, which were determined by secondary structure matching (SSM)43 as implemented in COOT, is 0.59 Å for 165 of 172 Cα atoms. This value is very similar to the RMSD values of the free protein (6VXS; 0.66 Å) and the AMP-bound form (6W6Y; 0.50 Å), indicating that no large conformational changes occur upon ligand binding. In fact, the only notable conformational changes occur in three surface-exposed loops in or near the ligand-binding pocket (Fig. 4C). These loops connect strand β2 with helix α2 (the β2-α2 loop), strand β4 with helix α4 (β4-α4 loop), and strand β5 with helix α5 (β5-α5 loop). The subtle change in conformation of the β4-α4 loop (purple in Fig. 4C) appears to be the result of crystal contacts and not the direct influence of ADP-ribose binding. The other two loops are more intimately involved in ligand binding. The main chain of the α2-β2 loop rotates 180° to allow the amide N atom of G252 to participate in a hydrogen bonding interaction with the 1’-hydroxyl of the ribose moiety of ADP-ribose. G252 corresponds to V33 residue in Chikungunya macrodomain, which Eckei et al. proposed is needed for de-ADP-ribosylation activity based on results for site directed mutagenesis.10 This loop also carries N244, which directly interacts with the ribose. The phenyl ring of F336 in the β5-α5 loop occupies the portion of the binding pocket in the unliganded structure that is occupied by the β-phosphate of ADP-ribose. Thus, without rearrangement of the β5-α5 loop, ADP-ribose would not be able to bind.
Conclusion—
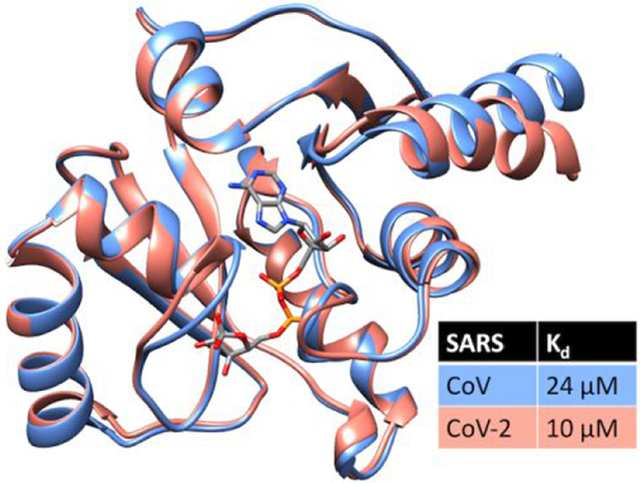
The significance of the study stems mainly from the demonstration that the SARS-CoV-2 Mac1 domain binds ADP-ribose. Although SARS-CoV and SARS-CoV-2 have 26% divergence in amino acid sequences, their structures are highly similar, which may explain its ability to bind ADP-ribose. This is the first step needed to justify screens for potential antivirals that bind in place of ADP-ribose. However, more work needs to be done to understand the antiviral potential of such compounds because the biological role for ADP-ribose binding is not completely understood. Some work with alpha coronaviruses suggest that ADP-ribose binding by the Mac1 domain is not required for viral replication.44 However, studies with other (+)RNA viruses suggest that macrodomains are essential for virulence.45 This work is also noteworthy because the synthetic codon-optimized plasmid reported here produces up to 100 mg of soluble Mac1 domain protein per liter of E. coli culture, and this protein retains high affinity for ADP-ribose. The protein could be used for structural studies and screening campaigns. Screening assays with the SARS-CoV-2 protein might be more efficient since the SARS-CoV-2 protein binds ADP-ribose somewhat more tightly (Kd = 10 μM) than the SARS-CoV protein (Kd = 24 μM). The recombinant protein reported here together with detailed structural information might also be useful to others developing SARS-CoV-2 diagnostics and/or therapeutics.
ACKNOWLEDGMENTS
The authors are grateful to Matt McCarty, Garrett Breit, Hayden Aristizabal, Trevor R. Melkonian, Dante A. Serrano and Prof. Ionel Popa for valuable technical assistance and helpful discussions.
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Funding Sources
This work was supported by National Institutes of Health Grant R01 AI088001 (to D.N.F.) and by Grant CHE-1903899 from the National Science Foundation (to N.R.S.).
Footnotes
REFERENCES
- (1).Wu F; Zhao S; Yu B; Chen YM; Wang W; Song ZG; Hu Y; Tao ZW; Tian JH; Pei YY; Yuan ML; Zhang YL; Dai FH; Liu Y; Wang QM; Zheng JJ; Xu L; Holmes EC; Zhang YZ (2020) A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Subissi L; Imbert I; Ferron F; Collet A; Coutard B; Decroly E; Canard B (2014) SARS-CoV ORF1b-encoded nonstructural proteins 12-16: replicative enzymes as antiviral targets. Antiviral Res. 101, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Neuman BW (2016) Bioinformatics and functional analyses of coronavirus nonstructural proteins involved in the formation of replicative organelles. Antiviral Res. 135, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kusov Y; Tan J; Alvarez E; Enjuanes L; Hilgenfeld R (2015) A G-quadruplex-binding macrodomain within the “SARS-unique domain” is essential for the activity of the SARS-coronavirus replication-transcription complex. Virology 484, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Saikatendu KS; Joseph JS; Subramanian V; Clayton T; Griffith M; Moy K; Velasquez J; Neuman BW; Buchmeier MJ; Stevens RC; Kuhn P (2005) Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1“-phosphate dephosphorylation by a conserved domain of nsP3. Structure 13, 1665–1675.’ [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Li C; Debing Y; Jankevicius G; Neyts J; Ahel I; Coutard B; Canard B (2016) Viral Macro Domains Reverse Protein ADP-Ribosylation. J. Virol 90, 8478–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fehr AR; Channappanavar R; Jankevicius G; Fett C; Zhao J; Athmer J; Meyerholz DK; Ahel I; Perlman S (2016) The Conserved Coronavirus Macrodomain Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fehr AR; Jankevicius G; Ahel I; Perlman S (2018) Viral Macrodomains: Unique Mediators of Viral Replication and Pathogenesis. Trends Microbiol. 26, 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Alhammad YMO; Fehr AR (2020) The Viral Macrodomain Counters Host Antiviral ADP-Ribosylation. Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Eckei L; Krieg S; Bütepage M; Lehmann A; Gross A; Lippok B; Grimm AR; Kümmerer BM; Rossetti G; Lüscher B; Verheugd P (2017) The conserved macrodomains of the non-structural proteins of Chikungunya virus and other pathogenic positive strand RNA viruses function as mono-ADP-ribosylhydrolases. Sci Rep 7, 41746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McPherson RL; Abraham R; Sreekumar E; Ong SE; Cheng SJ; Baxter VK; Kistemaker HA; Filippov DV; Griffin DE; Leung AK (2017) ADP-ribosylhydrolase activity of Chikungunya virus macrodomain is critical for virus replication and virulence. Proc. Natl. Acad. Sci. U S A 114, 1666–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Claverie JM (2020) A Putative Role of de-Mono-ADP-Ribosylation of STAT1 by the SARS-CoV-2 Nsp3 Protein in the Cytokine Storm Syndrome of COVID-19. Viruses 12, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Piotrowski Y; Hansen G; Boomaars-van der Zanden AL; Snijder EJ; Gorbalenya AE; Hilgenfeld R (2009) Crystal structures of the X-domains of a Group-1 and a Group-3 coronavirus reveal that ADP-ribose-binding may not be a conserved property. Protein Sci 18, 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Egloff MP; Malet H; Putics A; Heinonen M; Dutartre H; Frangeul A; Gruez A; Campanacci V; Cambillau C; Ziebuhr J; Ahola T; Canard B (2006) Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J. Virol 80, 8493–8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Clabbers MTB; Gruene T; Parkhurst JM; Abrahams JP; Waterman DG (2018) Electron diffraction data processing with DIALS. Acta Crystallogr D Struct Biol 74, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Winter G; Waterman DG; Parkhurst JM; Brewster AS; Gildea RJ; Gerstel M; Fuentes-Montero L; Vollmar M; Michels-Clark T; Young ID; Sauter NK; Evans G (2018) DIALS: implementation and evaluation of a new integration package. Acta Crystallogr D Struct Biol 74, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Potterton L; Agirre J; Ballard C; Cowtan K; Dodson E; Evans PR; Jenkins HT; Keegan R; Krissinel E; Stevenson K; Lebedev A; McNicholas SJ; Nicholls RA; Noble M; Pannu NS; Roth C; Sheldrick G; Skubak P; Turkenburg J; Uski V; von Delft F; Waterman D; Wilson K; Winn M; Wojdyr M (2018) CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr D Struct Biol 74, 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Evans PR; Murshudov GN (2013) How good are my data and what is the resolution Acta Crystallogr D Biol Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62, 72–82. [DOI] [PubMed] [Google Scholar]
- (21).Evans PR (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr 67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Emsley P; Lohkamp B; Scott WG; Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liebschner D; Afonine PV; Baker ML; Bunkóczi G; Chen VB; Croll TI; Hintze B; Hung LW; Jain S; McCoy AJ; Moriarty NW; Oeffner RD; Poon BK; Prisant MG; Read RJ; Richardson JS; Richardson DC; Sammito MD; Sobolev OV; Stockwell DH; Terwilliger TC; Urzhumtsev AG; Videau LL; Williams CJ; Adams PD (2019) Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Afonine PV; Grosse-Kunstleve RW; Echols N; Headd JJ; Moriarty NW; Mustyakimov M; Terwilliger TC; Urzhumtsev A; Zwart PH; Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lei J; Kusov Y; Hilgenfeld R (2018) Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res. 149, 58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Leung AKL; McPherson RL; Griffin DE (2018) Macrodomain ADP-ribosylhydrolase and the pathogenesis of infectious diseases. PLoS Pathog. 14, e1006864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Almeida MS; Johnson MA; Herrmann T; Geralt M; Wüthrich K (2007) Novel beta-barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. J. Virol 81, 3151–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Johnson MA; Chatterjee A; Neuman BW; Wüthrich K (2010) SARS coronavirus unique domain: three-domain molecular architecture in solution and RNA binding. J. Mol. Biol 400, 724–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Báez-Santos YM; Barraza SJ; Wilson MW; Agius MP; Mielech AM; Davis NM; Baker SC; Larsen SD; Mesecar AD (2014) X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem 57, 2393–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Serrano P; Johnson MA; Chatterjee A; Neuman BW; Joseph JS; Buchmeier MJ; Kuhn P; Wüthrich K (2009) Nuclear magnetic resonance structure of the nucleic acid-binding domain of severe acute respiratory syndrome coronavirus nonstructural protein 3. J. Virol 83, 12998–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Akaji K; Konno H; Mitsui H; Teruya K; Shimamoto Y; Hattori Y; Ozaki T; Kusunoki M; Sanjoh A (2011) Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J. Med. Chem 54, 7962–7973. [DOI] [PubMed] [Google Scholar]
- (33).Kirchdoerfer RN; Ward AB (2019) Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun 10, 2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Egloff MP; Ferron F; Campanacci V; Longhi S; Rancurel C; Dutartre H; Snijder EJ; Gorbalenya AE; Cambillau C; Canard B (2004) The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a single-stranded RNA-binding subunit unique in the RNA virus world. Proc. Natl. Acad. Sci. U S A 101, 3792–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ma Y; Wu L; Shaw N; Gao Y; Wang J; Sun Y; Lou Z; Yan L; Zhang R; Rao Z (2015) Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. U S A 112, 9436–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jia Z; Yan L; Ren Z; Wu L; Wang J; Guo J; Zheng L; Ming Z; Zhang L; Lou Z; Rao Z (2019) Delicate structural coordination of the Severe Acute Respiratory Syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 47, 6538–6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ricagno S; Egloff MP; Ulferts R; Coutard B; Nurizzo D; Campanacci V; Cambillau C; Ziebuhr J; Canard B (2006) Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc. Natl. Acad. Sci. U S A 103, 11892–11897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Decroly E; Debarnot C; Ferron F; Bouvet M; Coutard B; Imbert I; Gluais L; Papageorgiou N; Sharff A; Bricogne G; Ortiz-Lombardia M; Lescar J; Canard B (2011) Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2’-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 7, e1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cho CC; Lin MH; Chuang CY; Hsu CH (2016) Macro Domain from Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Is an Efficient ADP-ribose Binding Module: CRYSTAL STRUCTURE AND BIOCHEMICAL STUDIES. J. Biol. Chem 291, 4894–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (41).Xu Y; Cong L; Chen C; Wei L; Zhao Q; Xu X; Ma Y; Bartlam M; Rao Z (2009) Crystal structures of two coronavirus ADP-ribose-1”-monophosphatases and their complexes with ADP-Ribose: a systematic structural analysis of the viral ADRP domain. J. Virol 83, 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wojdyla JA; Manolaridis I; Snijder EJ; Gorbalenya AE; Coutard B; Piotrowski Y; Hilgenfeld R; Tucker PA (2009) Structure of the X (ADRP) domain of nsp3 from feline coronavirus. Acta Crystallogr D Biol Crystallogr 65, 1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Krissinel E; Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60, 2256–2268. [DOI] [PubMed] [Google Scholar]
- (44).Keep S; Bickerton E; Armesto M; Britton P (2018) The ADRP domain from a virulent strain of infectious bronchitis virus is not sufficient to confer a pathogenic phenotype to the attenuated Beaudette strain. J. Gen. Virol 99, 1097–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Abraham R; McPherson RL; Dasovich M; Badiee M; Leung AKL; Griffin DE (2020) Both ADP-Ribosyl-Binding and Hydrolase Activities of the Alphavirus nsP3 Macrodomain Affect Neurovirulence in Mice. mBio 11, e03253–19. [DOI] [PMC free article] [PubMed] [Google Scholar]