

Abstract
Objectives
One of the reasons as to why chimeric antigen receptors (CAR)‐T cell therapy for malignancies other than CD19‐ or BCMA‐positive tumors has yet to produce remarkable progress is the paucity of targetable antigens. NKp44 is only expressed by activated natural killer cells and detects a variety of transformed cells, while it reportedly does not react with normal tissues. The aim of this study is to develop CAR‐T cell that can target multiple types of tumor cells.
Methods
We created a series of novel CAR constructs in first‐generation (1G) and second‐generation (2G) CAR format with the extracellular immunoglobulin‐like domain of NKp44 (NKp44‐CAR).
Results
Transduction of the best 1G construct into human primary T cells led to specific cytotoxic effects and cytokine secretion upon encountering multiple types of neoplastic cells including AML, T‐ALL and childhood solid tumors. Replacement of the extracellular hinge domain of NKp44 with that of CD8α resulted in diminished CAR function. The 1G NKp44‐CAR‐T cells exhibited significantly better tumor control in long‐term co‐culture assays compared with activated NK cells, as well as with NK cells transduced with identical NKp44‐CAR. T cells transduced with the best 2G‐CAR construct with 4‐1BB co‐stimulatory domain proliferated at significantly higher levels upon single antigen exposure and showed significantly better tumor control compared with the 1G‐CAR and 2G‐CAR with CD28 co‐stimulatory domain.
Conclusions
NKp44‐based CAR endows T cells with NK cell‐like anti‐tumor specificity. The CAR gene created in this study will be useful for the development of novel gene‐modified T‐cell immunotherapy.
Keywords: acute myeloid leukaemia, chimeric antigen receptor, natural killer cell, NKp44, paediatric solid tumors, T‐cell acute lymphoblastic leukaemia
A challenge in the development of effective chimeric antigen receptor transduced T (CAR‐T) cell therapy is to identify an appropriate target on tumor cells. We created a series of novel CAR constructs with the extracellular immunoglobulin‐like domain of NKp44, one of the activating receptors expressed on activated natural killer cells. Human T cells transduced with optimised NKp44‐based CAR constructs exerted specific cytotoxic effects and cytokine secretion upon encountering multiple types of neoplastic cells, including those associated with acute myeloid leukaemia, T‐cell acute lymphoblastic leukaemia, osteosarcoma, rhabdomyosarcoma, Ewing sarcoma, neuroblastoma and glioblastoma cells.

Introduction
Recent progress in immunotherapy has generated unprecedented prospects in cancer treatment. Adoptive immunotherapy using chimeric antigen receptors (CARs) against lymphoid neoplasia has produced notable results. Anti‐CD19 CAR‐transduced T (CAR‐T) cell therapy yielded response rates of 70–90% in paediatric, adolescent and young adult patients with CD19‐positive relapsed or refractory B‐cell acute lymphoblastic leukaemia (ALL). 1 , 2 CAR‐T cells targeting B‐cell maturation antigen (BCMA) have strong cytotoxicity against multiple myeloma (MM) that was resistant to standard therapies. In fact, second‐generation CAR‐T cells targeting BCMA that had a CD28 co‐stimulatory domain yielded an 81% overall response rate and median event‐free survival (EFS) of 31 weeks in 16 MM patients. 3 Furthermore, anti‐BCMA CAR‐T cells equipped with a 4‐1BB co‐stimulatory domain achieved 85% response rates including 45% complete response and a median EFS rate of 11.8 months in 33 MM patients. 4 In contrast, CAR‐T cell therapy against acute myeloid leukaemia (AML) and solid tumors has yet to produce clinically meaningful outcomes, 5 , 6 although some important progress has been recently reported, such as GD2‐specific CAR‐T cells against paediatric neuroblastoma, 7 HER2‐specific CAR‐T cells against HER2‐positive sarcoma 8 and interleukin‐13 receptor alpha 2‐specific CAR‐T cells against disseminated glioblastoma. 9 CAR‐T cells targeting other molecules including carcinoembryonic antigen (CEA) and CD133 have been reported to be feasible, but significant tumor shrinkage has not yet been achieved. 10 , 11
One major obstacle in exploiting CAR‐T technology for the novel treatment of refractory solid tumors is a paucity of appropriate surface antigens expressed by tumor cells that can be safely targeted. Tumor‐associated antigens are usually expressed heterogeneously on the surface of solid tumors, and most are also expressed in healthy tissues at low levels. 6 Because of this nature, CAR‐T cells targeting solid tumors have a risk of severe complications that can be fatal. In fact, one patient who was infused with HER2‐specific third‐generation CAR‐T cells died of severe respiratory distress resulting from extraordinary activation of CAR‐T cells because of recognition of HER2 expressed in the lung at low levels. 12
Human natural killer (NK) cells have a major role in innate immunity, and unlike T cells, exert rapid and strong cytotoxicity against tumor cells and virus‐infected cells in the absence of specific immunisation. 13 NK cells can discriminate a wide variety of transformed cells from normal cells in various tissues. Activation of NK cells is well regulated by the integration of signals triggered by ligand binding to different activation and inhibitory receptors, and NK cells kill targets when activation signals are well above inhibitory signals. 14 NK cells express several activating receptors on their surface such as natural killer group 2 member D (NKG2D), activating killer immunoglobulin‐like receptors (KIR), NKp80, CD94/natural killer group 2 member C (NKG2C), DNAX accessory molecule‐1 (DNAM‐1) and natural cytotoxicity receptors (NCR). 15 NCRs consist of NKp30, NKp44 and NKp46, which have immunoglobulin‐like extracellular domains as a ligand‐binding domain. 16 The impact of NCRs on the cytotoxicity of NK cells against neoplastic cells is emphasised by reports demonstrating that deficient NCR expression in NK cells from patients with AML correlates with poor cytocidal function against AML cells and predicts poor patient outcome. 17 NKp44 is specifically expressed on activated NK cells and is linked to a homodimer of DNAX‐activating protein of 12kD (DAP12) adapter protein that contains a single immunoreceptor tyrosine‐based activation motif (ITAM). 16 , 18 Ligands for NKp44 have been reported to be expressed on the surface of a variety of cancer cells but are not expressed in normal tissues, except at very low levels in the lung and testis. 19 Infusion of autologous or allogeneic activated NK cells for patients with refractory or relapsed malignancies including leukaemia and solid tumors has been performed. However, serious damage to normal solid organs and haematological and immune systems has not been reported. 20 , 21 Adopting the extracellular domain of NKp44 as an antigen recognition site of CAR, therefore, will safely endow CAR‐transduced T cells with NK cell‐like specificity and function against a wide range of tumors.
We and others recently reported the construction of NKp44‐based CARs that could target multiple types of leukaemia and childhood solid tumors. 22 , 23 In this study, we aimed to create a series of novel CAR constructs that had an extracellular immunoglobulin‐like domain of NKp44 and extensively studied their functional diversities to develop a novel CAR‐T therapy against multiple types of haematological and solid tumors. We also compared the anti‐tumor effects of NKp44‐based CAR‐T cells with those of primary activated NK cells and NKp30‐based CAR‐T cells.
Results
We created several first‐generation NKp44‐based CARs. The CAR domains, except for the ligand‐binding domain, were derived from various components of human T cells and NK cells, such as NKp44, CD8α, CD28 and CD3ζ (Figure 1a). Transduction of wild‐type NCR2 (NKP44) gene did not induce NKp44 surface expression on T cells, while primary NK cells were able to express NKp44 protein on the cell surface, reflecting the absence (in T cells) and the presence (in NK cells) of adaptor protein DAP12 (WT in Figure 1b). Chimeric receptors consisting of wild‐type NKp44 followed by CD3ζ intracellular signalling domain did not show surface expression (1G‐a). However, interestingly, truncation of the NKp44 protein at the transmembrane domain led to strong expression on the cell surface in T cells (TR1). These observations clearly indicated that deletion of the intracellular domain of NKp44 (1G‐b to 1G‐f) is needed for chimeric receptor with NKp44 protein to be expressed on the surface in T cells in the absence of DAP12 expression. Although the association of NKp44 with DAP12 has been previously reported to occur at the transmembrane domain in NK cells, 24 our observations suggested that a site within NKp44 that is associated with DAP12 is located not only within the transmembrane domain but also within the intracellular domain or that another unknown mechanism operates in the pathophysiology of ectopic expression of NKp44 in T cells.
Figure 1.
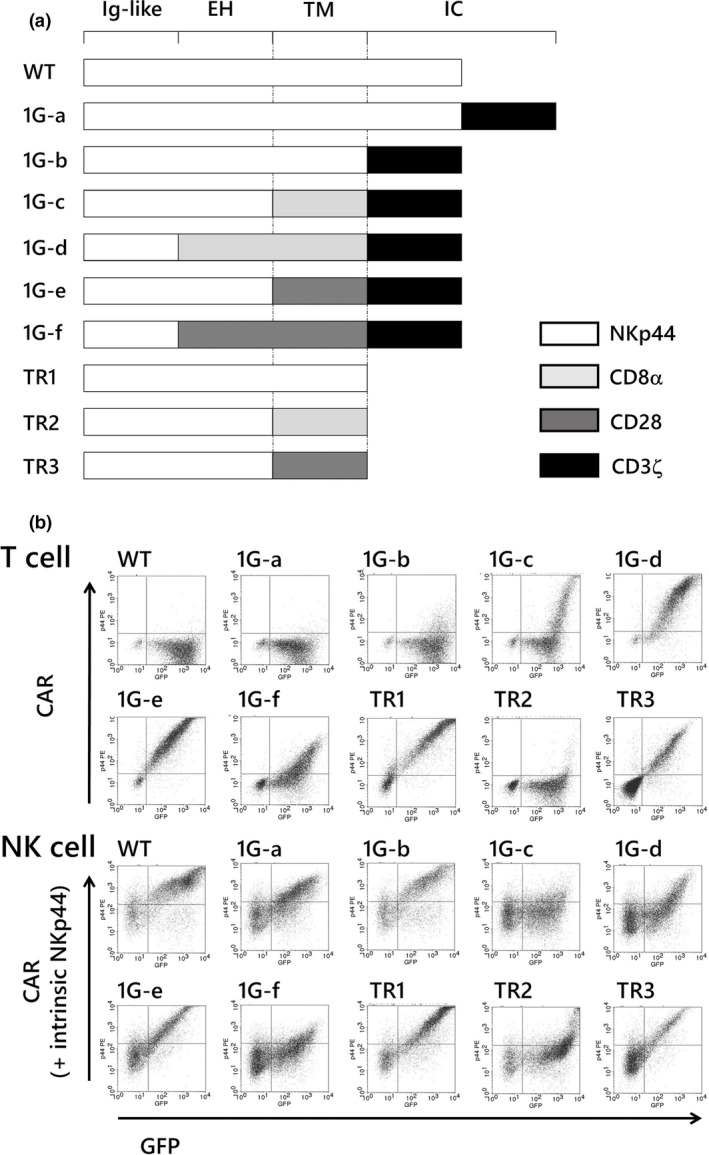
Gene constructs and surface expression on T and NK cells of first‐generation NKp44‐based CARs. (a) A series of first‐generation NKp44‐based CARs shared the ligand‐binding domain of NKp44. (b) Surface expression levels of the first‐generation NKp44‐based CARs in transduced T cells and NK cells are presented. The horizontal axis represents levels of GFP. The vertical axis represents the surface expression levels of NKp44‐based CAR demonstrated by PE‐conjugated anti‐NKp44 monoclonal antibody in transduced T cells. Same antibody detects NKp44‐based CAR expressed by transgene as well as intrinsic NKp44 in the transduced NK cells. Expression levels are much higher for NKp44‐based CAR (shown in the right upper quadrant) than for intrinsic NKp44 (the left upper and lower quadrant). The data are representative of at least three independent experiments using different peripheral blood donors. EH, extracellular hinge domain; IC, intracellular domain; Ig‐like, immunoglobulin‐like domain; TM, transmembrane domain.
When we added the CD3ζ intracellular signalling domain to ‘truncated NKp44 (TR1)’, the surface expression was severely disturbed (1G‐b). Replacement of the transmembrane domain of NKp44 with that of CD8α induced modest surface expression of the CAR (1G‐c). Replacement of both the hinge and transmembrane domains of NKp44 with those of CD8α caused an impressive increase in the surface expression levels in T cells (1G‐d). In contrast, replacement of the transmembrane domain of NKp44 alone with that of CD28 yielded the highest surface expression among this series of first‐generation CAR constructs created in this study, although replacement of both the hinge region and transmembrane domain of NKp44 with those of CD28 caused apparently inferior surface expression in both T cells and NK cells. A decrease in surface expression because of the introduction of the CD28 intracellular domain in a second‐generation CAR construct has been previously reported. 25 Our observations indicated that the CD28 hinge domain might also have deleterious effect on CAR expression on the cell surface, at least in NKp44‐based CAR. According to these results, we used a construct comprising the hinge domain of NKp44, the transmembrane domain of CD28 and the intracellular signalling domain of CD3ζ (Figure 1a, construct 1G‐e) in the subsequent experiments. NKp44‐based non‐signalling CARs were also constructed by replacing the transmembrane domain with that of CD8α or CD28. The former did not show surface expression; however, the latter showed moderate expression.
Surface expression of ligands for NKp44 has been reported in various tumor cell lines and primary tumor cells such as kidney and bladder cancer. 19 However, expression in paediatric solid tumors and sarcomas remains to be elucidated. Therefore, we examined the expression of ligands for NKp44 on the cell surface of various leukaemia and paediatric solid tumor cell lines using human NKp44‐Fc chimera protein. The ligands for NKp44 were detected in various types of leukaemia, paediatric solid tumor, sarcoma and brain tumor cells examined in this study. High expression of ligands was especially observed on the surface of solid tumor cell lines (Figure 2). Conversely, T cells, B cells, NK cells and monocytes obtained from healthy adults were negative for the ligands, as previously reported by others. 19 These data, together with those from other studies, suggest that ligands for NKp44 are reasonable targets for CAR‐mediated immune effector cell therapy against a wide range of malignant tumors in children, adolescents and adults. 19 , 26
Figure 2.
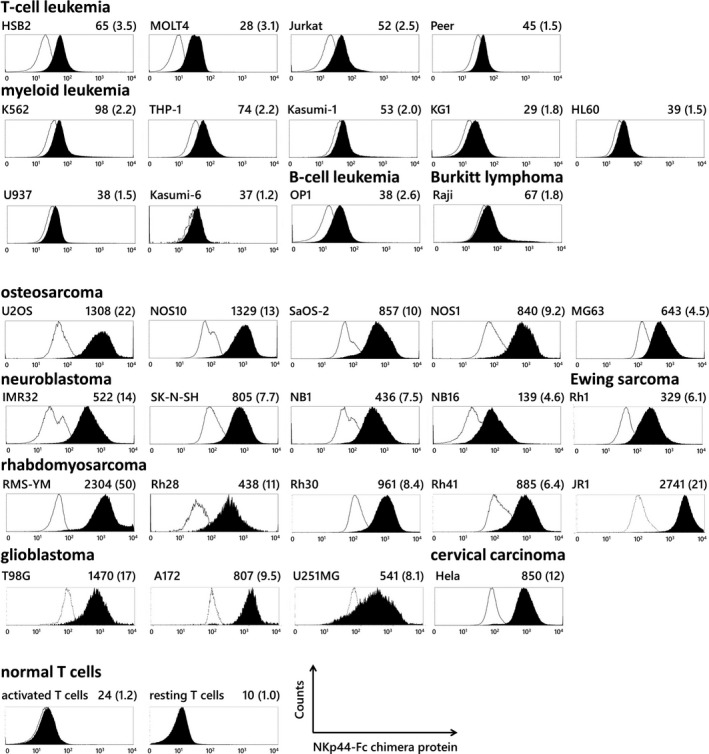
Surface expression of ligands for NKp44 in various leukaemia and solid tumor cell lines. Expression of NKp44 ligands was examined in a wide range of leukaemia cell lines and solid tumor cell lines. The ligands were not expressed by normal T cells in either resting or activated status. Each histogram shows the signal intensity of PE channels when stained with recombinant human NKp44 Fc chimera protein (filled area) or control Fc reagent (open area) followed by PE‐conjugated secondary antibodies. MFI of the filled histogram and the ratio of MFI of the filled/open histogram in parenthesis are presented at the top right of each histogram.
Primary T cells transduced with the first‐generation NKp44‐based CAR (construct 1G‐e) demonstrated significantly higher cytotoxic effects in a 4‐h cytotoxicity assay against various leukaemia cell lines including myeloid leukaemia (KG1, U937, THP‐1, K562) and T‐cell leukaemia (Jurkat, Peer, HSB2), and B‐cell leukaemia (OP‐1) than T cells transduced with NKp44 non‐signalling CARs (which binds to NKp44 ligands but lacks intracellular signals, TR1) (Figure 3a). The 1G‐e CAR‐T cells did not kill autologous or allogeneic T cells obtained from healthy adult volunteers (Figure 3b). The cytotoxic effect of the 1G‐e CAR‐T cells was significantly attenuated after blocking using an anti‐NKp44 monoclonal antibody (Figure 3c), confirming NKp44‐specific recognition. Although there are statistically significant differences in all comparison, a magnitude of blocking effects varied between the target cell lines, which might be a result of nature (e.g. target epitope) of blocking antibody used in this study or a result of the fact that there are multiple ligands for NKp44 receptor. The 1G‐e CAR‐T cells produced and secreted a large amount of interferon (IFN)‐γ following a single exposure to the target cells, whereas almost no IFN‐γ production was observed in T cells transduced with NKp44 non‐signalling CARs, as well as empty vector (mock) transduced T cells (Figure 3d and e). Further, the 1G‐e CAR‐T cells produced more cytotoxic granules after a single exposure to target cells than the T cells transduced with NKp44 non‐signalling CARs (Figure 3d and f).
Figure 3.
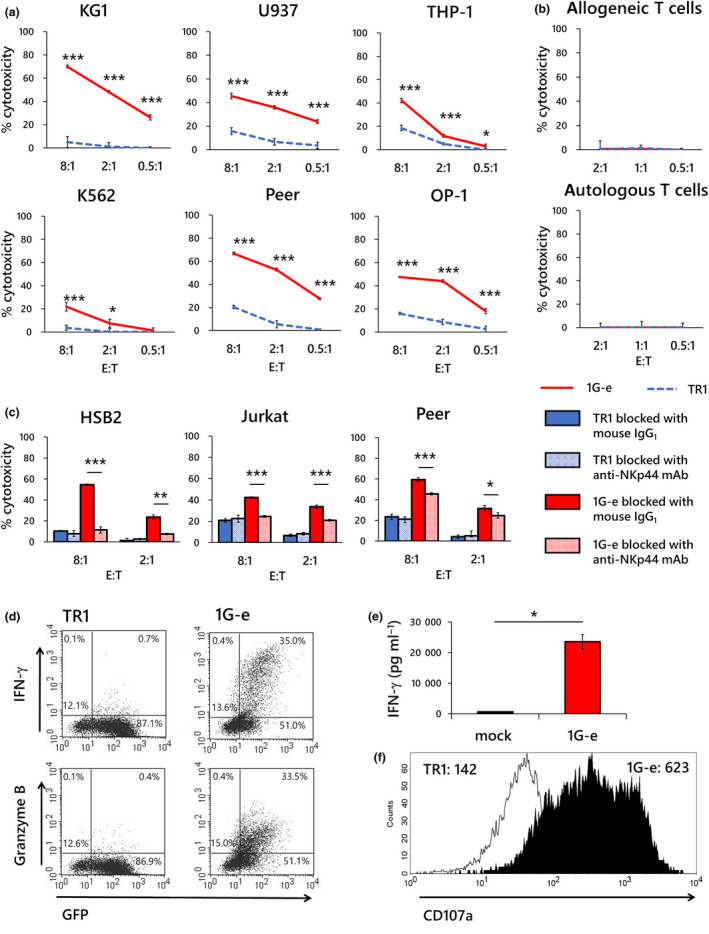
Specific response of NKp44‐based CAR‐transduced T cells to target exposure. (a) Specific cytotoxicity in a 4‐h killing assay against a variety of leukaemia cell lines was examined in first‐generation NKp44‐based CAR‐T cells (1G‐e; red solid line). T cells transduced with NKp44‐based CAR lacking signalling domain (TR1; blue dotted line) served as a control. The 1G‐e CAR‐T cells demonstrated significantly higher cytotoxic effects than T cells with non‐signalling CAR (TR1), which showed marginal or negligible cytotoxicity. (b) Neither the 1G‐e CAR‐T cells (red solid line) nor T cells with non‐signalling CAR (TR1; blue dotted line) showed cytotoxicity against autologous or allogeneic T cells. (c) A blocking assay was conducted in a 4‐h killing assay using anti‐NKp44 monoclonal antibodies or mouse IgG1, κ isotype control, with the 1G‐e CAR‐T cells and T cells with non‐signalling CAR (TR1), respectively. Cytotoxic effects were significantly attenuated after blocking with anti‐NKp44 monoclonal antibodies compared to the isotype control antibody in each assay. A blocking assay was conducted in a 4‐h killing assay using anti‐NKp44 monoclonal antibody, with T cells with NKp44‐based CAR (1G‐e) and T cells with non‐signalling control CAR, respectively. Cytotoxic effects were significantly attenuated after blocking in each assay. (d) The intracellular production of IFN‐γ and granzyme B was assessed using PE‐conjugated monoclonal antibodies and flow cytometry after 24‐h exposure to target cells (K562) in the 1G‐e CAR‐T cells and T cells with non‐signalling CAR (TR1). The NKp44‐based CAR‐T cells produced intracellular IFN‐γ and granzyme B, while non‐signalling control T cells did not. (e) Secretion of IFN‐γ was assessed by ELISA after 24‐h exposure to target cells (K562) in the 1G‐e CAR‐T cells (red square) and T cells transduced with empty vector (mock; open square). The 1G‐e CAR‐T cells produced significantly higher levels of IFN‐γ than mock‐transduced T cells. (f) Degranulation after exposure to target cells was assessed by CD107a mobilisation, which was detected using PE‐conjugated anti‐CD107a monoclonal antibodies and flow cytometry in the 1G‐e CAR‐T cells (filled area) and T cells with non‐signalling CAR (TR1; open area). The values of MFI are presented. NKp44‐based CAR‐T cells showed increased CD107a signal levels compared with the non‐signalling control. The experiments for a, b, c and e were performed in triplicates, and their results are shown as mean ± SD. The levels of statistical difference are shown using asterisks (*P < 0.05, **P < 0.01, ***P < 0.001). The data are representative of three independent experiments using different peripheral blood donors.
Next, we investigated the anti‐tumor effect of the NKp44‐based CAR‐T cells against various solid tumor and leukaemia cell lines in long‐term co‐culture with lower effector to target cell ratios. Control T cells transduced with NKp44 non‐signalling CAR (TR1) showed almost no reduction of tumor cells. In contrast, T cells transduced with the 1G‐e CAR exerted significant tumor cell reduction in all tumor cell lines, including osteosarcoma, neuroblastoma, Ewing sarcoma, rhabdomyosarcoma, glioblastoma, T‐cell ALL, AML and B‐cell ALL, whereas there were some inter‐individual variations (showing different levels of cytotoxicity among the three donors) (Figure 4a–e).
Figure 4.
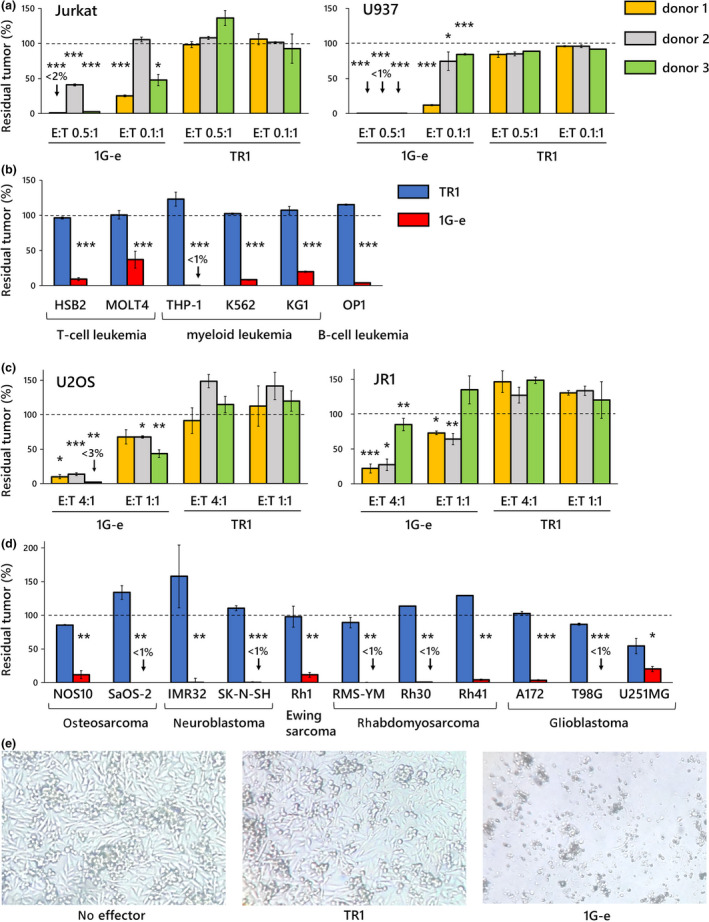
Long‐term anti‐tumor effects of first‐generation NKp44‐based CAR‐T cells against various tumor cells. (a) First‐generation NKp44‐based CAR‐T cells (1G‐e) were co‐cultured for 7 days with T‐cell leukaemia cells (Jurkat) and myeloid leukaemia cells (U937) at E:T ratios of 0.5:1 and 0.1:1. Although there were some inter‐individual variations showing different levels of cytotoxicity among the three donors, the 1G‐e CAR‐T cells showed significantly higher long‐term anti‐tumor effects (inhibition of tumor growth) than T cells with non‐signalling CAR (TR1), which induced no or essentially negligible inhibition of tumor growth. Statistical comparison was performed between 1G‐e and TR1 at the same E:T ratio in each donor. (b) The 1G‐e CAR‐T cells (red square) showed significantly higher anti‐tumor effects than T cells with non‐signalling CAR (TR1; blue square), in a 7‐day co‐culture assay (E:T ratios of 0.5:1) against various leukaemia cells. (c) The 1G‐e CAR‐T cells showed significantly higher anti‐tumor effects than T cells with non‐signalling CAR (TR1) in a 7‐day co‐culture assay against the osteosarcoma cell line (U2OS) and rhabdomyosarcoma (JR1) at E:T ratios of 4:1 and 1:1; among the samples from the three donors. Some inter‐individual variation was observed. (d) The 1G‐e CAR‐T cells (red square) showed significantly higher anti‐tumor effects in a 7‐day co‐culture assay against various solid tumor cells than T cells with non‐signalling CAR (TR1; blue square) at an E:T ratio of 4:1.The y‐axis in a–d shows percentage of residual tumors (as compared with tumor cells without effector cells). Data are mean ± SD of triplicate experiments. The data are representative of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. (e) Rhabdomyosarcoma cell line Rh30 was co‐cultured for 7 days with the 1G‐e CAR‐T cells or T cells with non‐signalling CAR (TR1), or it was cultured without any effector cells. In the culture with 1G‐e CAR‐T cells, no viable tumor cells were observed via inverted microscopy.
The sialic acid moiety at the hinge domain of NKp44 has been reported to be involved in ligand binding and recognition. 16 Therefore, we investigated the impact of replacement of the hinge domain of NKp44‐based CAR on CAR function. We compared two types of NKp44‐based CAR‐T cells, ‘1G‐e’ harbouring the hinge of NKp44 versus ‘1G‐d’ harbouring the hinge of CD8α, respectively. Levels of surface expression of the CAR‐T cells were comparable. The CAR‐T cells with the original NKp44‐hinge domain (1G‐e) produced larger amounts of IFN‐γ after a single antigen stimulation than those with CD8α‐hinge domain (1G‐d) (Figure 5a). In a 4‐h short‐term assay, the cytotoxic effects of CAR‐T cells with the original NKp44‐hinge domain (1G‐e) were significantly higher than those of CAR‐T cells with CD8α‐hinge domain (1G‐d) (Figure 5b). We also compared the long‐term anti‐tumor effects of these two types of CAR‐T cells. CAR‐T cells with the original NKp44‐hinge domain (1G‐e) showed significantly stronger effects against various leukaemia and solid tumor cells than CAR‐T cells with CD8α‐hinge domain (1G‐d) (Figure 4c). These observations suggested that, as reported previously, the NKp44‐hinge domain has an important functional role for ligand binding and the resulting functions.
Figure 5.
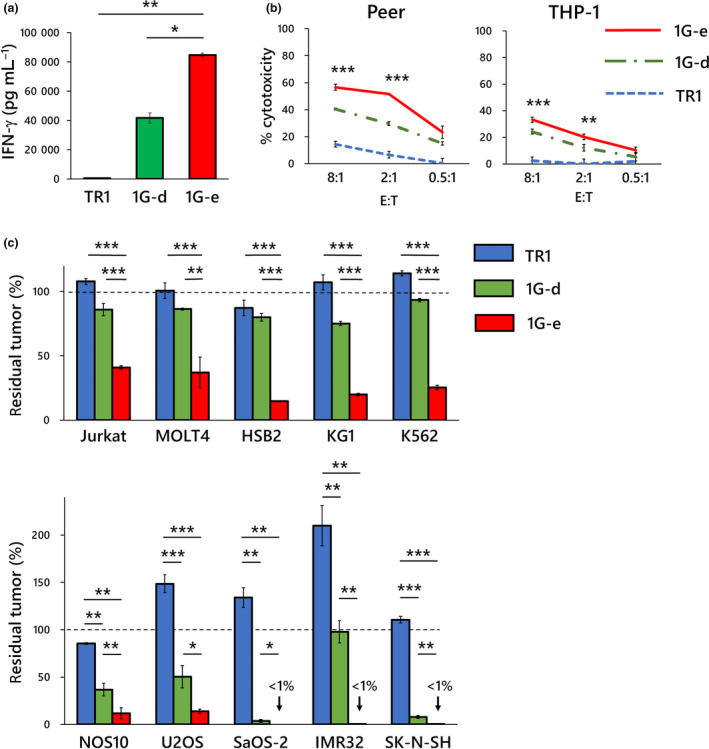
Hinge region of NKp44 is involved in ligand binding and recognition in NKp44‐based CAR. The impact of the hinge domain of NKp44‐based CAR on CAR function was examined. (a) NKp44‐based CAR‐T cells with alternative hinge domain (1G‐d; green square), in which NKp44‐original hinge was replaced by CD8α hinge, produced significantly lower levels of IFN‐γ than NKp44‐based CAR‐T cells with the original hinge (1G‐e; red square) after 24‐h exposure to target cells (KG‐1) as measured by cytometric beads array. In both CAR‐T cells used for the experiments, surface expression levels of CAR on gene‐modified T cells were similar (Figure 1b). (b) NKp44‐based CAR‐T cells with an alternative hinge domain (1G‐d; green chain line) showed inferior 4‐h cytotoxicity as compared with NKp44‐based CAR‐T cells with the original hinge (1G‐e; red solid line). As shown by the asterisks, statistical comparisons between 1G‐d and 1G‐e CAR‐T cells in both cell lines at E:T ratio of 8:1 and 2:1 were significantly different. (c) NKp44‐based CAR‐T cells with the alternative hinge domain (1G‐d; green square) showed significantly lower long‐term anti‐tumor effects in 7 days of co‐culture assay, as compared to NKp44‐based CAR‐T cells with the original hinge (1G‐e; red square) against various leukaemia (at an E:T ratio of 0.5:1; upper graph) and solid tumor cell lines (at an E:T ratio of 4:1). The y‐axis shows percentage of residual tumors (as compared with tumor cells without effector cells). These observations suggest an important role of the original NKp44 hinge domain in CAR function including antigen recognition and signalling. Data are means ± SD of three technical replicates. Experiments were independently repeated at least twice, and representative data are shown. *P < 0.05, **P < 0.01, ***P < 0.001.
We expected that primary T cells were able to exert NK cell‐like anti‐tumor activity when they were transduced with NKp44‐based CAR. NK cells can exert potent cytotoxicity against a variety of transformed cells by sensing several cancer‐associated or genostress‐induced ligands with an array of activating receptors including NKG2D, NKG2C, activating KIRs and NCRs. However, CAR‐T cells use a single CAR to sense the tumor cells. Thus, we wanted to explore how the anti‐tumor activities varied between NKp44‐based CAR‐T cells and activated NK cells. For this purpose, we used primary NK cells that had been ex vivo‐expanded and activated by K562‐mb15‐41BBL. 27 Indeed, activated NK cells exerted significantly more potent cytotoxic effects against leukaemic cells in a 4‐h short‐term cytotoxicity assay than NKp44‐based CAR‐T cells (1G‐e) (Figure 6a). However, in sharp contrast, NKp44‐based CAR‐T cells showed significantly better anti‐tumor effects against the identical tumor cells including leukaemia and solid tumors in prolonged co‐cultures (7 days) than activated NK cells (Figure 6b).
Figure 6.
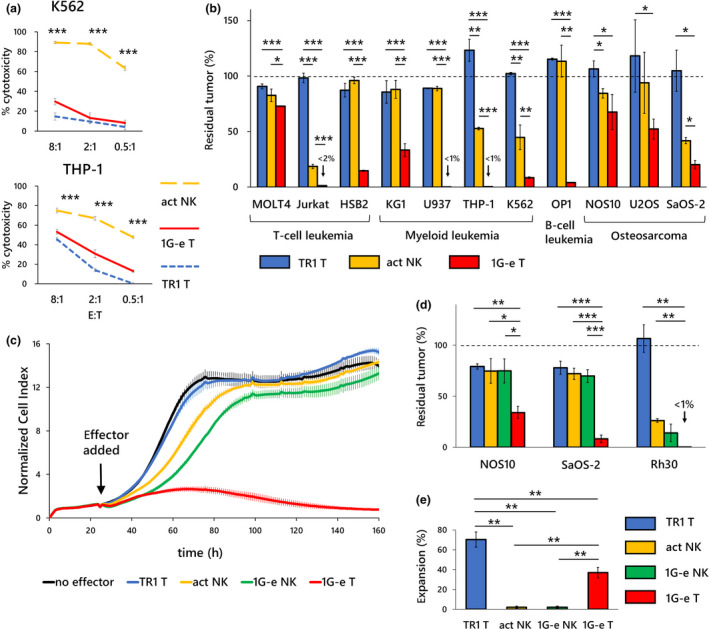
Comparisons with activated primary NK cells and NKp44‐based CAR‐NK cells. (a) Primary NK cells that were ex vivo‐activated by co‐culture with K562‐mb15‐41BBL (act NK; orange dashed line) showed significantly higher cytotoxicity against K562 and THP‐1 cells in a 4‐h killing assay than first‐generation NKp44‐based CAR‐T cells (1G‐e T: solid line). T cells with non‐signalling CAR (TR1 T; blue dashed line) served as control, although non‐specific effects at the E:T ratio of 8:1 are seen against THP‐1 cells. (b) On the contrary, in a long‐term (7 days) co‐culture assay, 1G‐e CAR‐T cells (1G‐e T; red square) exerted, in most of the cell lines, significantly stronger inhibitory effects on tumor growth than activated NK cells (act NK; orange square). Activated NK cells showed modest but significantly higher anti‐tumor effects than T cells with non‐signalling CAR (TR1 T; blue square) in only 4 of 11 cell lines. E:T ratios for leukaemia cell lines and solid tumor cell lines were 0.5:1 and 4:1, respectively. (c) In a long‐term cytotoxicity assay using iCELLigence real‐time cell analyser, both 1G‐e CAR‐NK cells (1G‐e NK; green line) and non‐transduced NK cells (act NK; orange line) showed enhanced inhibitory effects on tumor growth in the target cells (NOS10), as compared to T cells with non‐signalling CAR (TR1) or no effector cells, in the first 80 h. CAR‐NK showed more enhanced cytotoxicity than non‐transduced NK cells. However, the differences in inhibitory effects among CAR‐NK, non‐transduced NK and control T cells became negligible in the later stage (80‐160 h). In contrast, the 1G‐e CAR‐T cells (1G‐e T; red line) were able to continuously kill the tumor cells and completely abrogate tumor cell growth. (d) The 1G‐e CAR‐T cells (1G‐e T; red square) showed significantly higher anti‐tumor effects against osteosarcoma (NOS10 and SaOS‐2) and rhabdomyosarcoma (Rh30) cells than the 1G‐e CAR‐NK (1G‐e NK; green square) and non‐transduced NK (act NK; orange square) cells, as measured by the WST‐8 assay at an E:T ratio of 4:1. In this assay, inhibitory effects on tumor growth were not statistically different between CAR‐NK and non‐transduced NK cells. (e) Effector cell expansion (recovery) after 7 days of co‐culture with tumor cells was investigated to see whether a difference in persistence of effector cells might explain the difference in long‐term anti‐tumor effects. The 1G‐e CAR‐T cells (1G‐e T; red square) showed a significantly prolonged survival, after a single exposure to the target cells (50Gy‐irradiated THP‐1), as compared to 1G‐e CAR‐NK cells (1G‐e NK; green square) or non‐transduced NK cells (act NK; orange square). TR1 CAR‐T cells served as control, which indicated more survival than 1G‐e CAR‐T cells because of lack of activation‐induced cell death. Data are means ± SD of three technical replicates. Experiments were independently repeated at least twice, and representative data are shown. *P < 0.05, **P < 0.01, ***P < 0.001.
Next, long‐term anti‐tumor effects of CAR‐T cells (1G‐e), activated NK cells and NK cells transduced with the identical CAR (CAR‐NK) were compared using a real‐time cell‐analyser iCELLigence, which provides non‐invasive electrical impedance monitoring to quantify cell proliferation in a real‐time manner. Transduction of CAR gene into human primary NK cells provides antigen‐specific cytotoxicity in addition to natural cytotoxicity, which was shown for the first time by the use of a novel method for ex vivo expansion and genetic modification utilising K562‐mb15‐41BBL feeder cells. 27 Activated NK cells suppressed the tumor cell growth, as compared with control T cells with non‐signalling CAR (TR1), in the early stage (up to 80 h) of co‐culture with the target cells (NOS10); after 100 h, there was no difference in the cell number index. CAR‐NK cells showed slightly more suppressive effects, as compared to NK cells without genetic modification, in the early stage; however, the difference of the cell number index between them became negligible after 100 h. In contrast, CAR‐T cells, although transduced with first‐generation CAR, continued to kill target cells and completely inhibited the proliferation of tumor cells (Figure 6c). Additionally, these results were confirmed by the conventional endpoint assay using WST‐8. CAR‐T cells (1G‐e) showed significantly increased anti‐tumor effects against osteosarcoma cells (NOS10 and SaOS‐2) and rhabdomyosarcoma cells (Rh30), as compared to activated NK cells with or without the identical CAR (1G‐e), upon co‐culturing for 7 days (Figure 6d). Finally, we observed a significantly higher persistence of effector cells, after 7 days of exposure to the target cells (50 Gy‐irradiated THP‐1), in CAR‐T cells (1G‐e) than in activated NK cells without genetic modification or CAR‐NK cells with the identical CAR (Figure 6e). These observations suggested that T cells transduced with first‐generation CARs (not second‐generation CARs) could persist longer and undertake more serial killing of target tumor cells than primary ex vivo‐activated NK cells or CAR‐NK cells, suggesting intrinsic differences in T cells and NK cells.
NKp30 belongs to the natural cytotoxicity receptor family and recognises B7‐H6 and BAT3 (BAG6), which are not recognised by NKp44. 26 T cells genetically modified with CARs harbouring the extracellular domain of NKp30 as a target recognition site have been reported. 28 We compared the performance of NKp44‐based CAR‐T cells and NKp30‐based CAR‐T cells. We first examined ligands for NKp30 and NKp44 in the solid tumor cell lines using the NKp30‐Fc or NKp44‐Fc reagents, respectively (Figure 7a). We considered tumor cells were positive for the surface NCR ligands if the MFI ratio (MFI of NKp30‐Fc or NKp44‐Fc/MFI of control Fc) was more than 2. While we observed surface expression of the NKp44 ligands in all cell lines tested, only six tumors (out of 21) were positive for NKp30 ligands. Then, we created a series of first‐generation CARs with an extracellular antigen recognition site and the hinge domain of NKp30. The NKp30‐based CAR used for this experiment had a similar structure to the 1G‐e construct of NKp44‐based CAR. Surface expression of the two different CARs was comparable when transduced into human T cells (Figure 7b). Indeed, when we compared the anti‐tumor effect in the long‐term co‐cultures, NKp44‐based CAR‐T cells (1G‐e) showed significantly superior cytotoxic effects than NKp30‐based CAR‐T cells against most of the solid tumor cells, such as osteosarcoma, rhabdomyosarcoma, Ewing sarcoma, neuroblastoma and glioblastoma (Figure 7c). Of note, only neuroblastoma cell line NB1 showed similar surface expression levels of NKp44 and NKp30. Against this cell line, both NKp44‐based CAR‐T cells and NKp30‐based CAR‐T cells exerted similar levels of inhibition against tumor cell proliferation, suggesting that cytotoxicity would be comparable if the ligands of each CAR were equally expressed in tumor cells.
Figure 7.
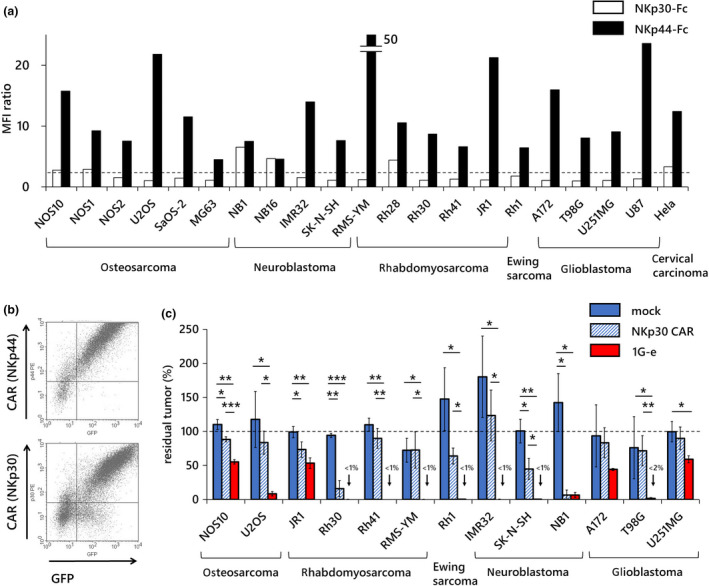
Comparisons with NKp30‐based CAR‐T cells. (a) Surface expression of ligands for NKp44 and NKp30 on various solid tumor cells was examined using NKp44‐Fc or NKp30‐Fc chimera protein, respectively, followed by staining with PE‐conjugated secondary antibodies. The vertical axis indicates the ratio of MFI of NKp44‐Fc/control Fc (filled square) or NKp30‐Fc/control Fc (open square). An MFI ratio of more than 2 was considered positive. While NKp44 was considered positive in 21 of 21 cell lines, NKp30 was considered positive in only six of 21 cell lines. (b) Surface expression levels of CAR in NKp44‐based CAR‐T cells (1G‐e; upper) and in NKp30‐based CAR‐T cells (lower) were similar. (c) NKp30‐based CAR‐T cells (NKp30 CAR; blue shaded square) showed no or only mild anti‐tumor effects in long‐term co‐culture assay at an E:T ratio of 1:1 against various solid tumor cell lines tested, except for a few cell lines (Rh30, SK‐N‐SH and NB1). NKp44‐based CAR‐T cells (1G‐e; red square) showed significantly stronger anti‐tumor effects than NKp30‐based CAR‐T cells against most cell lines. T cells transduced with an empty vector (mock; blue square) served as the control. The y‐axis shows the percentage of residual tumors (as compared with tumor cells without effector cells). Data are means ± SD of three technical replicates. Experiments were independently repeated at least twice, and representative data are shown. *P < 0.05, **P < 0.01, ***P < 0.001.
Finally, we created an array of second‐generation NKp44‐based CAR constructs, which incorporated an intracellular co‐stimulatory signalling domain derived from 4‐1BB (2G‐c, 2G‐d, 2G‐e, 2G‐f), and from CD28 (CD28 CAR: NKp44 immunoglobulin‐like and hinge domain, CD28 transmembrane and signalling domain, followed by CD3ζ signalling domain) that was reported by Eisenberg et al. 23 (Figure 8a). Surprisingly, introduction of this 4‐1BB signalling domain to the 1G‐c construct (2G‐c) apparently upregulated its surface expression, although the 1G‐c CAR showed modest surface expression. A CAR with the CD28 hinge, CD28 transmembrane domain, 4‐1BB signalling domains and CD3ζ (2G‐f) showed decreased surface expression in comparison with a CAR in which the hinge domain was replaced with that of NKp44 (2G‐e), indicating again that use of the CD28 hinge domain decreased the surface expression of the NKp44‐based CAR. Introduction of the 4‐1BB signalling domain to 1G‐e, the best construct among the first‐generation CARs, led to decreased surface expression (2G‐e). A CAR construct with a hinge and transmembrane domain derived from CD8α and an intracellular signalling domain from 4‐1BB and CD3ζ showed excellent surface expression on both human T cells and NK cells (2G‐d) (Figure 8b). T cells transduced with the 2G‐d construct showed significantly higher expansion rates in response to single antigen exposure than the truncated control T cells (TR1), first‐generation CAR‐T cells (1G‐e) or another version of second‐generation 4‐1BB CAR‐T cells (2G‐e), and the second‐generation CAR with the CD28 signalling domain (CD28 CAR) (Figure 8c). CAR‐T cells with the 2G‐d construct showed significantly increased cytotoxic effects against various paediatric solid tumor cell lines in 7‐day co‐culture assays compared with T cells transduced with non‐signalling CAR (TR1), first‐generation CAR‐T cells (1G‐e) and second‐generation CD28 CAR‐T cells (CD28 CAR) (Figure 8d).
Figure 8.
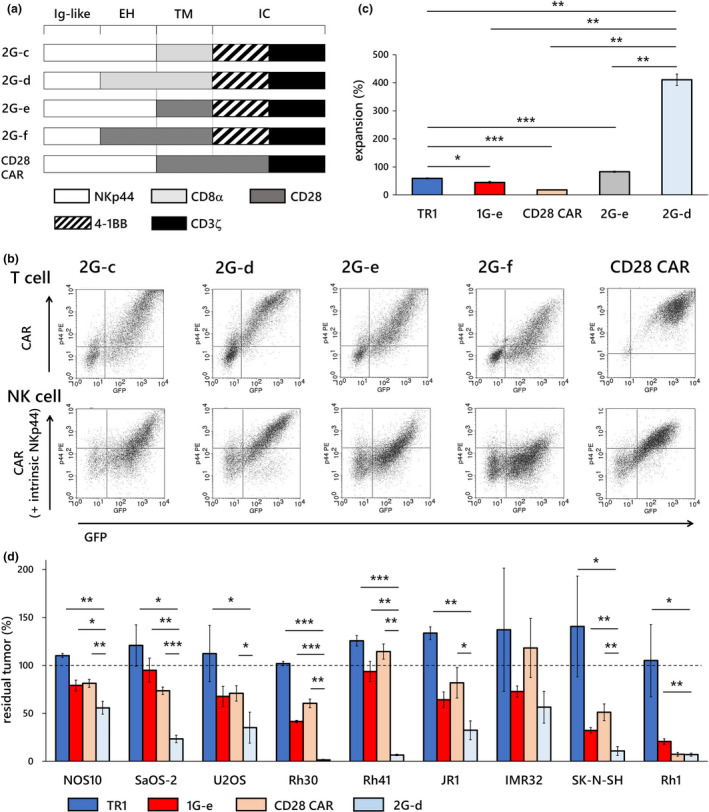
Second‐generation NKp44‐based CAR‐T cells with 4‐1BB co‐stimulation demonstrated excellent proliferation and enhanced anti‐tumor effects. (a) We created an array of second‐generation NKp44‐based CAR constructs that incorporated the 4‐1BB co‐stimulatory domain (2G‐c, 2G‐d, 2G‐e, 2G‐f). We also constructed NKp44‐based CAR with CD28 co‐stimulation that had been reported by Eisenberg et al. 23 (CD28 CAR). (b) Transduction of the CAR construct with CD8α hinge and transmembrane domain, 4‐1BB signalling domain, and CD3ζ (2G‐d) resulted in the highest surface expression on both T cells and NK cells among all the second‐generation 4‐1BB CARs generated. (c) T cells transduced with the construct 2G‐d (pale blue square) showed significantly higher expansion in response to target cell exposure than T cells with non‐signalling CAR (TR1; blue square), T cells transduced with first‐generation CAR (1G‐e; red square), other second‐generation CAR‐T cells with the 4‐1BB co‐stimulatory domain (2G‐e; grey square) and second‐generation CAR with the CD28 co‐stimulatory domain (CD28 CAR; beige square). (d) T cells transduced with our best construct (2G‐d; pale blue square) showed significantly stronger inhibitory effects in long‐term co‐culture assay at E:T ratios of 1:1 against various solid tumor cells than T cells (TR1; blue square), first‐generation NKp44‐based CAR‐T cells (1G‐e; red square) and second‐generation CD28 NKp44‐based CAR‐T cells (CD28 CAR; beige square). The y‐axis shows percentage of residual tumors (as compared with tumor cell without effector cells). Data are means ± SD of three technical replicates. Experiments were independently repeated at least three times, and representative data are shown. *P < 0.05, **P < 0.01, ***P < 0.001. EH, extracellular hinge domain; IC, intracellular domain; Ig‐like, immunoglobulin‐like domain; TM, transmembrane domain.
Discussion
In this study, we created several types of CAR that used an NKp44 immunoglobulin‐like domain as an antigen recognition site, and explored the best construct for novel gene‐modified T‐cell immunotherapy for multiple types of cancer, including AML, T‐cell ALL and lymphoma, and solid tumors, such as neuroblastoma, osteosarcoma, rhabdomyosarcoma, Ewing sarcoma and glioblastoma. Surface expression of the CARs was compared in human T cells and NK cells. Several patterns of hinge region and transmembrane domain combinations were tested in first‐ and second‐generation format because the substitution of these sites greatly affects the expression and function of CARs. 29 , 30 Investigation of the expression levels and function of different NKp44‐based CARs confirmed the importance of optimisation by changing the hinge domain and the transmembrane domain depending on the antigen recognition moiety. 6 Our optimal 4‐1BB‐based second‐generation construct was determined as that containing NKp44 immunoglobulin‐like domain, CD8α hinge, CD8α transmembrane, 4‐1BB signalling domain and CD3ζ signalling domain (construct 2G‐d).
Various types of ligands for NKp44 have been previously reported, 31 , 32 , 33 , 34 and new ligands are still being discovered, such as an isoform of the mixed‐lineage leukaemia (MLL)‐5 protein, 19 proliferating cell nuclear antigen, 35 platelet‐derived growth factor‐DD 36 and Nidogen‐1. 37 In particular, the MLL‐5 isoform has been demonstrated to be expressed on the surface of several cancer cell lines including cervical carcinoma, endothelial carcinoma and melanoma. Importantly, the MLL‐5 isoform was expressed on the surface of human primary tumors such as kidney cell carcinoma and bladder cell carcinoma. 19 We confirmed ligands for NKp44 were expressed on the surface of leukaemia and lymphoma cell lines and solid tumor cell lines of various origins. Indeed, NKp44‐based CAR conferred on human primary T cells, the ability to attack diverse tumor cell lines and produce IFN‐γ and granzyme B in response to antigen stimulation on target cells. However, we found that ligand for NKp44 was not expressed on the resting or activating T cells, and NKp44‐based CAR‐T cells did not attack autologous or allogeneic T cells (Figures 2 and 3b). Baychelier et al. 19 failed to detect the cellular ligand of NKp44, MLL‐5 truncated isoform, in any of the normal tissues such as brain, colon, heart, kidney, liver, muscle, placenta, intestine, spleen and stomach, except for very low expression in lung and testis. Based on these characteristics, NKp44‐based CAR‐T cells appeared to have the capacity to effectively and safely attack a wide range of leukaemia and solid tumors by use of a single CAR gene construct.
We assessed the anti‐tumor properties of activated NK cells and T cells transduced with first‐generation NKp44‐based CARs and NK cells with or without the identical CAR in short‐term, as well as long‐term, assays and found that the first‐generation CAR‐T cells were significantly better effector cells than ex vivo‐expanded activated NK cells with or without CAR in prolonged co‐cultures with tumor cells. NK cells are an attractive effector cell population for cancer therapy because they have a capacity to kill transformed cells of various origin with a favorable safety profile. Cancer therapy using autologous lymphokine‐activated killer cells (so‐called ‘LAK cells’), in which activated NK cells seemed to be representative for the capacity of killing cancer cells in vitro, have been explored in clinical trials against a variety of refractory cancers, but has rarely led to clinical success. 38 One major reason for the failure of autologous LAK cells to show clinical activity seems to have been caused by self‐HLA molecules that can transmit inhibitory signals in NK cells. 14 , 15 In this context, the use of allogeneic NK cells might be a better choice. In fact, infusion of allogeneic NK cells to treat refractory and/or relapsed AML in adults has been tested and showed transient clinical benefits in a subset of patients. 20 , 39 NK cells usually do not expand clonally as T cells do and may lead to activation‐induced cell death after killing of target cancer cells. Although T‐cell therapy transduced with first‐generation CARs did not show any clinical benefits, 40 , 41 the introduction of a co‐stimulatory receptor signalling domain into the CAR gene (second‐generation CARs) resulted in the unforeseen profound clinical success. In the successful CAR‐T trials with second‐generation CD19‐specific CARs, long‐term detection of the CAR‐T cells in the blood or bone marrow of the patients has been observed and correlated to the disease control, indicating the importance of persistence of the infused effector cells in the anti‐cancer cellular therapy. 1 , 2 Ex vivo‐expanded allogeneic NK cells, which had been activated and cultured with the use of an artificial feeder cell line (K562‐mb15‐41BBL), have been tested in patients with refractory and/or relapsed multiple myeloma in a phase I trial. Although safe, the responses observed and NK cell persistence after infusion have been modest. 21 Second‐generation CAR‐T cells with NK‐cell‐like specificity might show longer persistence and have a higher possibility of clinical benefits as compared with NK cell therapy, although activated NK cells appear to have better safety profiles than the CAR‐T cells, which could cause severe cardiopulmonary deterioration via profound cytokine release. Safety concerns might be addressed by the use of a suicide switch 42 and/or an artificial inhibitory receptor. 43 Conversely, for allogeneic NK cells to be clinically more effective, their persistence in vivo must be prolonged further. Unlike T cells, NK cells do not expand after recognising and killing target cells. Indeed, in the current study, we showed that genetic modification of NK cells with NKp44‐based CAR enhanced short‐term cytotoxicity but did not enhance the long‐term ability to suppress tumor growth, as compared with activated NK cells without CAR, suggesting that unlike T cells, CAR engagement does not ensure NK‐cell persistence. Infused allogeneic NK cells may require exogenous cytokine support (IL‐2 or IL‐15) or autologous expression of wild‐type or membrane‐bound IL‐15 for successful treatment of malignant tumors. 44 , 45 Recently, a phase I/II clinical trial of allogeneic cord blood NK cells, which had been stimulated with another K562‐based feeder cells and transduced with a lentiviral vector encoding using‐CD19 CAR, IL‐15 and a suicide gene iCAS9, has been conducted, targeting relapsed or refractory malignant lymphoma in adults. In this trial, 8 out of 11 patients treated had an objective response. CAR‐NK cells had been detected for up to a year in the peripheral blood of the recipients. 46 The high rate of response might be attributed to the high persistence of NK cells, which are presumed to be a result of IL‐15 self‐expression from the transgene.
NKp44 is reported to have the ability to recognise certain antigens, not only at the immunoglobulin‐like domain but also at the hinge domain. 16 As for the first‐generation NKp44‐based CAR generated in the current study, we demonstrated that the original hinge domain was important to efficiently produce cytokines in response to exposure to the target, to kill target cells, and to reduce tumor burden. However, in second‐generation NKp44‐based CARs that had an intracellular 4‐1BB co‐stimulatory domain, CAR construct with the CD8α hinge and transmembrane domain (2G‐d) had better function, despite some loss of function because of replacement of the original NKp44 hinge domain. The CAR‐T cells with this construct showed significantly superior proliferation and tumor control assessed in a long‐term co‐culture assay. We think that this outstanding function is probably related to combinatorial use of CD8α hinge, CD8α transmembrane, 4‐1BB signalling domain and CD3ζ. 47 , 48 . Eisenberg et al. 23 reported that T cells transduced with CAR harbouring the NKp44 extracellular domain (immunoglobulin‐like + hinge), CD28 transmembrane and signalling domain, and CD3ζ signalling domain provided in vivo anti‐tumor effects in a melanoma xenograft model. In the present study, we constructed a NKp44‐CAR gene with a CD28‐co‐stimulatory domain that was almost identical structure to Eisenberg's 23 and compared it with the second‐generation CARs with 4‐1BB signalling. We found that CAR‐T cells with the second‐generation construct with the 4‐1BB signalling domain (2G‐d) outperformed CAR‐T cells transduced with CAR‐T cells with the CD28 signalling domain. Therefore, NKp44‐based CAR with the 4‐1BB domain is expected to be useful for tumor control even in vivo.
Approaches using combinatorial antigen recognition have been recently developed to target the antigens expressed by solid tumors and not to injure normal cells. The split CAR system, in which the co‐stimulatory signal is transmitted after stimulation of the extracellular domain of an independent construct, 49 , 50 and a universal adaptor CAR approach, in which combining cell surface‐fixed structures and corresponding soluble parts, enable the recognition of diverse antigens. 51 , 52 While these approaches are highly promising in enhancing specificity to targets and sparing normal tissues, appropriate targets are limited. In this context, combinatorial use of CARs that use the recognition site of activating NK‐cell receptors will be a good option.
Conclusions
We created CAR constructs using NKp44 extracellular immunoglobulin‐like domain as an antigen recognition site and extensively studied the optimal structures of first‐ and second‐generation CAR formats. This CAR endowed T cells with an NK cell‐like capacity to attack a wide range of leukaemia and paediatric solid tumors, including AML, T‐ALL, B‐ALL, osteosarcoma, rhabdomyosarcoma, neuroblastoma, Ewing sarcoma and glioblastoma, and is expected to contribute to the development of novel immunotherapy for malignancies that are not currently a target of CAR‐T therapy.
Methods
Cells
The CD19+ human Philadelphia‐chromosome‐positive B‐lineage ALL cell line OP‐1 was a generous gift from Dr. Dario Campana (St. Jude Children's Research Hospital, Memphis, TN, USA). The Ewing sarcoma cell line Rh1 and rhabdomyosarcoma cell lines Rh28, Rh30, Rh41 and JR1 were a generous gift from Dr. Peter Houghton (St. Jude Children's Research Hospital). The Burkitt lymphoma cell line Raji, myeloid leukaemia cell lines K562, HL60, THP‐1 and U937, T‐lineage ALL cell lines MOLT4 and Jurkat, osteosarcoma cell lines U2OS and SaOS‐2, cervical carcinoma cell line HeLa and embryonic kidney fibroblast cell line 293T were obtained from the American Type Culture Collection (Rockville, MD, USA). The T‐lineage cell lines CCRF‐HSB2 and Peer, neuroblastoma cell lines SK‐N‐SH, NB1 and NB16, and rhabdomyosarcoma cell line RMS‐YM were obtained from RIKEN BRC cell bank (Tsukuba, Japan). The myeloid cell lines Kasumi‐1, Kasumi‐6 and KG1; neuroblastoma cell line IMR32; and glioblastoma cell lines T98G, A172 and U251MG were obtained from the JCRB cell bank of National Institutes of Biomedical Innovation, Health and Nutrition (Osaka, Japan). The osteosarcoma cell line MG63 was obtained from Health Science Research Resource Bank (Osaka, Japan). NOS1 and NOS10 were a generous gift from Dr. Akira Ogose (Division of Orthopedic Surgery, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan). HeLa and 293T were maintained in Dulbecco's modified Eagle's medium (Sigma‐Aldrich Japan, Tokyo, Japan) supplemented with 10% foetal bovine serum (FBS). The other leukaemia and solid tumor cell lines were maintained in RPMI‐1640 (Sigma‐Aldrich Japan) supplemented with 10% FBS.
CAR constructs and gene transduction
MSCV‐IRES‐GFP, pEQ‐PAM3(‐E) and pRDF plasmids were obtained from St. Jude Vector Development and Production Shared Resource. To generate NKp44‐based CAR genes, we used splicing by overlapping extension by PCR to assemble several genetic fragments. To generate RD114‐pseudotyped retrovirus, we used a lipofection method to transfect 3 × 106 293T cells (Fugene HD, Promega, Madison, WI, USA). Titre of retrovirus was measured using HeLa cells. Peripheral blood mononuclear cells (PBMCs) were collected from healthy volunteers after obtaining informed consent, and T cells were isolated using RosetteSep™ Human T Cell Enrichment Cocktail (STEMCELL technology, Vancouver, BC, Canada). This study was approved by the ethical committee of Niigata University School of Medicine (approval #2015‐2686). T cells were incubated for 48–72 h with Dynabeads human T‐activator CD3/CD28 (Thermo Fisher Scientific,Waltham, MA, USA) in the presence of 200 IU mL−1 of recombinant human interleukin‐2 (rhIL‐2; Peprotech, Cranbury, NJ, USA) in RPMI‐1640 with 10% FBS. The activated T cells were then transduced using Retronectin (Takara, Otsu, Japan) according to the manufacturer's instructions with slight modifications. The multiplicity of infection was almost identical in each experiment comparing different CARs. The transduced cells were maintained in RPMI‐1640, 10% FBS and 200 IU mL−1 rhIL‐2 added every 2–3 days until use. To obtain activated primary NK cells, we co‐cultured PBMCs obtained from healthy adult volunteers with irradiated K562‐mb15‐41BBL cells. 27 Retroviral transduction of primary NK cells was undertaken using the same approach as T cells, except that NK cells were maintained with 100 IU mL−1 rhIL‐2 every 2–3 days. Surface expression of CAR in T cells and NK cells was detected by using phycoerythrin (PE)‐conjugated anti‐NKp44 antibody (Beckman Coulter, Tokyo, Japan). Antibody staining was detected with a FACSCaliber flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). For detection of NKp44 ligands or NKp30 ligands on the cell surface, tumor cell lines were incubated with either recombinant human NKp44‐Fc chimera protein or recombinant human NKp30‐Fc chimera protein (R&D systems, Minneapolis, MN, USA), followed by staining with PE‐conjugated F(ab′)2 fragment goat‐anti‐human IgG secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA). Recombinant human IgG1 Fc protein (R&D systems) was used as the control.
Short‐term cytotoxicity assay
Effector cells that had expanded for 2–3 weeks, including T cells transduced with NKp44‐based CARs, activated NK cells without genetic modification, and NK cells transduced with NKp44‐based CAR were used in the assays. T cells transduced with CAR harbouring binding domain of NKp44 but lacking signalling domain (non‐signalling CAR) served as a control, unless otherwise stated. CAR‐T cells were co‐cultured with the target cells stained using CellTrace Calcein Red‐Orange, AM (Thermo Fisher Scientific) in 96‐well U‐bottom plates in a humidified incubator at 37°C under 5% CO2. After 4 h of co‐culture, the number of viable target cells was counted using flow cytometry, and cytotoxic effects were calculated as previously described. 27 , 48 Autologous or allogeneic primary T cells were collected from healthy adult volunteers and were immediately used as a target (resting T cells) or were used after activation with Dynabeads human T‐activator CD3/CD28 and rhIL‐2 (activated T cells). For blocking assay, CAR‐T cells were incubated in 200 μL PBS added with anti‐NKp44 monoclonal antibody [LEAF™ purified anti‐human CD336 (NKp44) antibody, BioLegend, San Diego, CA, USA] or LEAF™ purified mouse IgG1, κ isotype Ctrl Antibody (BioLegend) as a control for 30 min at room temperature. Then, the CAR‐T cells were washed twice with PBS and used in subsequent assays.
Cytokines, granzyme and CD107a assay
For cytokine secretion assay, 1 × 105 NKp44‐based CAR‐T cells were co‐cultured with target cells at an effector to target (E:T) ratio of 1:1 in 200 μL RPMI‐1640 supplemented with 10% FBS and 200 IU mL−1 of rIL‐2 in a 96‐well round bottom plate for 24 h. Interferon‐γ levels in the supernatant were measured by Enzyme‐Linked ImmunoSorbent Assay (ELISA BioSource Europe S.A., Nivelles, Belgium) or cytometric beads array (CBA, Becton Dickinson). For intracellular cytokine and granzyme production assays, CAR‐T cells were co‐cultured with target cells for 24 h in the presence of 5 μg mL−1 of Brefeldin A (BioLegend). Then, the CAR‐T cells were stained with PE‐conjugated antibodies after permeabilisation (FIX & PERM™ Cell Permeabilization Kit, Thermo Fisher Scientific). For the CD107a mobilisation assay, CAR‐T cells were co‐cultured with target cells for 6 h in the presence of 2 μm Monensin and PE‐conjugated anti‐CD107a antibodies (BioLegend), and the signal levels were determined by flow cytometry.
Long‐term cytotoxicity assay
For leukaemia cells, we co‐cultured CAR‐T cells, CAR‐NK cells or T cells transduced with non‐signalling CAR with the target cells in RPMI‐1640 medium containing 10% FBS supplemented with low‐dose rhIL‐2 (10 IU mL−1) at E:T ratios of 0.5:1 and 0.1:1. After 7 days, the number of residual leukaemia cells was counted using flow cytometry. For solid tumor cells, cells were seeded in a 96‐well flat‐bottom plate (2 × 103 for NOS10, SaOS‐2, U2OS, Rh1, JR1, Rh30, Rh41, A172 and T98G; 1 × 104 for U251MG, IMR32, SK‐N‐SH, NB1 and RMS‐YM) and effector cells were added at E:T ratios of 4:1 and 1:1, respectively. After 7 days, residual tumor cells were analysed using WST‐8 assay reagent (Cell Counting Kit‐8, Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. In particular experiments, to compare the persistence of each effector cells, recovery rates of effector cells after 7 days of co‐culture were analysed using flow cytometry.
iCELLigence real‐time killing assay
Long‐term cytotoxicity (anti‐tumor effect) was compared using a real‐time cell analyzer iCelligence™ (ACEA Biosciences, San Diego, CA, USA), which is able to measure cell proliferation by monitoring cellular impedance of cells attached to the plate in a non‐invasive way. Target cells (1 × 104) were seeded in triplicate and left to adhere and grow for 24 h in iCelligence™ E‐plate containing RPMI‐1640 medium constituting 10% FBS supplemented with low‐dose rhIL‐2 (10 IU mL−1). On the next day, 1 × 104 effector cells were added. Impedance measurements were performed every 15 min for up to 7 days, according to the manufacturer's instructions. 53 Changes in electrical impedance were expressed as a normalised cell index value, which was derived from the relative impedance changes of wells containing co‐culture normalised to baseline impedance values with wells containing solely the medium.
Ex vivo proliferation assay
To examine the proliferation rate of NKp44‐based CAR‐T cells after single antigen stimulation, we co‐cultured 4 × 104 CAR‐T cells and 1 × 104 50 Gy‐irradiated THP‐1 cells in a 96‐well flat‐bottom plate with RPMI‐1640 containing 10% FBS with low‐dose rhIL‐2 (10 IU mL−1). The proliferation rate of CAR‐T cells was calculated after 5 days.
Statistical analysis
Statistical evaluation was conducted with EZR software (version 1.40). 54 The significance of differences in each comparison in cytotoxicity, cytokine production and proliferation rate was analysed by the Student's t‐test. All experiments were performed in three technical replicates, and data are presented as mean ± standard deviation (SD). P‐values < 0.05 were considered statistically significant as follows: *P < 0.05, **P < 0.01 and ***P < 0.001.
Conflict of interest
Chihaya Imai reports patent royalties from Juno Therapeutics, and the other authors have no conflicts of interest to declare.
Author contributions
Yasushi Kasahara: Formal analysis; Investigation; Writing‐original draft. Chansu Shin: Investigation. Nobuhiro Kubo: Investigation. Keichiro Mihara: Formal analysis; Resources. Haruko Iwabuchi: Formal analysis. Takayuki Takachi: Investigation. Masaru Imamura: Formal analysis. Akihiko Saitoh: Formal analysis. Chihaya Imai: Conceptualization; Formal analysis; Funding acquisition; Project administration; Supervision; Writing‐original draft; Writing‐review & editing.
Acknowledgments
We gratefully acknowledge Dr Ko Kudo, Department of Pediatrics, Hirosaki University, for his valuable input. We also thank Ms Yuko Suzuki and Ms Ai Itoh for excellent technical assistance, and Ms Masami Yamagishi and Ms Nobuko Tsuchida for secretarial support. Finally, we thank H Nikki March, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript. This work was supported by Japan Society for the Promotion of Science (JSPS) Grants‐in‐Aid for Scientific Research, JSPS KAKENHI Grant Number 16K10015 and 19K08317 (to CI).
References
- 1. Maude SL, Laetsch TW, Buechner J et al Tisagenlecleucel in children and young adults with B‐cell lymphoblastic leukemia. N Engl J Med 2018; 378: 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maude SL, Frey N, Shaw PA et al Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371: 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brudno JN, Maric I, Hartman SD et al T cells genetically modified to express an anti‐B‐cell maturation antigen chimeric antigen receptor cause remissions of poor‐prognosis relapsed multiple myeloma. J Clin Oncol 2018; 36: 2267–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raje N, Berdeja J, Lin Y et al Anti‐BCMA CAR T‐cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med 2019; 380: 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hansrivijit P, Gale RP, Barrett J, Ciurea SO. Cellular therapy for acute myeloid Leukemia ‐ current status and future prospects. Blood Rev 2019; 37: 100578. [DOI] [PubMed] [Google Scholar]
- 6. Schmidts A, Maus MV. Making CAR T cells a solid option for solid tumors. Front Immunol 2018; 9: 2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Louis CU, Savoldo B, Dotti G et al Antitumor activity and long‐term fate of chimeric antigen receptor‐positive T cells in patients with neuroblastoma. Blood 2011; 118: 6050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahmed N, Brawley VS, Hegde M et al Human epidermal growth factor receptor 2 (HER2)‐specific chimeric antigen receptor‐modified T cells for the immunotherapy of HER2‐positive sarcoma. J Clin Oncol 2015; 33: 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown CE, Alizadeh D, Starr R et al Regression of glioblastoma after chimeric antigen receptor T‐cell therapy. N Engl J Med 2016; 375: 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Katz SC, Burga RA, McCormack E et al Phase I hepatic immunotherapy for metastases study of intra‐arterial chimeric antigen receptor‐modified T‐cell therapy for CEA+ liver metastases. Clin Cancer Res 2015; 21: 3149–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Y, Chen M, Wu Z et al CD133‐directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology 2018; 7: e1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vivier E, Raulet DH, Moretta A et al Innate or adaptive immunity? The example of natural killer cells. Science 2011; 331: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol 2012; 12: 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol 2011; 89: 216–224. [DOI] [PubMed] [Google Scholar]
- 16. Koch J, Steinle A, Watzl C, Mandelboim O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol 2013; 34: 182–191. [DOI] [PubMed] [Google Scholar]
- 17. Fauriat C, Just‐Landi S, Mallet F et al Deficient expression of NCR in NK cells from acute myeloid leukemia: evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 2007; 109: 323–330. [DOI] [PubMed] [Google Scholar]
- 18. Vitale M, Bottino C, Sivori S et al NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non‐major histocompatibility complex‐restricted tumor cell lysis. J Exp Med 1998; 187: 2065–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baychelier F, Sennepin A, Ermonval M et al Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood 2013; 122: 2935–2942. [DOI] [PubMed] [Google Scholar]
- 20. Miller JS, Soignier Y, Panoskaltsis‐Mortari A et al Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005; 105: 3051–3057. [DOI] [PubMed] [Google Scholar]
- 21. Szmania S, Lapteva N, Garg T et al Ex vivo‐expanded natural killer cells demonstrate robust proliferation in vivo in high‐risk relapsed multiple myeloma patients. J Immunother 2015; 38: 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kasahara Y. A novel NKp44‐based chimeric antigen receptor that targets multiple types of cancer. Blood 2016; 128: 3517. [Google Scholar]
- 23. Eisenberg V, Shamalov K, Meir S et al Targeting multiple tumors using T‐cells engineered to express a natural cytotoxicity receptor 2‐based chimeric receptor. Front Immunol 2017; 8: 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Campbell KS, Yusa S, Kikuchi‐Maki A, Catina TL. NKp44 triggers NK cell activation through DAP12 association that is not influenced by a putative cytoplasmic inhibitory sequence. J Immunol 2004; 172: 899–906. [DOI] [PubMed] [Google Scholar]
- 25. Nguyen P, Moisini I, Geiger TL. Identification of a murine CD28 dileucine motif that suppresses single‐chain chimeric T‐cell receptor expression and function. Blood 2003; 102: 4320–4325. [DOI] [PubMed] [Google Scholar]
- 26. Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol 2019; 10: 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005; 106: 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang T, Wu MR, Sentman CL. An NKp30‐based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo . J Immunol 2012; 189: 2290–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qin L, Lai Y, Zhao R et al Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J Hematol Oncol 2017; 10: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guest RD, Hawkins RE, Kirillova N et al The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother; 2005; 28: 203–211. [DOI] [PubMed] [Google Scholar]
- 31. Hecht ML, Rosental B, Horlacher T et al Natural cytotoxicity receptors NKp30, NKp44 and NKp46 bind to different heparan sulfate/heparin sequences. J Proteome Res 2009; 8: 712–720. [DOI] [PubMed] [Google Scholar]
- 32. Ho JW, Hershkovitz O, Peiris M et al H5‐type influenza virus hemagglutinin is functionally recognized by the natural killer‐activating receptor NKp44. J Virol 2008; 82: 2028–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Esin S, Batoni G, Counoupas C et al Direct binding of human NK cell natural cytotoxicity receptor NKp44 to the surfaces of mycobacteria and other bacteria. Infect Immun 2008; 76: 1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hershkovitz O, Rosental B, Rosenberg LA et al NKp44 receptor mediates interaction of the envelope glycoproteins from the West Nile and dengue viruses with NK cells. J Immunol 2009; 183: 2610–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosental B, Brusilovsky M, Hadad U et al Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J Immunol 2011; 187: 5693–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barrow AD, Edeling MA, Trifonov V et al Natural Killer cells control tumor growth by sensing a growth factor. Cell 2018; 172: 534–548.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gaggero S, Bruschi M, Petretto A et al Nidogen‐1 is a novel extracellular ligand for the NKp44 activating receptor. Oncoimmunology 2018; 7: e1470730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rosenberg SA, Lotze MT, Yang JC et al Prospective randomized trial of high‐dose interleukin‐2 alone or in conjunction with lymphokine‐activated killer cells for the treatment of patients with advanced cancer. J Natl Cancer Inst 1993; 85: 622–632. [DOI] [PubMed] [Google Scholar]
- 39. Curti A, Ruggeri L, D'Addio A et al Successful transfer of alloreactive haploidentical KIR ligand‐mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011; 118: 3273–3279. [DOI] [PubMed] [Google Scholar]
- 40. Till BG, Jensen MC, Wang J et al Adoptive immunotherapy for indolent non‐Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20‐specific T cells. Blood 2008; 112: 2261–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jensen MC, Popplewell L, Cooper LJ et al Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19‐specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant 2010; 16: 1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gargett T, Brown MP. The inducible caspase‐9 suicide gene system as a ‘safety switch’ to limit on‐target, off‐tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol 2014; 5: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fedorov VD, Themeli M, Sadelain M. PD‐1‐ and CTLA‐4‐based inhibitory chimeric antigen receptors (iCARs) divert off‐target immunotherapy responses. Sci Transl Med 2013; 5: 215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Imamura M, Shook D, Kamiya T et al Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane‐bound interleukin‐15. Blood 2014; 124: 1081–1088. [DOI] [PubMed] [Google Scholar]
- 45. Liu E, Tong Y, Dotti G et al Cord blood NK cells engineered to express IL‐15 and a CD19‐targeted CAR show long‐term persistence and potent antitumor activity. Leukemia 2018; 32: 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu E, Marin D, Banerjee P et al Use of CAR‐transduced natural killer cells in CD19‐positive lymphoid tumors. N Engl J Med 2020; 382: 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ying Z, Huang XF, Xiang X et al A safe and potent anti‐CD19 CAR T cell therapy. Nat Med 2019; 25: 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Imai C, Mihara K, Andreansky M et al Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004; 18: 676–684. [DOI] [PubMed] [Google Scholar]
- 49. Wilkie S, van Schalkwyk MC, Hobbs S et al Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol 2012; 32: 1059–1070. [DOI] [PubMed] [Google Scholar]
- 50. Roybal KT, Rupp LJ, Morsut L et al Precision tumor recognition by T cells with combinatorial antigen‐sensing circuits. Cell 2016; 164: 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 2018; 173: 1426–1438.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rodgers DT, Mazagova M, Hampton EN et al Switch‐mediated activation and retargeting of CAR‐T cells for B‐cell malignancies. Proc Natl Acad Sci USA 2016; 113: E459–E468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peper JK, Schuster H, Löffler MW et al An impedance‐based cytotoxicity assay for real‐time and label‐free assessment of T‐cell‐mediated killing of adherent cells. J Immunol Methods 2014; 405: 192–198. [DOI] [PubMed] [Google Scholar]
- 54. Kanda Y. Investigation of the freely available easy‐to‐use software ‘EZR’ for medical statistics. Bone Marrow Transplant 2013; 48: 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]