

Abstract
During an immune response to microbial infection, CD8+ T cells give rise to distinct classes of cellular progeny that coordinately mediate clearance of the pathogen and provide long-lasting protection against reinfection, including a subset of non-circulating tissue-resident memory (TRM) cells that mediate potent protection within non-lymphoid tissues. Here, we utilized single-cell RNA-sequencing to examine the gene expression patterns of individual CD8+ T cells in the spleen and small intestine intraepithelial lymphocyte (siIEL) compartment throughout the course of their differentiation in response to viral infection. These analyses revealed previously unknown transcriptional heterogeneity within the siIEL CD8+ T cell population at several stages of differentiation, representing functionally distinct TRM cell subsets as well as a subset of TRM cell precursors within the tissue early in infection. Taken together, these findings may inform strategies to optimize CD8+ T cell responses to protect against microbial infection and cancer.
One sentence summary:
Heterogeneity of small intestine intraepithelial CD8+ T cells suggests functionally distinct subsets of TRM cells and their precursors.
Introduction
CD8+ T cells responding to microbial challenge differentiate into distinct subsets of cellular progeny with unique migratory and functional properties that coordinately mediate clearance of the pathogen (effector cells) and provide long-lasting protection against reinfection (memory cells). Considerable heterogeneity has been previously described within the long-lived circulating memory T cell pool (1–3). While central memory (TCM) cells exhibit greater self-renewal and plasticity with the ability to rapidly proliferate and differentiate into secondary effector cells upon reinfection, effector memory (TEM) cells provide immediate pathogen control via rapid and potent ‘effector’ function. Moreover, recent studies have revealed additional heterogeneity within the classically defined TEM cell population, including long-lived effector (LLE) cells and peripheral memory (TPM) cells, which can be distinguished by distinct surface molecule expression and trafficking properties (1, 2, 4–6). In addition to these circulating memory T cell populations, a non-circulating subset, termed tissue-resident memory (TRM) cells, has recently been described (7). TRM cells are found in most tissues and positioned at key barrier surfaces, such as the skin and intestinal epithelium, where they play critical roles in limiting early pathogen spread and controlling infection, and also help to control the outgrowth of cancer cells (8–11). Whereas heterogeneity within the circulating CD8+ T cell memory population has been well characterized, it remains unclear whether the tissue-resident CD8+ T cell population might also be comprised of distinct subsets that play unique roles in mediating protective immunity.
Recent studies have begun to illuminate the mechanisms regulating TRM cell differentiation, function, and survival. Activation of naïve CD8+ T cells occurs in the spleen or draining lymph nodes, resulting in the upregulation of key transcription factors including Blimp-1 (12). Recruitment of activated CD8+ T cells to nonlymphoid tissue sites is mediated by chemokine receptors that promote tissue entry, such as CCR9 and CXCR3 (12–14). Upon entry to tissue, CD8+ T cells undergo transcriptional changes that enforce tissue residency, in part by dampening expression of receptors that promote return to circulation such as CCR7 and S1PR1 (14), and begin to direct the TRM cell differentiation program. These changes include upregulation of transcription factors such as Hobit, which, together with Blimp-1, repress genes associated with recirculation, including Klf2, S1pr1, and Ccr7; and downregulation of the T-box transcription factors T-bet and Eomes, enabling TGF-β responsiveness (12–15). TGF-β signals within the tissue induce expression of CD103, a key factor for tissue retention, while low levels of T-bet expression are required for IL-15 responsiveness, which plays an important role for long-term survival of TRM cells in some tissues (13, 15). However, additional unidentified regulators likely contribute to coordinating the TRM cell differentiation program. Moreover, although it has been shown that TRM cells preferentially arise from KLRG1lo precursors in the circulation (13, 16), it remains unknown whether all cells that enter the tissue are destined to persist and continue their differentiation into TRM cells, or whether TRM cells are derived from a specific subset of precursors within the tissue at early time points.
Several studies have described a core transcriptional signature that is shared among TRM cells from distinct tissues, highlighting several key transcriptional regulators of TRM cell differentiation, including Hobit, Blimp-1, and Runx3 (12, 16–19). However, most of these prior studies have focused on relatively late time points after the TRM cell population has been well established, providing only a snapshot of the gene-expression patterns utilized by TRM cells and potentially missing early events important for their differentiation. Moreover, since these studies relied on RNA sequencing of bulk cell populations, potential heterogeneity representing distinct functional subsets or intermediate states of differentiation may have been missed.
Single-cell RNA sequencing (scRNA-seq) is a powerful approach that can reveal heterogeneity within cell populations and has been used extensively in recent studies to probe the dynamic gene-expression patterns within a wide range of immune cell types in health and disease (20–29). This approach has allowed for the elucidation of new cell subsets and states, such as highly efficacious subpopulations of tumor-infiltrating lymphocytes and early states of differentiation for circulating effector and memory CD8+ T cells (20, 22, 23). Here, we utilized scRNA-seq to generate a single-cell transcriptomic dataset elucidating the dynamic gene expression patterns of individual CD8+ T cells in the spleen and small intestine intraepithelial lymphocyte (siIEL) compartment throughout the course of their differentiation in response to viral infection. These analyses demonstrate that the circulating and tissue-resident subtypes of memory CD8+ T cells utilize highly overlapping patterns of gene expression, revealing a core transcriptional program used by both subtypes throughout their differentiation. In addition, these analyses elucidated sets of genes with kinetics and expression patterns unique to circulating or tissue-resident memory CD8+ T cells that may contribute to the specification of each distinct subtype. Importantly, these data also revealed transcriptional heterogeneity within the siIEL CD8+ T cell population throughout their differentiation. We show that within the established TRM cell population, this molecular heterogeneity reflects functionally distinct subsets that were previously unknown. Moreover, we identify a distinct subset within the CD8+ T cell pool within the tissue early in infection that likely represents the precursors of TRM cells. These findings should inform future studies aimed at improving our understanding of TRM cell differentiation and function, which may contribute to our ability to better manipulate CD8+ T cell responses to protect against infection and cancer.
Results
Single-cell RNA sequencing analyses of circulating and siIEL CD8+ T cells responding to viral infection
We employed a single-cell RNA sequencing approach to investigate the transcriptional changes that occur in circulating and siIEL CD8+ T cells responding to viral infection. We adoptively transferred P14 CD8+CD45.1+ T lymphocytes, which have transgenic expression of a T cell receptor (TCR) that recognizes an immunodominant epitope of lymphocytic choriomeningitis virus (LCMV), into congenic CD45.2+ wild-type recipients that were infected with the Armstrong strain of LCMV one day later. We FACS-sorted naïve (CD62LhiCD44lo) P14 T cells from spleens of uninfected mice as well as activated donor P14 T cells (CD44hi) from the spleens and siIEL compartments of recipient mice at 11 time points post-infection, and performed scRNA-seq using the 10X Genomics Chromium platform (Fig. 1A and fig. S1).
Fig. 1. Single-cell RNA sequencing analyses of circulating and siIEL CD8+ T cells responding to viral infection.
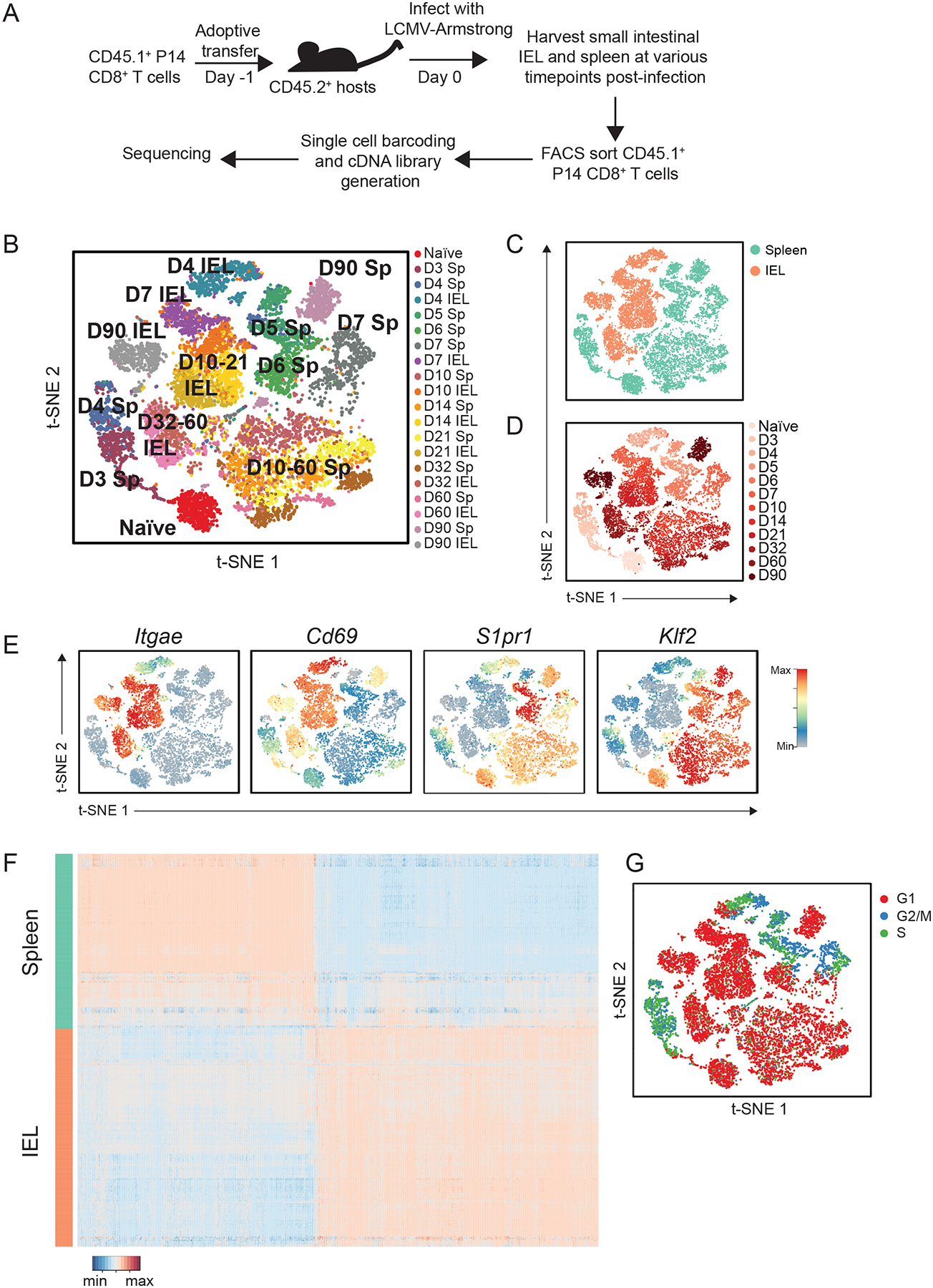
(A) Experimental setup. P14 CD45.1+CD8+ T cells were adoptively transferred into congenic CD45.2+ hosts one day prior to infection with LCMV-Armstrong. Splenocytes were harvested at 3, 5, and 6 days; splenocytes and siIEL CD8+ T cells were harvested at 4, 7, 10, 14, 21, 32, 60, and 90 days post-infection. Naïve T cells (CD44loCD62Lhi) were harvested from spleens of uninfected P14 TCR transgenic mice. Antigen-experienced P14 CD8+ T cells (CD45.1+Vα2+CD44hi) were sorted and processed for scRNA-seq with the 10X Genomics platform. (B to D) tSNE analysis of all scRNA-seq samples, where each individual sample (B), tissue (C), or time point (D) is represented by a unique color. (E) Relative expression of known regulators of circulating and tissue-resident memory CD8+ T cell differentiation superimposed on individual cells. (F) Differential gene expression in Day 4 splenic (teal) and siIEL (coral) CD8+ T cells, represented as expression relative to the mean expression among all cells; each row represents an individual cell and each column represents an individual gene. Threshold for differentially expressed genes was P < 0.05 *(two-sided Wilcoxon test) and an absolute fold-change > 2. (G) Cell cycle status of individual CD8+ T cells, inferred from transcriptional profiles.
In order to relate changes in CD8+ T cell populations over time and across tissues, we merged the data from all time points and anatomic sites and performed unsupervised t-distributed stochastic neighborhood embedding (tSNE) and uniform manifold approximation and projection (UMAP) analyses (Fig. 1, B to D, and fig. S2). CD8+ T cells from the siIEL compartment clustered distinctly from splenic CD8+ T cells at all time points (Fig. 1 B to D, and fig. S2), consistent with the distinct transcriptional profile of TRM cells that has been previously reported (12, 13, 16, 18, 19). For example, expression of genes previously associated with TRM cells, such as Cd69 and Itgae, was strongly enriched among siIEL CD8+ T cells compared to splenic CD8+ T cells, whereas expression of genes that promote tissue egress and recirculation, such as Klf2 and S1pr1, was significantly lower among siIEL cells compared to splenic cells (Fig. 1E). Strikingly, the divergence in gene-expression profiles between splenic and siIEL CD8+ T cells was evident as early as day 4 post-infection (Fig. 1, B to D and fig. S2), the earliest time point at which CD8+ T cells can be detected within the intestinal epithelium (30), indicating that CD8+ T cells begin to change their transcriptional profile rapidly upon entry into tissue. Differential expression analyses revealed that 928 genes were more highly expressed in day 4 splenic CD8+ T cells and 1103 genes were more highly expressed in day 4 siIEL CD8+ T cells, including Cd69 and Itgae (Fig. 1, E and F and fig. S3, and Table S1). Genes more highly expressed by siIEL CD8+ T cells included those associated with processes known to be important for establishment and maintenance of TRM cells, including integrins and cell adhesion molecules (Itga1, Itgb2, Itgal, Itgb7, Itgax, Jaml); regulators of cell trafficking (Ccr9, Cxcr3) and TGF-β signaling (Tgif1, Tgfbr2, Smad7, Skil, Smurf2); the tissue damage receptor P2xr7 (31); and fatty acid-binding proteins (Fabp1, Fabp2, Fabp6) (32) (Fig. 1F and fig. S3, and Table S1). Additionally, genes associated with other aspects of T cell function, including cytokines (Il2, Il10), cytokine receptors (Il4ra, Il2rg, Il10rb, Il21r), chemokines (Ccl3, Ccl4, Ccl5, Cxcl10, Cxcl11), effector molecules (Gzma, Gzmk, Fasl), and both costimulatory (Icos, Cd28) and inhibitory (Ctla4, Tigit, Lag3) receptors, were more highly expressed by siIEL CD8+ T cells. We also observed that genes encoding components and downstream mediators of the TCR signaling pathway (Zap70, Itk, Lats2, Rasgrp1, Fyb, Nr4a1, Nr4a2, Nfatc1, Irf4, Ikzf2, Cd5), regulators of intracellular calcium (Orai1, Orai2, Sri, Rrad), and regulators of NF-κB signaling (Nfkbia, Nfkbiz, Rel, Ikbkb, Pim1, Tnfaip3) were more highly expressed among siIEL CD8+ T cells. Notably, many of these differentially expressed genes were transcription factors with no previously reported role in TRM cells (Ikzf2, Ikzf3, Gata3, Irf4, Id2). Among the genes more highly expressed by splenic CD8+ T cells were Batf and Zeb2, transcription factors that have been shown to regulate effector CD8+ T cell differentiation (33, 34); genes encoding for transcription factors, such as Batf3, that have not been previously implicated in CD8+ T cell differentiation; and genes encoding the high-affinity IL-2 receptor (IL-2Rα) and the OX40 costimulatory receptor (Fig. 1F and fig. S3, and Table S1). Thus, these analyses reveal previously unappreciated signaling pathways and transcription factors that may represent early regulators of circulating versus siIEL CD8+ T cell differentiation.
Gene ontology (GO) analyses revealed that genes associated with DNA replication and cell cycle regulation were enriched among splenic CD8+ T cells, suggesting that siIEL CD8+ T cells may be less proliferative than splenic CD8+ T cells (Table S1). Indeed, assessment of cell cycle status inferred from transcriptional analyses suggested that while both splenic and siIEL CD8+ T cells were actively dividing at day 4 post-infection, siIEL CD8+ T cells had stopped proliferating by day 7 post-infection whereas splenic CD8+ T cells continued proliferating until 7–10 days post-infection (Fig. 1G). These findings indicated that siIEL CD8+ T cells become quiescent more rapidly following activation compared to splenic CD8+ T cells. Taken together, these data reveal that CD8+ T cells that enter the siIEL compartment receive signals that alter their transcriptional profile rapidly after arrival, indicating that the TRM cell fate may begin to be specified earlier than previously appreciated, and may serve as a useful resource for identifying early molecular determinants of TRM cell differentiation.
Shared and tissue-specific components of gene expression programs in circulating and siIEL CD8+ T cells
We next sought to elucidate changes in gene-expression programs over time in CD8+ T cells responding to viral infection and to understand how gene expression programs in circulating versus siIEL CD8+ T cells relate to one another. We analyzed splenic and siIEL CD8+ T cells separately and performed weighted gene co-expression network analyses on each set of cells, defining tissue-specific modules of genes that exhibited similar patterns of expression over time in splenic (fig. S4, A and B, and Table S2) or siIEL (fig. S4 C and D, and Table S2) CD8+ T cells. We identified 10 and 8 distinct gene-expression modules among splenic and siIEL CD8+ T cells, respectively (fig. S4A to D, and Table S2).
Although recent studies have primarily highlighted the differences in gene expression between circulating and tissue-resident CD8+ T cells (12, 13, 16, 18, 19), we notably found substantial overlap in the gene-expression patterns utilized by differentiating splenic and siIEL CD8+ T cells. For example, 38% of the genes in Spleen Module 1 were shared with IEL Module 2; genes in these early differentiation modules were characterized by a decrease in expression, relative to that by naïve cells, following T cell activation (fig. S4, B, D, and E). Similarly, 40% of the genes in IEL Module 4 were shared with Spleen Module 4; genes in these intermediate differentiation modules were characterized by high expression during the peak of infection, followed by a subsequent decrease. Lastly, 33% of the genes in IEL Module 7 were shared with Spleen Module 10; genes in these late differentiation modules were characterized by a reduction in expression after T cell activation, followed by increasing expression that was sustained at late time points (fig. S4, B, D, and E, and Table S2). These observations suggested that splenic and siIEL CD8+ T cells share a core transcriptional program throughout their differentiation, consistent with their common cytolytic lymphocyte functions (fig. S4E and Table S2), and highlight the potential value of this dataset to reveal how specific genes or pathways, including those known to play key roles in regulating circulating memory CD8+ T cells, may act similarly or disparately in regulating tissue-resident memory differentiation.
To elucidate distinct regulators of the circulating and tissue-resident memory differentiation programs, we again performed weighted gene co-expression network analyses, but this time analyzed splenic and siIEL CD8+ T cells together. This approach enabled us to directly compare the level of representation of each module in cells from each anatomic site at each time point. Fifteen modules representing distinct temporal patterns of gene expression were defined and annotated as Combined Modules 1 – 15 (Fig. 2, A to C, and Table S3). This analysis confirmed our observation that splenic and siIEL CD8+ T cells indeed share a core transcriptional program, as many Combined Modules were similarly represented in splenic and siIEL CD8+ T cells throughout most of their differentiation (Fig. 2, B and C). Moreover, this analysis confirmed our finding that siIEL CD8+ T cells were transcriptionally distinct from splenic CD8+ T cells as early as day 4 post-infection (Fig. 1, B to F), since we noted that many modules (Combined Modules 2, 3, 5, 7, 11, and 12) were differentially represented within splenic and siIEL CD8+ T cells at day 4 post-infection, but not at later time points (Fig. 2, B and C). For example, genes in one such module, Combined Module 12, were enriched in several pathways known to play roles in circulating CD8+ T cell memory, including autophagy, protein ubiquitination, and proteasome protein catabolism (35, 36) (Table S3). These findings suggested that differentiating siIEL CD8+ T cells may begin to acquire certain aspects of a memory-like transcriptional profile more rapidly than splenic CD8+ T cells.
Fig. 2. Identification of a siIEL CD8+ T cell-enriched gene-expression profile.
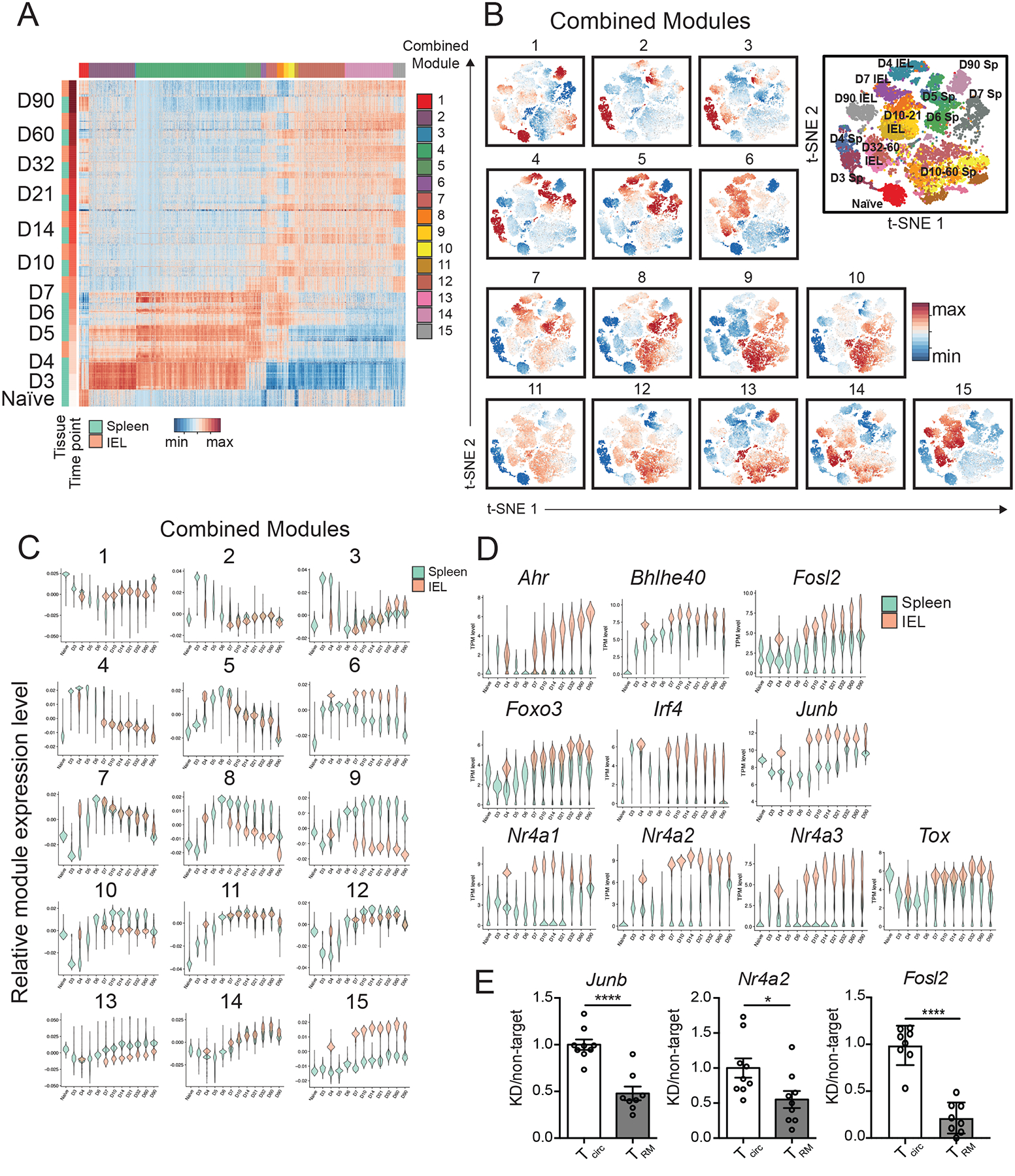
(A) Gene-expression patterns of single splenic and siIEL CD8+ T cells responding to infection, where each row represents an individual cell, grouped by tissue within each time point, and each column represents an individual gene, grouped by module, represented as expression relative to the mean expression among all cells. Weighted gene co-expression network analyses of splenic and siIEL CD8+ T cells considered together were performed to derive gene modules. (B and C) Representation of each module among single CD8+ T cells in both the spleen and siIEL compartment over time, relative to the mean representation among all cells, depicted as a representation score superimposed on individual cells (B) or as violin plots showing the relative representation of each module in individual splenic (teal) versus siIEL CD8+ T cells (coral) (C). (D) Violin plots depicting gene-expression patterns of known or putative regulators of TRM cell differentiation (represented as transcripts per million, TPM), selected from siIEL CD8+ T cell-enriched modules, among single splenic (teal) or siIEL (coral) CD8+ T cells over time. (E) CD45.1+ P14 T cells were transduced with retrovirus encoding shRNA targeting Nr4a2, Junb, or Fosl2 (knockdown, KD), and mixed with CD45.1.2+ P14 T cells transduced with shRNA encoding control (non-target) shRNA at a 1:1 ratio of KD: non-target cells prior to adoptive transfer into CD45.2+ hosts that were subsequently infected with LCMV. 22–26 days later, splenic and siIEL CD8+ T cells were analyzed by flow cytometry. Ratio of KD: non-target P14 T cells within transduced P14 CD8+ T cells in the spleen (Tcirc) or within CD69+CD103+ siIEL P14 CD8+ T cells (TRM), normalized to the ratio of KD: non-target cells at the time of transfer. Values are normalized to Tcirc. *P<0.05, ****P<0.0001 (Student’s two-tailed t-test). Data were pooled from two independent experiments, with mean and SEM of n=8 mice per gene, where each dot represents an individual mouse.
These analyses also defined several modules that were significantly and consistently differentially enriched in splenic vs. siIEL CD8+ T cells over time. For example, Combined Modules 6 and 15 were enriched in siIEL compared to splenic CD8+ T cells; genes in these modules were highly upregulated in siIEL CD8+ T cells throughout their differentiation (Fig. 2B and C). Combined Modules 6 and 15 contained 157 and 371 genes, respectively, some of which have been previously implicated in CD8+ TRM cell differentiation and function, such as Cd69, Itgae, Itga1, and Ccr9, which regulate homing and retention within the intestinal environment; transcription factors Nr4a1 and Ahr, which regulate the long-term maintenance of TRM (37, 38), Runx3, a transcriptional regulator of key genes that establish tissue-residency that is critical for TRM cell differentiation (16), and Bhlhe40, which sustains TRM cell metabolic fitness and promotes an epigenetic state permissive for expression of key tissue-residency genes (39); and Ccl3 and Ccl4, which are involved in the rapid recruitment of innate cells by TRM cells (8, 10) (Fig. 2D and fig. S5, Table S3). Additionally, GO analyses revealed enrichment of pathways known to play roles in TRM cell differentiation and maintenance, such as TGF-β signaling (15) (Skil, Smurf, Tgif1) and fatty acid homeostasis (Dgat1, Got1), consistent with the dependence of TRM cells on fatty acid metabolism for their maintenance and function (32) (Fig. 2D and fig. S5, and Table S3). These TRM cell-enriched modules also included genes encoding effector molecules and cytokines, such as Il2, Ifng, Tnf, Fasl, and Gzmb, sustained high expression of which may contribute to the rapid and potent recall responses of TRM cells. Moreover, other groups of genes represented in these modules implicated pathways with previously unexplored roles in TRM cell differentiation, some of which were already upregulated in siIEL CD8+ T cells as early as day 4 post-infection (Fig. 1F and fig. S3). These included inhibitory receptors (Ctla4, Lag3, Cd101, Tigit); factors involved in TCR signaling and costimulation (Zap70, Lat, Cd8a, Cd3e, Cd40lg, Nfatc1); regulators of cell survival (Bcl2a1b, Bcl2l11, Bcl2a1d) and NF-κB signaling (Nfkbia, Nfkbiz, Nfkbid, Rel); genes involved in cytokine responses (Stat3, Stat1, Irak2, Socs1, Jak2); and transcription factors with previously uncharacterized roles in TRM cell differentiation (Nr4a2, Nr4a3, Junb, Fosl2, Tox, Foxo3, Irf4) (Fig. 2D and fig. S5, and Table S3). In addition, genes involved in cholesterol biosynthesis (Fdps, Cyp51, Mvk, Fdft1, Hmgcr, Hmgcs) and steroid hormone mediated signaling pathways were enriched in Combined Modules 6 and 15, respectively, raising the possibility of a role for these processes in TRM cell differentiation.
By contrast, Combined Modules 8 and 9 were enriched in splenic relative to siIEL CD8+ T cells beginning at day 7 post-infection, and included 196 and 138 genes, respectively, some of which have been previously implicated in promoting differentiation of circulating memory CD8+ T cells at the expense of TRM cells (Fig. 2, B and C, and Table S3). For example, these spleen-enriched modules included Klf2, S1pr1, Ly6c1, and Ly6c2, which regulate trafficking and egress, and Eomes, a transcription factor that negatively regulates TRM cell differentiation (12, 14, 15, 30) (fig. S5 and Table S3). These spleen-enriched modules also included several cell surface markers, including Cx3cr1 and Klrg1, that have been associated with subsets of circulating effector CD8+ T cells. Additionally, the presence of genes encoding specific cytokine receptors (Il18r1, Il18rap, Il17ra) in these spleen-enriched modules suggests that signaling through these receptors might negatively regulate TRM cell differentiation and maintenance.
In order to demonstrate that genes identified by these analyses indeed represent functionally important regulators of TRM cell differentiation, we selected for further studies three genes found in Combined Module 15, Nuclear Receptor Subfamily 4 Group A Member 2 (Nr4a2) and two AP-1 transcription factor subunits, Junb proto-oncogene, (Junb) and FOS-Like 2 (Fosl2). These genes exhibited increased expression in siIEL relative to splenic CD8+ T cells at all time points (Fig. 2D). Nr4a2 is an orphan nuclear receptor that belongs to the Nr4a family of transcription factors, plays a central role in the development of regulatory T cells as well as pathogenic Th17 cells (40–43), and has been implicated as an important driver of exhaustion in CD8+ T cells (44). Junb and Fosl2 play important roles in regulating Th17 cell identity and pathogenicity (45–47), and Junb has also been implicated as part of the effector CD8+ T cell transcriptional program (34). Nr4a2, Junb, and Fosl2 have not been previously reported to regulate the differentiation of TRM cells. To investigate whether these genes play a role in TRM cell differentiation, we transduced P14 CD8+CD45.1+ T cells with retroviruses encoding shRNA targeting Nr4a2, Junb, or Fosl2, and transduced P14 CD8+CD45.1.2+ T cells with retroviruses encoding non-targeting shRNA. Cells were mixed at a 1:1 ratio and adoptively transferred into CD45.2 recipient mice that were subsequently infected with LCMV (fig. S6A). Relative to non-targeting controls, knockdown of Nr4a2, Junb, or Fosl2 resulted in a greater reduction in siIEL TRM cells than splenic memory CD8+ T cells (Fig. 2E and fig. S6B). Taken together, these results highlight key transcriptional differences between circulating and siIEL CD8+ T cells undergoing differentiation and demonstrate the potential value of this dataset in identifying previously unknown genes and pathways involved in specifying TRM versus circulating memory CD8+ T cell fates.
Single-cell RNA-sequencing analyses highlight circulating CD8+ T cell heterogeneity and reveal previously unappreciated heterogeneity within the siIEL CD8+ T cell pool
We next investigated potential heterogeneity of differentiating splenic and siIEL CD8+ T cells. We observed that individual splenic CD8+ T cells at certain time points expressed genes previously associated with CD8+ T cell subsets, such as terminal effector (TE)- and memory precursor (MP)- phenotype cells at the peak of infection, and TEM, TCM, and LLE cell subsets at later time points following infection (Fig. 3, A and B). For example, the majority of cells at days 6 and 7 post-infection expressed high levels of Klrg1, likely representing TE cells, whereas a smaller number of cells exhibited lower expression of Klrg1 and higher expression of Tcf7 and Bcl2, likely representing MP cells (48–50). We also observed that certain cells at later time points expressed genes previously associated with TCM (Il7r, Tcf7, Sell, Bcl2, Cxcr3), LLE (Klrg1, Cx3cr1, Zeb2), and TEM (intermediate levels of Cx3cr1, Zeb2, Il7r, Tcf7, and Bcl2) cells (1, 3) (Fig. 3, A and B). Indeed, mapping individual splenic CD8+ T cells to TE, MP, TCM, TEM, and LLE transcriptional signatures defined by bulk RNA-seq signatures of sorted cells from each subset revealed that heterogeneity observed among splenic CD8+ T cells might indeed represent cells differentiating into each of these memory cell subsets (Fig. 3, C and D). Notably, low, intermediate, or high expression of Cx3cr1 at day 6 post-infection generally corresponded to cells with TCM, TEM, or LLE profiles, respectively, consistent with previously reported CX3CR1hi, CX3CR1int, and CX3CR1lo subsets with distinct functional capacities and terminal differentiation potentials that can be found at the peak of infection (51) (Fig. 3, B and D). These results demonstrate the concurrent presence of these memory subsets within the circulating CD8+ T cell memory pool, consistent with previous studies (4–6, 52) and support findings suggesting that these memory CD8+ T cell subsets may begin to diverge in their differentiation pathways earlier than is widely appreciated (51). These analyses also uncovered a striking transcriptional divergence between cells at days 60 vs. 90 post-infection (Fig. 3A). Prior studies have shown that TCM cells are the predominant circulating memory subset at late time points, but whether and how TCM cells change transcriptionally at late time points has not been investigated in depth. Compared to Day 60 TCM cells, Day 90 TCM cells expressed higher levels of Eomes and lower levels of genes encoding cytolytic granules such as Gzma and Gzmb, suggesting a progressive loss of cytotoxic capability over time (Table S4). Moreover, these analyses highlighted a number of genes without known functions in TCM cell differentiation and/or maintenance, such as Hspa1a and Hspa1b, which encode members of a family of conserved heat shock proteins (HSP70) that plays a role in protecting against cellular stressors, including hypoxia, temperature aberrations, pH alterations, and oxidative stress (53).
Fig. 3. Single-cell RNA sequencing analyses highlight heterogeneity among circulating CD8+ T cells.
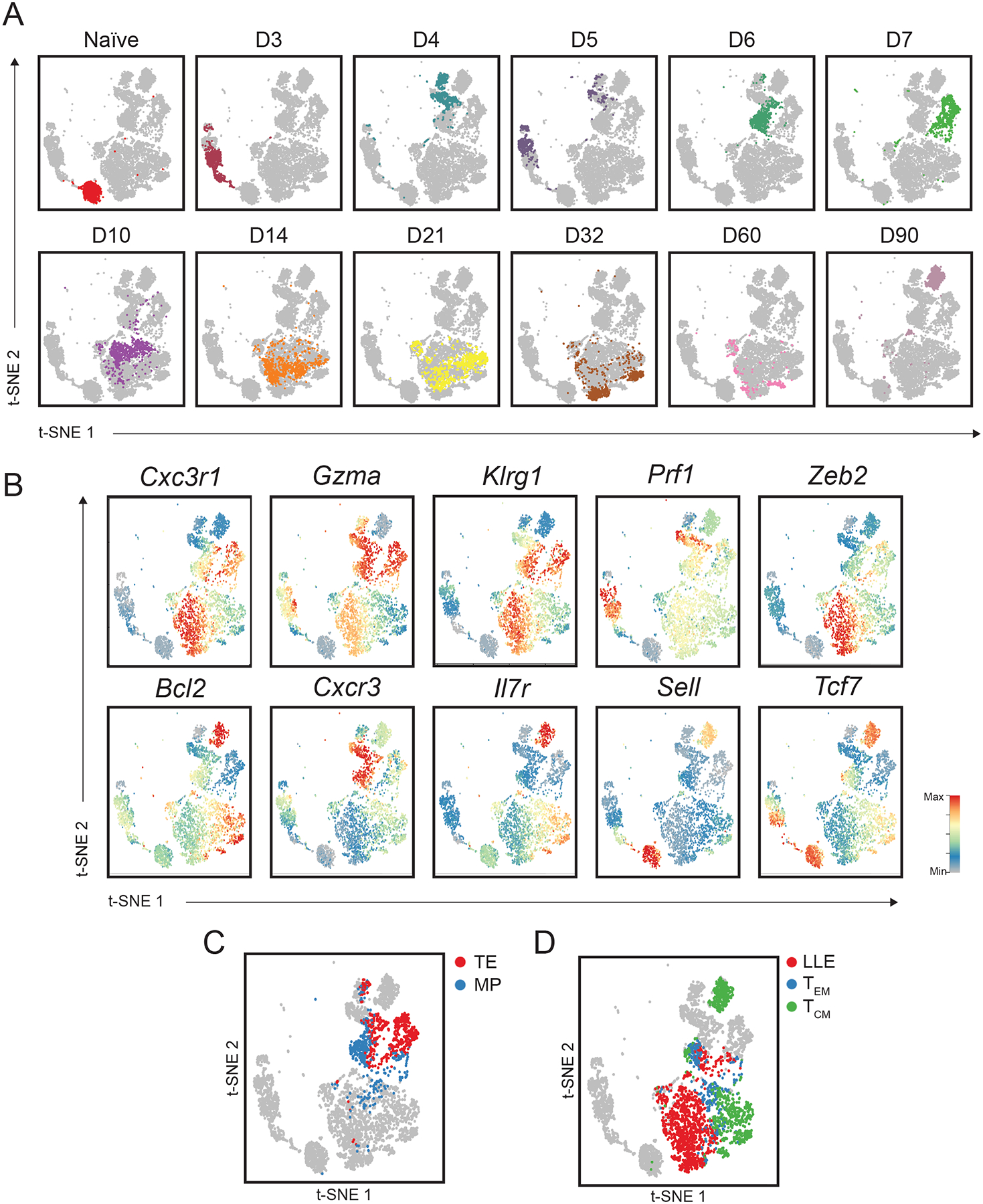
(A) tSNE analysis of splenic CD8+ T cells; each plot represents an individual time point with each color representing a specific time point, as in Fig. 1B. (B) Relative expression (compared to mean expression among all spleen cells) of known regulators of circulating CD8+ T cell differentiation superimposed on individual spleen cells. (C and D) Similarity of gene-expression programs among single spleen cells to transcriptional signatures (derived from bulk RNA-seq profiles from FACS-sorted cells) of previously defined circulating CD8+ T cell subsets. (C) Similarity of gene-expression programs among single spleen cells to terminal effector (TE) (D7 KLRG1hiIL-7Rαlo, red) or memory-precursor (MP) (D7 KLRG1loIL-7Rαhi, blue) transcriptional signatures. (D) Similarity of gene-expression programs among single spleen cells to LLE (D35 CD62LloIL-7Rαlo, red), TEM (D35 CD62LloIL-7Rαhi, blue), and TCM (D35 CD62LhiIL-7Rαhi, green) cell transcriptional signatures.
Although heterogeneity within circulating CD8+ T cells has been relatively well described, whether heterogeneity exists within tissue-resident CD8+ T cell populations remains unclear. To investigate potential heterogeneity within the siIEL CD8+ T cell compartment, we defined cell clusters across the entire dataset, revealing 40 distinct clusters, 17 of which were found in the siIEL compartment (Fig. 4A). We also performed partition-based graph abstraction (PAGA) trajectory analysis (54) to infer relationships between the siIEL clusters we defined (fig. S7). We observed that multiple clusters of siIEL CD8+ T cells were present within several individual time points, suggesting that distinct subsets might exist within the siIEL CD8+ T cell population (Fig. 4B). For example, at day 60 post-infection, two major cell clusters, Clusters 3 and 29, were observed. These clusters exhibited differential expression of 236 genes, including several transcription factors, suggesting that these clusters were regulated by distinct gene-expression programs (Table S5). For example, Cluster 3 cells had higher expression of Id3, Jun, Fos, Klf2, and Myc, while Cluster 29 cells had higher expression of transcription factors including Bcl6, Zeb2, and Klf3. Cluster 3 cells also exhibited higher expression of several genes associated with TRM cell function, including cytokines and chemokines (Ifng, Tnf, Ccl3, Ccl4), suggesting that Cluster 3 cells might be better poised to produce cytokines rapidly upon re-infection (Fig. 4C, Table S5). Notably, Cluster 3 cells also had higher transcript levels of the transcription factor Id3, and a recent study identified an Id3hi TRM cell subset with distinct functional capabilities including an enhanced capacity to produce cytokines (55). These analyses suggest that distinct costimulatory and other signaling pathways might be responsible for regulating each of these cell clusters and raised the possibility that these cell clusters might have distinct functional capacities. Consistent with this possibility, PAGA analysis revealed that while Cluster 3 cells were linked to day 90 clusters, Cluster 29 cells were not, suggesting that Cluster 29 cells might represent a terminal state (fig. S7). These findings are consistent with increased expression of the transcription factor Zeb2, which is known to promote the terminal differentiation of circulating CD8+ T cells (33), by Cluster 29, and the reciprocal increased expression among Cluster 3 cells of the transcription factor Id3, which is known to regulate the development of long-lived circulating memory CD8+ T cells (56, 57).
Fig. 4. Single-cell RNA sequencing analyses reveal functionally distinct subsets within the siIEL CD8+ T cell pool.
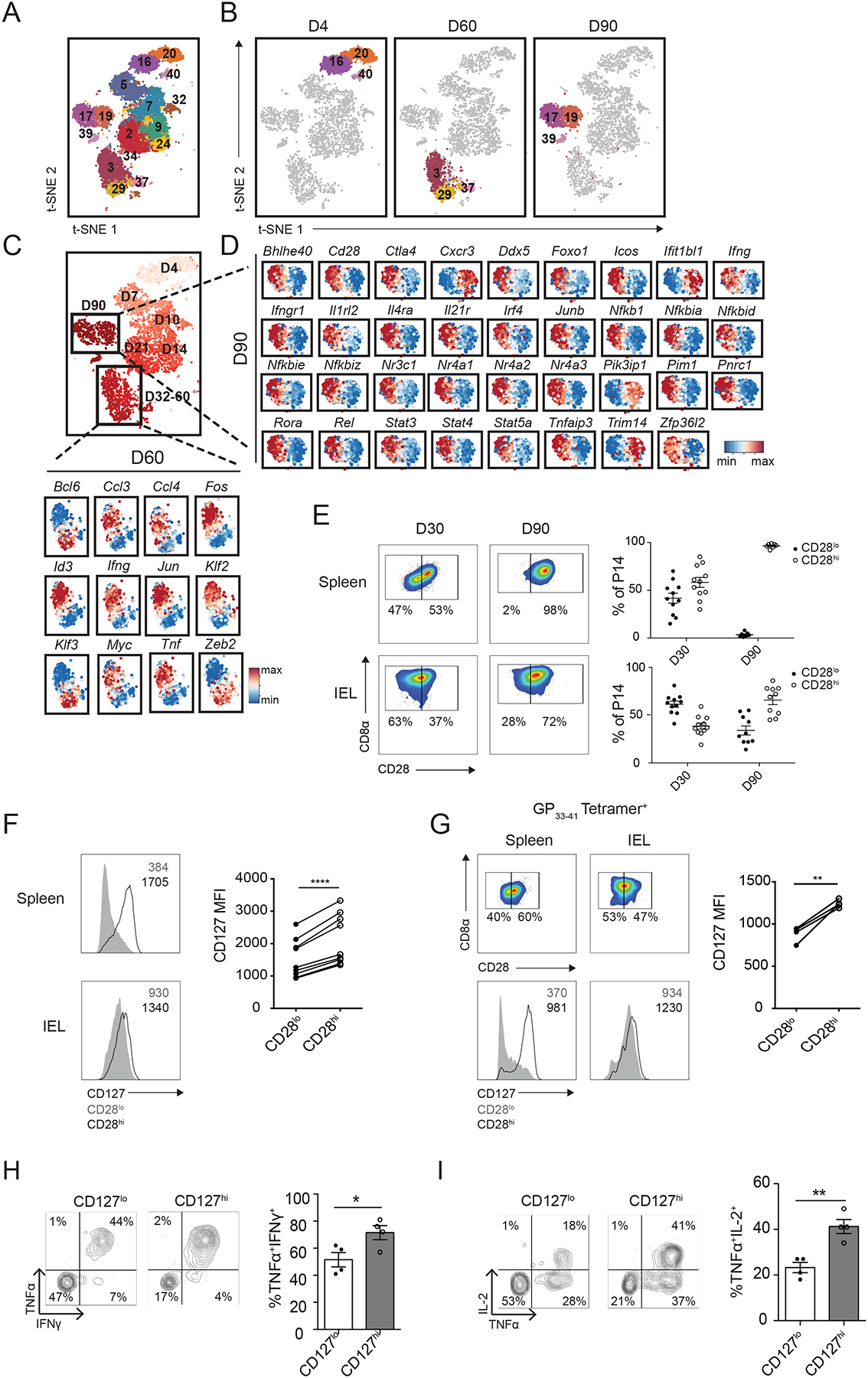
(A and B) Clustering analyses of siIEL CD8+ T cells from all time points (A) or cells at days 4, 60, and 90 post-infection (B); numbers represent different cluster annotations. (C and D) Expression of selected genes within single siIEL CD8+ T cells at day 60 (C) or 90 (D) post-infection, relative to the mean expression among siIEL CD8+ T cells at the indicated time point, demonstrating differential gene expression between Clusters 3 and 29 (C) or between Clusters 17 and 19 (D). (E to I) P14 CD8+ T cells were transferred into congenically distinct recipients 1 day prior to infection with LCMV. Splenic and siIEL CD8+ T cells were harvested at day 30 or 90 post-infection. (E) Representative flow cytometry plots displaying distribution of CD28 expression among total P14 T cells in the spleen (top) or CD69+CD103+ P14 T cells in the siIEL compartment (bottom). Numbers represent the percentage of total P14 T cells in each gate, and graphs to the right demonstrate the quantification of CD28lo (filled dots) and CD28hi (open circles) cells within the indicated population at each time point. Data are representative of (flow cytometry plots, left), or compiled from (graphs, right) 3 independent experiments where n=2–5 mice per experiment (10–11 total mice). (F) Quantification of CD127 (IL-7Rα) expression by CD28lo (gray) or CD28hi (black) subsets within the spleen (top) or siIEL (bottom) at day 30 post-infection, with representative flow cytometry plots shown on the left. Numbers represent Mean Fluorescence Intensity (MFI) of CD127 within the indicated population. Data are representative of (left) or compiled from (graph on right side) n=3 independent experiments with n=7 total mice, where each dot represents an individual mouse. (G) Representative flow cytometry plots displaying (top) distribution of CD28 expression at day 30 post-infection among total H-2Db GP33–41 tetramer+ endogenous CD8+ T cells in the spleen (left) or among H-2Db GP33–41 tetramer+ endogenous CD69+CD103+ CD8+ T cells in the siIEL compartment (right), or displaying (bottom) expression of CD127 by CD28lo (gray) or CD28hi (black) subsets within the indicated compartment. Numbers represent the percentage of H-2Db GP33–41 tetramer+ cells in each gate (top), or MFI of CD127 expression in the indicated population (bottom), quantified in the graph (right), where n=4 individual mice. (H and I) At day 30 post-infection, siIEL P14 T cells were sorted into CD127lo and CD127hi subsets and cultured in the presence of GP33–41 peptide in vitro. Quantification (right) of the proportion of CD127hi or CD127lo P14 siIEL CD8+ T cells producing IFN-γ and TNF-α (H), or IL-2 and TNF-α (I) as shown in representative flow cytometry plots (left). Data are representative of 2 independent experiments, with mean and SEM of n=4 wells of cultured cells per phenotype, derived from two separate pools of sorted cells plated in duplicate. *P<0.05, **P<0.01, ****P<0.0001 (Paired t-test, F and G, or Student’s two-tailed t-test, H and I).
Additionally, at day 90 post-infection, two major cell clusters, Clusters 17 and 19, were observed; these clusters exhibited differential expression of 361 genes (Table S6). Cluster 17 cells exhibited higher expression of 243 genes, including transcription factors (Bhlhe40, Ddx5, Foxo1, Irf4, Junb, Nr3c1, Nr4a1, Nr4a2, Nr4a3, Pnrc1, and Rora) and mediators of cytokine signaling (Ifngr1, Il1rl2 Il21r, Il4ra, Stat3, Stat5a, and Stat4). Cluster 17 cells also exhibited higher expression of Zfp36l2, which encodes for a zinc finger protein involved in the regulation of mRNA decay (58), and the transcript for IFN-γ, as well as genes associated with pathways that were highly represented within TRM cell-enriched modules, such as the regulation of NF-κB signaling (Nfkbia, Nfkbid, Tnfaip3, Pim1, Rel, Nfkb1, Nfkbie, Nfkbiz) and costimulatory and inhibitory molecules Icos and Ctla4 (Fig. 4D and Table S6). Cluster 17 cells also exhibited higher expression of the costimulatory receptor Cd28, suggesting that costimulation through this receptor could specifically influence the differentiation or responsiveness of a specific subset of TRM cells. By contrast, Cluster 19 exhibited higher expression of 117 genes, including the tissue-homing molecule Cxcr3, interferon response genes (Trim14, Ifit1bl1), and Pik3ip1, a negative regulator of T cell activation (Fig. 4D and Table S6). Taken together, these findings suggested that specific cytokine and/or costimulatory signals might be responsible for maintaining or directing the differentiation of Cluster 17 cells. Moreover, while Cluster 17 cells might be better poised to produce cytokines rapidly upon re-infection, Cluster 19 cells might be more likely to respond immediately to type I interferon signals within the tissue. These data suggest that multiple subsets with distinct transcriptional programs and functional capacities may exist within the TRM cell pool.
We next performed flow cytometry to determine whether the heterogeneity revealed by scRNA-seq analyses could be discerned at the protein level. CD45.1+ P14 CD8+ T cells were adoptively transferred into congenic CD45.2+ hosts that were infected with LCMV one day later. At days 30 and 90 post-infection, the spleens and siIEL compartments of recipient mice were harvested for analysis. At day 30 post-infection, both splenic and siIEL CD8+ T cells could be divided into CD28hi and CD28lo populations (Fig. 4E). In contrast, at day 90 post-infection, splenic P14 CD8+ T cells uniformly expressed high levels of CD28, but heterogeneity was retained within the siEL P14 CD8+ T cell population (Fig. 4E), in line with data revealing multiple clusters within the TRM cell population with differential expression of Cd28 at the mRNA level at day 90 post-infection (Fig. 4D). We also noted that high expression of IL-7Rα (CD127), which was observed in the functionally distinct Id3hi TRM cell subset described in a recent study (55), also correlated with high expression of CD28 (Fig. 4F). Notably, we also observed this heterogeneity among endogenous, siIEL CD8+ H-2Db GP33–41 tetramer+ cells, with CD28hi cells expressing higher levels of CD127 (Fig. 4G). To investigate whether heterogeneity in expression of these markers might indeed reflect functional heterogeneity, we sorted CD127hi and CD127lo populations from the siIEL CD8+ T cell pool at day 30 post-infection, and measured their capacities to produce cytokines when rechallenged with cognate antigen in vitro. We found that CD127hi cells produced higher levels of the cytokines IFN-γ, TNF-α, and IL-2 in response to restimulation than did CD127lo cells (Fig. 4, H and I), consistent with enhanced cytokine production in Id3hi TRM cells (55), and the higher level of Ifng transcript detected within CD28hi cells at day 90 post-infection (Fig. 4, C and D). Although it remains possible that differences in the ability to produce cytokine upon restimulation may reflect different responses to ex vivo conditions, the observation that these two populations respond differently suggests that they are functionally distinct. Taken together, these data demonstrate that the heterogeneity revealed by scRNA-seq analyses within the TRM cell pool at late time points after infection may be functionally important.
The observation that expression of a number of transcriptional regulators, including Ddx5, Junb, Nr4a2, and Pnrc1, as well as the RNA-binding protein Zfp36l2, correlated with Cd28 expression (Fig. 4D), raised the possibility that these genes might represent regulators of TRM cell heterogeneity. To investigate this possibility, we transduced P14 CD8+CD45.1+ T cells with retroviruses encoding shRNA targeting Junb, Nr4a2, Pnrc1, or Zfp36l2 prior to adoptive transfer into congenic CD45.2+ hosts that were subsequently infected with LCMV. Knockdown of Nr4a2 and Junb resulted in a loss of total TRM cells, while knockdown of Pnrc1 or Zfp36l2 did not affect the size of the total TRM cell population (fig. S6C). However, knockdown of each of these genes resulted in a reduction in the proportion of siIEL TRM (CD69+CD103+) cells expressing high levels of CD28 compared to congenically distinct, co-transferred control P14 CD8+ T cells transduced with retroviruses encoding non-targeting shRNA (Fig. 5A and fig. S6C). Additionally, we co-transferred P14 CD8+ T cells from mice with a T cell-specific deletion of Ddx5 (Ddx5fl/flCD4cre+: ‘Ddx5−/−’) and congenically distinct control P14 CD8+ T cells (Ddx5fl/flCD4cre−: ‘wild-type, WT’) into recipients infected with LCMV one day later, and found that while Ddx5 deletion resulted in a severe depletion of both the total circulating T cell and total TRM cell populations, the proportion of CD28hi cells was further reduced among the remaining Ddx5−/− TRM cell population (Fig. 5B and fig. S6C). Moreover, consistent with this decrease in the proportion of CD28hi cells, Ddx5−/− siIEL, but not splenic, CD8+ T cells, exhibited a reduced ability to produce IFN-γ and TNF-α upon restimulation with cognate antigen in vitro (Fig. 5C). Taken together, these findings suggest that, like Prdm1 and Id3 (55), Ddx5, Junb, Nr4a2, Pnrc1, and Zfp36l2 regulate the differentiation of transcriptionally and functionally distinct TRM cell subsets.
Fig. 5. Single-cell RNA sequencing analyses identify putative regulators of CD8+ TRM cell heterogeneity.
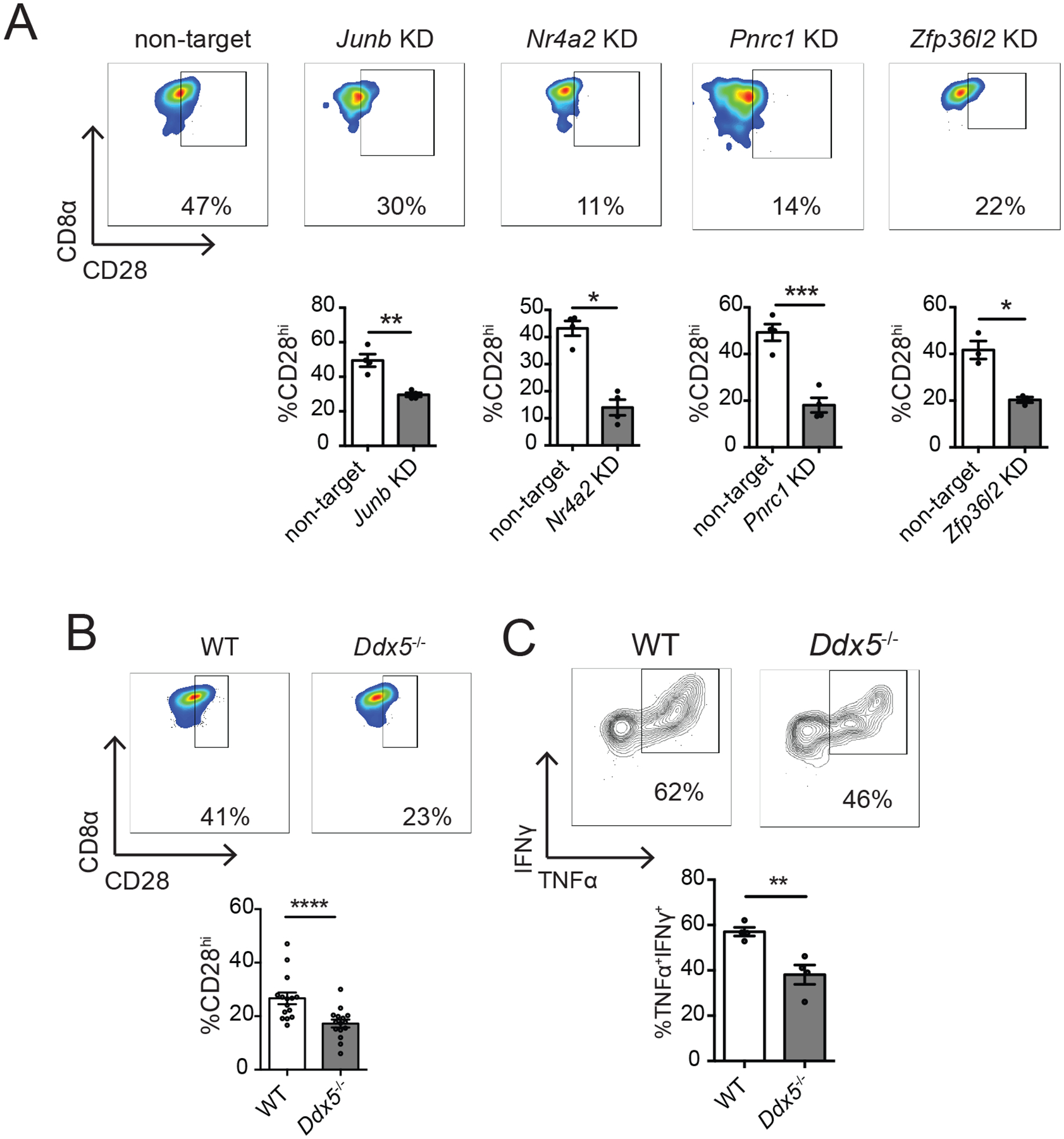
(A) CD45.1+ P14 T cells were transduced with retrovirus encoding shRNA targeting the indicated genes (knockdown, KD), and mixed with CD45.1.2+ P14 T cells transduced with shRNA encoding control (non-target) shRNA at a 1:1 ratio of KD: non-target cells prior to adoptive transfer into CD45.2+ hosts that were subsequently infected with LCMV, as in fig. S6. 22–23 days later, siIEL CD8+ T cells were analyzed by flow cytometry. Quantification of the percent of non-target or KD P14 CD8+ TRM cells (CD69+CD103+) expressing high levels of CD28 (bottom) as shown in representative flow cytometry plots (top). Data are representative of 1–2 experiments, with mean and SEM of n=4 mice per gene, where each dot represents an individual mouse. (B and C) P14 CD8+ T cells from mice with T cell-specific deletion of Ddx5 (Ddx5fl/fl CD4-Cre+: ‘Ddx5−/−’) were co-transferred at a 1:1 ratio with congenically distinct control P14 CD8+ T cells (Ddx5fl/fl CD4-Cre−: ‘WT’) into congenically distinct hosts that were infected with LCMV one day later. At day 30 post-infection, spleens and siIEL were harvested for flow cytometric analysis. (B) Quantification of the percent of Ddx5−/− or WT P14 CD8+ TRM cells (CD69+CD103+) expressing high levels of CD28 (bottom) as shown in representative flow cytometry plots (top). (C) Lymphocytes harvested from siIEL compartment were cultured in the presence of GP33–41 peptide in vitro prior to staining for flow cytometric analysis. Cytokine production in Ddx5−/− or WT siIEL P14 CD8+ T cells, shown as quantification of the proportion of cells producing TNF-α and IFN-γ (bottom), as shown in representative flow cytometry plots (top). Data are compiled from (B) or representative of (C) two independent experiments with mean and SEM of n=4–12 mice, where each dot represents an individual mouse. *P<0.05, **P<0.01, ***P<0.001 (Paired t-test).
Although it is known that two distinct populations with differing memory potentials, distinguished by high or low expression of the high-affinity IL-2 receptor, IL-2Rα (CD25) (59, 60), exist within the circulating CD8+ T cell pool early in infection, whether the CD8+ T cell pool that seeds the tissue early in infection is heterogeneous remained unknown. To determine whether heterogeneity of siIEL CD8+ T cells can be discerned early after infection, we next examined the earliest time point, day 4 post-infection, at which CD8+ T cells could be detected in the siIEL compartment. In addition to the heterogeneity observed at late time points following infection (Fig. 4B), two major clusters, Clusters 16 and 20, were evident at day 4 post-infection and exhibited differential expression of 332 genes (Fig. 4B, 5B and 6A, and Table S7). Only 17 of these genes were more highly expressed by Cluster 16 cells, such as interferon response genes Ifit1, Ifit1bl1, and Ifit3; Pik3ip1, a negative regulator of TCR signaling; Mxd4, a Myc-antagonist that promotes survival in T cells (61); and Kdm5b, a histone demethylase (Fig. 6A and Table S7). Gene Ontology analyses revealed that the genes expressed more highly by Cluster 20 cells were overrepresented in biologic processes such as DNA replication, mitotic spindle and nucleosome assembly, cytokinesis, and cell division; specific genes included Cdc7, Cdc25b, Cenpk, Cenpm, Cdc20, and Cdk1 (Fig. 6A and Table S7). Moreover, additional analyses confirmed that a greater proportion of Cluster 20 cells were in the G2/M phases of the cell cycle compared to Cluster 16 cells, indicating that cells in Cluster 20 were more actively proliferating (Fig. 6B). Cluster 20 cells also expressed higher levels of Dnmt1, a DNA methyltransferase that is critical for the expansion of CD8+ T cells during the effector phase of the immune response (62), along with several components of the Polycomb Repressive Complex 2, including Suz12 and Ezh2 (Fig. 6A), which negatively regulates gene expression via histone methylation and has been previously shown to promote effector CD8+ T cell differentiation by mediating the repression of memory-associated genes (20, 63). Consistent with these findings, PAGA trajectory analysis (fig. S7) revealed that while Cluster 16 cells were linked to day 7 clusters, Cluster 20 cells were not, suggesting that Cluster 20 cells might be undergoing terminal differentiation whereas Cluster 16 cells might represent precursors to siIEL CD8+ TRM cells.
Fig. 6. TRM precursors identified within the siIEL CD8+ T cell pool early in infection.
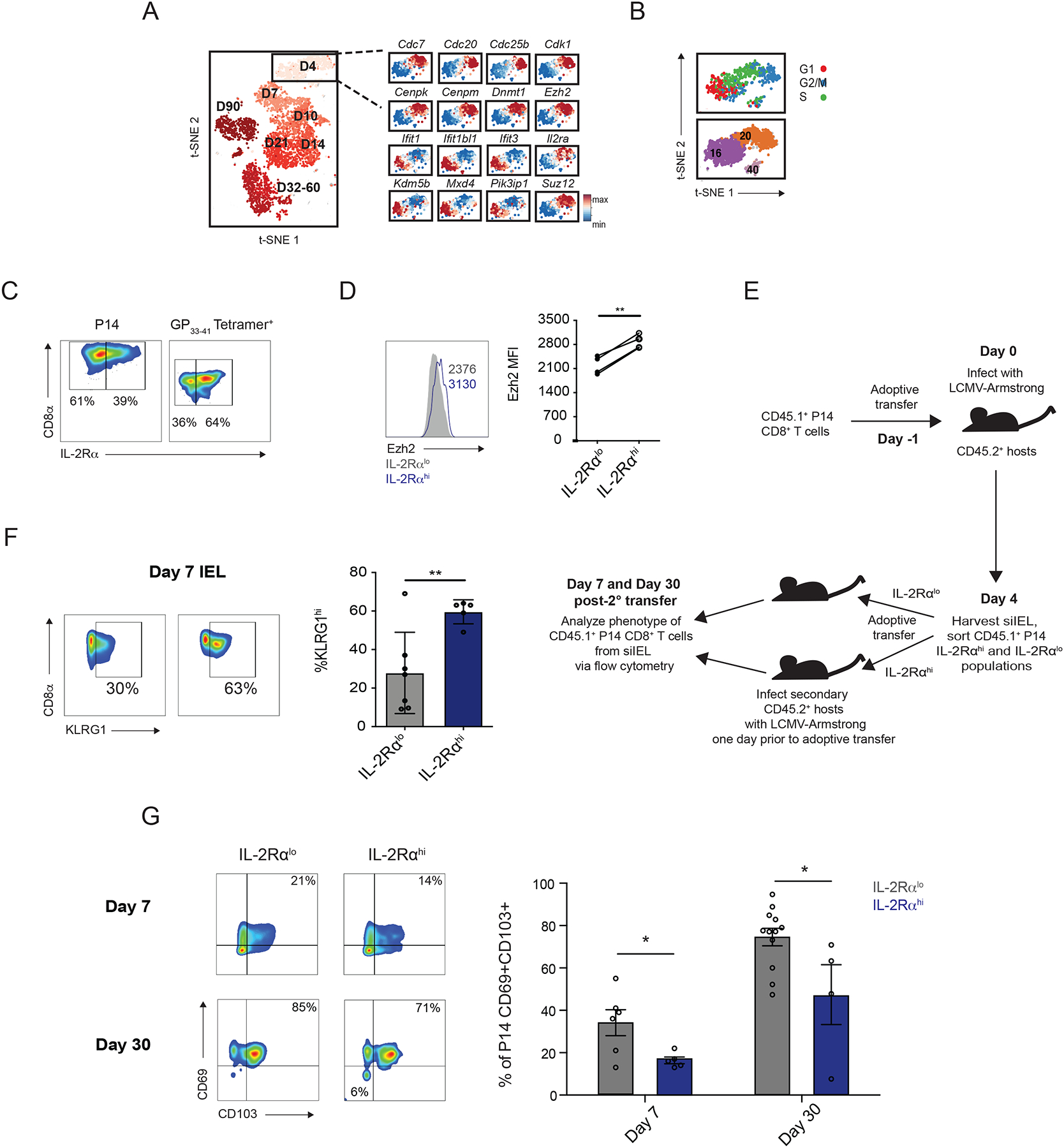
(A) Expression of selected genes within single siIEL CD8+ T cells at day 4 post-infection, relative to the mean expression among all cells, demonstrating differential gene expression between cells in Clusters 16 and 20. (B) Cell cycle status of siIEL CD8+ T cells at day 4 post-infection, inferred from transcriptional profiles. (C and D) P14 CD8+ T cells were adoptively transferred into congenic hosts one day prior to infection with LCMV. Splenic and siIEL CD8+ T cells were analyzed by flow cytometry at day 4 post-infection. Data are representative of two independent experiments, with n=2–3 mice per experiment. (C) Representative flow cytometry plots displaying distribution of IL-2Rα expression among total siIEL P14 CD8+ T cells at day 4 post-infection (left), or among total H-2Db GP33–41 tetramer+ endogenous siIEL CD8+ T cells at day 4 post-infection in mice that had not received adoptive transfer of P14 CD8+ T cells (right). (D) Quantification of Ezh2 expression among IL-2Rαlo (gray) and IL-2Rαhi (blue) P14 siIEL CD8+ T cells, as gated in (C), with representative flow cytometry plot (left) and quantification (right). Numbers represent Mean Fluorescence Intensity (MFI) of Ezh2 within the indicated population. Data are representative of (plot to left) or compiled from (graph to right) 2 independent experiments, with n=4 total mice, where each dot represents an individual mouse. (E) Schematic of experimental setup. CD45.1+ P14 CD8+ T cells were adoptively transferred into CD45.2+ congenic hosts one day prior to infection with LCMV. CD45.1+ P14 CD8+ T cells were collected from the siIEL compartment at day 4 post-infection and sorted based on expression of IL-2Rα as shown in (C). IL-2Rαhi and IL-2Rαlo populations were adoptively transferred into secondary hosts that had been infected with LCMV one day prior to transfer. 7 or 30 days later, siIEL P14 CD8+ T cells were analyzed by flow cytometry. (F) Quantification of the proportion of KLRGhi P14 CD8+ T cells present within the siIEL compartment at day 7 post-infection. Data were pooled from 2 independent experiments, with mean and SEM of n=7 (IL-2Rαlo) or 5 (IL-2Rαhi), where each dot represents an individual mouse. (G) Quantification of the percent of siIEL CD45.1+ P14 CD8+ CD69+CD103+ T cells derived from IL-2Rαlo (gray) or IL-2Rαhi (blue) day 4 siIEL cells at day 7 or day 30 post-transfer, with representative flow cytometry plots shown to the left. Day 7 data were pooled from 2 independent experiments, with mean and SEM of n=5 (IL-2Rαhi) or 6 (IL-2Rαlo) hosts, where each dot represents an individual mouse. Day 30 data were pooled from 3 independent experiments, with mean and SEM of n=4 (IL-2Rαhi) or 12 (IL-2Rαlo) hosts, where each dot represents an individual mouse. *P<0.05, **P<0.01 (Paired t-test, D, or Student’s two-tailed t-test, F and G).
In order to formally test this possibility, P14 CD8+CD45.1+ T cells were adoptively transferred into CD45.2+ hosts infected with LCMV one day later, and the siIEL compartment was harvested at day 4 post-infection for flow cytometric analysis. We found that, in addition to IL-2Rαhi and IL-2Rαlo subsets within the early circulating CD8+ T cell population, there was also heterogeneity in IL-2Rα expression among day 4 CD8+ T cells within the small intestinal epithelial tissue; notably, this heterogeneity was also evident among endogenous siIEL CD8+ H-2Db GP33–41 tetramer+ cells at day 4 post-infection (Fig. 6C). We also found that protein expression of IL-2Rα and Ezh2 were correlated (Fig. 6D) and took advantage of this observation to test whether these populations within the tissue have distinct differentiation potentials. CD45.1+ CD8+IL-2Rαhi or CD8+IL-2Rαlo cells from the siIEL compartment were FACS-sorted at 4 days post-infection and adoptively transferred intravenously into CD45.2+ recipient mice infected with LCMV one day prior to transfer; analysis of siIEL CD8+ T cells was performed 7 or 30 days later (Fig. 6E). P14 CD8+ T cells within the siIEL compartment of mice that had received IL-2Rαlo cells expressed lower levels of KLRG1 at day 7 post-infection (Fig. 6F), indicating a less terminally differentiated phenotype. Moreover, mice that had received IL-2Rαlo P14 siIEL CD8+ T cells had a higher proportion of CD69+CD103+ P14 CD8+ T cells within the siIEL compartment at both day 7 and at day 30 (Fig. 6G), suggesting that IL-2Rαlo siIEL CD8+ T cells at day 4 post-infection are more likely to give rise to bona fide TRM cells that persist long-term. Taken together, these data suggest that, analogous to IL-2Rαlo and IL-2Rαhi populations within the circulating CD8+ T cell pool early in infection (59, 60), two functionally distinct CD8+ T cell populations are present within the siIEL compartment at day 4 post-infection: a IL-2Rαhi subset with a more terminally differentiated phenotype that likely represents a transient tissue effector population, and a IL-2Rαlo subset with higher memory potential that likely contains the precursors of TRM cells. Cluster 20 cells, corresponding to the IL-2Rαhi population, are more proliferative, exhibit a transcriptional profile that includes factors that are associated with the effector CD8+ T cell fate (Ezh2, Dnmt1), and are more likely to give rise to terminally differentiated effector-like CD8+ T cells. In contrast, Cluster 16 cells, corresponding to the IL-2Rαlo subset, may be more quiescent and better poised to respond to type I interferons, have greater survival capacity, and have greater potential to give rise to long-lived tissue-resident memory CD8+ T cells. Taken together, these results demonstrate previously unappreciated transcriptional and functional heterogeneity within the siIEL CD8+ T cell pool at multiple states of differentiation.
Discussion
Recent studies have begun to elucidate key regulators of TRM cell differentiation, function, and survival, such as the transcription factors Blimp1, Hobit, and Runx3. Our single-cell RNA sequencing analyses have identified a number of additional putative regulators of TRM cell differentiation. For example, Nr4a2, Junb, and Fosl2 were among the 528 genes that were substantially enriched in siIEL CD8+ T cells relative to splenic cells at all time points following infection, and we found that knockdown of Nr4a2, Junb, or Fosl2 resulted in impaired TRM cell differentiation. Junb and Fosl2 encode for AP-1 dimerization partners that have been reported to repress T-bet expression in Th17 cells (46, 47). Given that downregulation of T-bet expression is important for early establishment of the TRM cell transcriptional program (15), it is tempting to speculate that Junb and Fosl2 may regulate TRM cell differentiation by cooperatively regulating T-bet expression. Since Fosl2 has been previously reported to positively regulate Smad3 (47), a key component of the TGF-β signaling pathway, Fosl2 may also promote TRM cell differentiation through effects on TGF-β signaling.
In addition to identifying specific putative regulators of TRM cell differentiation, our analyses have implicated new pathways that may control aspects of TRM cell biology. For example, we observed that siIEL CD8+ T cells exhibited increased levels of transcripts associated with TCR signaling, even after infection had been cleared and antigen was no longer present, suggesting that CD8+ T cells within tissues are transcriptionally poised for rapid responsiveness to TCR stimulation. Since TRM cells have been reported to exhibit a decreased ability to scan for antigens owing to reduced motility relative to circulating CD8+ T cells (64, 65), such enhanced TCR responsiveness might represent a potential mechanism enabling TRM cells to mount robust protective responses even in the face of limiting amounts of cognate antigen. Moreover, we observed that differentiating siIEL CD8+ T cells also exhibited sustained expression of genes encoding inhibitory receptors, including Ctla4, Tigit, and Lag3, consistent with prior reports (13, 18), which may provide a balance against higher basal TCR responsiveness and enable TRM cells to avoid excessive responses that might lead to autoimmune pathology (66).
Our analyses also provide new insights regarding TRM cell ontogeny, which has not been as well studied as that of circulating memory CD8+ T cells (20, 59, 67). Other than the observations that TRM cells are derived from circulating cells lacking high expression of KLRG1 (13, 16) and share a common clonal origin as TCM cells (68), very little is known about the precursors of TRM cells following their arrival in the tissue. We found that siIEL CD8+ T cells were transcriptionally distinct from splenic CD8+ T cells at 4 days post-infection, the earliest time point at which these cells can be detected within the small intestinal tissue (30). One interpretation of this finding is that transcriptional changes induced by the local tissue microenvironment occur rapidly upon tissue entry. Alternatively, CD8+ T cells that seed tissues may represent pre-committed TRM cell precursors that have already acquired some aspects of the TRM cell transcriptional program. Although there were distinct clusters of splenic CD8+ T cells observed at day 3 post-infection, none of these clusters appeared to be transcriptionally similar to siIEL CD8+ T cells. This finding suggests that the TRM cell transcriptional program may not be initiated until after the cells have entered the tissue and does not support the hypothesis that pre-committed TRM precursor cells are formed within the circulating CD8+ T cell pool. However, it has been previously shown that priming by DNGR1+ dendritic cells in the draining lymph nodes is an important step for TRM cell differentiation in vaccinia virus infection in the skin and influenza A infection in the lung (69). These findings suggest that the mechanisms underlying early TRM cell fate specification might differ for localized infections and might even be distinct among different tissues or pathogens. A priming step that specifically induces a TRM cell precursor population that is poised to home to a specific tissue could be particularly advantageous for a localized infection actively occurring within that tissue. In contrast, a more generalized mechanism of TRM cell specification, in which less committed precursor cells enter tissues and differentiate into TRM cells only in response to local tissue signals, might be more permissive for seeding of multiple, diverse tissues, as occurs in a systemic infection. Alternatively, it is possible that both models of TRM cell fate specification could occur simultaneously during an infection, whereby each pathway induces functionally distinct subsets of TRM cells.
In addition to finding substantial transcriptional differences between splenic and siIEL CD8+ T cells at day 4 post-infection, we also observed two transcriptionally distinct cell clusters within the siIEL CD8+ T cell pool at day 4 post-infection. Cells from one cluster expressed higher levels of genes associated with proliferation and effector differentiation, and were less likely to develop into long-lived CD69+CD103+ TRM cells when transferred into new recipients. By contrast, cells from the other cluster, marked by lower expression of IL-2Rα, exhibited a greater potential to give rise to TRM cells upon adoptive transfer. Although it remains unknown whether this heterogeneity is a specific feature of the siIEL compartment or generalizable to TRM cell differentiation in other non-lymphoid tissues, our analyses establish that analogous to the IL-2Rαlo population present within the circulating CD8+ T cell pool early in infection that contains the precursors of circulating memory cells (59, 60), bona fide TRM cell precursors can be found within the IL-2Rαlo cell subset present within the siIEL compartment at day 4 post-infection. This finding represents an important step in understanding the ontogeny of TRM cells after their arrival in the tissue that will likely inform future studies to identify early determinants of TRM cell fate specification.
Whereas the heterogeneity within the circulating memory CD8+ T cell pool is well characterized, heterogeneity among TRM cells is much less clear. Some studies have reported tissue-specific differences in CD69 and CD103 expression by CD8+ T cells, but it is unknown whether this heterogeneity represents functionally distinct subsets (18, 70–72). Our analyses identify two transcriptionally distinct cell clusters at days 60 and 90 post-infection that exhibit unique functional capacities. One population, distinguished by higher expression of CD28 and IL-7Rα, expressed high levels of transcripts encoding inflammatory cytokines and exhibited an enhanced ability to produce cytokines in response to restimulation. In addition, this population exhibited higher expression of Klf2, which promotes tissue egress and recirculation. By contrast, the other population, characterized by lower CD28 and IL-7Rα expression, exhibited higher expression of transcripts encoding molecules that promote tissue retention, such as Cxcr3 and Klf3 (13, 73). Recent studies have revealed that the ‘tissue-residency’ of TRM cells is not as permanent as previously thought; upon reactivation, TRM cells harbor the potential to leave the tissues and join the circulating memory pool or form TRM populations within secondary lymphoid organs (74, 75). Our findings suggest that this functionality may be restricted to a specific subset of TRM cells that are better poised to leave the tissue. Indeed, a recent study found that a subset of TRM cells with high expression of the transcription factor Id3, which we also found to be more highly expressed within the CD28hi TRM population, exhibits an enhanced capacity to survive and give rise to circulating memory cells when adoptively transferred into new hosts (55). By contrast, the second TRM cell subset might be more prone to remain within the tissue and mediate protective responses directly within the tissue microenvironment. Moreover, in addition to revealing functionally distinct subsets within the TRM cell pool, our transcriptional analyses have also elucidated a number of factors regulating the differentiation of these subsets. Analogous to our findings, a recent study provided evidence for distinct subsets of human TRM cells, one with a greater potential for cytokine production and another with a higher proliferative potential in response to TCR stimulation (76).
Overall, our work has resulted in a single-cell transcriptomic dataset encompassing the gene-expression patterns of circulating and siIEL CD8+ T cells in response to viral infection. Our study reveals a core transcriptional program that is shared between circulating memory and TRM cells in addition to key differences in the kinetics and magnitude of gene expression between these two memory cell subtypes, which should serve as a useful resource for elucidating new genes and pathways regulating TRM cell differentiation. Notably, our analyses demonstrate that CD8+ T cells within the siIEL compartment become transcriptionally distinct from circulating cells rapidly upon entry into tissue and identify a subset of early siIEL CD8+ T cells enriched for precursors of TRM cells. Moreover, our study reveals previously unappreciated molecular and functional heterogeneity within the TRM cell pool, underscoring the power and necessity of using a single-cell approach. This dataset should inform future studies aimed at improving our understanding of CD8+ T cell differentiation and function, which may lead to strategies to optimize CD8+ T cell responses to protect against microbial infection and cancer.
Materials and Methods
Study design
The purpose of this study was to gain a broader understanding of the gene expression patterns that regulate CD8+ T cell differentiation and heterogeneity in response to viral infection. To this end, we employed single-cell RNA sequencing on CD8+ T cells in the spleen and small intestinal epithelium at various timepoints following viral infection to create a dataset analyzing gene-expression patterns over time in individual CD8+ T cells. Analysis of this dataset revealed a subset of genes whose expression was enriched in CD8+ T cells from the tissue, and the role of several of these factors in regulating the differentiation of tissue-resident memory CD8+ T cells was validated by flow cytometric analysis of CD8+ T cells with shRNA-mediated knockdown of these genes. Additionally, validation of putative subsets of CD8+ T cells within the tissue was validated by flow cytometric analysis, including sorting of individual putative subsets and assessment of function following ex vivo restimulation or adoptive transfer into secondary hosts. Information regarding the sample size and number of replicates for each experiment can be found in the relevant figure legend. All findings were successfully reproduced. No sample size calculations were performed; sample sizes were selected based on previous studies performed in our lab. Mice were randomly allocated into groups prior to adoptive transfer of sorted IL-2Rαhi or IL-2Rαlo intraepithelial P14 T cells (Fig. 6). For single-cell RNA sequencing experiments, mice were randomly selected for P14 T cell harvesting at specific time points post-infection. Randomization and blinding are not relevant to shRNA knockdown experiments, or experiments involving co-transfer of Ddx5−/− and WT CD8+ T cells, since P14 T cells transduced with retrovirus encoding shRNA against genes of interest and congenically distinct P14 T cells transduced with control retrovirus, or Ddx5−/− and WT CD8+ T cells, were co-transferred into the same animals (Fig. 2 and Fig. 5). For assessment of phenotype of adoptively transferred sorted IL-2Rαhi or IL-2Rαlo intraepithelial P14 T cells (Fig. 6), the investigator was aware of the cell type transferred into each recipient. No data were excluded from analysis, except for recipient mice that had rejected adoptively transferred P14 CD8+ T cells.
Mice
All mice were housed under specific pathogen-free conditions in an American Association of Laboratory Animal Care-approved facility at the University of California, San Diego (UCSD), and all procedures were approved by the UCSD Institutional Animal Care and Use Committee. Wild-type C57BL6/J (CD45.2+) and P14 TCR transgenic (CD45.1 or CD45.1.2+, both maintained on a C57BL6/J background) mice were bred at UCSD or purchased from Jackson Laboratories. Ddx5fl/fl mice were obtained from Dr. Frances Fuller-Pace’s laboratory (University of Dundee) and have been previously described (77). To obtain congenically distinct P14 Ddx5fl/flCD4-Cre+ and P14 Ddxfl/flCD4-Cre− mice, Ddx5fl/fl mice were crossed to P14 CD4-Cre+ mice (either CD45.1+ or CD45.2+). All mice were used from 6–9 weeks of age, male mice were used as recipients, and male or female mice were used as donors in adoptive transfer experiments.
Antibodies, flow cytometry, and cell sorting
Cells were stained for 10 minutes on ice with the following antibodies: Vα2 (B20.1), CD8α (53–6.7), CD8β (YTS156.7.7), CD45.1 (A20), CD45.2 (104), CD44 (1M7), CX3CR1 (SA011F11), CD127 (A7R34), CD27 (LG.3A10), CD69 (H1.2F3), CD103 (2E7), CD28 (37.51), CD25 (PC61), and KLRG1 (2F1/KLRG1) all purchased from Biolegend. In some experiments, cells were stained with H-2Db GP33–41 tetramer (obtained from the NIH Tetramer Core Facility) for 1 hour at room temperature prior to staining with cell surface antibodies. Samples were then stained in Fixable Viability Dye eFluor780 (Thermo Fisher) or Ghost Violet 510 (Tonbo Biosciences) at 1:1000 on ice for 10 minutes. For experiments with retroviral transduction of P14 T cells, cells were then fixed in 2% paraformaldehyde (Electron Microscopy Services) on ice for 45 minutes. For staining for Ezh2, cells were fixed and permeabilized using the FoxP3/Transcription Factor Staining Buffer Kit (Thermo Fisher) following staining with viability dye, prior to incubation with anti-Ezh2 antibody (11/Ezh2, BD Pharmingen) for 8 hours at 4°C. For assessment of cytokine production, cells were cultured in the presence of LCMV GP33–41 peptide (GenScript) and Protein Transport Inhibitor Cocktail (ThermoFisher Scientific) for 3 hours at 37°C. Following cell surface and viability staining, cells were fixed and permeabilized using BD Cytofix/Cytoperm (BD Biosciences) for 30 minutes at room temperature prior to staining with anti-IFN-γ (XMG1.2), TNF-α (MP6-XT22), and IL-2 (JES6–5H4) antibodies (all from Biolegend) for 30 minutes on ice. For analysis, all samples were run on an Accuri C6, LSRFortessa, or LSRFortessa X-20 (BD Biosciences), or Novocyte (ACEA Biosciences). For sorting, all samples were run on an Influx, FACSAria Fusion, or FACSAria2 (BD Biosciences). BD FACS DIVA software (BD Biosciences) was used for data collection, and FlowJo software (BD Biosciences) was used for analysis of flow cytometry data.
Naïve T cell transfer and infection
Splenocytes were collected from naïve CD45.1+ or CD45.1.2+ P14 mice and stained with antibodies against Vα2, CD8, and CD45.1. 1×105 Vα2+CD8+CD45.1+ cells were adoptively transferred into congenically distinct wild-type recipients one day prior to infection with 2×105 plaque-forming units (PFU) of LCMV Armstrong, injected intraperitoneally. For experiments where IEL were collected at 4 days post-infection, 5×105 Vα2+CD8+CD45.1+ P14 T cells were transferred. To distinguish circulating CD8+ T cells in non-lymphoid tissues from tissue-resident CD8+ T cells, 3 μg CD8α was injected intravenously prior to sacrifice and dissection. Cells negative for injected CD8α (CD8α− > 98%) were analyzed further.
CD8+ T cell isolation
For isolation of CD8+ T cells from spleen, spleens were collected and dissociated to yield a cell suspension prior to treatment with Red Blood Cell Lysing Buffer Hybri-Max (Sigma). For isolation of CD8+ T cells from the small intestinal epithelium, Peyer’s patches were removed and the tissue was cut longitudinally and washed of luminal contents. The tissue was then cut into 1 cm pieces that were incubated while shaking in DTE buffer (1 μg/ml dithioerythritol (Thermo Fisher Scientific) in 10% HBSS and 10% HEPES bicarbonate) at 37°C for 30 minutes. Cells within the supernatant were collected and passed through a 44/67% Percoll density gradient to enrich for lymphocytes.
10X Genomics library preparation and sequencing
Activated P14 T cells (CD8+Vα2+CD45.1+CD44+) were sorted from the spleen or siIEL compartment and resuspended in PBS+0.04% (w/v) bovine serum albumin. Approximately 10,000 cells per sample were loaded into Single Cell A chips (10X Genomics) and partitioned into Gel Bead In-Emulsions (GEMs) in a Chromium Controller (10X Genomics). Single cell RNA libraries were prepared according to the 10x Genomics Chromium Single Cell 3’ Reagent Kits v2 User Guide, and sequenced on a HiSeq4000 (Illumina).
Generation of shRNA-encoding retrovirus, transduction of CD8+ T cells, and adoptive transfer for analysis of gene knockdown
shERWOOD-designed UltramiR sequences targeting Junb, Nr4a2, Pnrc1, Fosl2, or Zfp36l2 (knockdown, KD) or control (non-targeting) constructs in LMP-d Ametrine vector were purchased from transOMIC technologies. To generate retroviral particles, 293T HEK cells were plated in 10cm plates one day prior to transfection and individually transfected with 10ug of each shRNA retroviral construct and 5ug pCL-Eco using TransIT-LT1 (Mirus). Retroviral supernatant was collected and pooled at 48 and 72 hours post-transfection, and in vitro activated P14 T cells were transduced with retrovirus encoding KD shRNA, and congenically distinct in vitro activated P14 T cells were transduced with control retrovirus. To activate P14 T cells in vitro, lymphocytes were collected from spleens and lymph nodes of naïve CD45.1+ and CD45.1.2+ P14 TCR transgenic mice and negatively enriched for CD8+ T cells using the CD8a+ T Cell Isolation Kit and LS MACS Columns (Miltenyi Biotec). 1×106 CD8+ T cells were plated per well in 48 well flat-bottom plates, pre-coated with 100 μg/ml goat anti-hamster IgG (H+L, Thermo Fisher Scientific) followed by 5 μg/ml each anti-CD3 (clone 3C11, BioXCell) and anti-CD28 (clone 37.51, BioXCell). Eighteen hours after activation, cells were transduced with retroviral supernatant supplemented with 8 μg/ml polybrene (Millipore) by spinfection for 90 minutes at 900rcf at room temperature. Following spinfection, retroviral supernatant was removed and replaced with culture medium (Iscove’s modified Dulbecco’s medium+10% fetal bovine serum (v/v)+2mM glutamine+100U/ml penicillin+100ug/ml streptomycin+55mM β-mercaptoethanol) and cells were rested for 2 hours at 37°C. Cells transduced with each individual construct were then pooled, washed three times with PBS, counted, and mixed to obtain a 1:1 ratio of transduced KD and transduced control cells, based on previously tested transduction efficiency of each retrovirus. 5×105 total P14 T cells were then adoptively transferred into congenically distinct hosts that were infected with 2×105 PFU LCMV-Arm one hour later. 1×106 cells from this mixture were returned to culture with recombinant IL-2 (100U/ml), and analyzed 18 hours later by flow cytometry to determine the input ratio of transduced KD:control P14 T cells. 22–26 days post-infection, splenocytes and siIEL were collected as described above, and analyzed by flow cytometry to determine the ratio of KD:control cells within each subset of transduced P14 T cells.
Statistical analysis of flow cytometry data
Statistical analysis of flow cytometry data was performed using Prism software (GraphPad). P values of <0.05 were considered significant. Statistical details for each experiment are provided within the relevant figure legends and the raw data file.
Quantitative and statistical analysis of scRNA-seq data
Single-cell RNA-seq mapping
Reads from single-cell RNA-seq were aligned to mm10 and collapsed into unique molecular identifier (UMI) counts using the 10X Genomics Cell Ranger software (version 2.1.0). All samples had sufficient numbers of genes detected (>1000), a high percentage of reads mapped to the genome (>70%), and sufficient number of cells detected (>1000).
Cell and gene filtering
Raw cell-reads were loaded to R using the cellrangerRkit package. The scRNA-seq dataset was then further filtered based on gene numbers and mitochondria gene counts to total counts ratio. To ensure that the samples with more cells would not dominate the downstream analysis, we randomly selected a portion of the cells that passed filtering for downstream analysis. We randomly selected ~2000 cells from each library for downstream analysis. After cell filtering and sampling, we filtered genes by removing genes that did not express > 1 UMI in more than 1% of the total cells.
Single-cell RNA-seq dataset normalization and pre-processing
Five cell-gene matrices were generated:
Raw UMI matrix.
UPM matrix. The raw UMI matrix was normalized to get UMIs per million reads (UPM), and was then log2 transformed. All downstream differential analysis was based on the UPM matrix. The prediction models were also based on the UPM matrix, as other normalizations are very time-consuming for large datasets.
MAGIC matrix. The UPM matrix was further permuted by MAGIC (78). R package Rmagic 1.0.0 was used, and all options were kept as default. MAGIC aims to correct the drop-out effect of single-cell RNA-seq data; thus, we used the MAGIC-corrected matrix for visualizing the gene-expression pattern rather than using the UPM matrix. All gene-expression heatmaps and gene expression overlaid on tSNE plots were based on the MAGIC matrix.
Super cell matrix. We merged 50 cells to create a ‘super’ cell and used the super cell matrix as the input for WGCNA and cell type annotation analysis. This approach enabled us to bypass the issue of gene dropouts with scRNA-seq and is equivalent to performing WGCNA on thousands of pseudo-bulk samples. We first calculated the mutual nearest neighbor network with k set to 15, and then cells that were not mutual nearest neighbors with any other cells were removed as outliers. We randomly selected ‘n’ cells in the UPM matrix as the seed for super cells. The expression of each super cell was equal to the average expression of its seed and the 50 nearest neighbor cells of its seed. We derived 7400 super cells from the dataset, so each single cell was covered ~10 times.
Single-cell RNA-seq dataset dimension reduction
Top variable genes, PCA, and tSNE were calculated by Seurat version 2.3.4 functions: FindVariableGenes, RunPCA, and RunTSNE (79). Only the top 3000 genes were considered in the PCA calculation and only the top 25 principal components (PCs) were utilized in tSNE. Louvain clustering was performed by Seurat’s FindClusters function based on the top 25 PCs, with resolution set to 2. UMAP was calculated by R packages umap_0.2 with default setting.
Differential gene expression analysis
Differentially expressed (DE) genes were identified by performing pairwise comparison using two-sided Wilcox test and the FindAllMarkers function of Seurat. The threshold for DE genes was p-value < 0.05 and an absolute fold-change > 2.
WGCNA analysis
We performed WGCNA (version 1.63) analysis on spleen cells alone, siIEL cells alone, or all cells considered together. The super cell matrices were used as the input to boost performance. Only the top 5000 variable genes were considered in this analysis. SoftPower was set to 9 and the signed adjacency matrix was calculated for gene module identification. Genetree clustering and eigengene clustering were based on average hierarchical clustering. Module cut height was set to 0.1. Gene ontology (GO) analysis of the gene module was performed by compareClusterfunction from R package clusterProfiler 3.2.14. Reference database was org,Hs.eg.db 3.4.0 from Bioconductor and options were fun = ”enrichGO”, pAdjustMethod = “fdr”, pvalueCutoff = 0.01 and qvalueCutoff = 0.05.
Annotating single-cells with bulk RNA-seq signatures
The log2 TPM data from bulk RNA-seq datasets were compared with the scRNA-seq super cell matrix. Bulk cell population RNA-seq samples were first grouped into different sets according to their mutual similarities. For each bulk RNA-seq sample set, the mean expression was first calculated. The 1st correlation was calculated between all the super cells and the mean expression from the bulk RNA-seq dataset. Based on the distribution of the 1st correlation, we were able to identify a group of super cells that were most similar to the mean expression of the bulk sample. To further identify the small differences between bulk RNA-seq expression within a given set, we removed the set mean from the bulk RNA-seq and the mean from the most similar group of super cells, and then calculated the 2nd correlation between the super cells and bulk RNA-seq. Based on the 2nd correlation, we annotated the super cells with each bulk sample label.
Partition-based graph abstraction (PAGA) trajectory analysis
The single cell trajectories were constructed using the partition-based graph abstraction (PAGA) algorithm (54) implemented in the scanpy package (80). The UMI counts of the filtered cells and genes (filtered by the same approach mentioned above) were normalized by sequence depth before trajectory construction. In the construction, the knn-graphs were built with the scanpy.pp.neighbors function (n_neighbors=7, n_pcs=20) and PAGA graphs were constructed using the scanpy.tl.paga function with default settings. In the PAGA graphs, each node represents a cell partition (a group of cells) and the edges between nodes represent the connection between these nodes measured by PAGA while the strength of the edge describes the degree of connection (connectivity) which varies from 0 to 1. Only the edges with connectivity larger than 0.2 were shown in the final PAGA graphs and graphs were based on clusters defined with Louvain clustering.
Supplementary Material
Table S2. Genes in Spleen and IEL Modules.
Table S1. Genes differentially expressed by splenic and siIEL CD8+ T cells at day 4 post-infection.
Table S3. Genes in Combined Modules.
Table S4. Genes differentially expressed by D60 versus D90 TCM-like cells.
Table S5. Genes differentially expressed by Cluster 3 versus Cluster 29 cells.
Table S6. Genes differentially expressed by Cluster 17 versus Cluster 19 cells.
Table S7. Genes differentially expressed by Cluster 16 versus Cluster 20 cells.
Figure S1. Sorting of P14 CD8+ T cells from spleen and siIEL for scRNA-seq.
Figure S2. Circulating and siIEL CD8+ T cells are transcriptionally distinct at all time points post-infection.
Figure S3. Differential gene expression of circulating and siIEL CD8+ T cells at day 4 post-infection.
Figure S4. Shared core transcriptional program among splenic and siIEL CD8+ T cells.
Figure S5. Components of the TRM cell-enriched transcriptional signature.
Figure S6. shRNA knockdown of putative regulators of TRM cell differentiation.
Figure S7. PAGA trajectory analysis of TRM cell differentiation.
Table S8. Raw data file (Excel spreadsheet).
Acknowledgements:
We thank members of the Chang, Yeo, and Goldrath laboratories for technical advice, helpful discussion, and critical reading of the manuscript. Tetramer reagents were provided by the NIH Tetramer Core Facility. Current address for N.S.K. is Cancer Immunology Discovery, Pfizer Oncology Research and Development, La Jolla, CA 92121.
Funding: Single-cell RNA-sequencing using the 10X Genomics platform was conducted at the IGM Genomics Center, University of California, San Diego, La Jolla, CA, and supported by grants P30KC063491 and P30CA023100. The study was supported by the NIDDK-funded San Diego Digestive Diseases Research Center (P30DK120515). B.S.B and M.S.T. were supported by NIH grants 1KL2TR001444 and T32DK007202. Z.H. was supported by NIH grants AI082850 and AI0088. This work was funded by NIH grants AI123202, AI129973, and BX003424 (J.T.C.); AI132122 (A.W.G., G.W.Y., J.T.C.); GM124494-01 (W.J.H.); MH107367 (G.W.Y.).
Footnotes
Competing interests: G.W.Y. is co-founder, member of the Board of Directors, on the Scientific Advisory Board, equity holder, and paid consultant for Locana and Eclipse BioInnovations. G.W.Y. is a visiting professor at the National University of Singapore. G.W.Y. is a paid consultant for the Allen Institute of Immunology. Dr. Yeo’s interests have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. B.S.B. was a consultant for Abbvie, Prometheus Laboratories, and Pfizer in the past 3 years. The other authors declare that they have no competing interests.
Data and materials availability: The single-cell RNA sequencing data are available from the Gene Expression Omnibus under accession number GSE131847. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Chang JT, Wherry EJ, Goldrath AW, Molecular regulation of effector and memory T cell differentiation, Nat Immunol 15, 1104–1115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sallusto F, Lenig D, Forster R, Martin L, Lanzavecchia A, Two subsets of memory T lymphocytes with distinct homing potentials and effector functions, Nature 401, 708–712 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Jameson SC, Masopust D, Understanding Subset Diversity in T Cell Memory, Immunity 48, 214–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Omilusik KD, Nadjsombati MS, Shaw LA, Yu B, Milner JJ, Goldrath AW, Sustained Id2 regulation of E proteins is required for terminal differentiation of effector CD8+ T cells, Journal of Experimental Medicine 215, 773–783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE, Effector-like CD8+ T cells in the memory population mediate potent protective immunity, Immunity 38, 1250–1260 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hikono H, Kohlmeier JE, Takamura S, Wittmer ST, Roberts AD, Woodland DL, Activation phenotype, rather than central– or effector–memory phenotype, predicts the recall efficacy of memory CD8+ T cells, Journal of Experimental Medicine 204, 1625–1636 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mueller SN, Mackay LK, Tissue-resident memory T cells: local specialists in immune defence, Nat Rev Immunol 16, 79–89 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, Jacobs H, Haanen JB, Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert, Science 346, 101–105 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS, Skin infection generates non-migratory memory CD8, Nature 483, 227–231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D, Resident memory CD8 T cells trigger protective innate and adaptive immune responses, Science 346, 98–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, McBain N, Wagner T, Edwards J, McConville R, Wilmott JS, Scolyer RA, ting TTX, Palendira U, Gyorki D, Mueller SN, Huntington ND, Bedoui S, lzel MHX, Mackay LK, Waithman J, Gebhardt T, Tissue-resident memory CD8+ T cells promote melanoma-immune equilibrium in skin, Nature, 1–23 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Mackay LK, Minnich M, Kragten N, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Bell LS, Andrews S, Diaz-Munoz MD, Cook SJ, Corcoran A, Turner M, Freestone D, Braun A, Wynne-Jones E, Behr FM, Stark R, Pellici DG, Godfrey DI, Belz GT, Pellegrini M, Gebhardt T, Busslinger M, Shi W, Carbone FR, van Lier R, Kallies A, van Gisbergen K, Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes, Science 352, 459–463 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T, The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin, Nat Immunol 14, 1294–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Skon CN, Lee J-Y, Anderson KG, Masopust D, Hogquist KA, Jameson SC, Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells, Nat Immunol 14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, Carbone FR, T-box Transcription Factors Combine with the Cytokines TGF-β and IL-15 to Control Tissue- Resident Memory T Cell Fate, Immunity 43, 1101–1111 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, Wang W, Pipkin ME, Goldrath AW, Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumors, Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milner JJ, Goldrath AW, Transcriptional programming of tissue-resident memory CD8, Current Opinion in Immunology 51, 162–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, Bevan MJ, The Molecular Signature of Tissue Resident Memory CD8 T Cells Isolated from the Brain, The Journal of Immunology 189, 3462–3471 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, Lau LS, Mollard V, Cozijnsen A, Collins N, Li J, Davey GM, Kato Y, Devi S, Skandari R, Pauley M, Manton JH, Godfrey DI, Braun A, Tay SS, Tan PS, Bowen DG, Koch-Nolte F, Rissiek B, Carbone FR, Crabb BS, Lahoud M, Cockburn IA, Mueller SN, Bertolino P, McFadden GI, Caminschi I, Heath WR, Liver-Resident Memory CD8+ T cells form a front-line defense against malaria liver-stage infection, Immunity 45, 889–902 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, Yu B, Wehrens EJ, Lopez J, Kim SH, Zuniga EI, Goldrath AW, Chang JT, Yeo GW, Early transcriptional and epigenetic regulation of CD8+ T cell differentiation revealed by single-cell RNA sequencing, Nat Immunol 18, 422–432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, Choi K, Fromme RM, Dao P, McKenney PT, Wasti RC, Kadaveru K, Mazutis L, Rudensky AY, Pe’er D, Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment, Cell 174, 1293–1308.e36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, Byrne A, Wein L, Luen SJ, Poliness C, Nightingale SS, Skandarajah AS, Gyorki DE, Thornton CM, Beavis PA, Fox SB, Darcy PK, Speed TP, Mackay LK, Neeson PJ, Loi S, Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis, Nature Medicine, 1–16 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, Mavilio D, Alloisio M, Ferrari F, Lopci E, Novellis P, Veronesi G, Lugli E, High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors, Journal of Experimental Medicine 215, 2520–2535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, Pandolfi PP, Mak T, Satija R, Shalek AK, Kuchroo VK, Park H, Regev A, Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity, Cell 163, 1400–1412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng C, Zheng L, Yoo J-K, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, Liu Z, Dong M, Hu X, Ouyang W, Peng J, Zhang Z, Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing, Cell 169, 1342–1356.e16 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Karamitros D, Stoilova B, Aboukhalil Z, Hamey F, Reinisch A, Samitsch M, Quek L, Otto G, Repapi E, Doondeea J, Usukhbayar B, Calvo J, Taylor S, Goardon N, Six E, Pflumio F, Porcher C, Majeti R, Göttgens B, Vyas P, Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells, Nat Immunol 19, 1–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HRB, Schreuder J, Lum J, Malleret B, Zhang S, Larbi A, Zolezzi F, Renia L, Poidinger M, Naik S, Newell EW, Robson P, Ginhoux F, Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow, Nat Immunol 16, 718–728 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, Jacome-Galarza CE, Handler K, Klughammer J, Kobayashi Y, Gomez-Perdiguero E, Schultze JL, Beyer M, Bock C, Geissmann F, Specification of tissue-resident macrophages during organogenesis, Science 353, aaf4238–aaf4238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villani A-C, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, Jardine L, Dixon D, Stephenson E, Nilsson E, Grundberg I, McDonald D, Filby A, Li W, De Jager PL, Rozenblatt-Rosen O, Lane AA, Haniffa M, Regev A, Hacohen N, Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors, Science 356, eaah4573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R, Cutting Edge: Gut Microenvironment Promotes Differentiation of a Unique Memory CD8 T Cell Population, The Journal of Immunology 176, 2079–2083 (2006). [DOI] [PubMed] [Google Scholar]
- 31.da Silva HB, Beura LK, Wang H, Hanse EA, Gore R, Scott MC, Walsh DA, Block KE, Fonseca R, Yan Y, Hippen KL, Blazar BR, Masopust D, Kelekar A, Vulchanova L, Hogquist KA, Jameson SC, The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8+ T cells, Nature, 1–21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, Divito SJ, Fuhlbrigge R, Puigserver P, Krueger JG, Hotamisligil GS, Clark RA, Kupper TS, Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism, Nature 543, 252–256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Omilusik KD, Best JA, Yu B, Goossens S, Weidemann A, Nguyen JV, Seuntjens E, Stryjewska A, Zweier C, Roychoudhuri R, Gattinoni L, Bird LM, Higashi Y, Kondoh H, Huylebroeck D, Haigh J, Goldrath AW, Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection, Journal of Experimental Medicine 212, 2027–2039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roychoudhuri R, Clever D, Li P, Wakabayashi Y, Quinn KM, Klebanoff CA, Ji Y, Sukumar M, Eil RL, Yu Z, Spolski R, Palmer DC, Pan JH, Patel SJ, Macallan DC, Fabozzi G, Shih H-Y, Kanno Y, Muto A, Zhu J, Gattinoni L, O’Shea JJ, Okkenhaug K, Igarashi K, Leonard WJ, Restifo NP, BACH2 regulates CD8+ T cell differentiation by controlling access of AP-1 factors to enhancers, Nat Immunol 17, 851–860 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Widjaja CE, Olvera JG, Metz PJ, Phan AT, Savas JN, de Bruin G, Leestemaker Y, Berkers CR, de Jong A, Florea BI, Fisch K, Lopez J, Kim SH, Garcia DA, Searles S, Bui JD, Chang AN, Yates III JR, Goldrath AW, Overkleeft HS, Ovaa H, Chang JT, Proteasome activity regulates CD8+ T lymphocyte metabolism and fate specification, J. Clin. Invest 127, 3609–3623 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, Watson AS, Cerundolo V, Townsend AR, Klenerman P, Simon AK, Autophagy is a critical regulator of memory CD8+ T cell formation, eLife 3, 2516 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boddupalli CS, Nair S, Gray SM, Nowyhed HN, Verma R, Gibson JA, Abraham C, Narayan D, Vasquez J, Hedrick CC, Flavell RA, Dhodapkar KM, Kaech SM, Dhodapkar MV, ABC transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells, J. Clin. Invest 126, 3905–3916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, Manton JH, Heath WR, Mueller SN, Persistence of skin-resident memory T cells within an epidermal niche, Proceedings of the National Academy of Sciences 111, 5307–5312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, Ye Z, Beckermann KE, Wu Y, Jenkins JW, Siska PJ, Vincent BG, Prakash YS, Peikert T, Edelson BT, Taneja R, Kaplan MH, Rathmell JC, Dong H, Hitosugi T, Sun J, The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8+ T cell fitness and functionality, Immunity 51, 491–507.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sekiya T, Kashiwagi I, Yoshida R, Fukaya T, Morita R, Kimura A, Ichinose H, Metzger D, Chambon P, Yoshimura A, Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis, Nat Immunol 14, 230–237 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Sekiya T, Kashiwagi I, Inoue N, Morita R, Hori S, Waldmann H, Rudensky AY, Ichinose H, Metzger D, Chambon P, Yoshimura A, The nuclear orphan receptor Nr4a2 induces Foxp3 and regulates differentiation of CD4+ T cells, Nature Communications, 273–12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raveney BJE, Oki S, Yamamura T, Stangel M, Ed. Nuclear Receptor NR4A2 Orchestrates Th17 Cell-Mediated Autoimmune Inflammation via IL-21 Signaling, PLoS ONE 8, e56595 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doi Y, Oki S, Ozawa T, Hohjoh H, Miyake S, Yamamura T, Orphan nuclear receptor NR4A2 expressed in T cells from multiple sclerosis mediates production of inflammatory cytokines, Proceedings of the National Academy of Sciences 105, 1–6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott-Browne JP, López-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, Pereira RM, Dynamic Changes in Chromatin Accessibility Occur in CD8+ T cells responding to viral infection, Immunity 45, 1327–1340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carr TM, Wheaton JD, Houtz GM, Ciofani M, JunB promotes Th17 cell identity and restrains alternative CD4+ T cell programs during inflammation, Nature Communications 8, 1–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hasan Z, Koizumi S-I, Sasaki D, Yamada H, Arakaki N, Fujihara Y, Okitsu S, Shirahata H, Ishikawa H, JunB is essential for IL-23-dependent pathogenicity of Th17 cells, Nature Communications 8, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, Newberry KM, Meadows S, Greenfield A, Yang Y, Jain P, Kirigin FK, Birchmeier C, Wagner EF, Murphy KM, Myers RM, Bonneau R, Littman DR, A Validated Regulatory Network for Th17 Cell Specification, Cell 151, 289–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R, Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells, Nat Immunol 4, 1191–1198 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM, Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor, Immunity 27, 281–295 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R, Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates, Journal of Experimental Medicine 205, 625–640 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, von Andrian UH, The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis, Immunity 45, 1270–1284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R, Lineage relationship and protective immunity of memory CD8 T cell subsets, Nat Immunol 4, 225–234 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Murphy ME, The HSP70 family and cancer, Carcinogenesis 34, 1181–1188 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolf FA, PAGA: graph abstraction reconciles clustering with trajectory inference througha topology preserving map of single cells, Genome Biology 20, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milner JJ, Toma C, He Z, Kurd NS, Nguyen QP, McDonald B, Quezada L, Widjaja CE, Witherden DA, Crowl JT, Yeo GW, Chang JT, Omilusik KD, Goldrath AW, Functional delineation of tissue-resident CD8+ T cell heterogeneity during infection and cancer, bioRxiv 126, 3905. [Google Scholar]
- 56.Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D’Cruz LM, Watowich SS, Murre C, Goldrath AW, The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets, Nat Immunol 12, 1221–1229 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ji Y, Pos Z, Rao M, Klebanoff CA, Yu Z, Sukumar M, Reger RN, Palmer DC, Borman ZA, Muranski P, Wang E, Schrump DS, Marincola FM, Restifo NP, Gattinoni L, Repression of the DNA-binding inhibitor Id3 by Blimp-1 limits the formation of memory CD8+ T cells, Nat Immunol 12, 1230–1237 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salerno F, Engels S, Biggelaar M, Alphen FPJ, Guislain A, Zhao W, Hodge DL, Bell SE, Medema JP, Lindern M, Turner M, Young HA, Wolkers MC, Translational repression of pre-formed cytokine- encoding mRNA prevents chronic activation of memory T cells, Nat Immunol, 1–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arsenio J, Kakaradov B, Metz PJ, Kim SH, Yeo GW, Chang JT, Early specification of CD8+ T lymphocyte fates during adaptive immunity revealed by single-cell gene-expression analyses, Nat Immunol 15, 365–372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R, Prolonged Interleukin-2Rα Expression on Virus-Specific CD8+ T cells favors terminal-effector differentiation in vivo, Immunity 32, 91–103 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Vasilevsky NA, Ruby CE, Hurlin PJ, Weinberg AD, OX40 engagement stabilizes Mxd4 and Mnt protein levels in antigen-stimulated T cells leading to an increase in cell survival, Eur. J. Immunol 41, 1024–1034 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chappell C, Beard C, Altman J, Jaenisch R, Jacob J, DNA Methylation by DNA Methyltransferase 1 Is Critical for Effector CD8 T Cell Expansion, The Journal of Immunology 176, 4562–4572 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Gray SM, Amezquita RA, Guan T, Kleinstein SH, Kaech SM, Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8+ T cell terminal differentiation and loss of multipotency, Immunity 46, 596–608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, Carbone FR, Tscharke DC, Heath WR, Mueller SN, Mackay LK, Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses, Nat Immunol, 1–14 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, Fonseca R, Burbach BJ, Hickman HD, Vezys V, Fife BT, Masopust D, Intravital mucosal imaging of CD8+ resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory, Nat Immunol, 1–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheuk S, Schlums H, Sérézal IG, Martini E, Chiang SC, Marquardt N, Gibbs A, Detlofsson E, Introini A, Forkel M, Höög C, Tjernlund A, Michaëlsson J, Folkersen L, Mjösberg J, Blomqvist L, Ehrström M, Ståhle M, Bryceson YT, Eidsmo L, CD49a Expression Defines Tissue-Resident CD8+ T cells poised for cytotoxic function in human skin, Immunity 46, 287–300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang JT, Palaivel V, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, Killeen N, Orange JS, Russell SM, Weninger W, Reiner SL, Asymmetric T Lymphocyte Division in the Initiation of Adaptive Immune Responses, Science 315, 1687–1691 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Gaide O, Emerson RO, Jiang X, Gulati N, Nizza S, Desmarais C, Robins H, Krueger JG, Clark RA, Kupper TS, Common clonal origin of central and resident memory T cells following skin immunization, Nature Medicine 21, 647–653 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iborra S, Martínez-López M, Khouili SC, Enamorado M, Cueto FJ, Conde-Garrosa R, del Fresno C, Sancho D, Optimal Generation of Tissue-Resident but Not Circulating Memory T Cells during Viral Infection Requires Crosspriming by DNGR-1, Immunity 45, 847–860 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, Southern PJ, Masopust D, Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance, Cell 161, 737–749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bergsbaken T, Bevan MJ, Fink PJ, Local Inflammatory Cues Regulate Differentiation and Persistence of CD8+ Tissue-Resident Memory T Cells, Cell Reports 19, 114–124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bergsbaken T, Bevan MJ, Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection, Nat Immunol 16, 406–414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hart GT, Hogquist KA, Jameson SC, Krüppel-like Factors in Lymphocyte Biology, The Journal of Immunology 188, 521–526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, Scott MC, Fares-Frederickson NJ, Wijeyesinghe S, Thompson EA, da Silva HB, Vezys V, Youngblood B, Masopust D, Developmental plasticity allows outside-in immune responses by resident memory T cells, Nat Immunol, 1–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, Schenkel JM, Mitchell JS, Vezys V, Fife BT, Shen S, Masopust D, T Cells in Nonlymphoid Tissues Give Rise to Lymph- Node-Resident Memory T Cells, Immunity 48, 327–338.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kumar BV, Kratchmarov R, Miron M, Carpenter DJ, Senda T, Lerner H, Friedman A, Reiner SL, Farber DL, Functional heterogeneity of human tissue-resident memory T cells based on dye efflux capacities, JCI Insight 3, 1561 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bates GJ, Nicol SM, Wilson BJ, Jacobs AF, Bourdon JC, Wardrop J, Gregory DJ, Lane DP, Perkins ND, Fuller-Pace FV, The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor, EMBO J 24, 543–553 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, Bierie B, Mazutis L, Wolf G, Krishnaswamy S, Pe’er D, Recovering Gene Interactions from Single-Cell Data Using Data Diffusion, Cell 174, 716–729.e27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species, Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wolf FA, Angerer P, Theis FJ, SCANPY: large-scale single-cell gene expression data analysis, Genome Biology 19, 1–5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Genes in Spleen and IEL Modules.
Table S1. Genes differentially expressed by splenic and siIEL CD8+ T cells at day 4 post-infection.
Table S3. Genes in Combined Modules.
Table S4. Genes differentially expressed by D60 versus D90 TCM-like cells.
Table S5. Genes differentially expressed by Cluster 3 versus Cluster 29 cells.
Table S6. Genes differentially expressed by Cluster 17 versus Cluster 19 cells.
Table S7. Genes differentially expressed by Cluster 16 versus Cluster 20 cells.
Figure S1. Sorting of P14 CD8+ T cells from spleen and siIEL for scRNA-seq.
Figure S2. Circulating and siIEL CD8+ T cells are transcriptionally distinct at all time points post-infection.
Figure S3. Differential gene expression of circulating and siIEL CD8+ T cells at day 4 post-infection.
Figure S4. Shared core transcriptional program among splenic and siIEL CD8+ T cells.
Figure S5. Components of the TRM cell-enriched transcriptional signature.
Figure S6. shRNA knockdown of putative regulators of TRM cell differentiation.
Figure S7. PAGA trajectory analysis of TRM cell differentiation.
Table S8. Raw data file (Excel spreadsheet).