

Abstract
A promising approach in cancer therapy is to find ligands that directly bind ubiquitin (Ub) chains. However, finding molecules capable of tightly and specifically binding Ub chains is challenging given the range of Ub polymer lengths and linkages and their subtle structural differences. Here, we use total chemical synthesis of proteins to generate highly homogenous Ub chains for screening against trillion-member macrocyclic peptide libraries (RaPID system). De novo cyclic peptides were found that can bind tightly and specifically to K48-linked Ub chains, confirmed by NMR studies. These cyclic peptides protected K48-linked Ub chains from deubiquitinating enzymes and prevented proteasomal degradation of Ub-tagged proteins. The cyclic peptides could enter cells, inhibit growth and induce programmed cell death, opening new opportunities for therapeutic intervention. This highly synthetic approach, with both protein target generation and cyclic peptide discovery performed in vitro, will make other elaborate post-translationally modified targets accessible for drug discovery.
Graphical Abstract
Introduction
The initial step in ubiquitin (Ub) signaling is the modification of a protein substrate by Ub or polyUb chains1,2. This is achieved by the collaborative action of three enzymes, the E1, E2 and E3 ligases3. In ubiquitination, an isopeptide bond is formed between the C-terminus of a Ub molecule and a lysine residue from the protein substrate. PolyUb chains are then made by repeated isopeptide or amide bond formation, linking any of the seven lysines, or the N-terminus, of the last Ub in the chain and to the C-terminus of a new Ub4,5. Interestingly, Ub chains of different linkage types signal for different biological functions, likely due to their unique conformations which allows for distinct recognition by cellular proteins. The most studied polyUbs are the K48-linked chains, which are known to adopt a compact structure and label their protein substrate for proteasomal degradation6,7. Ubiquitination is a reversible process and a family of enzymes known as deubiquitinases (DUBs) cleave Ub from polyUb chains, or remove polyUb entirely5,8.
Ubiquitination is a major post-translational modification affecting many aspects of cell biology9,10. Several tumor suppressors or oncogenes are involved in ubiquitination or deubiquitination pathways, making the Ub system an excellent target for cancer therapy11. For example, the ubiquitin-proteasome system (UPS) has proven to be a valid target for anticancer drug development, as exemplified by the approval of the small molecule Bortezomib for the treatment of multiple myeloma and mantle cell lymphoma12,13,14. Drugs such as Bortezomib interfere with protein degradation by inhibiting the UPS proteolysis machinery15,16, whereas other strategies target different components of Ub signaling, such ubiquitination itself or the action of DUBs, with success yet to be translated to useful therapies5,17.
Another promising approach is targeting the Ub chain itself. In this regard, the small molecules ‘ubistatins’ fortuitously emerged from a chemical genetic screen, and were found to bind monomeric Ub as well as K48-linked chains, and thereby impair recognition by the proteasome and inhibit protein degradation18. However, ubistatins have weak (sub-μM to μM) binding and poor chain specificity19.
Targeting Ub chains of particular lengths and linkages could control different cellular processes. However, this represents a significant challenge for molecular recognition, given the number of possible Ub chains4 and the often subtle differences in their structure and dynamics20,21. Yet, we have envisioned that this could be achieved using macrocyclic peptides, which can tightly interact with proteins using extended interfaces22, similar to natural protein-protein interactions, but at the same time are small enough to emulate drug-like in vivo properties of small molecules23.
There are powerful methods to discover de novo binding peptide sequences, such as the RaPID system (Random non-standard Peptides Integrated Discovery). In the RaPID system, in vitro translation is modified to use reprogrammed genetic codes24, including nonproteinogenic amino acids that allow for spontaneous peptide macrocyclization25. RaPID can generate huge, trillion-member libraries of DNA-tagged cyclic peptides26, 27. Panning these libraries for target engagement, followed by DNA sequencing, allows for selection of highly specific, tight binding de novo cyclic peptide sequences.
We reasoned that the RaPID system could produce cyclic peptide ligands capable of discriminating between Ub chains. However, the success of such a selection would depend critically on the homogeneity, in length and linkage, of these target Ub polymers. This is difficult to achieve using biochemical and biological approaches for all the Ub chains. However, recently it has become possible to assemble the protein Ub and polyUb by total chemical synthesis. This allows for the construction of high-purity Ub chains, with defined lengths28 and linkage types, including any required modifications, suitable for use as targets in these challenging selections.
By combining the RaPID system and chemical protein synthesis of Ub chains, we discover for the first time de novo cyclic peptides that tightly bind the K48-linked Ub chains, with great selectivity towards the chain length (mono-, di-, and tetra-Ub) and linkage type (K48 vs K63 or K11). We also report the in vitro effects of the selected cyclic peptide on the behavior of these Ub chains, e.g. their interaction with DUBs and the proteasome, as well as their ability to inhibit cellular protein degradation.
Results
Choice of K48-linked target
We sought to determine whether selectivity towards different Ub oligomeric forms or a particular chain linkage is possible using cyclic peptides. We chose K48-linked Ub chains as targets for de novo cyclic peptide discovery, as chemical compounds that can bind K48-linked Ub have biological and therapeutic significance. In particular, the degradation of specific cellular proteins by the 26S proteasome, controlled using K48-linked polyUb tags, determines whether a cell proliferates or dies29. Cancer cells use degradation of certain proteins e.g. p53, Bax and p2730–32, to avoid the triggering of apoptosis. Therefore, K48-linked polyUb-binding cyclic peptides may elevate the levels of these short-lived proteins and might selectively restore apoptosis in these cells31,33,11.
Total chemical synthesis of biotin tagged K48-linked Ub chains
We applied our chemical toolbox to construct K48-linked Ub chains suitable for RaPID selection. This consists of orthogonally protected δ-mercaptolysine34, solid phase peptide synthesis (SPPS) of Ub monomers (modified as required), coupled with isopeptide chemical ligation and desulfurization to prepare mono Ub (Ub1), di-Ub (K48Ub2) and tetra-Ub (K48Ub4) chains35. The N-terminus of each distal Ub was further modified with biotin, to facilitate immobilization to a solid support (Fig. 1a). Highly homogenous Ub chains were obtained which could be reconstituted into folded proteins, and bound to streptavidin (Supplementary Figs 1–3).
Figure 1. RaPID selection of K48Ubn binding cyclic peptides.
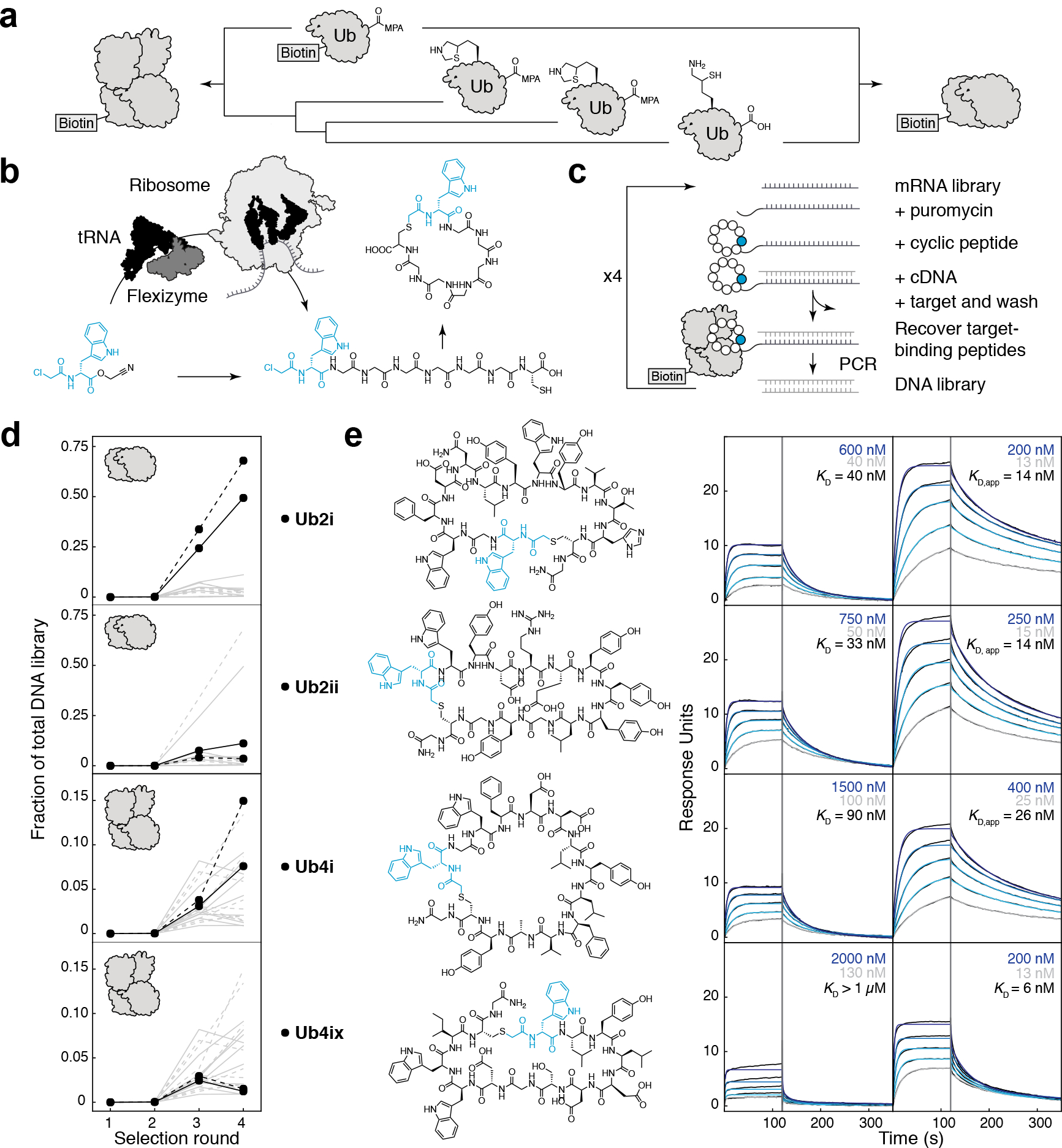
a) Chemical synthesis of biotin-K48-linked di/tetra-Ub chain. b) Flexizyme, an artificial tRNA aminoacylation ribozyme, can load nonproteinogenic amino acids onto tRNA (black), for use by the ribosome in in vitro translation. nonproteinogenic amino acids with an N-terminal ClAc group will spontaneously react with intramolecular cysteine and can be used to form macrocyclic peptides. c) Large libraries (~1013) of DNA-tagged cyclic peptides can be generated in the RaPID system, from which sequences can be isolated to tightly bind a protein target. d) Using K48Ub2 as a target, RaPID DNA libraries became enriched in certain peptide sequences (grey lines), with Ub2i and Ub2ii (black) as the dominant peptides (selection repeat shown as dashed lines). Using K48Ub4 as a target, Ub4i and Ub4ix (black) emerged amongst the enriched peptides. e) (top to bottom) Synthesized peptides Ub2i, Ub2ii, Ub4i, Ub4ix and their binding to K48Ub2 (left) and K48Ub4 (right) detected using SPR. Highest and lowest peptide SPR concentrations are shown in blue and grey respectively.
Discovery of de novo cyclic peptides for K48 Ub chains
The chemically synthesized Ub chains, biotin-K48Ub2 and biotin-K48Ub4, were used as targets for two separate RaPID selections (Fig. 1b). The initial cyclic peptide libraries contained ~1013 unique sequences, with 8–12 random amino acids per peptide. After the first round of selection an additional step was introduced, binding to biotin-Ub1, to remove cyclic peptides that might use Ub1 as the main recognition element. Deep sequencing of the DNA library after each round showed that a small number of peptide sequences, and their variants, came to dominate the library by round 4 (Fig. 1c, Supplementary Fig. 4). Rounds 2–4 were repeated and produced a similar pattern of enrichment (Supplementary Fig. 4). In the K48Ub2 selections the libraries became dominated by the same two sequences, Ub2i and Ub2ii (Fig. 1c, Supplementary Fig. 4). In the K48Ub4 selections, many of the top peptides had appreciable sequence identity to Ub2i (Supplementary Fig. 4); One of these high identity peptides (58 %), Ub4i, was chosen for further study together with a shorter, lower identity peptide (8 %), Ub4ix (Fig. 1c).
De novo cyclic peptides tightly bind K48-linked Ub chains
To test the binding and specificity of the Ub2i, Ub2ii, Ub4i and Ub4ix cyclic peptides, each was prepared by SPPS (Supplementary Fig. 5) for Surface Plasmon Resonance (SPR) analysis (Fig 1d). Each peptide was flowed over immobilized Ub chains: the selection targets K48Ub2 and K48Ub4, as well as Ub1, and alternatively linked K11Ub2, K63Ub2. Ub2i and Ub2ii bound tightly to their selection target K48Ub2, with low-nM Kd values (40 ± 12 nM, 33 ± 8 nM, respectively, Fig 1d). However, Ub2i and Ub2ii also strongly bound the longer chain K48Ub4, with a low apparent Kd and an increased SPR response (Fig 1d). This is consistent with a higher stoichiometry of binding to the tetramer, which could be explained by the fact that K48Ub2 structure can be superimposed twice on the K48Ub4 crystal structure (Supplementary Fig. 6)36.
Ub4i tightly bound its selection target K48Ub4, but also bound K48Ub2 similar to Ub2i and Ub2ii. This is perhaps unsurprising, given the high sequence identity between Ub4i and Ub2i. On the other hand, the cyclic peptide Ub4ix was highly specific for the K48-linked Ub tetramer: weakly binding K48Ub2 ( > 1 μM Kd) but tightly binding to K48Ub4 (Kd = 6 ± 1 nM).
None of the cyclic peptides showed detectable binding to Ub1 (Supplementary Methods). This was expected as the RaPID selection was designed to avoid recovering Ub1 binders. More interestingly, the cyclic peptides showed marked selectivity for the K48-linkage over alternative linkages: there was no detectable binding, for any of the cyclic peptides, to either K11-linked or K63-linked di-Ub (Supplementary Methods).
Mapping of cyclic peptide/K48-linked poly-Ub interface using NMR
We then became interested in understanding the binding mode of the discovered cyclic peptides with the K48-linked Ub chains. We used NMR to confirm physical interaction between Ub2ii and K48Ub2 and to map the residues/sites involved in the peptide binding. The peptide was titrated into solution of K48Ub2 where either distal or proximal Ub was 15N-enriched (Supplementary Fig. 7), and the binding was monitored by 1H-15N NMR. The titration revealed site-specific interactions with both Ub units (Figs. 2a,b and Supplementary Fig. 8). Interestingly, the NMR spectra followed a text-book example of slow exchange, where a gradual decrease in intensity of the “unbound” Ub signals was accompanied by appearance and increase of alternate (“bound”) signals. At a 1:1 molar ratio, practically all affected unbound signals vanished (Figs. 2a and Supplementary Fig. 8). This likely reflects slow off-rates, in full agreement with the SPR data (Supplementary Table S1) and strong, nM-Kd binding (Fig. 1e). By contrast, only minor spectral perturbations were observed upon addition of Ub2ii to mono-Ub and K63Ub2 (data not shown), corroborating high specificity of the peptide for K48Ub2.
Figure 2. Cyclic peptides bind to residues at the hydrophobic Ub-Ub interface in K48-linked di- and tetra-Ub.
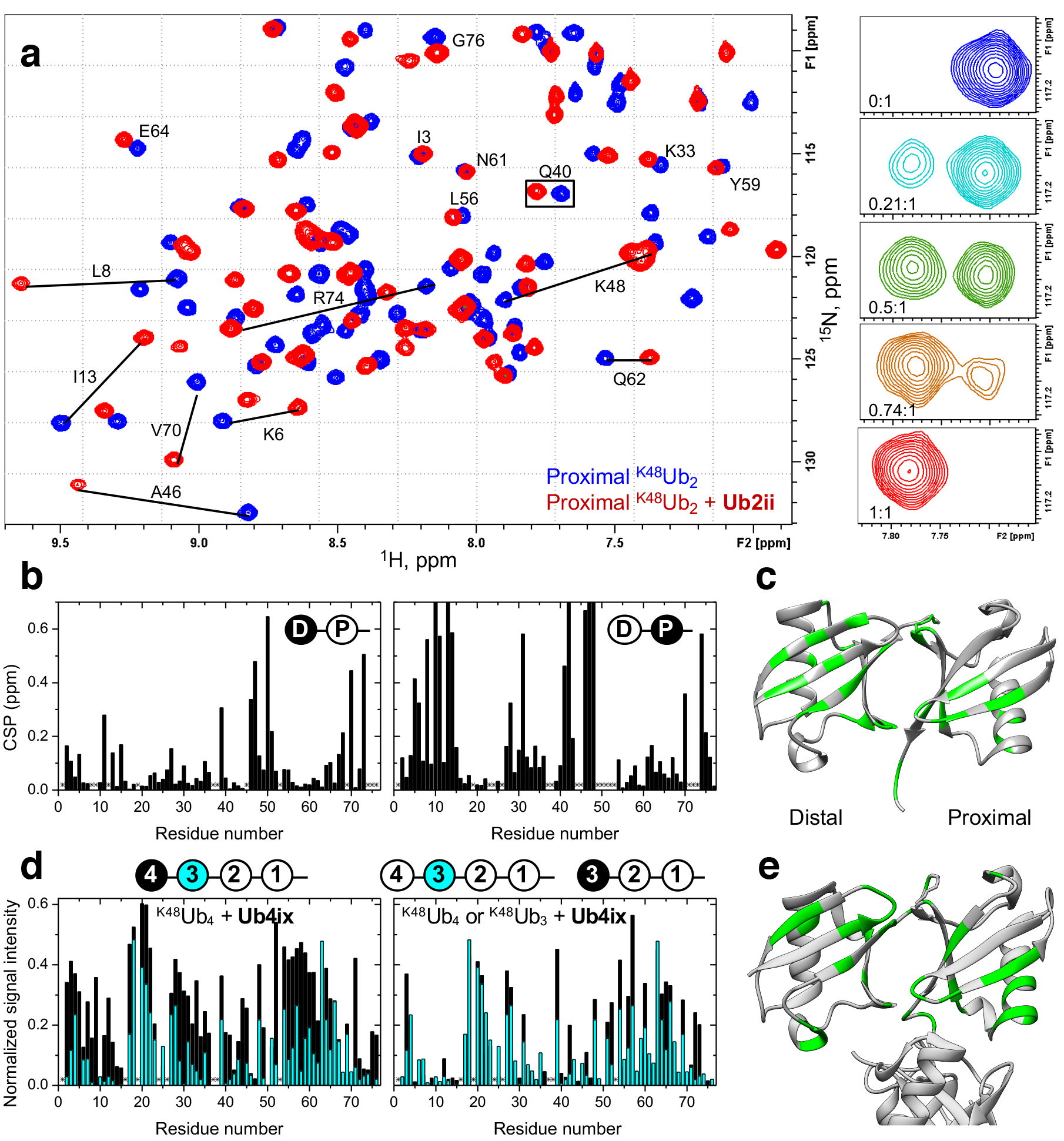
a) Overlay of the 1H-15N correlation spectra of K48Ub2 (proximal Ub) free (blue) and in the presence (red) of 1 molar equivalent of Ub2ii. Signal shifts for select residues are indicated. Insets on the right illustrate the behavior of the signal of Q40 (boxed) during the titration, at the indicated peptide:Ub2 molar ratios. b) Residue-specific chemical shift perturbations (CSP) at the endpoint of titration with Ub2ii in the distal (left) and proximal (right) Ub units in K48Ub2. c) The perturbed residues are mapped (CSP above the mean for each domain colored green) on the crystal structure of the closed state of K48Ub2 (PDB 1AAR) d) (Left) Comparison of the intensities of the unbound signals of the most distal (unit 4, black bars) and the next to distal (unit 3, cyan bars) Ub units in K48Ub4 upon addition of Ub4ix in 2:1 and 1.5:1 molar ratio, respectively. The signal intensity for each residue was normalized to the corresponding intensity in the absence of peptide. (Right) A similar comparison of the signal intensities in K48Ub4 unit 3 (cyan) versus the distal Ub in K48Ub3 (unit 3, black), both at the 1.5:1 molar ratio (peptide:polyUb). The drawings on top indicate which Ub units in K48Ub4 and K48Ub3 are analyzed. Asterisks in (b) and (d) indicate residues that could not be detected or could not be reliably quantified due to signal overlap. e) Map of the residues showing strong signal attenuations (normalized signal intensity < 0.2 colored green) in the two distal Ub units (4 and 3) of K48Ub4 on the crystal structure of the closed form of K48Ub4 (PDB 2O6V).
The K48Ub2 sites affected by Ub2ii binding were mapped to residues in and around the L8-I44-V70 hydrophobic patch on both Ub units (Fig. 2b–c), indicating that the cyclic peptide intercalates between the two Ub units in K48Ub2. Simultaneous binding of both Ub units to the same cyclic peptide is supported by the fact that the saturation was reached at the 1:1 molar ratio indicating a 1:1 stoichiometry of the resulting Ub2/Ub2ii complex. The observed interface is similar to that involved in K48Ub2 interactions with ubistatins18,19 as well as K48-specific protein receptors37. Interestingly, despite the general symmetry between the two Ub units in Ub2, the chemical shift perturbations (CSPs) and the directions of the signal shifts are drastically different between the distal and the proximal Ubs. Combined with the fact that only a single set of “bound” NMR signals is observed for each Ub unit, this indicates that Ub2ii binds K48Ub2 unidirectionally and in a single conformation.
Using NMR, we also monitored the interaction between Ub4ix and K48Ub4. As with K48Ub2/Ub2ii, the binding was in slow-exchange regime, and the interface was mapped primarily to the hydrophobic surface patch on each Ub unit studied (Figs. 2d,e and Supplementary Figs. 9–11). Interestingly, the most distal Ub exhibited relatively weaker spectral perturbations compared to the other units in the chain, and the disappearance of the unbound signals occurred at higher peptide concentrations than for Ub unit 3 or the proximal Ub, suggesting relatively weaker Ub4ix interaction with the most distal Ub in the chain. To examine if this was related to the length of the chain or the external (distal) position of the Ub unit, we performed similar binding studies for K48Ub3 (Supplementary Figs. 12–14). The spectral perturbations in the distal Ub of this tri-Ub were comparable with those in unit 3 of K48Ub4 but different from the most distal Ub in K48Ub4 (Figs. 2d, Supplementary Fig. 15). Also the proximal Ub units in K48Ub4 and K48Ub3 exhibited similar patterns of residue-specific signal shifts (Supplementary Fig. 16). Together, these results suggest that the cyclic peptide Ub4ix primarily binds to/across the first three Ub units in K48Ub4. Consistent with this conclusion and the SPR results, the addition of Ub4ix to K48Ub2 caused lesser spectral perturbations than in the case of Ub2ii peptide at the same (1:1) molar ratio (data not shown), indicating weaker binding.
Cyclic peptide binding to Ub chains protects these chains against DUB cleavage in vitro.
It is possible that the binding of these cyclic peptides to Ub chains prevents their recognition by interacting proteins such as DUBs. We chose to examine the effect of these cyclic peptides on two DUBs: OTUB138, a K48-linkage chain specific DUB, and USP2, which cleaves most Ub chains without specificity39, using K48Ub2 and K48Ub4 chains as substrates (Fig. 3). All peptides were able to inhibit OTUB1 with K48Ub2 as the substrate (Fig. 3a), at ratio of 1:1 cyclic peptide: substrate, even the weakest binding peptide Ub4ix was able to fully inhibit OTUB1 (Fig. 3c). This suggests that binding to Ub chains by cyclic peptides can interfere with recognition by the DUB. However, with USP2 using K48Ub2 as the substrate, we observed only ~40% inhibition in the presence of Ub2i, Ub2ii or Ub4i, and only minimal inhibition by Ub4ix (Fig. 3b). Excess Ub4ix over substrate concentration was required to completely inhibit the activity of USP2 (Fig. 3d). Using a K48Ub4 chain as the substrate instead, both OTUB or USP2 could cleave down to monomer Ub units, and this cleavage was inhibited in the presence of the cyclic peptides (Fig. 3e, f). However, USP2 in the presence of the cyclic peptides Ub2i, Ub4ix, and Ub4i could only remove the distal Ub of K48Ub440, shortening the chain to the trimer K48Ub3, the peptides show strong inhibition of further disassembly of the chain (Fig. 3e, f). For Ub4ix this can be explained by the NMR result that suggested that the cyclic peptide only weakly interacts with the distal Ub, which may be sterically accessible for cleavage by USP2. Interestingly, the same cyclic peptides did not protect K63Ub2 chains from USP2 cleavage, nor the structurally similar K11Ub2, from Cezanne cleavage41 (Supplementary Fig. 17), highlighting the specificity of the cyclic peptides towards the K48-linkage, supporting by the NMR and SPR studies (see above) .
Figure 3. Cyclic peptides inhibit the DUB cleavage of K48-linked di/tetra-Ub.
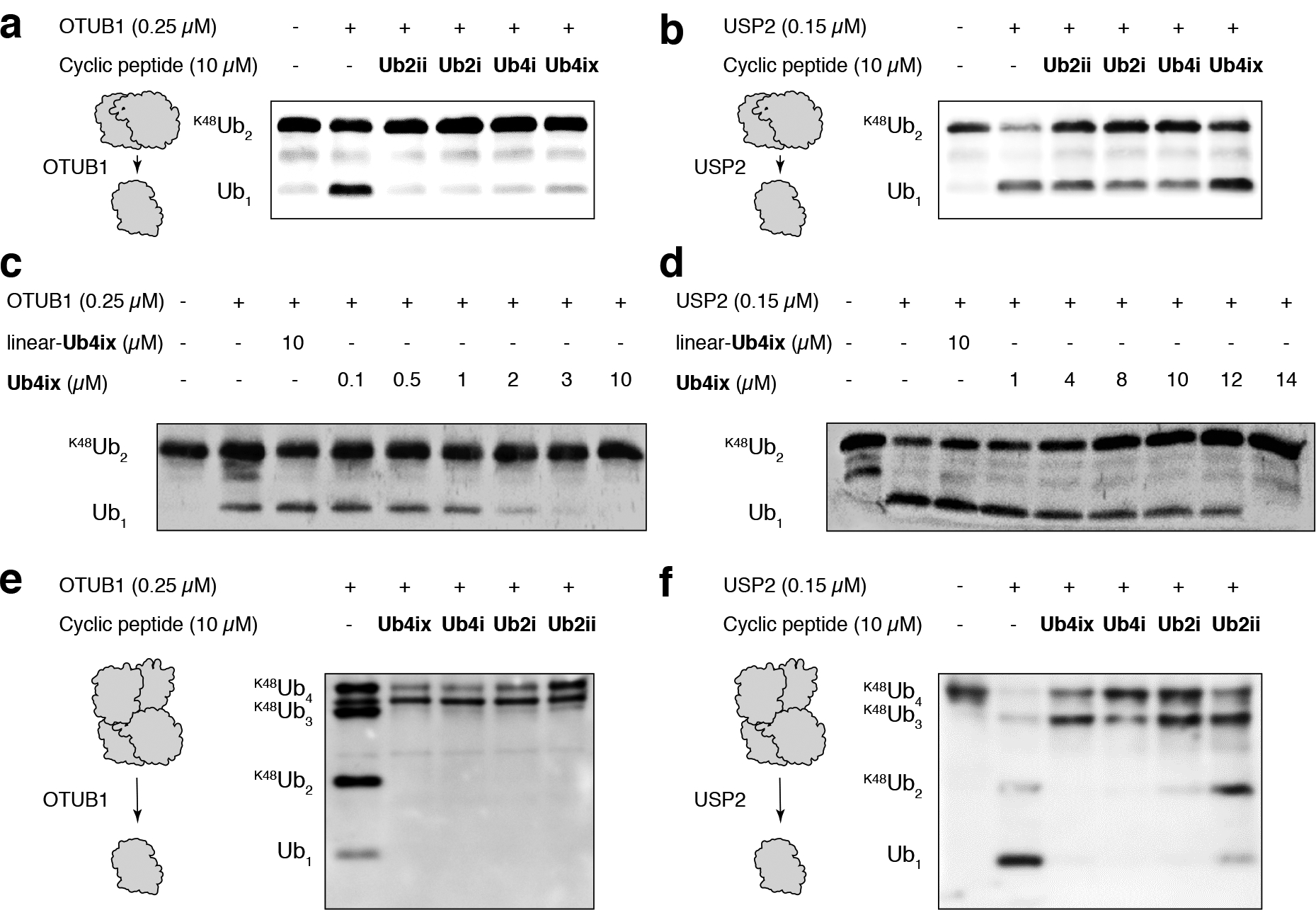
a) K48Ub2 incubated with the K48-linked specific DUB OTUB1 for 1 hour resulted in cleavage to Ub1. This reaction was inhibited by all cyclic peptides. b) K48Ub2 incubated with the non-linkage-specific DUB USP2 resulted in cleavage to Ub1, cleavage could be inhibited by all peptides. c) Ub4ix inhibits OTUB1 in a dose dependent manner, with an IC50 approximately the same as the substrate concentration (K48Ub2 initially at 2 μM). d) Excess of Ub4ix (relative to K48Ub2 substrate) was required to fully inhibit the activity of USP2 over a 1 hour incubation. e) K48Ub4 incubated for 2 hours with OTUB1 resulted in cleavage to K48Ub3, K48Ub2 and Ub1. In the presence of cyclic peptides, cleavage to K48Ub3 occurred, but further cleavage was inhibited. f) Similar for OTUB1, K48Ub4 incubated for 1 hour with USP2 could cleave to K48Ub3, K48Ub2 and Ub1, and this cleavage was inhibited by the cyclic peptides. In all experiments, equal amounts were loaded on SDS-PAGE gel, electro-blotted to nitrocellulose membrane, and probed with an anti-Ub antibody. Initial concentrations of K48Ub2 or K48Ub4 substrate were 2 μM in each case.
Cyclic peptide binding to Ub chains prevents ubiquitinated proteins from proteasomal degradation
It is possible that the binding of cyclic peptides to K48-linked Ub chains could prevent the recognition by the proteasome, and inhibit the degradation of proteins tagged with these Ub chains. To test this we incubated synthetic α-globin-K48Ub4 with 26S proteasome, with and without Ub4ix. Without Ub4ix, the α-globin-K48Ub4 substrate was degraded by the proteasome. However, with equimolar amounts of Ub4ix relative to α-globin-K48Ub4, degradation was inhibited, a similar effect to that of the direct proteasome inhibitor, MG132 (Fig. 4). Uncyclized linear Ub4ix showed no inhibition, highlighting the requirement of the cyclic topology for binding to Ub.
Figure 4. Ub4ix cyclic peptide inhibits 26S proteasomal activity in vitro.
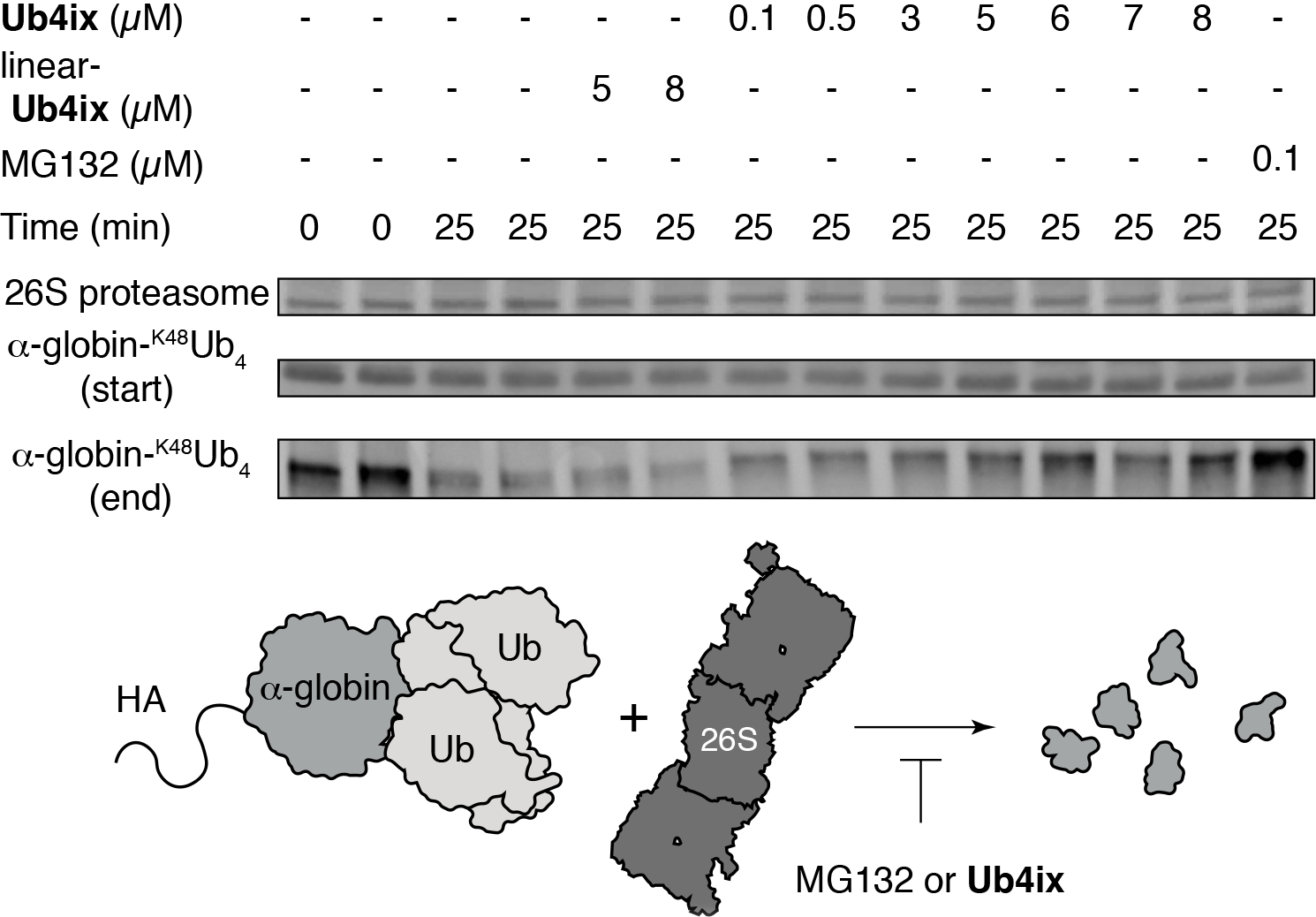
HA-α-globin-K48Ub4 (5 μM) and 26S proteasome (150 nM) were combined in the presence and absence of cyclic peptide Ub4ix, linear-Ub4ix or MG132. Before and after incubation, products were separated on SDS-PAGE gel, electro-blotted and probed with anti-HA antibody. Linear-Ub4ix offers no protection to HA-α-globin-K48Ub4 from the activity of the proteasome, resulting in degradation. Cyclic peptide Ub4ix, at a similar concentration to the HA-α-globin-K48Ub4 substrate (5 μM) can protect it from degradation by the proteasome, a similar result to the adding the direct proteasome inhibitor MG132.
Ub4ix is able to enter into HeLa cells.
In order to assess the feasibility of using Ub4ix in cultured cells, we next tested whether Ub4ix is able to cross the cellular membrane of human cells. We synthesized Ub4ix labeled with fluorescein (Fig. 5a, Supplementary Fig. 18), and used live cell imaging to monitor its entrance into HeLa cells. The uptake of labeled Ub4ix was apparent as early as 4 hours (Fig. 5b). Following its addition to the culture medium, with further intracellular marked accumulation, throughout the following 48 hours (Fig. 5b). Comparing the uptake of fluorescein-Ub4ix with fluorescein alone in three cell lines (HeLa, U2OS osteosarcoma and U87 primary glioblastoma cells), fluorescein-Ub4ix exhibited considerable cell permeability (Fig. 5c). In addition, we screened the cellular uptake fluorescein-Ub4ix compared to free fluorescein, after incubation for 16 hr (Fig. 5d). Ub4ix was quickly and efficiently distributed within cells, allowing tests in more representative biological models without further modification or special delivery systems.
Figure 5. Uptake of Ub4ix by living cells and effect on ubiquitination.
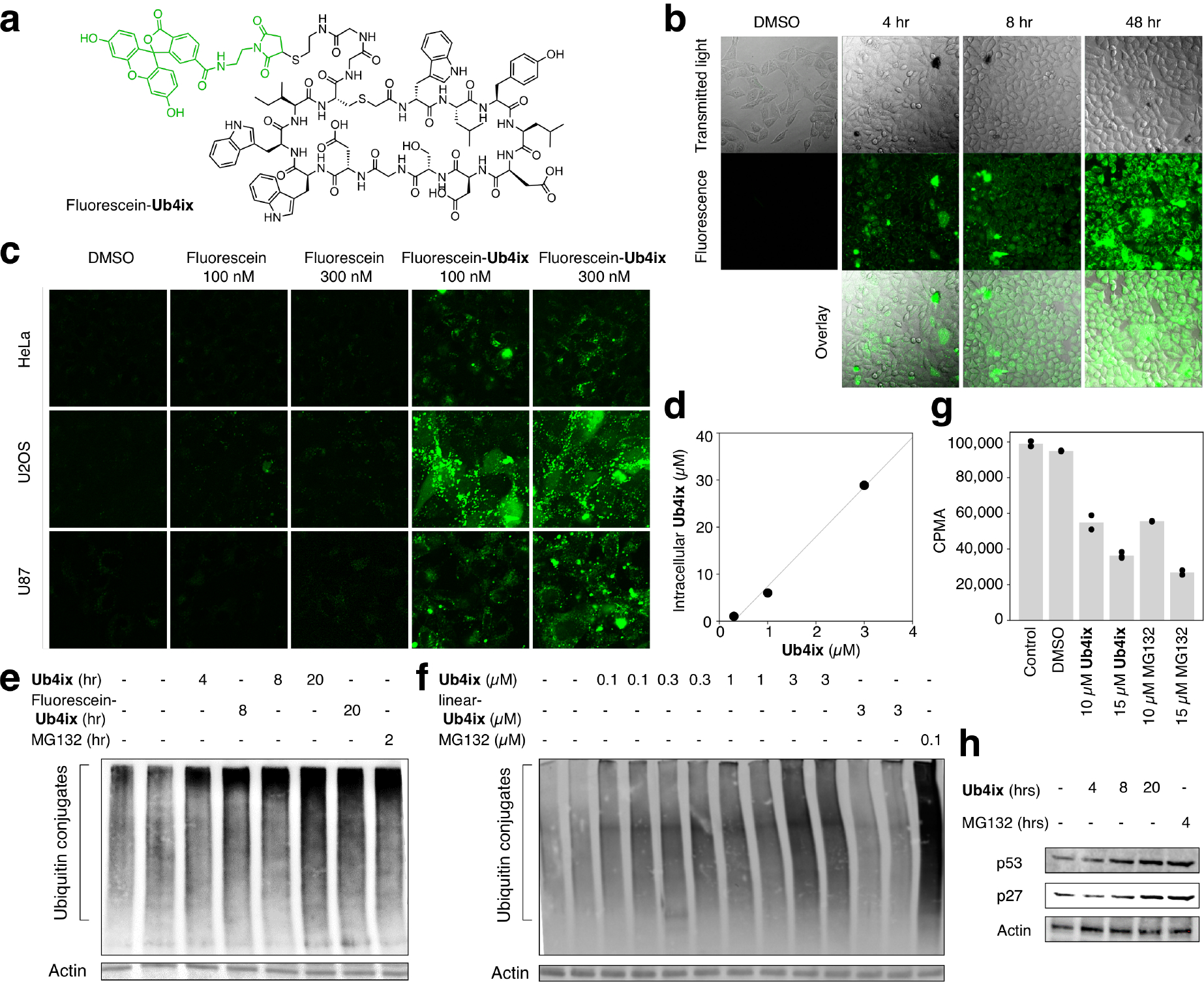
a) Fluorescein-Ub4ix structure. b) Ub4ix can enter cells. HeLa cells were incubated with fluorescein-Ub4ix cyclic peptide (20 μM). Internalization of the peptide into the cells was monitored at the indicated time points. c) In three cell lines, HeLa, U2OS osteosarcoma and U87 primary glioblastoma cells, fluorescein-Ub4ix is retained in cells after washing, whereas fluorescein alone is lost. d) Screening of the cellular uptake fluorescein-Ub4ix after incubation for 16 hr shows significant retention inside cells. e) Ub4ix induced the accumulation of Ub-conjugates. HeLa cells were incubated in the presence of Ub4ix, Fluorescein-Ub4ix, or MG132 for the indicated time-periods. f) Ub4ix induced the accumulation of Ub-conjugates in a dose dependent manner. Cells were lysed, resolved using SDS-PAGE, proteins were transferred to a nitrocellulose membrane, and the membrane was blotted using an antibody against Ub-conjugates and Actin. g) Ub4ix inhibits protein breakdown inside cells. HeLa cells were labeled with radioactive 35S-Met and 35S-Cys, and were then chased for 4 hr in “cold” media. Cells were either non-treated, or incubated in the presence of DMSO, Ub4ix, or MG132. Radioactivity levels (counts per min, CPMA) from protein breakdown during the chase phase, show that Ub4ix and MG132 inhibit. Bars represent mean values. h) Ub4ix causes the accumulation of proteins that are normally turned over by the UPS. HeLa cells were incubated in the presence of 10 μM Ub4ix or 10 μM MG132 for the indicated times. Cells were lysed, resolved using SDS-PAGE, proteins were transferred to a nitrocellulose membrane, and the membrane was blotted using an antibody against p53, p27 and Actin.
Ub4ix activity causes accumulation of Ub-conjugates
Under basal conditions, the levels of Ub-conjugates reflect a dynamic steady state between the enzymatic tagging of Ub to proteins on the one hand, with the removal of conjugated Ub moieties by DUBs, and subsequent degradation by the proteasome, on the other. We have shown in vitro that the cyclic peptide Ub4ix can protect Ub-chains from cleavage by DUBs, and also prevent recognition by the proteasome. Both effects would be predicted to prevent degradation, and lead to the accumulation, of proteins tagged with K48-Ub chains in cells. First we confirmed that Ub4ix does not affect protein synthesis (Supplementary Fig. 19). Then, we monitored the cellular level of Ub-conjugates in cells upon treatment with Ub4ix. We were able to demonstrate a time and dose dependent elevation in Ub-conjugates following treatment with Ub4ix, both in its free and fluorescein labeled forms (Fig. 5e and f), a similar result to treating with the direct proteasome inhibitor MG132 (Fig. 5e and f).
In order to directly measure the effect of Ub4ix on proteolysis, we radiolabeled cellular proteins, followed by a chase experiment in the presence of either Ub4ix or the proteasome inhibitor MG132. When compared to non-treated cells, or ones treated with DMSO, protein breakdown in cells treated with Ub4ix was markedly reduced, a similar result to those observed in the presence of MG132 (Fig. 5g). Both agents show a dose-dependent effect on cellular protein degradation. We validated this inhibitory effect on the breakdown of proteins known to be proteasomal substrates; the proteins p2742 and p5343. In the presence of the Ub4ix, p53 and p27 were accumulated over time, up to a similar level to those observed in the presence of MG132 (Fig. 5h).
Cyclic peptides induce apoptosis
Given that Ub4ix has a pronounced inhibitory effect on the UPS system, it may also be able to inhibit cell growth and induce apoptosis in cancer cells. To test this, HeLa cells were treated with each cyclic peptide, and cell growth was found to be dramatically suppressed when viability was assessed using an MTT (3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) assay. We noted that Ub4ix was the most effective in suppressing growth (Fig. 6a). The effects of the cyclic peptides on apoptosis were also evaluated using fluorescence-activated cell sorting (FACS) analysis. Intriguingly, Ub4ix at 10 μM exhibited clear increase in apoptosis after 24 hr and 48 hr treatment, similar to the direct proteasome inhibitor MG132 (Fig. 6b).
Figure 6. Ub4ix reduces cell viability and induces apoptosis.
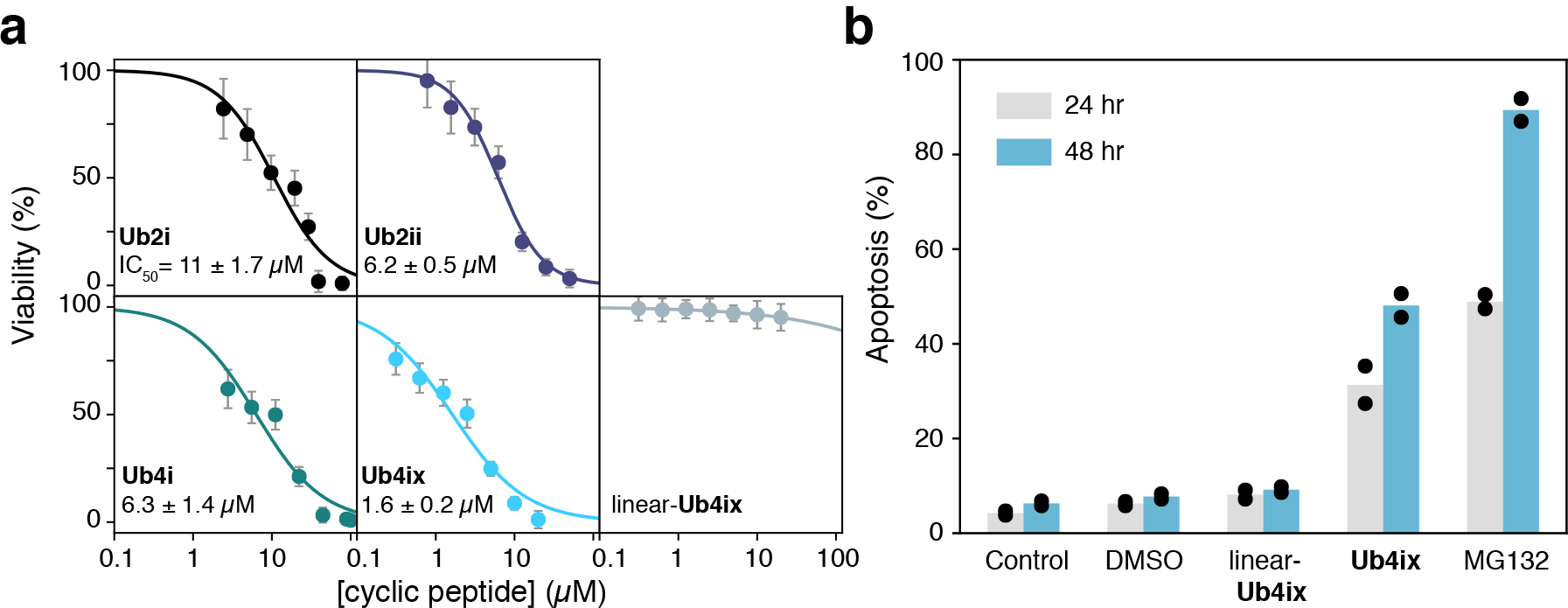
a) Cyclic peptides reduce HeLa cells viability, as measured by MTT assay. Ub4ix inhibits the cell viability of HeLa cells with an IC50 of 1.6 μM after 48 hr incubation. Viability was measured after 72 hr for Ub2i, Ub2ii, Ub4i and linear-Ub4ix. Error bars represent SD of technical replicates (n=16). b) Ub4ix induces apoptosis in HeLa cells. Cells were exposed to the Ub4ix cyclic or linear peptide, MG132 (all at 10 μM) for 24 and 48 hr, FACS was used to follow apoptosis and cumulative cell death, bars represent mean values.
Discussion
We have discovered de novo cyclic peptides that bind tightly and specifically to K48-linked Ub chains. Ub-binding domains with a similar function exist in biology, but typically these bind with weak affinities (μM Kd, values44), mainly to the L8-I44-V70 hydrophobic patch on the Ub surface, and display a narrower range of affinities for different Ub chain lengths and linkages. By contrast, antibodies have been discovered that can bind strongly (nM Kd), and specifically to polyUb chains by directly interacting with the isopeptide bond and its surrounding residues45. Surprisingly, the de novo macrocyclic peptides exhibit remarkably high affinity and linkage specificity like the antibodies. Yet, they interact with the hydrophobic surface of Ub frequently used by the weak-binding, poorly-selective natural Ub-binding domains. The affinity and selectivity profiles of these cyclic peptides are even more impressive given their significantly lower molecular masses relative to the protein domains and antibodies. Importantly, it is this smaller size that permits entry into cells46.
These cyclic peptides show in vitro and cellular activity comparable to a potent proteasome inhibitor, and can similarly trigger apoptosis in cancer cells. However, they act through a different mechanism. By binding to Ub chains directly and strongly, they interfere with the recognition and subsequent proteasomal processing of these chains. This different mechanism may have therapeutic advantages, as it can avoid the innate and acquired resistance associated with direct proteasome inhibitors47: it is difficult to mutate the Ub target, given its essentiality and its short, highly conserved sequence48. Importantly, unlike generic Ub-binding small molecules19, these cyclic peptides do not bind Ub1, K11- or K63-linked Ub chains strongly or specifically. Therefore, many Ub-directed cellular processes can continue unimpeded in the presence of these cyclic peptides. Our ability to synthesize different Ub chains with various lengths and linkage types, coupled with RaPID system, could lead to the discovery of novel cyclic peptides that interfere with specific cellular responses associated with these different Ub chains, to shed light on the full range of Ub signals in health and disease.
The combination of high-fidelity total protein synthesis and the cyclic peptide discovery RaPID system has produced de novo ligands capable of distinguishing between subtly different oligomers of the same protein domain. The control offered by this highly synthetic, in vitro approach will make other elaborate, post-translationally modified targets proteins accessible for ligand discovery. In particular, other Ub chains with different linkages and lengths, ubiquitinated proteins and Ub-like conjugates can be targeted to interfere with a wide range of cellular processes28. These cyclic peptides will be valuable tools to understand Ub signaling in health and disease, and open new opportunities in drug discovery related to the Ub system.
Supplementary Material
Acknowledgments:
A. Brik holds The Jordan and Irene Tark Academic Chair. H. Sun is supported at the Technion by a Technion-Guangdong Fellowship. This work was supported by the Japan Agency for Medical Research and Development, Basic Science and Platform Technology Program for Innovative Biological Medicine (JP18am0301001) to H. Suga, and by NIH grant GM065334 to D. Fushman. J.M.R was supported by Grants-in-aid for JSPS Fellows (P13766), Joint ANR-JST grant (ANR-14-JITC-2014-003 and JST-SICORP). We thank Ananya Majumdar for help with triple-resonance NMR experiments. A.C. is supported by the Dr. Miriam and Sheldon Adelson Medical Research Foundation (AMRF), the Israel Science Foundation (ISF), the German-Israeli Foundation for Research and Development (GIF) and a Professorship funded by the Israel Cancer Research Fund (ICRF).
Footnotes
Competing Interests: The authors declare no competing interests.
Data Availability: The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Glickman MH & Ciechanover A The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev 82, 373–428 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Hersko A & Ciechanover A The ubiquitin system. Annu. Rev. Biochem 67, 425–479 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Pickart CM Mechanisms underlying ubiquitination. Annu. Rev. Biochem 70, 503–33 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Komander D & Rape M The Ubiquitin Code. Annu. Rev. Biochem 81, 203–229 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Gopinath P, Ohayon S, Nawatha M & Brik A Chemical and semisynthetic approaches to study and target deubiquitinases. Chem. Soc. Rev 45, 4171–4198 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Ikeda F & Dikic I Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 9, 536–42 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finley D Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem 78, 477–513 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reyes-Turcu FE & Wilkinson KD Polyubiquitin binding and disassembly by deubiquitinating enzymes. Chem. Rev 109, 1495–1508 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang X & Dixit VM Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 26, 484–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickart CM & VanDemark AP Opening doors into the proteasome. Nat. Struct. Biol 7, 999–1001 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Adams J The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5, 417–421 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Goldberg AL Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol 199, 583–588 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groll M, Berkers CR, Ploegh HL & Ovaa H Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 14, 451–456 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Richardson PG et al. (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352, 2487–2498. [DOI] [PubMed] [Google Scholar]
- 15.Deshaies RJ Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 12, 94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee DH & Goldberg AL Proteasome inhibitors : valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Harrigan JA, Jacq X, Martin NM & Jackson SP Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov 17, 57–77 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verma R et al. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 306, 117–120 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Nakasone MA et al. Structural Basis for the Inhibitory Effects of Ubistatins in the Ubiquitin-Proteasome Pathway. Structure. 25, 1839–1855 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye Y et al. Ubiquitin chain conformation regulates recognition and activity of interacting proteins. Nature 492, 266–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castañeda CA et al. Linkage-specific conformational ensembles of non-canonical polyubiquitin chains. Phys. Chem. Chem. Phys 18, 5771–5788 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jongkees S. a. K., Hipolito CJ, Rogers JM & Suga H Model foldamers: applications and structures of stable macrocyclic peptides identified using in vitro selection. New J. Chem 39, 3197–3207 (2015). [Google Scholar]
- 23.Zorzi A, Deyle K & Heinis C Cyclic peptide therapeutics: past, present and future. Curr. Opin. Chem. Biol 38, 24–29 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Goto Y, Katoh T & Suga H Flexizymes for genetic code reprogramming. Nat. Protoc 6, 779–790 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Goto Y et al. Reprogramming the initiation event in translation for the synthesis of physiologically stable cyclic peptides. ACS Chem. Biol 3, 120–129 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Yamagishi Y et al. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol 18, 1562–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Jongkees SAK et al. Rapid Discovery of Potent and Selective Glycosidase-Inhibiting De Novo Peptides. Cell Chem. Biol 24, 381–390 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Mali SM, Singh SK, Eid E & Brik A Ubiquitin signaling: chemistry. comes to the rescue. J. Am. Chem. Soc 139, 4971–4986 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Orlowski RZ The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ. 6, 303 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Maki CG, Huibregtse JM & Howley PM In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res. 56, 2649–2654 (1996). [PubMed] [Google Scholar]
- 31.Li B & Dou QP Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc. Natl. Acad. Sci. U. S. A 97, 3850–5 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lloyd RV et al. P27Kip1: a Multifunctional Cyclin-Dependent Kinase Inhibitor With Prognostic Significance in Human Cancers. Am. J. Pathol 154, 313–23 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaziri SAJ et al. Inhibition of proteasome activity by bortezomib in renal cancer cells is p53 dependent and VHL independent. Anticancer Res. 29, 2961–2969 (2009). [PMC free article] [PubMed] [Google Scholar]
- 34.Ajish Kumar KS, Haj-Yahya M, Olschewski D, Lashuel HA & Brik A Highly efficient and chemoselective peptide ubiquitylation. Angew. Chem. Int. Ed 48, 8090–8094 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Bavikar SN et al. Chemical synthesis of ubiquitinated peptides with varying lengths and types of ubiquitin chains to explore the activity of deubiquitinases. Angew. Chem. Int. Ed 51, 758–763 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Eddins MJ, Varadan R, Fushman D, Pickart CM & Wolberger C Crystal Structure and Solution NMR Studies of Lys48-linked Tetraubiquitin at Neutral pH. J. Mol. Biol 367, 204–211 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Varadan R, Assfalg M, Raasi S, Pickart C & Fushman D Structural determinants for selective recognition of a Lys48-linked polyubiquitin chain by a UBA domain. Mol. Cell 18, 687–698 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Mevissen TET et al. OTU Deubiquitinases Reveal Mechanisms of Linkage Specificity and Enable Ubiquitin Chain Restriction Analysis. Cell 154, 169–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Renatus M et al. Structural Basis of Ubiquitin Recognition by the Deubiquitinating Protease USP2. Structure 14, 1293–1302 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stanley M & Virdee S Chemical ubiquitination for decrypting a cellular code. Biochem. J 473, 1297–1314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bremm A, Freund SMV & Komander D Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat. Struct. Mol. Biol 17, 939–947 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pagano M et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 269, 682–5 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Devine T & Dai M-S Targeting the ubiquitin-mediated proteasome degradation of p53 for cancer therapy. Curr. Pharm. Des 19, 3248–62 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raasi S, Varadan R, Fushman D & Pickart CM Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat. Struct. Mol. Biol 12, 708–714 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Newton K et al. Ubiquitin Chain Editing Revealed by Polyubiquitin Linkage-Specific Antibodies. Cell 134, 668–678 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Pye CR et al. Nonclassical Size Dependence of Permeation Defines Bounds for Passive Adsorption of Large Drug Molecules. J. Med. Chem 60, 1665–1672 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lü S & Wang J The resistance mechanisms of proteasome inhibitor bortezomib. Biomark. Res 1, 13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roscoe BP, Thayer KM, Zeldovich KB, Fushman D & Bolon DNA Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J. Mol. Biol 425, 1363–1377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.