

Summary
Regulatory T (Treg) cells play a pivotal role in suppressing auto-reactive T cells and maintaining immune homeostasis. Treg cell development and function are dependent on the transcription factor Foxp3. Here, we performed a genome-wide CRISPR loss-of-function screen to identify Foxp3 regulators in mouse primary Treg cells. Foxp3 regulators were enriched in genes encoding subunits of the SWI/SNF nucleosome-remodeling and SAGA chromatin-modifying complexes. Among the three SWI/SNF-related complexes, the Brd9-containing non-canonical (nc) BAF complex promoted Foxp3 expression, whereas the PBAF complex was repressive. Chemical-induced degradation of Brd9 led to reduced Foxp3 expression and reduced Treg cell function in vitro. Brd9 ablation compromised Treg cell function in inflammatory disease and tumor immunity in vivo. Furthermore, Brd9 promoted Foxp3 binding and expression of a subset of Foxp3 target genes. Our findings provide an unbiased analysis of the genetic networks regulating Foxp3 and reveal ncBAF as a target for therapeutic manipulation of Treg cell function.
Keywords: Foxp3, Treg, Brd9, ncBAF, GBAF, SWI/SNF, CRISPR, screen
Graphical Abstract
Highlights
-
•
Genome-wide CRISPR screen identifies Foxp3 regulators in primary regulatory T cells
-
•
A SWI/SNF variant, the Brd9-containing ncBAF complex, regulates Foxp3 expression
-
•
Foxp3 binding to a subset of its target genes is dependent on co-binding of Brd9
-
•
Brd9 deficiency in Treg exacerbates IBD and enhances anti-tumor immunity
Regulation of Foxp3 expression is central to Treg cell development and function. Loo et al. performed a genome-wide CRISPR screen and identified the Brd9-containing ncBAF complex as a key regulator of Foxp3 and a subset of its target genes, which could be targeted to cripple Treg cell function and improve anti-tumor immunity.
Introduction
Regulatory T (Treg) cells play a crucial role in maintaining immune system homeostasis by suppressing over-reactive immune responses (Josefowicz et al., 2012; Sakaguchi et al., 2008). Defects in Treg cells lead to autoimmune disorders and immunopathology, whereas certain tumors are enriched with Treg cells that suppress anti-tumor immune responses (Tanaka and Sakaguchi, 2017). Foxp3, a member of the Forkhead transcription factor family, is a critical regulator that orchestrates the molecular processes involved in Treg cell differentiation and function (Zheng and Rudensky, 2007). Therefore, understanding the regulation of Foxp3 expression could reveal novel therapeutic targets to potentially change Treg cell numbers or alter their function. T cell receptor (TCR) and interleukin-2 (IL-2) signaling pathways play critical roles in Foxp3 induction (Chinen et al., 2016; Lee et al., 2012). Transforming growth factor β (TGF-β) signaling is also essential for Foxp3 induction in periphery-derived Treg cells and in-vitro-derived induced Treg (iTreg) cells, although its role in thymus-derived Treg cell development is still under debate (Chen et al., 2003; Liu et al., 2008; Ouyang et al., 2010). Accordingly, a number of downstream transcription factors regulate Foxp3 induction in vitro or in vivo, including Stat5a/b, Cbf-β/Runx1/3, Nfat1, Smad3/4, cRel, and Creb (Burchill et al., 2007; Kim and Leonard, 2007; Kitoh et al., 2009; Long et al., 2009; Rudra et al., 2009; Tone et al., 2008; Yang et al., 2008). Compared with the large number of studies focused on the mechanism of Foxp3 induction, relatively less is known about the factors that maintain Foxp3 expression in mature Treg cells. An intronic enhancer in Foxp3 called CNS2 (conserved non-coding sequence 2), also known as TSDR (Treg-specific demethylated region), is a key cis-regulatory element required for stable Foxp3 expression (Polansky et al., 2008; Zheng et al., 2010). CNS2 is heavily methylated in naive and activated conventional T cells by DNA methyltransferase 1 (Dnmt1), and deletion of Dnmt1 leads to aberrant expression of Foxp3 in conventional T cells (Josefowicz et al., 2009). When Foxp3 expression is induced during Treg cell development, the CNS2 region is rapidly demethylated, opening it up for binding of transcription factors (Polansky et al., 2008). Foxp3 can bind to CNS2 as well as an to additional upstream enhancer called CNS0 (Kitagawa et al., 2017) and stabilize its own expression in a positive feedback loop (Feng et al., 2014; Li et al., 2014b).
Post-translational modifications (PTM) of the Foxp3 protein, including phosphorylation, acetylation, and ubiquitination, are also a crucial part of the regulatory circuit that controls Foxp3 function and stability (van Loosdregt and Coffer, 2014). For example, a pair of enzymes, the ubiquitin ligase Stub1 and the ubiquitin hydrolase Usp7, promote and inhibit degradation of Foxp3 via ubiquitination, respectively (Chen et al., 2013; van Loosdregt et al., 2013). Finally, intracellular metabolism, specifically the metabolic regulator mTOR (mammalian target of rapamycin), has emerged as a key regulator of Foxp3 expression and Treg cell function. Weakened mTOR signaling increases Foxp3 expression in iTreg in vitro (Delgoffe et al., 2009), whereas complete ablation of mTOR in Treg cells using genetic models compromises effector Treg cell homeostasis and function (Chapman et al., 2018; Sun et al., 2018). Despite these and other significant advances in understanding the molecular mechanisms regulating Foxp3, we lack a comprehensive picture of the regulatory networks that control Foxp3 expression.
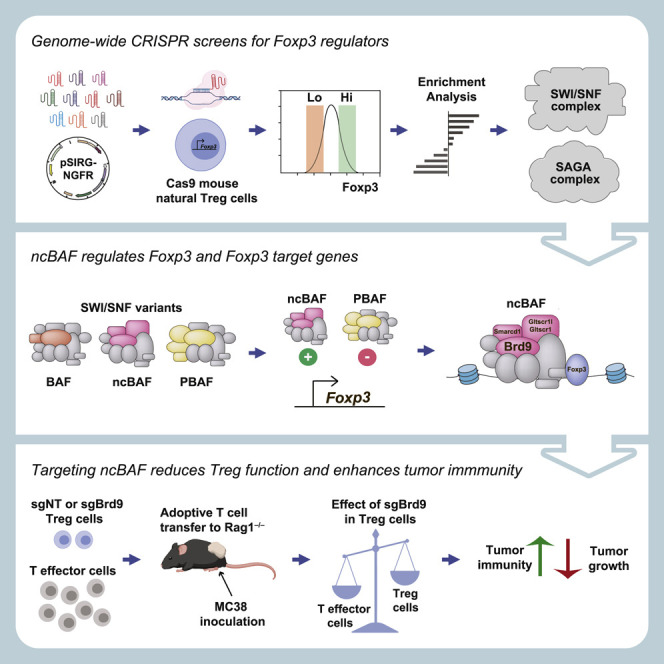
In this study, we performed a genome-wide clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 loss-of-function screen to identify regulators of Foxp3 in mouse primary natural Treg cells. Gene Ontology analysis showed that Foxp3 regulators are highly enriched in genes encoding subunits of the SAGA (Spt-Ada-Gcn5 acetyltransferase) chromatin-modifying and SWI/SNF (switch/sucrose non-fermentable) chromatin-remodeling complexes, which we further validated by single-guide RNA (sgRNA) CRISPR targeting and flow cytometry analysis. Of the three related complexes (BAF [BRG1/BRM-associated factors], PBAF [polybromo-associated BAF], and non-canonical BAF [ncBAF]) in the SWI/SNF family of nucleosome remodeling complexes, we found that the Brd9-containing ncBAF complex promoted transcription of Foxp3, whereas the PBAF complex repressed Foxp3 expression. Deletion of Brd9 or the PBAF component Pbrm1 in Treg cells reduced and enhanced Treg cell suppressor activity in in vitro assays, respectively, suggesting divergent regulatory roles of ncBAF and PBAF complexes in controlling Foxp3 expression and Treg cell function. Consistent with this model, we found that chemically induced degradation of Brd9 by dBRD9 led to reduced Foxp3 expression and compromised Treg cell function. Genome-wide binding studies revealed that Brd9 co-localized with Foxp3, including at the CNS0 and CNS2 enhancers at the Foxp3 locus. Furthermore, targeting Brd9 by sgRNA or dBRD9 reduced Foxp3 binding at the Foxp3 locus and a subset of Foxp3 binding sites genome-wide, which resulted in differential expression of many Foxp3-dependent genes, indicating that Brd9 participates in regulation of the Foxp3-dependent transcriptional program. Finally, deletion of Brd9 in Treg cells reduced suppressor activity in an in vivo model of T cell transfer-induced colitis and improved anti-tumor immune responses in an MC38 colorectal cancer cell-induced cancer model. Our findings reveal genes and networks regulating Foxp3 expression and identify the ncBAF complex as a potential target for manipulation of Treg function in vitro and in vivo.
Results
Genome-wide CRISPR Screen in Natural Treg Cells Identifies Regulators of Foxp3
To screen for genes that regulate Foxp3 expression, we developed a pooled retroviral CRISPR sgRNA library by subcloning an optimized mouse genome-wide lentiviral CRISPR sgRNA library (lentiCRISPRv2-Brie) (Doench et al., 2016) into a newly engineered retroviral vector pSIRG-NGFR, which allowed us to efficiently transduce mouse primary T cells and perform intracellular staining for Foxp3 without losing the transduction marker NGFR (nerve growth factor receptor) after cell permeabilization (Figure S1). Using this library, we performed a CRISPR loss-of-function screen on Treg cells to identify genes that regulate Foxp3 expression. We activated CD4+Foxp3+ Treg cells isolated from Rosa-Cas9/Foxp3Thy1.1 reporter mice (Liston et al., 2008; Platt et al., 2014) with CD3 and CD28 antibodies and IL-2 (Figure 1 A). Treg cells were transduced 24 h post-activation with the pooled retroviral sgRNA library at a multiplicity of infection of less than 0.2 to ensure that only one sgRNA was transduced per cell. NGFR+ transduced Treg cells were collected on day 3 and day 6 to identify genes that are essential for cell proliferation and survival. In addition, the bottom quintile (NGFR+Foxp3lo) and top quintile (NGFR+Foxp3hi) populations were collected on day 6 to identify genes that regulate Foxp3 expression. We validated the screen conditions by transducing Treg cells with sgRNAs targeting Foxp3 itself as well as previously reported positive (Cbfb) (Rudra et al., 2009) and negative (Dnmt1) (Lal et al., 2009) regulators of Foxp3 (Figures 1B–1D). Guide RNA sequences integrated within the genomic DNA of sorted cells were recovered by PCR amplification, constructed into amplicon libraries, and sequenced with a NextSeq sequencer.
Figure 1.

A Genome-wide CRISPR Screen in Treg Cells
(A) Workflow of the CRISPR screen in Treg cells.
(B–D) Validation of the CRISPR screen conditions.
(B) Fluorescence-activated cell sorting (FACS) plots showing Foxp3 expression in Treg cells after sgRNA targeting of Foxp3 (sgFoxp3), the positive regulator Cbfb (sgCbfb), and the negative regulator Dnmt1 (sgDnmt1). Red and green gates were set based on Foxp3 low 20% and high 20% in sgNT Treg cells, respectively.
(C and D) Mean fluorescence intensity (MFI) of Foxp3 (C) and relative log2 fold change of the cell count (D), comparing Foxp3lo with Foxp3hi after deletion of the indicated target gene (n = 3 per group; data represent mean ± SD).
See also Figures S1 and S2.
The relative enrichment of sgRNAs between samples and hit identification were computed by MAGeCK (model-based analysis of genome-wide CRISPR-Cas9 knockout), which generates a normalized sgRNA read count table for each sample, calculates the fold change of sgRNA read counts between two cell populations, and further aggregates information of four sgRNAs targeting each gene to generate a ranked gene list (Li et al., 2014a). Prior to hit calling, we evaluated the quality of screen samples by measuring the percentage of mapped reads to the sgRNA library and total read coverage, which showed a high mapping rate (79.8%–83.4%) with an average of 236X coverage and a low number of missing sgRNAs (0.625%–2.5%) (Figure S2). With the cutoff criteria of log2 fold change (LFC) of more than ± 0.5 and a p value of less than 0.01, we identified 254 potential positive Foxp3 regulators enriched in the Foxp3lo population and 490 potential negative Foxp3 regulators enriched in the Foxp3hi population (Figures 2A and 2B; Table S1). In a parallel analysis, we also identified 22 and 1,497 genes that affect cell expansion and contraction, respectively (p < 0.002, LFC > 1; Figure S3; Table S2). As expected, we identified genes belonging to pathways known to regulate Foxp3 expression transcriptionally (Cbfb and Runx3) (Rudra et al., 2009) and post-transcriptionally through regulation of Foxp3 protein stability (Usp7 and Stub1) (Chen et al., 2013; van Loosdregt et al., 2013; Figure 2C).
Figure 2.

Identification of Foxp3 Regulators in Treg Cells
(A and B) A scatterplot of the Treg cell screen results, showing positive regulators (A) and negative regulators (B). Genes that met the cutoff criteria (p < 0.01 and LFC > ±0.5) are shown as red dots for positive regulators and as green dots for negative regulators.
(C) Distribution of sgRNA LFC comparing Foxp3lo with Foxp3hi. Red stripes represent sgRNAs from positive Foxp3 regulators, whereas green stripes represent sgRNAs from negative Foxp3 regulators.
(D) Venn diagram showing the overlap of Foxp3 regulators with genes involved in cell contraction or expansion.
(E and F) Gene Ontology analysis of positive Foxp3 regulators (E) and negative Foxp3 regulators (F).
We next compared the potential positive and negative regulators with genes involved in cell contraction and expansion to exclude hits that might affect Foxp3 expression indirectly by affecting cellular fitness in general, leaving 197 positive Foxp3 regulators and 327 negative Foxp3 regulators (Figure 2D; Table S3). Gene Ontology analysis of positive Foxp3 regulators revealed a number of notable functional clusters, including SAGA-type complex, negative regulation of T cell activation, RNA polymerase II holoenzyme, positive regulation of histone modification, and SWI/SNF complex (Figure 2E; Table S4). Among negative Foxp3 regulators, genes were highly enriched in clusters related to negative regulation of mTOR signaling, transcriptional repressor complex, mRNA decay and metabolism, and hypusine synthesis from eIF5A-lysine (Figure 2F; Table S4). Several of these pathways, including mTOR signaling, Foxp3 ubiquitination and deubiquitination, and transcriptional regulation, have been implicated previously in Foxp3 regulation, suggesting that our screen is robust for validation of known pathways and discovery of additional regulators of Foxp3. Specifically, we identified many genes encoding subunits of the SAGA (Ccdc101, Tada2b, Tada3, Usp22, Tada1, Taf6l, Supt5, and Supt20) and SWI/SNF (Arid1a, Brd9, and Smarcd1) complexes (Table S4), strongly suggesting that these complexes could have indispensable roles in Foxp3 expression. We thus further validated and characterized the SAGA and SWI/SNF-related complexes to understand their roles in Foxp3 expression and Treg cell function.
Validation of the SAGA Complex as a Regulator of Foxp3 Expression and Treg Cell Suppressor Activity
The SAGA complex possesses histone acetyltransferase (HAT) and histone deubiquitinase (DUB) activity and functions as a transcriptional co-activator through interactions with transcription factors and the general transcriptional machinery (Helmlinger and Tora, 2017; Koutelou et al., 2010). We identified Ccdc101, Tada2b, and Tada3 in the HAT module; Usp22 in the DUB module; and Tada1, Taf6l, Supt5, and Supt20 from the core structural module among positive Foxp3 regulators that do not affect cell expansion or contraction (Figure S4A). We sought to validate the potential regulatory function of SAGA complex subunits by using sgRNAs to target individual subunits in Treg cells and measure Foxp3 expression (Figures S4B and S4C). We found that deletion of every subunit tested resulted in a significant and 19%–29% reduction in Foxp3 mean fluorescence intensity (MFI). We then further tested the function of the SAGA subunit Usp22 in an in vitro suppression assay, which measures suppression of T cell proliferation when conventional T cells are co-cultured with Treg cells at increasing ratios. We found that Treg cells transduced with sgRNAs targeting Usp22 had compromised Treg cell suppressor activity compared with Treg cells transduced with a non-targeting control sgRNA, with significantly more proliferation of T effector (Teff) cells at every ratio of Treg to Teff cells tested (Figure S4D). These results provide independent validation of our genome-wide screen analyses for this class of chromatin regulators and demonstrate that disrupting the SAGA complex with sgUsp22 reduces Foxp3 expression and Treg cell suppressor function.
Identification of the Brd9-Containing ncBAF Complex as a Specific Regulator of Foxp3 Expression
We next wanted to characterize the role of SWI/SNF complex variants (BAF, ncBAF, and PBAF complexes) in Foxp3 expression. Apart from uniquely incorporating Brd9, the ncBAF complex also contains Gltscr1 or the paralog Gltscr1l and lacks the BAF- and PBAF-specific subunits Arid1a, Arid1b, Arid2, Smarce1, Smarcb1, Smarcd2, Smarcd3, Dpf1–Dpf3, Pbrm1, Brd7, and Phf10 (Figure 3 A). The distinct biochemical compositions of these three SWI/SNF complex assemblies suggest functional diversity. However, it is not known which SWI/SNF complex assemblies are expressed in Treg cells, and the potential roles of specific SWI/SNF variants in regulating Foxp3 expression and Treg cell development have not been studied. Therefore, we performed co-immunoprecipitation assays to probe the composition of SWI/SNF-related complexes in Treg cells. As expected, immunoprecipitation of Smarca4, a core component of all three SWI/SNF complexes, revealed an association of the common subunits Smarcc1 and Smarcb1 as well as the specific subunits Arid1a, Brd9, and Pbrm1. Immunoprecipitations against Arid1a, Brd9, and Phf10 revealed a specific association of these subunits with BAF, ncBAF, and PBAF complexes, respectively (Figure 3A). These results established that all three SWI/SNF complexes are present with the expected composition in Treg cells.
Figure 3.

The Three SWI/SNF Complex Assemblies Have Distinct Regulatory Roles for Foxp3 Expression in Treg Cells
(A) A diagram showing three different variants of SWI/SNF complexes: BAF, ncBAF, and PBAF. BAF-specific subunits (Arid1a and Dpf1–Dpf3) are colored blue, ncBAF-specific subunits (Brd9, Smarcd1, Gltscr1l, and Gltscr1) are colored orange, and PBAF-specific subunits (Pbrm1, Arid2, Brd7, and Phf10) are colored green. Shared components among complexes are colored gray. Also shown is an immunoprecipitation assay of Arid1a, Brd9, Phf10, and Smarca4 in Treg cells. The co-precipitated proteins were probed for shared subunits (Smarca4, Smarcc1, and Smarcb1), BAF-specific Arid1a, ncBAF-specific Brd9, and PBAF-specific Pbrm1.
(B) FACS histogram of Foxp3 expression in Treg cells after sgRNA targeting of the indicated SWI/SNF subunits.
(C) MFI of Foxp3 after sgRNA targeting of the indicated SWI/SNF subunits. Data represent mean and standard deviation of biological replicates (n = 3–21).
(D) Principal-component analysis of RNA-seq data collected from Treg cells transduced with guides against the indicated SWI/SNF subunits. In cases where two independent guides were used to target a gene, the second guide for targeting the gene is indicated as “-2.”
(E) MFI of Foxp3 expression in Treg cells after treatment with DMSO or 0.16–10 μM dBRD9 for 4 days.
Data represent mean ± SD. Statistical analyses were performed using unpaired two-tailed Student’s t test (non-significant [ns], p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001). See also Figure S5.
In our screen, we identified Brd9, Smarcd1, and Arid1a among positive regulators of Foxp3, whereas the SWI/SNF shared subunits Smarca4, Smarcb1, Smarce1, and Actl6a were identified in cell contraction (Table S3). This suggests a potential regulatory role of ncBAF and/or BAF complexes. To explore the specific function of BAF, ncBAF, and PBAF complexes in Foxp3 expression, we cloned independent sgRNAs to target unique subunits for each complex and measured Foxp3 MFI in sgRNA-transduced Treg cells. We observed an essential role of the ncBAF complex in Foxp3 expression in Treg cells. Specifically, sgRNA targeting of ncBAF-specific subunits, including Brd9 and Smarcd1, significantly diminished Foxp3 expression by nearly 40% in Treg cells (Figures 3B and 3C). sgRNA targeting of the ncBAF-specific paralogs Gltscr1 and Gltscr1l individually resulted in a slight reduction in Foxp3 expression, which was further reduced by Gltscr1/Gltscr1l double deficiency, suggesting that these two paralogs can compensate in the regulation of Foxp3 expression (Figure 3C). In contrast, sgRNA targeting of PBAF-specific subunits, including Pbrm1, Arid2, Brd7, and Phf10, significantly enhanced Foxp3 expression by as much as 17% (Figure 3C, green). sgRNA targeting of the BAF-specific subunits Arid1a, Arid1b, Dpf1, and Dpf2 did not significantly affect Foxp3 expression (Figure 3C, blue). To determine whether Arid1a and Arid1b could be compensating for one another, we performed Arid1a/Arid1b double deletion and found that deletion of either or both Arid paralogs resulted in a slight but non-significant reduction in Foxp3 MFI (Figure 3C, blue). These data suggest that ncBAF and PBAF have opposing roles in the regulation of Foxp3 expression. To further explore the role of different SWI/SNF complexes in Treg cell genome-wide transcription, we performed RNA sequencing from Treg cells with sgRNA targeting of variant-specific subunits with one or two independent guide RNAs and conducted a principal-component analysis, which showed that ncBAF, PBAF, and BAF also have distinct effects at the whole-transcriptome level in Treg cells (Figure 3D).
We then made use of a recently developed chemical Brd9 protein degrader (dBRD9; Remillard et al., 2017) as an orthogonal method to probe Brd9 function. dBRD9 is a bifunctional molecule that links a small molecule that specifically binds to the bromodomain of Brd9 and another ligand that recruits the cereblon E3 ubiquitin ligase. We confirmed that treatment of Treg cells with dBRD9 resulted in reduced Brd9 protein (Figure S5A). Similar to sgRNA depletion of Brd9, dBRD9 treatment significantly decreased Foxp3 expression in Treg cells in a concentration-dependent manner without affecting cell viability or proliferation (Figure 3E; Figure S5B). These data demonstrate the requirement for Brd9 in maintenance of Foxp3 expression using genetic and chemically induced proteolysis methods.
Brd9 Regulates Foxp3 Binding at the CNS0 and CNS2 Enhancers and a Subset of Foxp3 Target Sites
To dissect the molecular mechanism of how ncBAF and PBAF complexes regulate Foxp3 expression in Treg cells, we performed chromatin immunoprecipitation followed by genome-wide sequencing (ChIP-seq) in Treg cells using antibodies against the ncBAF-specific subunit Brd9, the PBAF-specific subunit Phf10, and the shared enzymatic subunit Smarca4. Data generated from these ChIP-seq experiments revealed that Brd9, Smarca4, and Phf10 co-localize at CNS2 in the Foxp3 gene locus and at CNS0 found within the Ppp1r3f gene immediately upstream of Foxp3 (Figure 4 A). Because CNS2 has been shown previously to regulate stable Foxp3 expression through a positive feedback loop involving Foxp3 binding (Feng et al., 2014; Li et al., 2014b), and Foxp3 is additionally bound at CNS0 in Treg cells (Kitagawa et al., 2017), we rationalized that ncBAF and/or PBAF complexes might affect Foxp3 expression by regulating Foxp3 binding at CNS2 and CNS0. We therefore performed Foxp3 ChIP-seq in Treg cells transduced with sgNT, sgFoxp3, sgBrd9, or sgPbrm1. We observed a dramatic reduction in Foxp3 binding at CNS2/CNS0 in sgFoxp3-transduced cells, as expected, and there was also a marked reduction in Foxp3 binding at CNS2/CNS0 in Brd9-depleted Treg cells (Figure 4A). In contrast, we observed a subtle increase in Foxp3 binding at CNS2/CNS0 in Pbrm1 sgRNA-transduced Treg cells, which could explain why Pbrm1 emerged as a negative regulator of Foxp3 expression in our validation studies (Figure 4A). These data suggest that Brd9 positively regulates Foxp3 expression by promoting Foxp3 binding to its own enhancers.
Figure 4.

Brd9 Deletion Reduces Foxp3 Binding at CNS0 and CNS2 Enhancers and a Subset of Foxp3 Target Sites
(A) Genome browser tracks of Smarca4, Brd9, and Phf10 ChIP-seq and ATAC-seq signals as well as Foxp3 ChIP-seq in sgNT, sgFoxp3, sgBrd9, and sgPbrm1 Treg cells and Foxp3 in DMSO- and dBRD9-treated Treg cells (2.5 μM dBRD9 for 4 days). The Foxp3 locus is shown with CNS0 and CNS2 enhancers, indicated as gray ovals.
(B) Heatmap of Foxp3, Brd9, Smarca4, and Phf10 ChIP-seq and ATAC-seq signals ± 3 kb, centered on Foxp3-bound sites in Treg, ranked according to Foxp3 read density.
(C) Venn diagram of the overlap between ChIP-seq peaks in Treg cells for Brd9, Foxp3, and Phf10 (hypergeometric p of Brd9:Foxp3 overlap = e−27665, hypergeometric p of PHF10:Foxp3 overlap = e−17185, hypergeometric p of Brd9:PHF10 overlap = e−14217).
(D) Heatmap of Foxp3 ChIP-seq signals in sgNT, sgFoxp3, sgBrd9, and sgPbrm1 Treg cells ± 3 kb, centered on Foxp3-bound sites in sgNT, ranked according to read density.
(E) Venn diagram of the overlap (hypergeometric p = e−11,653) between sites that significantly lose Foxp3 binding (FC 1.5, Poisson p < 0.0001) in sgFoxp3 and sgBrd9, overlaid on all Foxp3-bound sites in sgNT (gray).
(F) Histogram of Foxp3 ChIP read density ± 1 kb surrounding the peak center of sites that significantly lose Foxp3 binding in sgFoxp3 and sgBrd9 (n = 1,699) in sgNT, sgFoxp3, sgBrd9, and sgPbrm1.
(G) As in (E) but for sites that lose H3K27ac (FC 1.5, Poisson p < 0.0001, hypergeometric p of overlap = e−7,938).
(H) As in (F) but for H3K27ac ChIP read density.
(I) As in (D) but for Foxp3 ChIP-seq signals in DMSO- and dBRD9-treated Treg cells at all Foxp3-bound sites in DMSO.
(J) As in (E), but for sites that significantly lose Foxp3 binding in dBRD9 treated Treg cells versus DMSO (FC 1.5, Poisson p < 0.0001).
(K) As in (F) but for DMSO- and dBRD9-treated cells.
(L) As in (F) but for Treg cells transduced with sgNT or sgBrd9, with ectopic expression of the MIGR vector control or Foxp3.
See also Figure S6.
We then extended this analysis to examine the cooperation between Brd9 and Foxp3 genome-wide. Notably, we found co-binding of Brd9, Smarca4, and Phf10 with Foxp3 at a subset of Foxp3-bound sites (Figures 4B and 4C). All four factors localized to promoters and intronic and intergenic regions of the genome, and their binding correlated well with chromatin accessibility, as measured by an assay of transposase-accessible chromatin with sequencing (ATAC-seq) (Figures 4B and S6A). Motif analysis of Foxp3-bound sites revealed an enrichment for motifs recognized by Ets and Runx transcription factors, consistent with what has been shown previously(Samstein et al., 2012). Ets and Runx motifs were also among the most significant motifs at both Brd9-bound sites, along with enrichment of the Ctcf motif, as we and others reported previously (Gatchalian et al., 2018; Michel et al., 2018; Figure S6B). These results demonstrate that ncBAF and PBAF complexes are co-localized with Foxp3 at Foxp3 binding sites genome-wide.
To assess the requirement for Brd9 or Pbrm1 in Foxp3 targeting genome-wide, we analyzed Foxp3 binding in Treg cells transduced with sgNT, sgFoxp3, sgBrd9, or sgPbrm1 at all Foxp3 binding sites (Figure 4D). As expected, we found that Foxp3 binding was lost at over 85% of its binding sites in sgFoxp3-transduced Treg cells (Figure 4E). Foxp3 binding at a subset of these sites was also significantly reduced in sgBrd9-transduced Treg cells (fold change 1.5, Poisson p < 0.0001) (Figure 4E; Figures S6C–S6F). This was a specific function of Brd9 because Foxp3 binding did not change in Pbrm1-depleted Treg cells at these Brd9-dependent sites (Figure 4F; Figure S6C). ChIP-seq for the active histone mark H3 lysine 27 acetylation (H3K27ac) revealed that Brd9 and Foxp3 cooperate to maintain H3K27ac at over 1,800 shared sites (Figure 4G). At Brd9-dependent Foxp3 sites, for example, we observed a reduction in H3K27ac in sgFoxp3 and sgBrd9-transduced Treg cells but not in sgPbrm1-transduced Treg cells (Figure 4H). Using dBRD9, we further recapitulated our observation that Brd9 loss resulted in diminished Foxp3 binding to chromatin at a subset of Foxp3 target sites (Figures 4I–4K Figure S6C), including at CNS2 and CNS0 (Figure 4A). To determine whether ncBAF complexes maintain chromatin accessibility for Foxp3 binding, we performed ATAC-seq on sgBrd9 and sgNT Treg cells (Figure S6G). Only 61 of 1,699 (3.5%) of Brd9-dependent Foxp3 binding sites had a significant reduction in chromatin accessibility in sgBrd9 Treg cells, suggesting that chromatin remodeling may only minimally contribute to ncBAF-dependent maintenance of Foxp3 binding.
Because Brd9 deficiency leads to reduced Foxp3 expression, we next wanted to find out whether reduced Foxp3 binding to its target regions in sgBrd9 Treg cells is due to reduced Foxp3 protein or whether Brd9 plays an additional role in facilitating Foxp3 binding to a subset of its targets. To this end, we ectopically expressed Foxp3 or MIGR (MSCV-IRES-GFP) vector control in sgNT- and sgBrd9-transduced Treg cells and performed Foxp3 ChIP-seq in these cells. Analysis of the Foxp3 ChIP-seq result showed that ectopic Foxp3 expression partially restored Foxp3 binding in sgBrd9 Treg cells but not to the level of sgNT alone or sgNT with ectopic Foxp3 expression (Figure 4L). Further analysis revealed that, although ectopic Foxp3 expression restored Foxp3 binding to a portion of Brd9-dependent Foxp3 binding sites (e.g., CD44 intergenic, Tigit intergenic, and Ctla2a promoter), binding to the majority of Brd9-dependent sites (~71%) (e.g., Icos intergenic, Ctla4 intergenic, and Ctla4 promoter) was not rescued by simply restoring Foxp3 expression (Figures S6H and S6I). These data demonstrate that Brd9 co-binds with Foxp3 at the Foxp3 locus to positively reinforce its expression. Brd9 additionally promotes Foxp3 binding and H3K27ac at a subset of Foxp3 target sites by potentiating Foxp3 expression and through epigenetic regulation at Brd9/Foxp3 co-bound sites.
Brd9 Co-regulates the Expression of Foxp3 and a Subset of Foxp3 Target Genes
Based on co-binding of Brd9 and Foxp3 at Foxp3 target sites, we assessed the effects of Brd9 ablation on transcription of Foxp3 target genes. We performed RNA sequencing (RNA-seq) in Treg cells transduced with sgFoxp3, sgBrd9, or sgNT. Consistent with Foxp3’s role as transcriptional activator and repressor, we observed 793 genes with reduced expression and 532 genes with increased expression in sgFoxp3-transduced Treg cells, which are enriched in “cytokine production,” “regulation of defense response,” and “regulation of cell adhesion” (Figures 5A and 5B). Of these, 72% were directly bound by Foxp3 in our ChIP-seq dataset, and 56% were co-bound by Foxp3 and Brd9 (Figure 5C). Based on this co-binding, we next examined whether Brd9 regulates Foxp3 target gene expression by positively affecting Foxp3 binding to its targets. Gene set enrichment analysis (GSEA) demonstrated that sgBrd9-increased genes are significantly enriched among genes that increase upon sgFoxp3 targeting, whereas sgBrd9-decreased genes are enriched among genes that decrease in sgFoxp3 Treg cells (Figure 5D). We also performed RNA-seq for Treg cells treated with vehicle or the dBRD9 degrader and observed a similar significant enrichment for dBRD9-affected genes among the Foxp3-regulated genes (Figure 5E). To determine how Brd9 control of Foxp3 binding affects gene expression, we divided Foxp3 binding sites into quartiles based on most affected (Brd9-dependent) to least affected (Brd9-independent) by sgBrd9 transduction and compared fold changes in gene expression in sgBrd9 versus sgNT Treg cells. Indeed, gene expression of Brd9-dependent Foxp3 target genes was significantly more affected upon sgBrd9 targeting than expression of Brd9-independent Foxp3 target genes (Figure S6I). Furthermore, gene expression was significantly more affected in sgSmarcd1-transduced (an ncBAF subunit) Treg cells, but not in sgPbrm1-transduced (a PBAF subunit) Treg cells, at Brd9-dependent Foxp3 target genes (Figure S6I). Thus, ncBAF complexes regulate Foxp3 target genes through potentiation of Foxp3 binding at its target sites. Notably, the Brd9-dependent target gene sets generated from our RNA-seq data were among the most significantly enriched datasets of 9,229 immunological, Gene Ontology, and curated gene sets when analyzed against the sgFoxp3-transduced Treg expression data (Figure 5F). In addition, both datasets were significantly enriched for genes that are differentially expressed between Treg cells and conventional T cells (Feuerer et al., 2010) and between Foxp3 mutant Treg cells from scurfy mice and wild-type Treg cells (Hill et al., 2007). These data define a role of Brd9 in Treg cells by specifically regulating the expression of Foxp3 itself and a subset of Foxp3 target genes.
Figure 5.

Brd9 Co-regulates the Expression of Foxp3 and a Subset of Foxp3 Target Genes
(A) Volcano plot of log2 fold change RNA expression in sgFoxp3 versus sgNT Treg cells versus adjusted p value (Benjamini-Hochberg). The numbers of down and up genes are indicated and colored blue and red, respectively.
(B) Significance of enrichment of Foxp3-dependent genes in each Gene Ontology.
(C) Pie chart of Foxp3 and Brd9 binding by ChIP-seq for Foxp3-dependent genes.
(D) Gene set enrichment analysis (GSEA) enrichment plot for up and down genes in sgBrd9 versus sgNT compared with RNA-seq data of genes that significantly change in sgFoxp3 versus sgNT Treg cells. ES, enrichment score; NES, normalized enrichment score; FWER, family-wise error rate.
(E) As in (D) but for up and down genes in dBRD9 versus DMSO Treg cells.
(F) GSEA of the sgFoxp3 versus sgNT RNA-seq data; the plot shows the FWER p value versus the NES.
The ncBAF Complex Is Required for Normal Treg Cell Suppressor Activity In Vitro and In Vivo
The divergent roles of ncBAF and PBAF complexes in regulating Foxp3 expression suggest that these complexes might also differentially affect Treg suppressor function. We performed sgRNA targeting of ncBAF-specific Brd9 and Smarcd1 or PBAF-specific Pbrm1 and Phf10 in Treg cells and measured their function by conducting an in vitro suppression assay. Treg cells depleted of Brd9 or Smarcd1 exhibited significantly reduced suppressor function, whereas depletion of Pbrm1 or Phf10 resulted in significantly enhanced suppressor function (Figure 6 A; Figure S7A). These data demonstrate that the opposing regulation of Foxp3 expression by ncBAF and PBAF complexes results in decreased and increased Treg suppressor activity upon ncBAF or PBAF subunit deletion, respectively. Similar to sgRNA depletion of Brd9, Treg cells treated with dBRD9 also showed significantly and specifically compromised Treg cell suppressor function in vitro (Figure S7B). We next determined whether the reduced suppressor activity in sgBrd9 Treg cells could be rescued by overexpression of Foxp3. We found that ectopic expression of Foxp3 in sgBrd9 Treg cells partially restored Treg cell suppressor activity to a level comparable with sgNT controls but still lower compared with sgNT Treg cells with ectopic Foxp3 expression (Figure 6B; Figure S7C). These results underscore the requirement for Brd9 in Foxp3 expression maintenance and optimal Treg cell suppressor activity and further demonstrate that dBRD9 reduces Treg cell suppressor activity without impairing Teff cell responses in vitro.
Figure 6.

The ncBAF Complex Regulates Treg Cell Suppressor Function In Vitro and In Vivo
(A) In vitro suppression assay of Treg cells with sgRNA targeting of Brd9, Smarcd1, Pbrm1, and Phf10. sgNT was used as a non-targeting control (n = 3 per group; data represent ± SD).
(B) In vitro suppression assay of sgBrd9 or sgNT with ectopic expression of Foxp3 or the control vector MIGR (n = 3 per group; data represent ± SD).
(C–F) Experiment to measure function of sgNT or sgBrd9 Treg cells relative to “no Treg” cells in a T cell transfer-induced colitis model.
(C) Experimental procedure.
(D) Body weight loss.
(E) Colon histology (left) and colitis scores (right).
(F) Percentage of Foxp3+ cells in the transferred CD45.2+CD4+ Treg population at the endpoint (n = 4–7 per group; data represent mean ± SEM).
Statistical analyses were performed using unpaired two-tailed Student’s t test (ns, p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). See also Figure S7.
To test whether Brd9 also affects Treg function in vivo, we utilized a T cell transfer-induced colitis model. In this model, Rag1 −/− mice received CD45.1+ CD4+ CD25−CD45RBhi Teff cells only or Teff cells along with CD45.2+ Treg cells transduced with sgBrd9 or control sgNT (Figure 6C). Mice that received Teff cells alone lost body weight progressively because of development of colitis. Co-transfer of Treg cells transduced with sgNT protected recipient mice from weight loss, whereas co-transfer of sgBrd9-transduced Treg cells failed to protect recipients from losing weight (Figure 6D). Mice that received Brd9-depleted Treg cells showed significant colitis pathology at 7 weeks compared with mice that received control Treg cells (Figure 6E). Furthermore, Brd9 depletion also led to compromised Treg cell stability after transfer, as manifested by reduced Foxp3+ cell frequencies within the CD45.2+CD4+ transferred Treg cell population (Figure 6F). These results demonstrate that Brd9 is an essential regulator of normal Foxp3 expression and Treg cell function in a model of inflammatory bowel disease in vivo.
In addition to their beneficial role in preventing autoimmune diseases, Treg cells also function as a barrier to anti-tumor immunity. We therefore wondered whether we could exploit the compromised suppressor function shown in Brd9-deficient Treg to disrupt Treg-mediated immune suppression in tumors. We used the MC38 colorectal tumor cell line to induce cancer because of the prominent role of Treg cells in this cancer model (Delgoffe et al., 2013). Rag1 −/− mice were used as recipients for adoptive transfer of Treg cell-depleted CD4 and CD8 T cells (Teff cells) only or co-transfer of Teff cells with Treg cells transduced with either sgBrd9 or sgNT. MC38 tumor cells were implanted subcutaneously on the following day (Figure 7 A). Transfer of sgNT Treg cells allowed significantly faster tumor growth compared with mice that received Teff cells only (“no Treg”) because of suppression of the anti-tumor immune response by Treg cells (Figures 7B and 7C). Furthermore, tumor growth in mice that received sgBrd9-transduced Treg cells was significantly slower than in mice that received sgNT Treg cells, consistent with our findings that Brd9 deficiency reduced Treg cell suppressor activity (Figures 7B and 7C). CD4 and CD8 T cell tumor infiltration significantly increased in mice that received sgBrd9-transduced Treg cells compared with sgNT Treg cells (Figures 7D and 7E). Additionally, the percentage of IFN-γ producing intra-tumor CD4 and CD8 T cells in mice that received sgBrd9-transduced Treg cells was significantly greater than the sgNT Treg condition and comparable with transfer of Teff cells alone (“no Treg”) (Figures 7F and 7G). Consistent with our finding that Brd9 is required for Treg cell persistence in vivo (Figure 6E), the percentage of transferred Treg cells was reduced in mice that received sgBrd9-transduced Treg cells relative to sgNT Treg cells (Figure 7H). Overall, a 2- to 3-fold increase in the ratio of CD8 T cells to Treg cells in tumors and spleen was observed under the sgBrd9 versus the sgNT condition, consistent with the enhanced anti-tumor immune response in mice that received sgBrd9-transduced Treg cells (Figure 7I). To examine whether Brd9 deficiency promotes generation of inflammatory ex-Treg cells, we measured Foxp3 and interferon γ (IFN-γ) expression within the transferred sgBrd9 or sgNT Treg cell population marked with a GFP reporter. Ablation of Brd9 led to an increase in the GFP+Foxp3– ex-Treg cell population compared with sgNT Treg cells (Figures 7J; Figure S7D). More importantly, a higher percentage of sgBrd9 ex-Treg cells produced IFN-γ compared with sgNT ex-Treg cells (Figure 7K; Figure S7D), contributing to slower tumor growth in mice that received sgBrd9 Treg cells. This experiment demonstrates that Brd9 promotes Treg lineage stability and suppressive function in MC38 tumors and that Brd9 deficiency in Treg improves anti-tumor immunity in this context.
Figure 7.

Targeting Brd9 in Treg Cells Improves Anti-tumor Immunity
(A) Experimental procedure to measure the function of sgNT or sgBrd9 Treg cells relative to “no Treg” cells in the MC38 tumor model.
(B) Tumor growth curve.
(C) Tumor weight at the endpoint.
(D and E) Bar graph of total CD4 T cell (D) and CD8 T cell (E) percentage in the CD45+ immune cell population.
(F and G) Bar graph of the IFN-γ+ cell percentage in CD4 T cells (F) and CD8 T cells (G).
(H) Bar graph of the CD4+GFP+Foxp3+ donor cell percentage in CD4 T cells.
(I) Ratio of CD8:Treg cells.
(J) Bar graph of the Foxp3– ex-Treg cell percentage in the transferred Treg population marked by the GFP reporter.
(K) Bar graph of Foxp3–IFN-γ+ cell percentage in the transferred Treg population (n = 5–7 per group; data represent mean ± SEM).
Statistical analyses were performed using unpaired two-tailed Student’s t test (ns, p ≥ 0.05; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). See also Figure S7.
Discussion
In this study, we performed a genome-wide CRISPR screen to identify positive and negative regulators of Foxp3 expression in mouse natural Treg cells. Among positive regulators, we identified Cbfb and Runx3, consistent with a requirement for Cbf-β and Runx3 in Foxp3 expression and Foxp3-dependent target gene expression (Kitoh et al., 2009; Rudra et al., 2009). Among the positive regulators, we discovered subunits from two chromatin remodeling complexes, the Brd9-containing ncBAF and SAGA complexes. Independent validation and functional assays demonstrated an essential role of the ncBAF complex and SAGA complex in Foxp3 expression and Treg cell suppressor function. A recent study using a CRISPR screen of 489 nuclear factors also identified Usp22, a subunit of the SAGA complex, as a positive regulator of Foxp3 expression (Cortez et al., 2020). The ncBAF subunits Brd9, Gltscr1, Gltscr1l, and Smarcd1 were not identified in this study because of exclusion of these genes from the sgRNA library.
Our screens also confirmed several known negative regulators of Foxp3, including the DNA methyltransferase Dnmt1 and the ubiquitin ligase Stub1. Additionally, we identified multiple negative regulators of the mTOR pathway as Foxp3 negative regulators (Tsc2, Flcn, Ddit4, Sesn2, and Nprl2), confirming an essential role of mTOR in homeostasis and function of activated Treg cells (Chapman et al., 2018; Sun et al., 2018). Among negative Foxp3 regulators, we uncovered genes encoding regulators of RNA metabolism, which have no previously reported function in Foxp3 expression. For example, Mettl3 and Mettl14 form a methyltransferase complex that is essential for m6A methylation of RNA, which is recognized as an important regulatory mechanism for a wide range of biological processes, including RNA stability, protein translation, stem cell self-renewal, cell lineage determination, and oncogenesis (Yue et al., 2015). Our screen suggests a potential role of RNA m6A methylation in post-transcriptional regulation of Foxp3. Our genome-wide screen provides the first comprehensive picture of the complex regulatory network controlling Foxp3 expression and reveals previously unknown pathways and factors that warrant further investigation.
Following identification of SWI/SNF subunit genes among Foxp3 regulators, we endeavored to characterize the roles of the three SWI/SNF-related complexes by deleting subunits unique to each of the ncBAF, BAF, and PBAF complexes. We observed specific and divergent roles of the ncBAF and PBAF complexes in regulating Foxp3 expression in Treg cells. In contrast, deletion of BAF-specific subunits had a slight but non-significant effect on Foxp3 expression. Nevertheless, several SWI/SNF core subunits were recovered in our screen, among genes that regulate Treg cell contraction, suggesting that BAF complexes may regulate Treg cell activation or proliferation in response to the TCR stimulation used to activate and culture Treg cells in our screen. This is consistent with the fact that genetic deletion of Smarca4 in Treg cells results in development of a fatal inflammatory disorder reminiscent of Foxp3 mutant scurfy mice (Chaiyachati et al., 2013). Although Treg cell development and Foxp3 expression are normal in Smarca4-deficient Treg cells, Treg cell function is nevertheless compromised because of impaired activation of TCR target genes; for example, chemokine receptor genes (Chaiyachati et al., 2013). Thus, deletion of Smarca4 or other BAF complex subunits likely results in overall defects in Treg cell fitness, whereas deletion of ncBAF subunits appears to have a selective effect on Foxp3 expression and its target genes. Mechanistically, we found that the ncBAF complex co-bound and cooperated with Foxp3 to potentiate its binding to the CNS2 and CNS0 enhancers of the Foxp3 locus. In addition to the Foxp3 locus itself, our ChIP-seq analysis revealed that ncBAF also colocalized with Foxp3 at regulatory elements in a subset of Foxp3 target genes to regulate their gene expression. Thus, we favor a model where reduced Foxp3 expression and loss of epigenetic regulation by ncBAF complexes upon sgBrd9 transduction results in less Foxp3 binding at Brd9/Foxp3 co-bound sites, affecting Foxp3 target gene expression.
Finally, we tested the in vivo relevance of our findings by disrupting the ncBAF subunit Brd9 in Treg cells in mouse models of inflammatory bowel disease and cancer. Brd9 deficiency in Treg cells weakened their suppressor function in a model of T cell-induced colitis, leading to exacerbated disease progression. In the context of cancer, we found that transfer of Brd9-deficient Treg cells failed to restrict anti-tumor immune responses in the MC38 cell induced cancer model, leading to slower tumor growth. Currently, there is a concerted effort to develop compounds targeting a number of SWI/SNF complex subunits to modulate their function. Our data show that bromodomain-directed degradation of Brd9 by dBRD9 recapitulated the effects of Brd9 genetic deletion, suggesting that the ncBAF complex can be targeted with small molecules to control Foxp3 expression and Treg cell function. Thus, through an unbiased screen of Foxp3 regulators, we identify proteins that can potentially be targeted to manipulate Treg cell homeostasis and function in autoimmune diseases and cancer.
Limitations of Study
Future studies are geared toward understanding the mechanism by which ncBAF facilitates expression of Foxp3 and its target genes. Although coronavirus disease 2019 (COVID-19) restrictions prevented us from directly repeating our analysis of Foxp3 ChIP binding in sgBrd9-transduced Treg cells, additional evidence comparing transduction of sgBrd9 versus sgNT in MIGR-transduced Treg cells confirmed our finding that Brd9 promotes Foxp3 binding to the Foxp3 locus and a subset of its target sites. One possibility is that ncBAF complexes maintain chromatin accessibility for Foxp3 binding; however, we detected minimal changes in chromatin accessibility upon sgBrd9 targeting by ATAC-seq. We speculate that alternative methods for mapping nucleosome dynamics and/or chromatin accessibility are necessary to determine the role of chromatin remodeling in this setting. It should also be noted that the methods employed to transduce primary Treg cells for the screen and validation analyses involve activating Treg cells and culturing them in vitro. Thus, Treg cell-specific conditional deletion approaches will be necessary to further study the functional relevance of candidates identified in this screen. Finally, we utilized adoptive transfer of Treg cells into Rag1 –/– mice to test the in vivo function of sgBrd9 Treg cells in models of colitis and tumor immunity. Although this approach is commonly used, we acknowledge that the rapid homeostatic proliferation of Treg cells in recipient mice could impose additional influence on their immune suppressor function.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD4-Alexa fluor 700 | Thermo Fisher | Cat#56-0042-82; RRID: AB_494000 |
| Anti-CD4-PerCP-Cy5.5 | TONBO | Cat#65-0042-U100; RRID: AB_2621876 |
| Anti-CD8-PE | Thermo Fisher | Cat#12-0081-85; RRID: AB_465532 |
| Anti-CD8-BV510 | Biolegend | Cat#100752; RRID: AB_2563057 |
| Anti-CD45.1-BV605 | Biolegend | Cat#110735; RRID:AB_11124743 |
| Anti-CD45.2-Alexa 700 | Biolegend | Cat#109822; RRID:AB_493731 |
| Anti-Foxp3-eFluor 450 | Thermo Fisher | Cat#48-5773-82; RRID:AB_1518812 |
| Anti-NGFR-PE | Biolegend | Cat#345106; RRID:AB_2152647 |
| Anti-NGFR-APC | Biolegend | Cat#345108; RRID:AB_10645515 |
| Anti-Thy1.1-PE | Thermo Fisher | Cat#12-0900-83; RRID:AB_465774 |
| Anti-CD44-BV650 | Biolegend | Cat#103049; RRID:AB_2562600 |
| Anti-CD62L-BV605 | Biolegend | Cat#104438; RRID:AB_2563058 |
| Anti-IFNg-APC | Thermo Fisher | Cat#17-7311-82; RRID:AB_469504 |
| Ghost Viability Dye Red 780 | TONBO | Cat#13-0865-T100 |
| Anti-Foxp3 | In-house | n/a |
| Anti-BRG1/SMARCA4 | Abcam | Cat#110641; RRID:AB_10861578 |
| Anti-BAF155/SMARCC1 | Santa Cruz | Cat#sc-10756; RRID:AB_2191997 |
| Anti-BAF47/SMARCB1 | Santa Cruz | Cat#sc-166165; RRID:AB_2270651 |
| Anti-Brd9 | Active Motif | Cat#61537; RRID:AB_2614970 |
| Anti-Pbrm1 | Bethyl | Cat#A301-591A; RRID:AB_1078808 |
| Anti-Phf10 | Thermo Fisher | Cat#PA5-30678; RRID:AB_2548152 |
| Anti-Arid1a | Santa Cruz | Cat#sc-32761; RRID:AB_673396 |
| Anti-Histone H3K27ac | Abcam | Cat#ab4729; RRID:AB_2118291 |
| Anti-IgG | Cell Signaling | Cat#2729S; RRID:AB_1031062 |
| Anti-mouse secondary | Thermo Fisher | Cat#A21058; RRID:AB_2535724 |
| Anti-rabbit secondary | Thermo Fisher | Cat#SA535571; RRID:AB_2556775 |
| Anti-mouse IL2 | BIO-X-CELL | Cat#BE0043 RRID:AB_1107702 |
| Recombinant DNA | ||
| pSIR-dsRed-Express2 | Addgene | Cat#51135; RRID:Addgene_51135 |
| pSIRG-NGFR | This paper | n/a |
| pSIRG-GFP | This paper | n/a |
| pCL-Eco | Addgene | Cat#12371; RRID:Addgene_12371 |
| lentiCRISPRv2-Brie library | Addgene | Cat#73632 |
| pSIRG-NGFR-Brie library | This paper | n/a |
| pSIRG-NGFR-sgFoxp3 Target: TCTACCCACAGGGATCAATG | This paper | n/a |
| pSIRG-NGFR-sgCbfb Target: GCCTTGCAGATTAAGTACAC | This paper | n/a |
| pSIRG-NGFR-sgDnmt1 Target: TAATGTGAACCGGTTCACAG | This paper | n/a |
| pSIRG-NGFR-sgArid1a Target: GCAGCTGCGAAGATATCGGG | This paper | n/a |
| pSIRG-NGFR-sgArid1a-2 Target: TACCCAAATATGAATCAAGG | This paper | n/a |
| pSIRG- NGFR-sgArid1b Target: TGAGTGCAAAACTGAGCGCG | This paper | n/a |
| pSIRG-NGFR-sgArid1b-2 Target: CAGAACCCCAACATATAGCG | This paper | n/a |
| pSIRG-NGFR-sgDpf1 Target: TCTTCTACCTCGAGATCATG | This paper | n/a |
| pSIRG-NGFR-sgDpf2 Target: GAAGATACGCCAAAGCGTCG | This paper | n/a |
| pSIRG-NGFR-sgPbrm1 Target: AAAACACTTGCATAACGATG | This paper | n/a |
| pSIRG-NGFR-sgPbrm1-2 Target: CAATGCCAGGCACTACAATG | This paper | n/a |
| pSIRG-NGFR-sgArid2 Target: ACTTGCAGTAAATTAGCTCG | This paper | n/a |
| pSIRG-NGFR-sgBrd7 Target: CAGGAGGCAAGCTAACACGG | This paper | n/a |
| pSIRG-NGFR-sgPhf10 Target: GTTGCCGACAGACCGAACGA | This paper | n/a |
| pSIRG-NGFR-sgBrd9 Target: ATTAACCGGTTTCTCCCGGG | This paper | n/a |
| pSIRG-NGFR-sgBrd9-2 Target: GGAACACTGCGACTCAGAGG | This paper | n/a |
| pSIRG-NGFR-sgGltscr1 Target: GTTCTGTGTAAAATCACACT | This paper | n/a |
| pSIRG-NGFR-sgGltscr1l Target: ATGGCTTTATGCAACACGTG | This paper | n/a |
| pSIRG-NGFR-sgSmarcd1 Target: CAATCCGGCTAAGTCGGACG | This paper | n/a |
| pSIRG-NGFR-sgEny2 Target: AGAGCTAAATTAATTGAGTG | This paper | n/a |
| pSIRG-NGFR-sgAtxn7l3 Target: GCAGCCGAATCGCCAACCGT | This paper | n/a |
| pSIRG-NGFR-sgUsp22 Target: GCCATCGACCTGATGTACGG | This paper | n/a |
| pSIRG-NGFR-sgCcdc101 Target: CCAGGTTTCCCGATCCAGAG | This paper | n/a |
| pSIRG-NGFR-sgTada3 Target: GAAGGTCTGTCCCCGCTACA | This paper | n/a |
| pSIRG-NGFR-sgTada1 Target: TTTCCTTCTCGACACAACTG | This paper | n/a |
| pSIRG-NGFR-sgTaf6l Target: TCATGAAACACACCAAACGA | This paper | n/a |
| pSIRG-NGFR-sgSupt20 Target: TTAGTAGTCAATCTGTACCC | This paper | n/a |
| pSIRG-NGFR-sgSupt5 Target: GATGACCGATGTACTCAAGG | This paper | n/a |
| pSIRG-NGFR-sgNT Target: AAAAAGTCCGCGATTACGTC | This paper | n/a |
| pSIRG-GFP-sgBrd9 Target: ATTAACCGGTTTCTCCCGGG | This paper | n/a |
| pSIRG-GFP-sgNT Target: AAAAAGTCCGCGATTACGTC | This paper | n/a |
| MSCV-IRES-GFP (MIGR) | Addgene | Cat#27490; RRID:Addgene_27490 |
| MIGR-Foxp3 | The laboratory of Alexander Rudensky | n/a |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human IL-2 | Peprotech | Cat#200-02 |
| Mouse IL-2 | Biolegend | Cat#575408 |
| dBRD9 | Tocris | Cat#6606 |
| NEBuilder HIFI assembly | NEB | Cat#E2621S |
| BbsI-HF | NEB | Cat#R3539S |
| Q5 High-Fidelity DNA polymerase | NEB | Cat#M0491 |
| Ficoll-Paque 1.084 | GE Health | Cat#17-5446-02 |
| FuGENE 6 HD transfection reagent | Promega | Cat#E2311 |
| Foxp3 Fix/Perm buffer | Thermo Fisher | Cat#00-5523-00 |
| CellTrace Violet | Thermo Fisher | Cat#C34571 |
| Cell Lines | ||
| HEK293T | ATCC | Cat#CRL-11268 |
| MC38 | The laboratory of Susan Keach | n/a |
| Mouse Strains | ||
| Rosa26-LSL-Cas9 mice | The Jackson Laboratory | Cat#024857 |
| Foxp3Thy1.1 reporter mice | Liston et al., 2008 | n/a |
| C57BL.6 Ly5.1+ congenic mice | The Jackson Laboratory | Cat#002014 |
| Rag1−/− mice | The Jackson Laboratory | Cat#002216 |
| Software and Algorithms | ||
| MAGeCK | https://sourceforge.net/p/mageck/wiki/Home/; Li et al., 2014a | n/a |
| MAGeCK-VISPR | https://bitbucket.org/liulab/mageck-vispr; Li et al., 2015 | n/a |
| EnhancedVolcano R script | https://github.com/kevinblighe/EnhancedVolcano | n/a |
| sgRNA distribution histogram R script | Shifrut et al., 2018 | n/a |
| Metascape | Zhou et al., 2019 | n/a |
| HOMER | http://homer.ucsd.edu/homer/ | n/a |
| Cluster 3.0 | http://bonsai.hgc.jp/∼mdehoon/software/cluster/software.htm | n/a |
| Gene Set Enrichment Analysis (GSEA) | https://www.gsea-msigdb.org/gsea/index.jsp; Mootha et al., 2003; Subramanian et al., 2005 | n/a |
| Cutadapt | http://cutadapt.readthedocs.io/en/stable/ | n/a |
| Samtools | http://htslib.org; Li et al., 2009 | n/a |
| Picard | http://broadinstitute.github.io/picard | n/a |
| BWA Aligner | http://bio-bwa.sourceforge.net; Li and Durbin, 2009 | n/a |
| Macs2 | http://pypi.org/project/MACS2 | n/a |
Lead Contact and Materials Availability
Lead Contact
Additional information and requests for resource and/or reagent should be addressed to Diana Hargreaves (dhargreaves@salk.edu) or Ye Zheng (yzheng@salk.edu).
Materials Availability
The pSIRG-NGFR vector and the pSIRG-NGFR-Brie sgRNA library are available from the Lead Contact with a completed Material Transfer Agreement (MTA).
Data and Code Availability
RNA-seq, ChIP-seq, and ATAC-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus under the accession code GEO Database: GSE129846 [https://www.ncbi.nlm.gov/geo/query/acc.cgi?acc=GSE129846]. The current study did not generate any code.
Experimental Model and Subject Details
Mice
C57BL/6 Rosa-Cas9/Foxp3Thy1.1 mice were generated by crossing Rosa26-LSL-Cas9 mice (Platt et al., 2014) (The Jackson Laboratory #024857) with Foxp3Thy1.1 reporter mice (Liston et al., 2008). Male Cas9/Foxp3Thy1.1 mice at 8-12 weeks age were used to isolate Treg cells for the CRISPR screen, and no gender preference was given for other experiments. C57BL.6 Ly5.1+ congenic mice and Rag1−/− mice purchased from the Jackson Laboratory were used for Treg suppression assay and adoptive T cell transfer in colitis and tumor models. All mice were bred and housed in the specific pathogen-free facilities at the Salk Institute for Biological Studies and were conducted under the regulation of the Institutional Animal Care and Use Committee (IACUC) and institutional guidelines.
Method Details
Retroviral vectors and sgRNA library construction
Self-inactivating retroviral vector pSIRG-NGFR was generated by modifying pSIR-dsRed-Express2 (Fujita and Fujii, 2014) (Addgene #51135), which enables us to clone sgRNA as efficient as lentiCRISPRv2, to enrich transduced cells via magnetic beads isolation, and to perform intracellular staining without losing transduced reporter marker. We first mutated all BbsI sites in pSIR-dsRed-Express2, then inserted a sgRNA expressing cassette containing the U6 promoter, guide RNA scaffold and a 500bp filler embedded with BbsI cloning site. The dsRed cassette was replaced by cDNA sequence of human nerve growth factor receptor (NGFR) with truncated intracellular domain. We also generated pSIRG vector with GFP (pSIRG-GFP) for the purpose of T cells transfer in tumor study, minimizing potential immune rejection. The pSIRG-GFP was generated by cutting pSIRG-NGFR with XcmI to remove NGFR cassette and replaced by GFP cDNA by Gibson cloning. For cloning single guide RNA into the pSIRG vector, an annealed sgRNA oligos can be directly inserted into BbsI-digested pSIRG-NGFR by T4 ligation similar to the cloning method utilized by lentiCRISPRv2 (Sanjana et al., 2014). To create a pooled sgRNA library in pSIRG-NGFR, we first amplified sgRNA sequences from an optimized mouse CRISPR sgRNA library lentiCRISPRv2-Brie (Addgene #73632). A total of eight 50 μL PCR reactions were performed to maximize coverage of sgRNA complexity. Each 50 μL PCR reaction contained Q5 High-Fidelity DNA polymerase and buffer (NEB #M0491), 15ng of lentiCRISPRv2-Brie, and targeted primers (Forward: GGCTTTATATATCTTGTGGAAAGGACGAAACACCG, Reverse: CTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC). PCR was performed at 98°C denature, 67°C annealing, 72°C extension for 12 cycles. The sgRNA library amplicons were then combined and separated in 2% agarose gel, and purified by the QIAquick Gel Extraction Kit (QIAGEN #28704). The purified sgRNA amplicons was inserted into the BbsI-digested pSIRG-NGFR by NEBuilder HIFI assembly (NEB #E2621S). The sgRNA representative of the retroviral CRISPR library (pSIRG-NGFR-Brie) was validated by deep sequencing and comparing to the original lentiCRISPRvs-Brie. The coverage of the new pSIRG-NGFR sgRNA library was evaluated by the PinAPL-Py program (Spahn et al., 2017; see Extended Data Figure 1).
T cell isolation and culture
For large scale Treg culture, we first expanded Treg in Rosa-Cas9/Foxp3Thy1.1 mice by injecting IL-2:IL-2 antibody immune complex according protocol described in Webster et al. (2009). Spleen and lymph node Treg cells were labeled with PE-conjugated Thy1.1 antibody and isolated by magnetic selection using anti-PE microbeads (Mitenyl #130-048-801). All isolated Treg cells were activated by plate bound anti-CD3 and anti-CD28 antibodies and cultured with X-VIVO 20 media (LONZA #04-448Q) supplemented by 1X Pen/Strep, 1X Sodium pyruvate, 1X HEPES, 1X GlutaMax, 55 μM beta-mercaptoethanol in the presence of IL-2 at 500 U/mL. For experiments with Brd9 degradation, Treg cells were treated at day 0 with 2.5 μM dBRD9 (Tocris #6606) and cultured for four days for RNA- and ChIP-seq and 0.16-10 μM treated at day 0 and cultured dBRD9 for four days for Foxp3 MFI, cell viability and cell proliferation assays. Live cells were enriched by Ficoll-Paque 1.084 (GE Health 17-5446-02) for RNA-seq and ChIP-seq.
Retroviral production and T cell transduction
HEK293T cells were seeded in 6-wells plate at 0.5 million cells per 2mL DMEM media supplemented by 10% FBS, 1% Pen/Strep, 1X GlutaMax, 1X Sodium Pyruvate, 1X HEPES, and 55 μM beta-mercaptoethanol. One day later, cells from each well was transfected with 1.2 μg of targeting vector pSIRG-NGFR and 0.8 μg of packaging vector pCL-Eco (Addgene, #12371) by using 4 μL of FuGENE HD transfection reagent (Promega #E2311) according manufactured protocol. Cell culture media was replaced by 3 mL fresh DMEM complete media at 24 hours and 48 hours after transfection. The retroviral supernatant was collected at 48 and 72 hours post transfection for T cell infection. For experiments with CRISPR sgRNA targeting, Cas9+ Treg cells were first seeded in 24-wells plate coated with CD3 and CD28 antibodies. At 24 hour post-activation, 70% of Treg media from each well was replaced by retroviral supernatant, supplemented with 4 μg/mL Polybrene (Milipore # TR-1003-G), and spun in a benchtop centrifuge at 1,258 x g for 90 minutes at 32°C. After centrifugation, Treg media was replaced with fresh media supplemented with IL-2 and cultured for another three days. Transduced cells were analyzed for Foxp3 and cytokine expression in eBioscience Fix/Perm buffer (eBioscience #00-5523-00) using flow cytometry. Transduced NGFR+ cells were FACS-sorted for subsequent RNA- and ChIP-seq experiments.
Genome-wide CRISPR screen in Treg
Approximately 360 million Treg cells isolated from Rosa-Cas9/Foxp3Thy1.1 mice were used for the Treg screen. On day 0, Treg cells were seeded at 1x106 cells/mL into 24-wells plate coated with anti-CD3/28 and cultured with X-VIVO complete media with IL-2 (500 U/ml). On day 1, sgRNA retroviral library transduction was performed with a MOI < 0.2. On day 3, approximately 4 million (~50X coverage) NGFR+ transduced cells were collected in three replicates as the starting state sgRNA input. Treg cells reached confluence on day 4. NGFR+ transduced cells were isolated via magnetic selection by anti-PE beads (Mitenyl #130-048-801), and then plated onto new 24-wells plates coated with anti-CD3/CD28, and cultured in X-VIVO complete media with IL-2 (500 U/ml). On day 6, approximately 4 million NGFR+ transduced cells were collected in three replicates as the ending state sgRNA output. The remaining cells were fixed, permeabilized, and stained for intracellular Foxp3. Approximately 2 million Foxp3hi (top 20%) and 2 million Foxp3lo (bottom 20%) cell populations were sorted in three replicates by a FACS Aria cell sorter for genomic DNA extraction and library construction.
Preparation of sgRNA amplicons for Next-Generation Sequencing
To extract genomic DNA, we first lysed cells with homemade digestion buffer (100mM NaCl, 10mM Tris, 25mM EDTA, 0.5% SDS, 0.1mg/mL Proteinase K) overnight in 50°C. On the following day, the lysed sample was mixed with phenol: chloroform: isoamyl alcohol (25:24:1, v/v) in 1:1 ratio, and spun at 6000rpm for 15 min at room temperature. The supernatant containing genomic DNA was transferred into a new tube and mixed with twice volume of 100% ethanol, then spun at 12,500 rpm for 5 min in room temperature to precipitate DNA. Supernatant was removed, and the precipitated DNA was dissolved in ddH2O. DNA concentration was measured by Nanodrop. To generate sgRNA amplicons from extracted genomic DNA, we used a two-step PCR protocol which was adopted from the protocol published by Shalem et al. (2014). We performed eight 50 μL PCR reactions containing 2 μg genomic DNA, NEB Q5 polymerase, and buffer, and targeted primers (Forward: GGCTTTATATATCTTGTGGAAAGGACGAAACACCG, Reverse: CTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC). PCR was performed at 98°C denature, 70°C annealing, 15 s extension for 20 cycles. The products from the first PCR were pooled together, and purified by AMPure XP SPRI beads according to manufacturer’s protocol, and quantified by Qubit dsDNA HS assay. For the second round PCR, we performed eight 50 μL PCR reactions containing 2 ng purified 1st round PCR product, barcoded primer (see primer set from Shalem et al., 2014; Priming site of reverse primer was changed to CTTCCCTCGACGAATTCCCAAC), NEB Q5 polymerase, and buffer. PCR was performed at 98°C denature, 70°C annealing, 15 s extension for 12 cycles. The 2nd round PCR products were pooled, purified by AMPure XP SPRI beads, quantified by Qubit dsDNA HS assay, and sequenced by NEXTSeq sequencer at single end 75 bp (SE75).
In vitro Treg suppression assay
Treg cells were transduced by retrovirus expressing sgRNA targeting gene of interest and cultured in X-VIVO complete media supplemented with IL-2 (500 U/ml). Four days after transduction, transduced cells were sorted and mixed with FACS sorted CD45.1+ naive CD4 T cells (CD4+ CD25– CD44lo CD62Lhi) labeled with CellTrace Violet (Thermo Fisher Scientific #C34571) in different ratio in the presence of irradiated T cell depleted spleen cells as antigen-presenting cells (APC). Three days later, Treg suppression function was measured by the percentage of non-dividing cells within the CD45.1+ effector T cell population. For dBRD9 treatment experiment, dBRD9 was first dissolved in DMSO (10 mM stock) and added into Treg:Teff:APC mixture at 2.5 μM. For Foxp3 overexpression rescue experiment, Treg cells were first transduced with sgNT or sgBrd9 at 24 hour post-activation, and then transduced with MIGR empty vector or MIGR-Foxp3 at 48 hour post-activation. Double transduced Treg cells were FACS sorted on day 4 based on NGFR+ and GFP+ markers and then mixed with CellTrace labeled effector T cells in the presence of APC. Treg suppression readout was measured after three days of co-culture.
Adoptive T cells transfer-induced colitis model
Treg cells were transduced by retrovirus expressing sgRNA targeting gene of interest, and cultured in X-VIVO complete media and IL-2 (500 U/ml). Four days after transduction, the NGFR+ transduced Treg cells were FACS sorted before transferred into recipient mice. To induce colitis, 2 million effector T cells (CD45.1+ CD4+ CD25– CD45RBhi) and 1 million sgRNA transduced Treg cells (CD45.2+ CD4+ Thy1.1+ NGFR+) were mixed together and transferred into Rag1 –/– recipient mice. The body weight of recipient mice was monitored weekly for signs of wasting symptoms. Mice were harvested 7 weeks after T cell transfer. Spleens were used for profiling immune cell populations by FACS. Colons were collected for histopathological analysis.
Adoptive T cells transfer and MC38 tumor model
Similar to the “Adoptive T cells transfer-induced colitis model,” Treg cells were activated in vitro and transduced with pSIRG-GFP expressing sgNT or sgBrd9. Four days after transduction, the GFP+ transduced Treg were FACS sorted. Concurrently, Treg depleted CD4 and CD8 T cells isolated from Rosa-Cas9/Foxp3Thy1.1 mice were used as effector T cells. A total of 1 million pSIRG-sgRNA transduced GFP+ Treg cells, 1 million effector CD8 T cells, and 2 million Treg-depleted CD4 T cells were mixed and transferred into Rag1 –/– recipient mice. on the following day, mice were implanted with 0.5 million MC38 cells (a kind gift from the laboratory of Dr. Susan Kaech) by subcutaneous injection on the flank of mouse. When palpable tumor appeared, tumor size was measured every two day by electronic calipers (volume = width2 x length x 0.5). At the end point, spleen and tumor were collected for immune profiling. For tumor processing, tumor tissues were minced into small pieces and digested with 0.5 mg/mL Collagenase IV (Sigma #C5138) and DNAase I (Roche #4716728001) for 20 minutes and passed through 0.75 μm cell strainer to collect single cell suspension. Isolated cells were stimulated with PMA/Ionomycin and Golgi plug for 5 hours, and then were subjected to Foxp3 and cytokines staining with eBioscience Fix/Perm buffer (eBioscience #00-5523-00).
Nuclear protein extraction
Nuclear lysates were collected from Treg cells following a revised Dignam protocol (Andrews and Faller, 1991). After cellular swelling in Buffer A (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl) supplemented with 1 mM DTT, 1 mM PMSF, 1 μM pepstatin, 10 μM leupeptin and 10 μM chymostatin, cells were lysed by homogenization using a 21-gauge needle with six to eight strokes. If lysis remained incomplete, cells were treated with 0.025 - 0.05% Igepal-630 for ten minutes on ice prior to nuclei collection. Nuclei were spun down at 700 x g for five minutes then resuspended in Buffer C (20 mM HEPES pH 7.9, 20% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA) supplemented with 1 mM DTT, 1 mM PMSF, 1 μM pepstatin, 10 μM leupeptin and 10 μM chymostatin. After thirty minutes of end-to-end rotation at 4°C, the sample was clarified at 21,100 x g for ten minutes. Supernatant was collected, flash frozen in liquid nitrogen and stored in the −80°C freezer.
Co-Immunoprecipitation
Nuclear lysates were thawed on ice then diluted with two-thirds of original volume of 50 mM Tris-HCl pH 8, 0.3% NP-40, EDTA, MgCl2 to bring down the NaCl concentration. Proteins were quantified using Biorad DC Protein Assay (Cat #5000112) according to manufacturer’s instructions. For the co-IP reaction, 200-300 μg of proteins were incubated with antibody against normal IgG, Smarca4, Brd9, Arid1a or Phf10 overnight at 4°C, with end-to-end rotation. Precipitated proteins were bound to 50:50 Protein A: Protein G Dynabeads (Invitrogen) for one to two hours and washed extensively with IP wash buffer (50 mM Tris pH 8, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% Triton X-100). Proteins were eluted in SDS-PAGE loading solution with boiling for five minutes and analyzed by western blotting.
Western blot
Protein samples were run on 4%–12% Bis-Tris gels (Life Technologies). After primary antibody incubation which is typically done overnight at 4°C, blots were probed with 1:20,000 dilution of fluorescently-labeled secondary antibodies in 2% BSA in PBST (1X Phospho-buffered saline with 0.1% Tween-20) for an hour at room temperature (RT). Fluorescent images were developed using Odyssey and analyzed using Image Studio 2. Protein quantitation was performed by first normalizing the measured fluorescence values of the proteins of interest against the loading control (TBP) then normalizing against the control sample (vehicle treated).
RNA-seq sample preparation
RNA from 1-3 × 106 cells was extracted and purified with TRIzol reagent (Thermo Fisher) according to manufacturer’s instructions. RNA-seq libraries were prepared using Illumina TruSeq Stranded mRNA kit following manufacturer’s instructions with 5 μg of input RNA.
ChIP-seq sample preparation
Treg cells were collected and cross-linked first in 3 mM disuccinimidyl glutarate (DSG) in 1X PBS for thirty minutes then in 1% formaldehyde for another ten minutes, both at RT, for chromatin binding protein ChIP or in 1% formaldehyde only for histone modification ChIP. After quenching the excess cross-linker with a final concentration of 125 mM glycine, the cells were washed in 1X PBS, pelleted, flash-frozen in liquid nitrogen, and stored at −80°C. Cell pellets were thawed on ice and incubated in lysis solution (50 mM HEPES-KOH pH 8, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP40, 0.25% Triton X-100) for ten minutes. The isolated nuclei were washed with wash solution (10 mM Tris-HCl pH 8, 1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl) and shearing buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris-HCl pH 8) then sheared in a Covaris E229 sonicator for ten minutes to generate DNA fragments between ~200-1000 base pairs (bp). After clarification of insoluble material by centrifugation, the chromatin was immunoprecipitated overnight at 4°C with antibodies against Foxp3, Smarca4, Brd9, Phf10 or H3K27ac. The next day, the antibody bound DNA was incubated with Protein A+G Dynabeads (Invitrogen) in ChIP buffer (50 mM HEPES-KOH pH 7.5, 300 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% DOC, 0.1% SDS), washed and treated with Proteinase K and RNase A. Cross-linking was reversed by incubation at 55°C for two and a half hours. Purified ChIP DNA was used for library generation (NuGen Ovation Ultralow Library System V2) according to manufacturer’s instructions for subsequent sequencing.
ATAC-seq sample preparation
ATAC-seq was performed according to previously published protocol (Corces et al., 2017). Briefly, Tregs transduced with either sgNT or sgBrd9 were subjected to Ficoll gradient purification to remove dead cells and ensure capture of cells that were 99% viable. 50,000 Treg cells were collected in duplicates per genotype and washed first with cold 1X PBS then with Resuspension buffer (RSB; 10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2). Cells were lysed in 50 μL of RSB supplemented with 0.1% NP40, 0.01% Digitonin and 0.1% Tween 20 for 3 minutes on ice then diluted with 1 mL of RSB with 0.1% Tween 20. Nuclei were isolated by centrifugation at 500 x g for ten minutes then resuspended in 50 μL of transposition mix (25 μL 2x Illumina Transposase buffer, 2.5 μL Illumina Tn5 Transposase, 16.5 μL PBS, 0.5 μL 1% Digitonin, 0.5 μL 10% Tween 20, 5 μL water) for 30 minutes at 37C in a thermomixer with shaking at 1,000 rpm. Reactions were cleaned up with QIAGEN Min-Elute columns. ATAC-seq libraries were prepared as described previously (Buenrostro et al., 2013). Briefly, purified DNA was ligated with adapters and amplified to a target concentration of 20 μL at 4 nM. Libraries were size selected using AMPure XP beads (Beckman) and sequenced using NextSeq for paired end 42 bp (PE42) sequencing.
Quantification and Statistical Analysis
Data analysis of pooled CRISPR screen
The screening hit identification and quality control was performed by MAGeCK-VISPR program (Li et al., 2014a, 2015). The abundance of sgRNA from a sample fastq file was first quantified by MAGeCK “Count” module to generate a read count table. For hit calling, we used MAGeCK “test” module to generate a gene-ranking table that reporting RRA gene ranking score, p value, and log2 fold change. The size factor for normalization was adjusted according to1000 non-targeting control assigned in the screen library. All sgRNAs that are zero read were removed from RRA analysis. The log2 fold change of a gene was calculated from a mean of 4 sgRNA targeting per gene. The scatterplots showing the screen results were generated by using the R script EnhancedVolcano (https://github.com/kevinblighe/EnhancedVolcano). The R script that generated the sgRNA distribution histogram was provided by E. Shifrut and A. Marson (UCSF) (Shifrut et al., 2018). A gene list from Foxp3 regulators (either positive or negative) without affecting cell proliferation was subjected to Gene Ontology analysis using Metascape (Zhou et al., 2019). Genes were analyzed for enrichment for Functional Set, Pathway, and Structural Complex.
Colon histopathological analysis
Histopathological analysis was performed in a blinded manner and scored using the following criteria. Eight parameters were used that include (i) the degree of inflammatory infiltrate in the LP (0-3); (ii) Goblet cell loss (0–2); (iii) reactive epithelial hyperplasia/atypia with nuclear changes (0–3); (iv) the number of IELs in the epithelial crypts (0–3); (v) abnormal crypt architecture (distortion, branching, atrophy, crypt loss) (0–3); (vi) number of crypt abscesses (0–2); (vii) mucosal erosion to frank ulcerations (0–2) and (viii) submucosal spread to transmural involvement (0-2). The severity of lesion was scored independently in 3 regions (proximal, middle and distal colon) over a maximal score of 20. The overall colitis score was based as the average of each regional score (maximal score of 20).
RNA-seq analysis
Single-end 50 bp reads were aligned to the mouse genome mm10 using STAR alignment tool (V2.5) (Dobin et al., 2013). RNA expression was quantified as raw integer counts using analyzeRepeats.pl in HOMER (Heinz et al., 2010) using the following parameters: -strand both -count exons -condenseGenes -noadj. To identify differentially expressed genes, we performed getDiffExpression.pl in HOMER, which uses the DESeq2 R package to calculate the biological variation within replicates. Cut-offs were set at log2 FC = 0.585 and FDR at 0.05 (Benjamin-Hochberg). Principal Component Analysis (PCA) was performed with the mean of transcript per million (TPM) values using Cluster 3.0 with the following filter parameters: at least one observation with absolute value equal or greater than two and gene vector of four. TPM values were log transformed then centered on the mean.
Gene Set Enrichment Analysis
GSEA software (Mootha et al., 2003; Subramanian et al., 2005) was used to perform the analyses with the following parameters: number of permutations = 1000; enrichment statistic = weighted; and metric for ranking of genes = difference of classes (Input RNA-seq data was log-transformed). For Figure 5G, input RNA-seq data contained the normalized log-transformed reads of the 1,325 differentially expressed genes (DEGs) in sgFoxp3/sgNT Treg cells. The compiled gene list included GSEA Gene Ontology, Immunological Signature, Curated Gene, and the up and down DEGs in sgBrd9/sgNT Treg cells. The resulting normalized enrichment scores and FWER p values were combined to generate the graph.
ChIP-seq analysis
Single-end 50 bp or paired-end 42 bp reads were aligned to mouse genome mm10 using STAR alignment tool (V2.5)(Dobin et al., 2013). ChIP-Seq peaks were called using findPeaks within HOMER using parameters for histone (-style histone) or transcription factor (-style factor) (Christopher Benner, HOMER; http://homer.ucsd.edu/homer/index.html; 2018). Peaks were called when enriched > two-fold over input and > four-fold over local tag counts, with FDR 0.001 (Benjamin-Hochberg). For histone ChIP, peaks within a 1000 bp range were stitched together to form regions. Differential ChIP peaks were found by merging peaks from control and experiment groups and called using getDiffExpression.pl with fold change ≥ 1.5 or ≤ −1.5, Poisson p value < 0.0001.
For k-means clustering analysis in Figure S6D, Foxp3 ChIP-seq tags were quantified at the sites that significantly lose Foxp3 binding in sgBrd9, MIGR compared to sgNT, MIGR using the annotatePeaks.pl command in HOMER with -size given. Log2FC values were calculated for sgBrd9, MIGR/sgNT, MIGR and sgBrd9, Foxp3/sgNT, MIGR. k-means clustering was performed using Gene Cluster 3.0 and visualized using Java TreeView.
For gene expression analysis in Figure S6F, Foxp3 ChIP-seq tags were quantified at the union of sites bound by Foxp3 in sgNT and sgBrd9 using the annotatePeaks.pl command in HOMER with size -given and each site was annotated to a gene by mapping to the nearest TSS. Sites were ranked from least to largest Foxp3 ChIP-seq Log2FC in sgBrd9 versus sgNT and divided into quartiles. Gene expression for the genes in the top and bottom quartiles (Brd9-dependent and -independent, respectively) was then plotted using RNA-seq data from Treg cells transduced with sgBrd9, sgSmarcd1, or sgPbrm1 compared to sgNT. Statistical analyses were performed using unpaired two-tailed Student’s t test (ns: p ≥ 0.05, ∗p < 0.05, ∗∗p < 0.01) in Graphpad Prism.
Motif analysis
Sequences within 200 bp of peak centers were compared to motifs in the HOMER database using the findMotifsGenome.pl command using default fragment size and motif length parameters. Random GC content-matched genomic regions were used as background. Enriched motifs are statistically significant motifs in input over background by a p value of less than 0.05. P values were calculated using cumulative binomial distribution.
ATAC-seq analysis
ATAC-seq data analysis used the following tools and versions: cutadapt (v2.4), samtools (v1.9), Picard (v1.7.1), BWA (v0.7.12), macs2 (v2.1.2), and HOMER (v4.11). Paired end 42 bp reads were trimmed using cutadapt to remove Nextera adaptor sequences then aligned to the reference mouse genome mm10 using BWA. The following were filtered out using Picard and samtools: duplicate reads, mitochondrial reads, low quality reads (Q < 20), and improperly paired or unpaired reads. Quality was assessed by calculating Fraction of Reads In Peaks (FRIP Score) which were > 40% for all samples. TSS enrichment was determined using mm10 Refseq TSSs. Broad and narrow peaks were called using macs2 using the following parameters:–slocal 1000 –qvalue 0.05 -f BAMPE. Differentially accessible sites were determined using getDifferentialPeaksReplicates.pl command in HOMER using the union of peaks in sgNT and sgBrd9 with the following parameters: edgeR, fold change cutoff 1.5, adjusted p value < 0.05.
Data and Software Availability
Data availability
RNA-seq, ChIP-seq, and ATAC-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus under the accession code GEO Database: GSE129846 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129846].
Acknowledgments
We would like to thank C. Gordon for mouse colony management; B. Kuo and X. Hu for assistance with plasmid extraction and mouse dissection; N. Hah and G. Chou for assistance with RNA-seq, ChIP-seq, and CRISPR screen experiments; E. Shifrut and A. Marson (UCSF) for providing the R script to generate the sgRNA distribution histograms; A. Williams and M. Shokhirev for bioinformatics assistance; M. Downes and R.M. Evans for helpful discussions; and A. Cao for graphic art assistance. C.-S.L. was partly supported by the Albert G. and Olive H. Schlink Foundation. J.G. was supported by Salk Institute T32 Cancer Training Grant T32CA009370 and NIGMS NRSA F32 GM128377-01. D.C.H. was supported by the National Institutes of Health (GM128943, AI151123, CA228211, and CA184043), the Pew-Stewart Scholar Award for Cancer Research, the V Foundation for Cancer Research (V2016-006), and the Leona M. and Harry B. Helmsley Charitable Trust. Y.Z. was supported by the NOMIS Foundation, the Crohn's and Colitis Foundation, the Leona M. and Harry B. Helmsley Charitable Trust, and the National Institutes of Health (AI107027, AI151123, and OD023689). This work was also supported by the National Cancer Institute funded Salk Institute cancer core facilities (CA014195).
Author Contributions
Conceptualization, C.-S.L., J.G., D.C.H., and Y.Z.; Methodology, C.-S.L.; Investigation, C.-S.L., J.G., Y.L., M.X., J.H., and B.V.; Resources, D.C.H. and Y.Z.; Formal Analysis, C.-S.L., J.G., and M.L.; Data Curation, C.-S.L. and J.G.; Supervision, D.C.H. and Y.Z.; Funding Acquisition, D.C.H. and Y.Z.; Writing – Original Draft, C.-S.L., J.G., D.C.H., and Y.Z.; Writing – Review & Editing, C.-S.L., J.G., D.C.H., and Y.Z.
Declaration of Interests
A patent application has been filed based on the findings described in this study.
Published: July 7, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.immuni.2020.06.011.
Contributor Information
Diana C. Hargreaves, Email: dhargreaves@salk.edu.
Ye Zheng, Email: yzheng@salk.edu.
Supplemental Information
List of Genes Comparing Foxp3-Low with Foxp3-High Populations Identified in the CRISPR-Cas9 Screen of Treg Cells, Related to Figure 2 This excel file consists of four tabs. A complete gene list with statistical analysis comparing Foxp3 low and high population is shown in the first tab labeled “Foxp3 LowVsHigh.” Genes that met the cutoff criteria are shown in the second tab labeled “Foxp3 Pos Regulator” and in the third tab labeled “Foxp3 Neg Regulator,” representing Foxp3 positive and negative regulators, respectively. In the fourth tab, a complete list of raw read counts is available for each sgRNA for all three replicates of the screen.
List of Genes Comparing Day 6 Cells with Day 3 Cells for Identifying Genes Involved in Cell Proliferation and Survival, Related to Figure 2 A complete gene list with statistical analysis comparing cells on day 6 and day 3 is shown in the first tab labeled “Day6 Vs Day3.” Genes that met cutoff criteria are shown in the second tab labeled “Contraction” and in the third tab labeled “Expansion,” representing genes whose deletion led to cell population contraction or expansion, respectively. In the fourth tab, a complete list of raw read counts is available for each sgRNA for all three replicates of the screen.
List of Genes in Different Categories Identified in the CRISPR-Cas9 Screen, Related to Figure 2 A summary of unique or shared genes identified in the Treg screen that regulate Foxp3 expression and/or cell contraction/expansion.
Gene Ontology Analysis of Positive and Negative Foxp3 Regulators Identified in the CRISPR-Cas9 Screen of Treg Cells, Related to Figure 2 Gene Ontology analysis of positive and negative Foxp3 regulators that do not affect Treg cell survival and proliferation (in tab 1 and tab 2, respectively) identified in the Treg screen was performed using Metascape.
GSEA of Foxp3-Dependent Genes in Treg Cells, Related to Figure 5 List of the Gene Ontology, C2, Immunology, and BRD9-dependent gene lists that were probed against the RNA-seq expression data of Foxp3-dependent genes in sgFoxp3 and sgNT transduced Treg cells.
References
- Andrews N.C., Faller D.V. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchill M.A., Yang J., Vogtenhuber C., Blazar B.R., Farrar M.A. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- Chaiyachati B.H., Jani A., Wan Y., Huang H., Flavell R., Chi T. BRG1-mediated immune tolerance: facilitation of Treg activation and partial independence of chromatin remodelling. EMBO J. 2013;32:395–408. doi: 10.1038/emboj.2012.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman N.M., Zeng H., Nguyen T.M., Wang Y., Vogel P., Dhungana Y., Liu X., Neale G., Locasale J.W., Chi H. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat. Commun. 2018;9:2095. doi: 10.1038/s41467-018-04392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Jin W., Hardegen N., Lei K.J., Li L., Marinos N., McGrady G., Wahl S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Barbi J., Bu S., Yang H.Y., Li Z., Gao Y., Jinasena D., Fu J., Lin F., Chen C. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity. 2013;39:272–285. doi: 10.1016/j.immuni.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinen T., Kannan A.K., Levine A.G., Fan X., Klein U., Zheng Y., Gasteiger G., Feng Y., Fontenot J.D., Rudensky A.Y. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016;17:1322–1333. doi: 10.1038/ni.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez J.T., Montauti E., Shifrut E., Gatchalian J., Zhang Y., Shaked O., Xu Y., Roth T.L., Simeonov D.R., Zhang Y. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature. 2020;582:416–420. doi: 10.1038/s41586-020-2246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe G.M., Kole T.P., Zheng Y., Zarek P.E., Matthews K.L., Xiao B., Worley P.F., Kozma S.C., Powell J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe G.M., Woo S.R., Turnis M.E., Gravano D.M., Guy C., Overacre A.E., Bettini M.L., Vogel P., Finkelstein D., Bonnevier J. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501:252–256. doi: 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Arvey A., Chinen T., van der Veeken J., Gasteiger G., Rudensky A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell. 2014;158:749–763. doi: 10.1016/j.cell.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M., Hill J.A., Kretschmer K., von Boehmer H., Mathis D., Benoist C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc. Natl. Acad. Sci. USA. 2010;107:5919–5924. doi: 10.1073/pnas.1002006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T., Fujii H. Identification of proteins associated with an IFNγ-responsive promoter by a retroviral expression system for enChIP using CRISPR. PLoS ONE. 2014;9:e103084. doi: 10.1371/journal.pone.0103084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchalian J., Malik S., Ho J., Lee D.S., Kelso T.W.R., Shokhirev M.N., Dixon J.R., Hargreaves D.C. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun. 2018;9:5139. doi: 10.1038/s41467-018-07528-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D., Tora L. Sharing the SAGA. Trends Biochem. Sci. 2017;42:850–861. doi: 10.1016/j.tibs.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J.A., Feuerer M., Tash K., Haxhinasto S., Perez J., Melamed R., Mathis D., Benoist C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Josefowicz S.Z., Lu L.F., Rudensky A.Y. Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz S.Z., Wilson C.B., Rudensky A.Y. Cutting edge: TCR stimulation is sufficient for induction of Foxp3 expression in the absence of DNA methyltransferase 1. J. Immunol. 2009;182:6648–6652. doi: 10.4049/jimmunol.0803320. [DOI] [PubMed] [Google Scholar]
- Kim H.P., Leonard W.J. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J. Exp. Med. 2007;204:1543–1551. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa Y., Ohkura N., Kidani Y., Vandenbon A., Hirota K., Kawakami R., Yasuda K., Motooka D., Nakamura S., Kondo M. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat. Immunol. 2017;18:173–183. doi: 10.1038/ni.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitoh A., Ono M., Naoe Y., Ohkura N., Yamaguchi T., Yaguchi H., Kitabayashi I., Tsukada T., Nomura T., Miyachi Y. Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity. 2009;31:609–620. doi: 10.1016/j.immuni.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Koutelou E., Hirsch C.L., Dent S.Y. Multiple faces of the SAGA complex. Curr. Opin. Cell Biol. 2010;22:374–382. doi: 10.1016/j.ceb.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal G., Zhang N., van der Touw W., Ding Y., Ju W., Bottinger E.P., Reid S.P., Levy D.E., Bromberg J.S. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 2009;182:259–273. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.M., Bautista J.L., Scott-Browne J., Mohan J.F., Hsieh C.S. A broad range of self-reactivity drives thymic regulatory T cell selection to limit responses to self. Immunity. 2012;37:475–486. doi: 10.1016/j.immuni.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Köster J., Xu H., Chen C.H., Xiao T., Liu J.S., Brown M., Liu X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015;16:281. doi: 10.1186/s13059-015-0843-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Liang Y., LeBlanc M., Benner C., Zheng Y. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell. 2014;158:734–748. doi: 10.1016/j.cell.2014.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston A., Nutsch K.M., Farr A.G., Lund J.M., Rasmussen J.P., Koni P.A., Rudensky A.Y. Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc. Natl. Acad. Sci. USA. 2008;105:11903–11908. doi: 10.1073/pnas.0801506105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhang P., Li J., Kulkarni A.B., Perruche S., Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- Long M., Park S.G., Strickland I., Hayden M.S., Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009;31:921–931. doi: 10.1016/j.immuni.2009.09.022. [DOI] [PubMed] [Google Scholar]
- Michel B.C., D’Avino A.R., Cassel S.H., Mashtalir N., McKenzie Z.M., McBride M.J., Valencia A.M., Zhou Q., Bocker M., Soares L.M.M. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018;20:1410–1420. doi: 10.1038/s41556-018-0221-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Ouyang W., Beckett O., Ma Q., Li M.O. Transforming growth factor-beta signaling curbs thymic negative selection promoting regulatory T cell development. Immunity. 2010;32:642–653. doi: 10.1016/j.immuni.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polansky J.K., Kretschmer K., Freyer J., Floess S., Garbe A., Baron U., Olek S., Hamann A., von Boehmer H., Huehn J. DNA methylation controls Foxp3 gene expression. Eur. J. Immunol. 2008;38:1654–1663. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- Remillard D., Buckley D.L., Paulk J., Brien G.L., Sonnett M., Seo H.S., Dastjerdi S., Wühr M., Dhe-Paganon S., Armstrong S.A., Bradner J.E. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew. Chem. Int. Engl. 2017;56:5738–5743. doi: 10.1002/anie.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra D., Egawa T., Chong M.M., Treuting P., Littman D.R., Rudensky A.Y. Runx-CBFbeta complexes control expression of the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 2009;10:1170–1177. doi: 10.1038/ni.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S., Yamaguchi T., Nomura T., Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Samstein R.M., Arvey A., Josefowicz S.Z., Peng X., Reynolds A., Sandstrom R., Neph S., Sabo P., Kim J.M., Liao W. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151:153–166. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E., Carnevale J., Tobin V., Roth T.L., Woo J.M., Bui C.T., Li P.J., Diolaiti M.E., Ashworth A., Marson A. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell. 2018;175:1958–1971.e15. doi: 10.1016/j.cell.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn P.N., Bath T., Weiss R.J., Kim J., Esko J.D., Lewis N.E., Harismendy O. PinAPL-Py: A comprehensive web-application for the analysis of CRISPR/Cas9 screens. Sci. Rep. 2017;7:15854. doi: 10.1038/s41598-017-16193-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun I.H., Oh M.H., Zhao L., Patel C.H., Arwood M.L., Xu W., Tam A.J., Blosser R.L., Wen J., Powell J.D. mTOR Complex 1 Signaling Regulates the Generation and Function of Central and Effector Foxp3+ Regulatory T Cells. J. Immunol. 2018;201:481–492. doi: 10.4049/jimmunol.1701477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A., Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–118. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tone Y., Furuuchi K., Kojima Y., Tykocinski M.L., Greene M.I., Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- van Loosdregt J., Coffer P.J. Post-translational modification networks regulating FOXP3 function. Trends Immunol. 2014;35:368–378. doi: 10.1016/j.it.2014.06.005. [DOI] [PubMed] [Google Scholar]
- van Loosdregt J., Fleskens V., Fu J., Brenkman A.B., Bekker C.P., Pals C.E., Meerding J., Berkers C.R., Barbi J., Gröne A. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 2013;39:259–271. doi: 10.1016/j.immuni.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster K.E., Walters S., Kohler R.E., Mrkvan T., Boyman O., Surh C.D., Grey S.T., Sprent J. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 2009;206:751–760. doi: 10.1084/jem.20082824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.O., Nurieva R., Martinez G.J., Kang H.S., Chung Y., Pappu B.P., Shah B., Chang S.H., Schluns K.S., Watowich S.S. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y., Liu J., He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343–1355. doi: 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Josefowicz S., Chaudhry A., Peng X.P., Forbush K., Rudensky A.Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Rudensky A.Y. Foxp3 in control of the regulatory T cell lineage. Nat. Immunol. 2007;8:457–462. doi: 10.1038/ni1455. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of Genes Comparing Foxp3-Low with Foxp3-High Populations Identified in the CRISPR-Cas9 Screen of Treg Cells, Related to Figure 2 This excel file consists of four tabs. A complete gene list with statistical analysis comparing Foxp3 low and high population is shown in the first tab labeled “Foxp3 LowVsHigh.” Genes that met the cutoff criteria are shown in the second tab labeled “Foxp3 Pos Regulator” and in the third tab labeled “Foxp3 Neg Regulator,” representing Foxp3 positive and negative regulators, respectively. In the fourth tab, a complete list of raw read counts is available for each sgRNA for all three replicates of the screen.
List of Genes Comparing Day 6 Cells with Day 3 Cells for Identifying Genes Involved in Cell Proliferation and Survival, Related to Figure 2 A complete gene list with statistical analysis comparing cells on day 6 and day 3 is shown in the first tab labeled “Day6 Vs Day3.” Genes that met cutoff criteria are shown in the second tab labeled “Contraction” and in the third tab labeled “Expansion,” representing genes whose deletion led to cell population contraction or expansion, respectively. In the fourth tab, a complete list of raw read counts is available for each sgRNA for all three replicates of the screen.
List of Genes in Different Categories Identified in the CRISPR-Cas9 Screen, Related to Figure 2 A summary of unique or shared genes identified in the Treg screen that regulate Foxp3 expression and/or cell contraction/expansion.
Gene Ontology Analysis of Positive and Negative Foxp3 Regulators Identified in the CRISPR-Cas9 Screen of Treg Cells, Related to Figure 2 Gene Ontology analysis of positive and negative Foxp3 regulators that do not affect Treg cell survival and proliferation (in tab 1 and tab 2, respectively) identified in the Treg screen was performed using Metascape.
GSEA of Foxp3-Dependent Genes in Treg Cells, Related to Figure 5 List of the Gene Ontology, C2, Immunology, and BRD9-dependent gene lists that were probed against the RNA-seq expression data of Foxp3-dependent genes in sgFoxp3 and sgNT transduced Treg cells.
Data Availability Statement
RNA-seq, ChIP-seq, and ATAC-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus under the accession code GEO Database: GSE129846 [https://www.ncbi.nlm.gov/geo/query/acc.cgi?acc=GSE129846]. The current study did not generate any code.
Data availability
RNA-seq, ChIP-seq, and ATAC-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus under the accession code GEO Database: GSE129846 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129846].
RNA-seq, ChIP-seq, and ATAC-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus under the accession code GEO Database: GSE129846 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129846].