

Abstract
The failures in Alzheimer's disease (AD) therapy strongly suggest the importance of reconsidering the research strategies analyzing other mechanisms that may take place in AD as well as, in general, in other neurodegenerative dementias. Taking into account that in AD a variety of defects result in neurotransmitter activity and signaling efficiency imbalance, neuronal cell degeneration and defects in damage/repair systems, aberrant and abortive cell cycle, glial dysfunction, and neuroinflammation, a target may be represented by the intracellular signaling machinery provided by the kinome. In particular, based on the observations of a relationship between cancer and AD, we focused on cancer kinases for targeting neurodegeneration, highlighting the importance of targeting the intracellular pathways at the intersection between cell metabolism control/duplication, the inhibition of which may stop a progression in neurodegeneration.
Keywords: Alzheimer's disease, c‐Abl, c‐kit, cancer kinases, dementia, Fyn, GSK‐3β, kinase inhibitors, neurodegeneration, p38 MAPK
1. OBJECTIVE
The present review aims to dissect the burgeoning landscape of druggable kinases in Alzheimer's disease (AD), focusing on selected cancer kinases currently under investigation in clinical trials as therapeutic targets. The present review intends to: (1) examine the dysregulation of intracellular signaling pathways, regulated by protein kinases, involved in the activation/inhibition of either pro‐survival or cell death pathways, playing a central role both in cancer and neurodegeneration; (2) pinpoint the most relevant druggable kinases to counteract neurodegeneration in AD, with strong implications also in other dementias; (3) discuss cancer kinases inhibition as a therapeutic approach for AD treatment, repurposing existing anti‐cancer drugs for non‐oncological indications; and (4) summarize current challenges and discuss future limitations of such a rapidly evolving field. Groundbreaking understating of kinase signaling networks at molecular level may lead to major advances in repurposing existing drugs for new targets or disease indications.
2. BACKGROUND
The current knowledge on the pathogenesis of AD, as well as the existing models of etiology, have been unable to provide an effective therapeutic option for the treatment of AD. As an example, therapeutic approaches targeting amyloid beta (Aβ), on which a great effort has been spent by the scientific and clinical communities, have so far largely failed to reach a significant clinical outcome. Several thousands of patients have been treated with anti‐Aβ drugs, ranging from strategies targeting the levels of Aβ peptides, either by interfering with Aβ production (eg, β‐ and γ‐secretase inhibitors), by promoting Aβ clearance, or neutralizing it with humanized monoclonal antibodies. However, although, using the latter, plaques may be cleared, so far no convincing and significant clinical advantages in affecting the ongoing degenerative processes have been reported. Notably, results from trials involving anti‐Aβ antibodies, such as gantenerumab, solanezumab, and aducanumab, suggested that to appreciate cognitive improvements in AD patients the treatment should probably be started at the very early stages of the disease. 1 Accordingly, to avoid the challenges associated with prevention trials design in late‐onset sporadic AD, the pioneering DIAN‐TU (Dominantly Inherited Alzheimer Network Trials Unit) was launched. DIAN‐TU is phase 2/3 trial based on a primary prevention of the autosomal dominant form of AD, which has been shown to be linked to Aβ dysfunction and to cause cognitive impairment at a younger and predictable age. 1 Unfortunately, a topline analysis of the trial reported that both of the investigational anti‐amyloid drugs, Roche's gantenerumab and Lilly's solanezumab, missed the primary endpoint, consisting of a composite of four cognitive tests (ie, DIAN‐Multivariate Cognitive Endopoint). Several considerations (small sample size, heterogeneity of the disease stage, secondary outcomes still under scrutiny) suggest caution in interpreting these preliminary disappointing data. Some encouragement derives from the application in October 2019 to the U.S. Food and Drug Administration (FDA) for the marketing approval of aducanumab 2 after that the reanalysis of the phase 3 studies, originally discontinued after a futility analysis showing no clinical advantage of the treatment, revealed some significant results. 2
The discouraging results observed in AD therapy emphasize the need to redirect the research strategies by better rethinking the biological mechanisms and intracellular signaling machinery involved in AD, as well as, more in general, in other neurodegenerative dementias. Even if the pathological profile of neurodegenerative disorders is different, common biological traits are present including neuronal cell degeneration, defects in damage/repair systems, aberrant and abortive cell cycle events, and neuroinflammation. The further observations of a relationship between cancer and neurodegenerative disorders, such as AD and Parkinson's disease (PD), 3 may direct to cancer kinases for targeting neurodegeneration. The field of cancer kinase inhibition for non‐oncological indications, such as AD, is emerging as a challenging area to develop disease‐modifying therapies. Indeed, tyrosine kinase inhibition provides a double‐edged swordby manipulating autophagy to inhibit cell division and tumor growth in cancer, and by inducing toxic protein degradation as well as neuronal survival in neurodegeneration on the other hand.
3. NEW OR UPDATED HYPOTHESIS
Over the past decades, kinases have emerged as one of the most intensively investigated drug targets in current pharmacological research, due to their pivotal roles in modulating a wide array of cellular processes. A great effort has been directed toward the development of molecules specifically targeting the human kinome. 4 To date, the majority of molecules show a spectrum of kinase inhibitors, with >250 currently in clinical trials and 48 approved by U.S. FDA, mostly to treat malignancies. 5 The therapeutic potential of kinase manipulation, as well as the functions of kinases as tumor biomarkers for diagnosis, prognosis, and treatment, have widely been characterized in oncology. Several kinase inhibitors have revolutionized the treatment of malignancies driven by a single oncogenic kinase, such as chronic myeloid leukemia and gastrointestinal stromal tumors. Initially focused on cancer therapy, kinase drug discovery has recently broadened its focus to include an expanded range of therapeutic areas, such as autoimmune and inflammatory diseases, as well as neurodegenerative disorders (reviewed by Ferguson and Gray 6 ), including AD. However, the contribution of the dysregulation of human kinome to neurodegeneration has not been clarified so far and the field of kinase‐directed therapies is still immature compared to their application in cancer therapy. The neuronal functions of many kinases are still largely uncharacterized, with a sparse indication of how these targets influence the major signaling pathways involved in AD. Further investigations on human brains are needed to profile the changes in protein kinase activity in the different brain areas during aging and the progression of neurodegeneration.
Highlights
New therapeutic strategies have to be considered in dementias.
Pathways controlling cell metabolism/duplication are relevant targets.
Kinases may represent modulable druggable targets in neurodegenerative diseases.
Tyrosine kinase inhibition appears promising in treating neurodegenerative diseases.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (eg, PubMed, ClinicalTrials.gov) sources.
Interpretation: Data from literature suggest that the dysregulation of intracellular signaling pathways at the crossroad between pro‐survival and cell death pathways, regulated by protein kinases, play a key role both in cancer and neurodegeneration. In particular, the inhibition of kinases presently targeted in cancer is emerging as a challenging strategy to counteract neurodegenerative dementias.
Future directions: The present work emphasizes the urgent need to refocus the research strategies by rethinking the biological mechanism and intracellular signaling machinery, regulated by protein kinases, involved in neurodegenerative dementias. The authors aim to propose a frame of reference for the generation of new hypotheses in the field of neurodegenerative diseases.
Unlike cancer, for which the identification of the specific kinase target led to the development of successful treatments, such lack of knowledge complicates the identification of single or clusters of specific kinases as drug target to counteract AD. The recognition of AD complexity suggests that addressing more than one target might be needed to set up a successful AD treatment. Accordingly, the complex and multifactorial pathophysiology of AD would suggest a multi‐pharmacological approach rather than single target therapy, also in the context of kinase‐directed drug discovery. Therefore, targeting multiple kinases rather than inhibiting any single kinase, by using either single drugs binding multiple proteins or cocktails of highly selective inhibitors, might be a promising strategy. In particular, some of the investigated protein kinase inhibitors show a “target promiscuity” profile. Owing to the fact that all kinases share a high degree of sequence conservation as well as common substrate recognition motifs, profiling the kinome selectivity of these inhibitors represents a fundamental step to attain the selectivity necessary for pharmacological target validation, as well as to predict and avoid off‐target adverse effects. Such aspects may also present positive implications by allowing the identification of novel drug targets for already approved drugs and their repurposing for new targets or clinical indications. However, such wide diversity of interaction patterns shows a number of limitations. For instance, Karaman et al. screened 38 kinase inhibitors against a panel of 317 distinct human kinases, by using an in vitro competition binding assay, and identified a total of 3175 potential binding interactions, 7 with several kinase inhibitors showing higher affinity for their secondary targets rather than for their primary recognized targets. Such wide diversity of interaction patterns strongly suggests the importance of fully characterizing the target spectrum of kinase inhibitors to better interpret their biological activity observed in preclinical and clinical studies. Furthermore, some kinase inhibitors exhibit a paradoxical effect, thus resulting in the activation of the same target kinase or different kinases. As an example, c‐Raf (rapidly accelerated fibrosarcoma) inhibitors have been reported to trigger a reactivation of c‐Raf, without affecting other targets involved in the same signaling pathway, such as MKK1 (mitogen‐activated protein kinase kinase 1) or p42 MAPK (mitogen‐activated protein kinase)/ERK2 (extracellular signal‐regulated kinase 2). 8 In addition, some Bcr (break point cluster)‐Abl (Abelson) inhibitors possess off‐target activity against Raf and stimulate paradoxical activation of BRAF and CRAF in a Ras‐dependent manner. 9
Based on these observations, in the following sections, we will focus on some selected kinase inhibitors, repurposed in AD and other dementias, that are currently under investigation in clinical trials as therapeutic tools (Table 1), highlighting the strength and weakness of their use.
TABLE 1.
Clinical trials of cancer kinase inhibitors in Alzheimer's Disease
| Agent | Mechanism of action | Therapeutic purpose | Clinicaltrials.gov ID | Phase/status | Sponsor |
|---|---|---|---|---|---|
|
Saracatinib (AZD0530) |
Selective Src tyrosine kinase inhibitor | Effect on cerebral metabolic rate for glucose | NCT02167256 |
Phase 2 Completed |
Yale University |
|
Masitinib (AB1010) |
Tyrosine kinase inhibitor targeting c‐Kit, platelet‐derived growth factor receptors, and, a lesser extent, Lyn, Fyn, and the FAK pathway | Activity on mast cells, modulation of inflammatory processes | NCT01872598 |
Phase 3 Active, not recruiting |
AB Science |
| NCT00976118 |
Phase 2 Completed |
AB Science | |||
| Neflamapimod (VX‐745) | Selective p38 MAPK inhibitor | Affects multiple cellular processes including inflammation and cellular plasticity; reduces amyloid plaque burden (DMT) |
Phase 2 Completed Phase 2 Recruiting |
EIP Pharma Inc, VU University EIP Pharma Inc, Toulouse University, Foundation Plan Alzheimer |
|
| Nilotinib (AMN107) | Abl Tyrosine kinase inhibitor | Reduce amyloid and tau phosphorylation (DMT) | NCT02947893 |
Phase 2 Active, not recruiting |
Georgetown University |
| DNL747 | RIPK1 inhibitor | Reduce cytokines and other inflammatory factors (DMT) | NCT03757325 |
Phase 1 Active, not recruiting |
Denali Therapeutics Inc |
|
Tideglusib (NP031112) |
GSK inhibitor | Reduce tau phosphorylation | NCT00948259 | Phase 1 and 2 Completed | Noscira SA |
| NCT01350362 |
Phase 2 Completed |
Noscira SA |
4. KINASE INHIBITORS TO COUNTERACT TAU AND Aβ‐DRIVEN NEUROTOXICITY
4.1. Saracatinib
Saracatinib (also known as AZD0530), a Fyn kinase inhibitor, has been largely investigated for its inhibitory effect on cell growth. Originally developed by AstraZeneca as a therapy for solid tumors to counteract tumor cell adhesion, migration, invasion, and cell proliferation, 10 saracatinib was deprioritized due to its limited benefits as a single agent for oncological conditions and is currently being investigated in clinical and preclinical programs for a variety of non‐oncological conditions, including AD, pain, and psychosis. In particular, it has been repurposed as a disease‐modifying therapy in AD, as Fyn modulates both Aβ‐induced synaptic dysfunctions and neurotoxicity, as well as tau phosphorylation. In particular, the capability of extracellular oligomeric Aβ to bind with nanomolar affinity to cellular prion protein (PrPC) on neuronal cell surface and to activate the downstream signaling pathway involving Fyn kinase has been demonstrated (Figure 1). 11 , 12 Notably, Aβ binding to PrPC has been shown to be highly specific for the soluble oligomeric form, with low or no affinity for fibrillary or monomeric Aβ peptides. 13 Such connection between oligomeric Aβ–PrPC complexes at the cell surface and intracellular Fyn kinase has been found to require the participation of the metabotropic glutamate receptor 5 (mGluR5). 14 Fyn activation by oligomeric Aβ–PrPC has been reported to activate N‐methyl‐D‐aspartate receptors (NMDA‐R) by phosphorylating the intracellular segment of the NR2B subunit at Y‐1472 11 , 15 and to induce dendritic spine loss. 11 In addition, Fyn kinase has been demonstrated to induce the downstream phosphorylation of tau. Accordingly, Fyn has been found to directly associate with tau and to phosphorylate tyrosine residues near the amino terminus. 16 , 17 , 18
FIGURE 1.
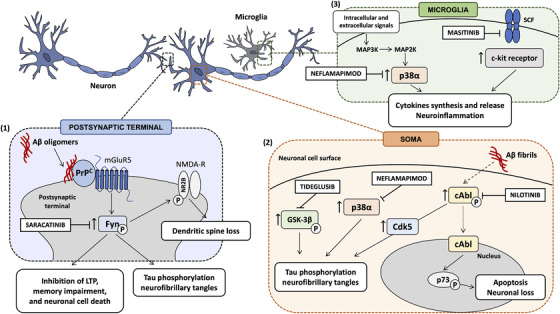
Targeting cancer kinases with inhibitors in dementias. (1) The intracellular pathway involving Fyn kinase has been demonstrated to be altered in Alzheimer's disease (AD), in which Fyn modulates both amyloid beta (Aβ)‐driven synaptic dysfunction and neurotoxicity. At postsynaptic terminal, the extracellular soluble Aβ oligomers bind with nanomolar affinity to cellular prion protein (PrPC) on neuronal cell surface, thus triggering the activation of the downstream intracellular signaling pathway involving Fyn kinase. This activation of Fyn kinase by oligomeric Aβ‐PrPC, which requires the participation of mGluR5, leads to the activation of N‐methyl‐D‐aspartate receptors (NMDA‐Rs) by phosphorylating their intracellular segment NR2B subunit, inducing dendritic spine loss. In addition, Fyn triggers the downstream phosphorylation of tau, by possibly contributing to neurofibrillary tangles formation. Saracatinib, a Fyn inhibitor, has been repurposed as disease‐modifying therapy in AD. (2) At neuronal cell surface, Aβ fibrils increase c‐Abl kinase activity, thus stimulating the nuclear translocation of c‐Abl and inducing apoptosis and neuronal loss through c‐Abl‐mediated p73 phosphorylation. Furthermore, the activation of c‐Abl kinase by Aβ fibrils promotes tau phosphorylation, both directly and indirectly, by activating the tau kinase Cdk5. c‐Abl has been found to be hyperactivated in human AD and PD brains, as well as in Lewy body dementia (LBD), and its inhibitor nilotinib has been repurposed for PD, LBD, and AD. Moreover, in neurons, the overactivation of GSK‐3β and p38α contributes to tau phosphorylation. Tideglusib, a GSK‐3 inhibitor, and neflamapimod, a p38α inhibitor, have been repurposed in AD as potential disease‐modifying therapies. Moreover, the U.S. Food and Drug Administration recently granted fast‐track designation to neflamapimod for the treatment of LBD. (3) In microglia, several extracellular and intracellular signals trigger the consequential activation of MAPK3, MAPK2, and p38α, stimulating the synthesis and release of pro‐inflammatory cytokines, thus promoting neuroinflammatory processes. Neflamapimod has been investigated as therapeutic approach to counteract neuroinflammation in AD. Finally, the activation of the stem cell factor (SCF)/c‐kit pathway mediates neuroinflammatory responses and the c‐kit inhibitor, masitinib, has been tested in clinical trials for the treatment of rheumatoid arthritis, asthma, and as add‐on therapy to riluzole in amyotrophic lateral sclerosis
Taken together, these data prompted intense investigations on targeting Fyn kinase for the treatment of AD. Accordingly, Kaufman et al. demonstrated that AZD0530 at a dose of 5mg/kg/d for 4 weeks fully rescued both spatial learning and memory deficits in 11/12‐month‐old APP/PS1 transgenic mice. 19 In addition, after 6 weeks of AZD0530 or vehicle treatment, the AZD0530‐treated APP/PS1 mice exhibited performance equal to wild type mice in novel object recognition, 19 demonstrating that AZD0530 was capable of reversing the age‐dependent memory impairment produced by the transgene. Moreover, AZD0530 at a dose of 5 mg/kg/day for 5 weeks decreased total tau and phosphorylated tau in 11‐month‐old APP/PS1/Tau transgenic mice. 19 However, a recent multicenter randomized clinical trial (NCT02167256) of 159 participants with mild AD, whose primary outcome was the reduction in relative CMRgl (cerebral metabolic rate for glucose) measured by 18F‐fluorodeoxyglucose (18F‐FDG) PET (positron emission tomography), reported non‐statistically significant effects of AZD0530 treatment on the relative cerebral metabolic rate for glucose or on secondary clinical or biomarker measures. 20 AZD0530 treatment did not slow cerebral metabolic decline and did not improve cognitive function, compared to placebo. In particular, the treatment groups did not significantly differ in secondary clinical outcomes, such as rates of change in ADAS‐Cog11 (Alzheimer's Disease Assessment Scale–Cognitive Subscale), ADCS‐ADL (Alzheimer's Disease Cooperative Study–Activities of Daily Living), CDR‐SB (Clinical Dementia Rating–Sum of Boxes), NPI (Neuropsychiatric Inventory), or MMSE (Mini‐Mental State Examination) scores. 20 Moreover, in patients receiving saracatinib, consistent trends in worsening cognitive, functional, as well as clinical global outcome, have been observed compared to placebo as measured by ADAS‐Cog, ADCS‐ADL, CDR‐SB, respectively. 20 It is likely that such negative trends did not reach statistical significance due to the limited sample size. In addition, almost two‐fold number of dropouts in the group receiving saracatinib (n = 21) has been reported, compared to placebo group (n = 12), mostly due to adverse events. 20 In detail, 73 participants (92.4%) treated with saracatinib and 65 participants (81.2%) receiving placebo have been reported to experience at least one adverse event. 20 In particular, the most frequent adverse events were diarrhea and other gastrointestinal disorders that occurred in 38 participants (48.1%) receiving saracatinib and 23 participants (28.8%) receiving placebo. 20 On the other hand, trends for slowing the decrease in hippocampal volume and entorhinal thickness were observed. 20
Therefore, although such results are discouraging, Fyn kinase cannot be excluded as a potential therapeutic target in AD. First of all, further optimization of selective Fyn inhibition is required to achieve a complete target engagement that can give us clues on Fyn kinase as a target of disease modification in AD. Moreover, given the well‐established effect of saracatinib on glutamatergic transmission, it can be speculated that the drug does not modify the cognitive ability, but it may affect other behavioral disturbances. Notably, the inhibition of Fyn kinase may be addressed to specific subpopulations of AD patients. The identification of kinase‐based molecular signatures in AD patients may allow us to identify patients more prone to respond to the therapy. In particular, AD patients carrying a specific Tyr682 APP phosphorylation might be more likely to respond to Fyn kinase inhibitor therapy. This hypothesis arises from literature data showing that Fyn binds to amyloid‐β precursor protein (APP) on the 682YENPTY687 domain in human AD neurons and mediates APP phosphorylation on the Tyr682 residue, in turn altering APP trafficking and sorting 21 , 22 and these effects are completely prevented by the Src tyrosine kinase inhibitor PP2. 21 Specific investigation of saracatinib on AD subpopulations carrying Tyr682 APP phosphorylation may solve this problem and help to better select the responsive patients. Notably, the identification of specific molecular signatures and biomarkers may be useful to better select and stratify subpopulations of AD patients for an appropriate drug treatment. However, this approach, yet theoretical, has to be investigated to practically translate it from the bench to the bedside and, to date, no clinical data are available to substantiate this hypothesis.
4.2. Nilotinib
Nilotinib (Tasigna, AMN107, Novartis, Switzerland), a Bcr‐Abl tyrosine kinase inhibitor, was approved by EMA in 2007 and by the U.S. FDA in 2010 and authorized for the treatment of adults with Philadelphia chromosome positive chronic myeloid leukemia. 23 Nilotinib has been recently repurposed in a number of neurodegenerative diseases, such as PD, Lewy body dementia (LBD), and AD. The rationale for c‐Abl inhibition as treatment for neurodegenerative diseases relies on the hyperactivation of such kinase in human AD and PD brains, as well as in a variety of tauopathies. 24 , 25 , 26 , 27 Accordingly, Schlatterer et al. reported an increased activation of the tyrosine kinase c‐Abl both in in vivo and in vitro transgenic AD models. 28 Notably, the activation of c‐Abl signaling has been reported as a crucial event mediating the synaptic damage induced by Aβ. 29 The exposure of rat hippocampal neurons to 5 μM Aβ fibrils has been found to increase c‐Abl activity, thus inducing apoptosis through c‐Abl‐mediated p73 phosphorylation (Figure 1). 30 The neuronal death of hippocampal neurons exposed to Aβ fibrils was prevented by the treatment with the c‐Abl inhibitor STI571 (imatinib mesylate, Gleevec). Moreover, the intraperitoneal administration of STI571 has been shown to reduce rat cognitive impairment on spatial memory performance, induced by the bilateral hippocampal injection of 5 μM Aβ fibrils, and to ameliorate spatial learning and memory impairment in 11‐month‐old APP/PS1 transgenic mice. 31 Furthermore, the activation of c‐Abl by Aβ has been found not only to stimulate proapoptotic signaling pathway through p73, but also to promote tau phosphorylation, 29 by activating the tau kinase Cdk5 (cyclin‐dependent kinase 5) and by directly phosphorylating tau at tyrosine 394. 29 , 32 Notably, tau phosphorylated at tyrosine 394 has been shown to be present in pre‐tangle neurons in AD brains, supporting the hypothesis that c‐Abl may contribute to neurofibrillary tangle formation and to their associated cognitive deficits. 26 , 32 It can be speculated that nilotinib, via c‐Abl inhibition, may prevent Aβ‐driven apoptosis and neurodegeneration by reducing both the activation of c‐Abl/p73 proapoptotic signaling pathway and c‐Abl/Cdk5‐mediated tau phosphorylation, possibly preventing neurofibrillary tangle formation. However, Aβ‐driven effects on c‐Abl activity and its downstream intracellular pathways require further investigations.
Taken together, these findings suggest that c‐Abl abnormal activation may contribute to neuronal dysfunction and support the use of c‐Abl inhibitors as potential AD treatments.
On the basis of preclinical data, nilotinib has been considered for a clinical application. A randomized, double‐blind, and placebo‐controlled phase 2 study (NCT02947893) is currently evaluating the impact of low doses of nilotinib in 42 patients with mild to moderate AD and ended in February 2020. Safety has been assessed as primary endpoint based on the number of participants who experienced adverse effects or had abnormal laboratory values after 12 months of treatment, whereas cerebrospinal fluid (CSF) biomarkers (eg, levels of Aβ and tau), clinical outcomes, as well as target engagement and proof of mechanism (c‐Abl inhibition) have been evaluated as secondary endpoints. Despite the strong limitations related to the study design, some preliminary results came from Pagan etal. open label pilot study, enrolling only 12 participants, that evaluated the safety and tolerability of nilotinib in patients with advanced PD with dementia or LBD, exposed to once daily oral dose of nilotinib for 6 months. 33 Nilotinib has been reported to be safe and well tolerated, to penetrate the blood brain barrier (BBB), as well as to significantly reduce CSF total tau and p‐tau. 33 Moreover, positive trends for cognitive improvement, measured by MMSE and the Scales for Outcomes in Parkinson's Disease‐Cognition, were observed. 33 In addition, c‐Abl target engagement was demonstrated, with an observed 30% reduction in c‐Abl activation. 33 Such decrease in c‐Abl phosphorylation may account, at least in part, for the observed reduction in CSF p‐tau. Beyond c‐Abl inhibition, however, nilotinib showed the capability to interfere with other signaling pathways. In particular, in a variety of lines expressing oncogenic RAS, nilotinib has been found to possess the spectrum of weak RAF inhibitor, and to lead to the formation of BRAF: CRAF dimers, thus stimulating paradoxical activation of the pathway. 9 Moreover, in a recent phase 2 placebo‐controlled randomized clinical trial testing the safety and tolerability of nilotinib in 75 patients with PD, doses of 150 or 300 mg nilotinib have been found reasonably safe, although serious adverse effects (eg, cardiovascular, gastrointestinal, renal, neurological, pulmonary) have been observed in 24% and 48% of the nilotinib‐150 mg and nilotinib‐300 mg groups, respectively, compared to 16% of the placebo group. 34 However, further larger and long‐term studies are required to assess the safety and tolerability of nilotinib in PD patients. In addition, nilotinib‐150 mg, but not nilotinib‐300 mg, treatment has been shown to significantly reduce the levels of oligomeric α‐synuclein, with no change of CSF total α‐synuclein at 12 months. 34 This result is consistent with previous findings reporting a higher reduction of α‐synuclein levels upon treatment with lower dose of nilotinib (1 mg/kg) compared to higher dose (10 mg/kg) in animal models of α‐synucleinopathies. 35
5. TIDEGLUSIB
Tideglusib (NP‐12, NP031112), a selective non‐ATP competitive GSK‐3 inhibitor, is repurposed for the treatment of AD. GSK‐3 represents a therapeutic node at the intersection of multiple disorders, ranging from cancer to neurodegenerative disorders. According to “the GSK‐3 hypothesis” of AD postulated by Hooper et al., the overactivation of GSK‐3β accounts for cognitive impairment, tau hyperphosphorylation, increased Aβ production, and neuronal death in AD. 36 Tideglusib has been reported to reduce a range of disease markers (Figure 1), including tau hyperphosphorylation, amyloid deposition, neuron loss, and gliosis in mouse entorhinal cortex and hippocampus, and to reverse a spatial memory deficit in AD transgenic mice. 37 , 38 , 39 Furthermore, GSK‐3β inhibition has been shown to reduce Aβ production and to ameliorate the AD‐like neuropathology and behavioral deficits in the water maze in hAPP transgenic mice. 40
A pilot, double‐blind, randomized phase II trial (NCT00948259) evaluated the safety and efficacy of tideglusib in 30 patients with mild to moderate AD, reporting good tolerability and positive trends for cognitive benefits in MMSE, ADAS‐cog, GDS (Geriatric Depression Scale), and GCA (Global Clinical Assessment). 41 A subsequent double‐blind, randomized, placebo‐controlled phase II trial (NCT01350362), testing the efficacy of tideglusib in a cohort of 306 mild to moderate AD patients, reported to have missed its primary cognitive endpoint. 42 Recently, Matsunaga et al. proposed a systematic review and meta‐analysis of randomized controlled trials testing the efficacy and safety of GSK‐3 inhibitors in mild cognitive impairment and AD patients. 43 Among the five trials included in study, no significant differences in cognitive function scores between GSK‐3 inhibitors and placebo groups were observed, further corroborating data demonstrating the ineffectiveness of such a therapeutic approach. 43 A better focused analysis might be useful to understand whether a marker, a subgroup of patients, and/or other different parameters may refine the effectiveness of such a therapeutic approach.
6. KINASE INHIBITORS TO COUNTERACT NEUROINFLAMMATION
6.1. Neflamapimod
Neflamapimod (previously code‐named VX‐745) is an oral selective small molecule initially tested for rheumatoid arthritis and then repurposed as a disease‐modifying drug for AD. 44 It is classified as an inhibitor of the intracellular signal transduction enzyme p38 MAPKα (p38α), a key modulator of microglia regulation and neuroinflammation (Figure 1). 45 Indeed, p38α is expressed in microglia where it mediates inflammatory responses stimulating the release of pro‐inflammatory cytokines such as tumor necrosis factor α (TNFα) and Interleukin‐1β (IL‐1β) 46 , and in neurons where it modulates memory formation through effects on long‐term potentiation (LTP)/depression. 47 Moreover, neuronal p38α has been implicated in tau phosphorylation 48 and in Aβ oligomer‐induced neurotoxicity, 49 and its role has been investigated as a therapeutic target for neuroinflammatory conditions, including AD. 50 In AD transgenic models, p38α inhibition has been found to reverse Aβ induced synaptic dysfunction and loss, 51 , 52 and p38α gene knockout improves synaptic function and memory as well as reduces Aβ production in AD transgenic mice. 53 , 54
Preclinical studies demonstrated that neflamapimod improved performance in the Morris water maze test and significantly reduced hippocampal IL‐1β protein levels in cognitively impaired aged (20 to 22 months) rats. 55 Such effects appear to be independent because the behavioral improvement in the Morris water maze was evident at a lower dose than that required to reduce IL‐1β. 55 A blinded and placebo‐controlled Phase 2b study (REVERSE‐SD), enrolling 161 people with mild AD, compared a 6‐month course of neflamapimod group with placebo on change in total and delayed recall on the Hopkins Verbal Learning Test, Revised (HVLT‐R). 56 The REVERSE‐SD trial failed to meet its primary endpoint of improving episodic memory at the end of the study period, as measured by HVLT‐R and, secondarily, by the Wechsler Memory Scale (WMS) immediate and delayed recall (https://www.prnewswire.com/news-releases/eip-pharma-announces-clinical-trial-results-of-reverse-sd-a-phase-2b-study-of-neflamapimod-in-early-stage-alzheimers-disease-300953422.html). Notably, a pharmacokinetics‐pharmacodynamic analysis showed positive trends toward improvement in the HVLT‐R and WMS in patients with the highest plasma drug concentrations, suggesting that the clinical outcome may be dose‐dependent. Thus, the observed effects of neflamapimod on CSF biomarkers, associated with those on episodic memory in patients with the highest blood concentrations, highlight the need to further investigate neflamapimod at higher doses and for long‐term exposure. Such promising results are currently under investigation. In the REVERSE‐SD trial, neflamapimod met its secondary objectives of target engagement and proof‐of‐mechanism, demonstrating statistically significant reductions in the CSF biomarkers phospho‐tau and total tau. Moreover, the CSF levels of the postsynaptic protein neurogranin have been measured as biomarker of AD correlating with cognitive decline 57 , and a trend toward reduced CSF neurogranin has been reported. 56 Notably, the observed reduction in CSF phospho‐tau and total tau by neflamapimod provides the rationale for the extended application of neflamapimod to tauopathies, such as LBD. Consistently, the FDA recently granted fast track designation for the treatment of LBD to neflamapimod, which is currently being studied in separate Phase 2 trials in patients with LBD.
Another Phase 2 study (NCT03435861), enrolling 40 people with prodromal AD and with cerebral amyloidopathy (as measured by CSF analysis or amyloid PET), is currently monitoring brain inflammation in response to a 12‐week course of treatment with neflamapimod or placebo, by using the microglial activation tracer DPA‐714. Three DPA‐714 SUV (standard uptake value) measures accounting for microglia activation and neuroinflammation represent the primary outcome. Secondary outcomes span 35 different parameters ranging from neuropsychological assessments to blood and CSF markers of inflammation. The trial is expected to run until January 2021.
6.2. Masitinib
Originally approved for veterinary therapeutics for the treatment of mast cell tumors in dogs 58 , 59 , masitinib (also known as AB1010) is a tyrosine kinase inhibitor with a wide spectrum of targets, among which are c‐kit; platelet‐derived growth factor receptors; and, to a lesser extent, lymphocyte‐specific kinase (Lck), Lck/Yes‐related protein (Lyn), Fyn, and FAK (focal adhesion kinase) pathways. 58 By combined targeting of c‐Kit and Lyn, masitinib is particularly efficient in controlling the survival, differentiation, and degranulation of mast cells, thus indirectly controlling the release of proinflammatory and vasoactive mediators. 58 Promising results come from human clinical trials testing masitinib for the treatment of mastocytosis, 60 rheumatoid arthritis, 61 and asthma, 62 and as add‐on therapy to riluzole in patients with amyotrophic lateral sclerosis. 63 Recently, the potential clinical application of masitinib in neurodegenerative diseases, such as AD, has emerged due to the involvement of the stem cell factor (SCF, the c‐kit natural agonist) receptor/c‐kit in mast cells‐mediated neuroinflammation (Figure 1). A randomized, placebo‐controlled, phase 2 study (NCT00976118) was performed in patients with mild to moderate AD, receiving masitinib as an adjunct to cholinesterase inhibitor and/or memantine. 64 Compared to placebo, masitinib administration as an adjunct to standard treatments slowed the rate of cognitive decline at 24 weeks, as evident from the sustained and statistically significant response in ADAS‐Cog. 64 Moreover, significant improvement in cognitive function and functional capacity was evident through the mean change in ADAS‐Cog, MMSE, and ADCS‐ADL values relative to baseline. 64 However, the weaknesses of this study concern the small sample size, with only 34 participants, and the high rate of discontinuation, with 17 dropouts (65.4%) in the group receiving masitinib versus 2 dropouts in the placebo group (25.0%). Moreover, the effective penetration of the BBB by masitinib was not assessed and, consequently, the mechanism underlying the positive cognitive outcome remains to be fully elucidated. One can speculate that, in the event that masitinib passes through BBB and accumulates to a sufficient high therapeutic concentration, it can reduce neuroinflammation by directly modulating microglial activity via disruption of SCF/c‐Kit signaling pathway. 58 Moreover, masitinib may also reduce tau phosphorylation via targeting Fyn and FAK intracellular pathway, thus providing a dual benefit in AD.
Currently, masitinib is under phase 3 clinical trial (NCT01872598) to test its efficacy and safety as add‐on therapy in patients with mild to moderate AD treated for a minimum of 6 months with a stable dose of cholinesterase inhibitor and/or memantine. As reported by the investor communication, the interim results showed positive trends of masitinib efficacy at one of the doses tested. However, the status of the trial is currently unknown.
7. CONCLUDING REMARKS
Among the drug discovery programs currently testing disease‐modifying strategies in AD, the field of protein kinase inhibition is emerging as a challenging area, with several molecules originally developed for oncological indications recently repurposed for the treatment of neurodegenerative diseases (Table 1). Notably, protein kinases represent key nodes at the intersection of multiple intracellular pathways, also acting as critical regulators of divergent signaling cascades. As well as in cancer, where mutated cells have to be counteracted, in neurodegenerative diseases the target is represented by dysfunctional cells. In both conditions, a common dysfunctional process is represented by an imbalance in the intracellular pathways regulating cell metabolism control and duplication, the inhibition of which may hence contribute to stop the disease progression (Figure 2). Therefore, kinases may represent modulable druggable targets in neurodegenerative diseases (Figure 1). However, despite promising preclinical results obtained in animal models, all the clinical trials testing kinase inhibitors in AD have ended in failure, with only few potential and still unconfirmed positive trends, further indicating that animal models cannot completely recapitulate the complexity of human biology and this is especially evident in the context of neurodegenerative diseases. The discouraging results may be justified by the fact that still few attempts have been made and few therapeutic strategies have been so far explored. In particular, addressing a single target and its related signaling pathway may not be an appropriate therapeutic strategy for AD, whose etiology is complex and multifactorial. The recognition of AD complexity suggests that using either single drugs binding multiple protein kinases or cocktails of highly selective inhibitors might be more effective, pending the assessment of their tolerability by frail elderly patients. The toxicity burden associated to kinase inhibitors and, in particular, substantial side effects due to off‐target effects (eg, cardiovascular, gastrointestinal, and hematologic toxicity) cannot be neglected. 65
FIGURE 2.
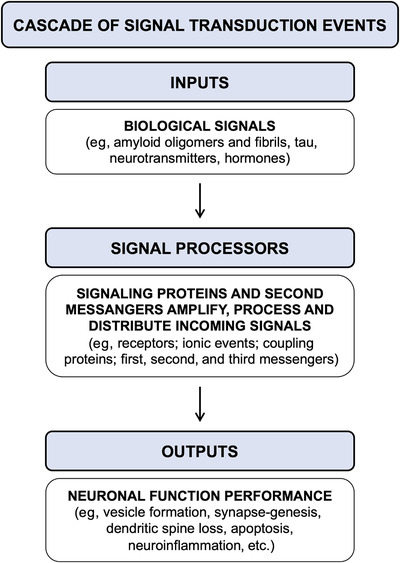
Cascade of signal transduction events: a schematic representation. Several biological signals, such as amyloid oligomers and fibrils, tau, neurotransmitters as well as hormones, trigger the activation of signaling proteins and cellular messengers that amplify, process, and distribute the incoming signals. The activation of intracellular signal processors induces neuronal responses, including vesicles trafficking, synapse‐genesis, dendritic spine loss, apoptosis, and neuroinflammation, as outputs affecting neuronal performance.
Moreover, another major weakness related to the field of kinase inhibition in AD is that most of the preclinical studies testing kinase inhibitors in AD‐like models investigated their impact on Aβ‐centered pathways. However, this vision is too limited and, considering the failures of the anti‐amyloid strategies, including the DIAN‐TU trial on familial cases, may not be optimal in addition to be limited to AD among the neurodegenerative diseases (1; https://www.reuters.com/article/us-roche-alzheimers/roche-lilly-drugs-fail-to-halt-gene-driven-alzheimers-disease-idUSKBN2040JQ). However, as well as in cancer, in neurodegeneration it is important to target the drug to the dysfunctional cells and to differentiate them from the healthy ones. A differential mapping of the kinases is fundamental to selectively identify the right target in the affected tissue depending on the disease to be treated. To date, it cannot be discounted that we still have a partial knowledge regarding the functions of protein kinases in the major signaling pathways in neurodegenerative processes and several key questions have yet to be addressed (Box 1). In particular, an accurate profile of degenerative modifications of protein kinase expression and activity in different brain areas, associated with aging and neurodegenerative processes, is still lacking. To this end, the recent advancements in proteomic technologies will facilitate a detailed profiling of the human brain kinome.
BOX 1. Emerging key questions while studying cancer kinase role in neurodegenerative diseases.
1.
|
|

CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
Fagiani F, Lanni C, Racchi M, Govoni S. Targeting dementias through cancer kinases inhibition. Alzheimer's Dement. 2020;6:e12044 10.1002/trc2.12044
Funding information
This work was supported by the University of Pavia (grant from FR&G 2019, Fondo Ricerca & Giovani) to CL.
Francesca Fagiani and Cristina Lanni contributed equally to the study.
REFERENCES
- 1. Bateman RJ, Benzinger TL, Berry S, et al. The DIAN‐TU next generation Alzheimer's prevention trial: adaptive design and disease progression model. Alzheimer's Dement. 2017;13:8‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schneider L. A resurrection of aducanumab for Alzheimer's disease. Lancet Neurol. 2020;19:111‐112. [DOI] [PubMed] [Google Scholar]
- 3. Houck AL, Seddighi S, Driver JA. At the crossroads between neurodegeneration and cancer: a review of overlapping biology and its implications. Curr Aging Sci. 2018;11:77‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klaeger S, Heinzlmeir S, Wilhelm M, et al. The target landscape of clinical kinase drugs. Science. 2017;358(6367):eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu P, Nielsen TE, Clausen MH. FDA‐approved small‐molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422‐439. [DOI] [PubMed] [Google Scholar]
- 6. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17:353‐377. [DOI] [PubMed] [Google Scholar]
- 7. Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127‐132. [DOI] [PubMed] [Google Scholar]
- 8. Hall‐Jackson CA, Eyers PA, Cohen P, et al. Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol. 1999;6:559‐568. [DOI] [PubMed] [Google Scholar]
- 9. Packer LM, Rana S, Hayward R, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug‐resistant chronic myeloid leukemia. Cancer Cell. 2011;20:715‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hennequin LF, Allen J, Breed J, et al. N‐(5‐chloro‐1,3‐benzodioxol‐4‐yl)‐7‐[2‐(4‐ methylpiperazin‐1‐yl)ethoxy]‐5‐ (tetrahydro‐2H‐pyran‐4‐yloxy)quinazolin‐4‐amine, a novel, highly selective, orally available, dual‐specific c‐Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465‐6488. [DOI] [PubMed] [Google Scholar]
- 11. Um JW, Nygaard HB, Heiss JK, et al. Alzheimer amyloid‐beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15:1227‐1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Larson M, Sherman MA, Amar F, et al. The complex PrPc‐Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer's disease. J Neurosci. 2012;32:16857‐16871. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13. Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid‐β (Aβ) oligomers: role of N‐terminal residues. J Biol Chem. 2010;285:26377‐26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Um JW, Kaufman AC, Kostylev M, et al. Metabotropic glutamate receptor 5 is a coreceptor for alzheimer Aβ oligomer bound to cellular prion protein. Neuron. 2013;79:887‐902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salter MW, Kalia L V. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317‐328. [DOI] [PubMed] [Google Scholar]
- 16. Bhaskar K, Hobbs GA, Yen SH, Lee G. Tyrosine phosphorylation of tau accompanies disease progression in transgenic mouse models of tauopathy. Neuropathol Appl Neurobiol. 2010;36:462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhaskar K, Yen SH, Lee G. Disease‐related modifications in tau affect the interaction between Fyn and tau. J Biol Chem. 2005;280:35119‐35125. [DOI] [PubMed] [Google Scholar]
- 18. Lee G, Thangavel R, Sharma VM, et al. Phosphorylation of tau by Fyn: implications for Alzheimer's disease. J Neurosci. 2004;24:2304‐2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaufman AC, Salazar S V, Haas LT, et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. 2015;77:953‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Dyck CH, Nygaard HB, Chen K, et al. Effect of AZD0530 on cerebral metabolic decline in Alzheimer disease. JAMA Neurol. 2019;76:1219‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poulsen ET, Iannuzzi F, Rasmussen HF, et al. An aberrant phosphorylation of amyloid precursor protein tyrosine regulates its trafficking and the binding to the clathrin endocytic complex in neural stem cells of Alzheimer's disease patients. Front Mol Neurosci. 2017;10:59. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Hoe HS, Minami SS, Makarova A, et al. Fyn modulation of Dab1 effects on amyloid precursor protein and apoe receptor 2 processing. J Biol Chem. 2008;283:6288‐6299. [DOI] [PubMed] [Google Scholar]
- 23. Deremer DL, Ustun C, Natarajan K. Nilotinib: a second‐generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin Ther. 2008;30(11):1956‐75. [DOI] [PubMed] [Google Scholar]
- 24. Jing Z, Caltagarone J, Bowser R. Altered subcellular distribution of c‐Abl in Alzheimer's disease. J Alzheimer's Dis. 2009;17:409‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ko HS, Lee Y, Shin JH, et al. Phosphorylation by the c‐Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc Natl Acad Sci U S A. 2010;107:16691‐16696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tremblay MA, Acker CM, Davies P. Tau phosphorylated at tyrosine 394 is found in Alzheimer's disease tangles and can be a product of the Abl‐related kinase, Arg. Arg J Alzheimer's Dis. 2010;19:721‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Imam SZ, Zhou Q, Yamamoto A, et al. Novel regulation of Parkin function through c‐Abl‐Mediated tyrosine phosphorylation: implications for Parkinson's disease. J Neurosci. 2011;31:157‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schlatterer SD, Acker CM, Davies P. C‐Abl in neurodegenerative disease. J Mol Neurosci. 2011;45:445‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cancino GI, Perez de Arce K, Castro PU, Toledo EM, von Bernhardi R, Alvarez AR. C‐Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol Aging. 2011;32:1249‐1261. [DOI] [PubMed] [Google Scholar]
- 30. Alvarez AR, Sandoval PC, Leal NR, Castro PU, Kosik KS. Activation of the neuronal c‐Abl tyrosine kinase by amyloid‐beta‐peptide and reactive oxygen species. Neurobiol Dis. 2004;17:326‐336. [DOI] [PubMed] [Google Scholar]
- 31. Cancino GI, Toledo EM, Leal NR, et al. STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer's beta‐amyloid deposits. Brain. 2008;131:2425‐2442. [DOI] [PubMed] [Google Scholar]
- 32. Derkinderen P, Scales TME, Hanger DP, et al. Tyrosine 394 is phosphorylated in Alzheimer's paired helical filament tau and in fetal tau with c‐abl as the candidate tyrosine kinase. J Neurosci. 2005;25:6584‐6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pagan F, Hebron M, Valadez EH, et al. Nilotinib effects in Parkinson's disease and dementia with Lewy bodiess. J Parkinsons Dis. 2016;6:503‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pagan FL, Hebron ML, Wilmarth B, et al. Nilotinib effects on safety, tolerability, and potential biomarkers in Parkinson disease. JAMA Neurol. 2019;77:309‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hebron ML, Irina L, Paul O, Selby ST, Fernando P, Moussa CE‐H. Tyrosine kinase inhibition regulates early systemic immune changes and modulates the neuroimmune response in α‐Synucleinopathy. J Clin Cell Immunol. 2014;30:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433‐1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Serenó L, Coma M, Rodríguez M, et al. A novel GSK‐3β inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359‐367. [DOI] [PubMed] [Google Scholar]
- 38. Morales‐Garcia JA, Luna‐Medina R, Alonso‐Gil S, et al. Glycogen synthase kinase 3 inhibition promotes adult hippocampal neurogenesis in vitro and in vivo. ACS Chem Neurosci. 2012;3:963‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang H, Huang S, Yan K, et al. Tideglusib, a chemical inhibitor of GSK3β, attenuates hypoxic‐ischemic brain injury in neonatal mice. Biochim Biophys Acta ‐ Gen Subj. 2016;1860:2076‐2085. [DOI] [PubMed] [Google Scholar]
- 40. Rockenstein E, Torrance M, Adame A, et al. Neuroprotective effects of regulators of the glycogen synthase kinase‐3β signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27:1981‐1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Del Ser T, Steinwachs KC, Gertz HJ, et al. Treatment of Alzheimer's disease with the GSK‐3 inhibitor tideglusib: a pilot study. J Alzheimer's Dis. 2013;33:205‐215. [DOI] [PubMed] [Google Scholar]
- 42. Lovestone S, Boada M, Dubois B, et al. A phase II trial of tideglusib in Alzheimer's disease. J Alzheimer's Dis. 2015;45:75‐88. [DOI] [PubMed] [Google Scholar]
- 43. Matsunaga S, Fujishiro H, Takechi H. Efficacy and safety of glycogen synthase kinase 3 inhibitors for Alzheimer's disease: a systematic review and meta‐analysis. J Alzheimer's Dis. 2019;69:1031‐1039. [DOI] [PubMed] [Google Scholar]
- 44. Duffy JP, Harrington EM, Salituro FG, et al. The discovery of VX‐745: a novel and selective p38α kinase inhibitor. ACS Med Chem Lett. 2011;2:758‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ. Microglial p38α MAPK is a key regulator of proinflammatory cytokine up‐regulation induced by toll‐like receptor (TLR) ligands or beta‐amyloid (Aβ). J Neuroinflammation. 2011;8:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bachstetter AD, Van Eldik LJ. The p38 MAP kinase family as regulators of proinflammatory cytokine production in degenerative diseases of the CNS. Aging Dis. 2010;1:199‐211 [PMC free article] [PubMed] [Google Scholar]
- 47. Barrientos RM, Frank MG, Watkins LR, Maier SF. Aging‐related changes in neuroimmune‐endocrine function: implications for hippocampal‐dependent cognition. Horm Behav. 2012;62:219‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Y, Liu L, Barger SW, Griffin WST. Interleukin‐1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38‐MAPK pathway. J Neurosci. 2003;23:1605‐1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long‐term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B‐containing NMDA receptors. J Neurosci. 2011;31:6627‐6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Corrêa SAL, Eales KL. The role of p38 MAPK and its substrates in neuronal plasticity and neurodegenerative disease. J Signal Transduct. 2012;2012:649079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Watterson DM, Grum‐Tokars VL, Roy SM, et al. Development of novel in vivo chemical probes to address CNS protein kinase involvement in synaptic dysfunction. PLoS One. 2013;8:e66226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Munoz L, Ranaivo H, Roy SM, et al. A novel p38α MAPK inhibitor suppresses brain proinflammatory cytokine up‐regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation. 2007;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Colié S, Sarroca S, Palenzuela R, et al. Neuronal p38α mediates synaptic and cognitive dysfunction in an Alzheimer's mouse model by controlling β‐amyloid production. Sci Rep. 2017;7:45306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schnöder L, Hao W, Qin Y, et al. Deficiency of neuronal p38α MAPK attenuates amyloid pathology in Alzheimer disease mouse and cell models through facilitating lysosomal degradation of BACE1. J Biol Chem. 2016;291:2067‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alam JJ. Selective brain‐targeted antagonism of p38 MAPKβ reduces hippocampal IL‐1β levels and improves morris water maze performance in aged rats. J Alzheimer's Dis. 2015;48:219‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alam J, Blackburn K, Patrick D. Neflamapimod: clinical phase 2b‐Ready oral small molecule inhibitor of p38α to reverse synaptic dysfunction in early Alzheimer's disease. J Prev Alzheimer's Dis. 2017;4:273‐278. [DOI] [PubMed] [Google Scholar]
- 57. Wang L. Association of cerebrospinal fluid neurogranin with Alzheimer's disease. Aging Clin Exp Res. 2019;31:185‐191. [DOI] [PubMed] [Google Scholar]
- 58. Dubreuil P, Letard S, Ciufolini M, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One. 2009;4:e7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marech I, Patruno R, Zizzo N, et al. Masitinib (AB1010), from canine tumor model to human clinical development: where we are?. Crit Rev Oncol Hematol. 2014;91:98‐111. [DOI] [PubMed] [Google Scholar]
- 60. Lortholary O, Chandesris MO, Livideanu CB, et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo‐controlled, phase 3 study. Lancet. 2017;389:612‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tebib J, Mariette X, Bourgeois P, et al. Masitinib in the treatment of active rheumatoid arthritis: results of a multicentre, open‐label, dose‐ranging, phase 2a study. Arthritis Res Ther. 2009;11:R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Humbert M, De Blay F, Garcia G, et al. Masitinib, a c‐kit/PDGF receptor tyrosine kinase inhibitor, improves disease control in severe corticosteroid‐dependent asthmatics. Allergy Eur J Allergy Clin Immunol. 2009;64:1194‐1201. [DOI] [PubMed] [Google Scholar]
- 63. Mora JS, Genge A, Chio A, et al. Masitinib as an add‐on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph Lateral Scler Front Degener. 2019;7:1‐10. [DOI] [PubMed] [Google Scholar]
- 64. Piette F, Belmin J, Vincent H, et al. Masitinib as an adjunct therapy for mild‐to‐moderate Alzheimer's disease: a randomised, placebo‐controlled phase 2 trial. Alzheimer's Res Ther. 2011;3:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Caldemeyer L, Dugan M, Edwards J, Akard L. Long‐term side effects of tyrosine kinase inhibitors in chronic myeloid leukemia. Curr Hematol Malig Rep. 2016;11:71‐79. [DOI] [PubMed] [Google Scholar]