

Abstract
Total synthesis of isatindigotindoline C, a 3,3’-spiropyrrolidine oxindole alkaloid, is achieved in two steps using an exo-selective decarboxylative [3+2]-dipolar cycloaddition as the key step. The synthesis verifies the originally assigned relative anti-stereochemistry for the bis-oxindole core of isatindigotindoline C.
Graphical Abstract
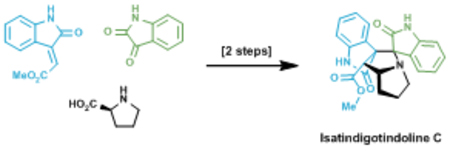
Total synthesis of isatindigotindoline C is achieved in two steps using a biomimetic 1,3-dipolar cycloaddition reaction as the key step.
Introduction
Isatindigotindolines are a structurally unique family of 3,3’-spiropyrrolidine oxindole alkaloids isolated by Huang and Song et. al. in 2018 from the leaves of Isatis indigotica plant used in traditional and folk medicine in Asia.1 The isatindigotindolines caught our attention as potential synthesis targets due to our continued interest in the synthesis of bioactive nitrogenous compounds.2 During the isolation studies, several isatindigotindolines were shown to inhibit β-amyloid aggregation.1 Alongside with their potential use in biology, the isatindigotindoline alkaloids are the first examples of secondary metabolites with a 3,3’-spiropyrrolidine oxindole core (Figure 1B), this is a sharp contrast to the abundance of alkaloids with the isomeric 2,3’-spiropyrrolidine oxindole structure (Figure 1A). In addition, the isatindigotindolines constitute a rare example of an entire alkaloid family occurring naturally as racemates.3
Figure 1.

A: The 3,2’-spiropyrrolidine oxindole scaffold is a common motif in many alkaloids (256 hits on Reaxys database). B: The isomeric 3,3’-spiropyrrolidine oxindole scaffold is present only in the 4 solitary isatindigotindoline alkaloids. C: Two diastereomeric structures originally considered for isatindigotindoline C (anti-1 and syn-1) differ in the relative configuration of the bisoxindole unit.1 D: Retrosynthesis of the proposed structure of 1 based on a 1,3-dipolar cycloaddition.
The structurally most complex member of this alkaloid family, (±)-isatindigotindoline C (1), has a bis-oxindole core. The relative stereochemistry of the bis-oxindole core proved non-trivial to assign during the original isolation and structure determination studies of 1. Correlating experimental 13C shifts with computed values gave very similar regressions for both anti and syn bisoxindole diastereomers of 1 (R2 = 0.9989 vs. R2 = 0.9984 respectively). The NMR shift correlations, in combination with comparisons to calculated ECD curves allowed the relative configuration to be assigned anti (Figure 1C).1 In order to verify this computation-assisted stereochemistry assignment, and to gain synthetic access to the chemically and biologically intriguing family of isatindigotindolines, we embarked on the total synthesis of the proposed structure of 1.
Overall isatindigotindoline C (1) displays a formidable synthetic challenge with four contiguous stereogenic centers, including a quaternary all-carbon stereogenic center, and six rings all compacted around a central pyrrolidine ring C (Figure 1D). As a corollary, retrosynthetically disconnecting the principal ring C of 1 through a 1,3-dipolar cycloaddition would result in marked overall simplification in complexity (Figure 1D). The resulting azomethine ylide 3 can be formed by a decarboxylative condensation between isatin (4) and proline (5).4 The chosen approach is also in line with the proposed biosynthetic route, and agrees with the general tendency of racemic natural products to arise from the facile cyclizations of achiral precursors.1,3
Results and Discussion
With this plan at hand, we prepared the α,β-unlsaturated methyleneindoline dipolarophile (E)-2 from isatin (2) and methyl (triphenylphosphoranylidene)acetate according to a previously reported literature procedure.7 In order to clear the methyl ester α-stereogenic center (C1’’’) of isatindigotindoline C (1), the (E)-methyleneindoline ester dipolarophile (E)-2 would have to be isomerized to (Z)-2 prior to the key cycloaddition step. However, attempts at isomerizing (E)-2 to (Z)-2 in a Michael–retro-Michael reaction with phenol, thiophenol, or pyridine as nucleophiles led to severe decomposition with (Z)-2 isolatable only in low yields (<5%).5
As we could not prepare synthetically useful quantities of (Z)-2, we envisioned that a exo-1,3-dipolar cycloaddition between (E)-2 and the azomethine ylide 3 would furnish 1’’’-epi-1, which could be late-stage epimerized to natural Isatindigotindoline C (1) with a suitable base.6 The late-stage epimerization was also supported by a short computational study at B97D3/DEF2TZVP-level. Isatindigotindoline C (1) lies 1.9 kcal/mol lower than 1’’’-epi-isatindigotindoline (1’’’-epi-1) for minimum energy conformers (See SI for details).
The three-component reaction between (E)-2, proline (5) and isatin (4) (1:1:1 molar ratio) readily precipitated the desired exo cycloaddition product 1’’’-epi-isatindigotindoline C (1’’’-epi-1) as a white flocculent solid. The reaction was readily scalable, allowing us to produce 1.7 g (87%) of 1’’’-epi-1. The relative anti stereochemistry of the bisoxindole core, and the configuration of the methyl ester α-stereogenic center of the thus formed 1’’’-epi-1 were both reliably established from single crystal x-ray data (Scheme 1).7
Scheme 1:

Total Synthesis of Isatindigotindoline C (1) and 1’’’-epi-Isatingigotindoline C (1’’’-epi-1). Reagents and conditions: a) methyleneidonoline ester 2 (1.0 equiv.), proline (5) (1.0 equiv.), isatin (4) (1.0 equiv.), MeOH, 60 °C, 3 h, 87% b) NaOMe (10 equiv.), MeOH, 97% recovery, dr 36:64 (epi-1 to 1). Crystalline water is omitted from the single-crystal x-ray diffraction structure of 1 for clarity.
The late-stage epimerization was then addressed. 1’’’-epi-1 was only partially epimerized with K2CO3 (96:4) and NaH (92:8) at rt in THF. Under the same conditions KOtBu and KHMDS led to decomposition of 1’’’-epi-1. With NaOMe in MeOH, a synthetically viable dr (36:64, favoring 1) was obtained with 97% mass recovery. Careful flash chromatographic purification of the diastereomeric mixture furnished an analytically pure sample of 1. The 1H and 13C NMR data of synthetic Isatindigotindoline C (1) were in full agreement with those reported for the natural product (see SI).1 Furthermore, synthetic isatindigotindoline C (1) could be crystallized and the relative anti-stereochemistry of the bis-oxindole core unambiguously verified by single crystal x-ray analysis.
Conclusions
In summary, we have achieved the first total synthesis of 3,3’-spiropyrrolidine oxindole alkaloid isatindigotindoline C (1) in two steps from the known ester 2. The single crystal x-ray structure and NMR data of our synthetic material corroborates the original computation-assisted anti stereochemistry assignment of the bisoxindole core of 1. The synthetic route also supports the postulated biosynthetic pathway, and suggests that the key dipolar cycloaddition forging the isatindigotindoline core takes place in an exo fashion. The approach discussed herein is readily applicable to the preparation of other members of the isatindigotindoline family as well. Also, considering the ease at which the key dipolar cycloaddition proceeds, it is likely that additional secondary metabolites containing the elusive 3,3’-spiropyrrolidine oxindole scaffold remain to be discovered in Nature.
Supplementary Material
Acknowledgements
L. K. acknowledges financial support of Rice University, the National Institutes of Health (R01 GM-114609-04), the National Science Foundation (CAREER:SusChEM CHE-1546097), the Robert A. Welch Foundation (grant C-1764), Amgen (2014 Young Investigators’ Award), and Biotage (2015 Young Principal Investigator Award). J. H. S acknowledges the support from the Wiess Teaching Postdoctoral fellowship and the Osk. Huttunen foundation.
Notes and References
- 1.Liu SF, Lin B, Xi YF, Zhou L, Lou LL, Huang XX, Wang XB and Song SJ, Org. Biomol. Chem, 2018, 16, 9430–9439. [DOI] [PubMed] [Google Scholar]
- 2.:a) Kattamuri PV, Bhakta U, Siriwongsup S, Kwon DH, Alemany LB, Yousufuddin M, Ess DH and Kürti L, J. Org. Chem, 2019, 84, 7066–7099;b) Kattamuri PV, Yin J, Siriwongsup S, Kwon DH, Ess DH, Li Q, Li G, Yousufuddin M, Richardson PF, Sutton SC and Kürti L, J. Am. Chem. Soc, 2017, 139, 11184–11196;c) Zhou Z, Cheng QQ and Kürti L, J. Am. Chem. Soc, 2019, 141, 2242–2246.
- 3.Zask A and Ellestad G, Biomimetic Syntheses of Racemic Natural Products. Chirality. John Wiley and Sons Inc; 2018, p. 157–164. [DOI] [PubMed] [Google Scholar]
- 4.:a) Angyal A, Demjén A, Harmat V, Wölfling J, Puskás LG and Kanizsai I, J. Org. Chem, 2019, 84, 4273–4281;b) Wang Y, Wang JL, Burgess KS, Zhang JW, Zheng QM, Pu YD, Yan LJ and Chen XB, RSC Adv, 2018, 8, 5702–5713;c) Kasaboina S, Bollu R, Ramineni V, Gomedhika PM, Korra K, Basaboina SR, Holagunda UD, Nagarapu L, Dumala N, Grover P, Bathini R and Vijjulatha M, J. Mol. Struct, 2019, 1180, 355–362.
- 5.Song Z, Chen CP, Liu J, Wen X, Sun H and Yuan H, Eur. J. Med. Chem, 2016, 124, 809–819; [DOI] [PubMed] [Google Scholar]; b) Grigg R, Donegan G, Gunaratne HQN, Kennedy DA, Malone JF, Sridharan V and Thianpatanagul S, Tetrahedron, 1989, 45, 1723–1746. [Google Scholar]
- 6.Rossetti A, Sacchetti A, Bonfanti M, Roda G, Rainoldi G and Silvani A, Tetrahedron, 2017, 73, 4584–4590. [Google Scholar]
- 7.a) Liu J, Sun H, Liu X, Ouyang L, Kang T, Xie Y, Wang X, 2012, 53, 2336–2340;b) He J, Ouyang G, Yuan Z, Tong R, Shi J, Ouyang L, Molecules, 2013, 18, 5142–5154;c) Murugan R, Raghunathan R, Narayanan SS, Synth. Commun, 2010, 40, 3135–3151.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.