

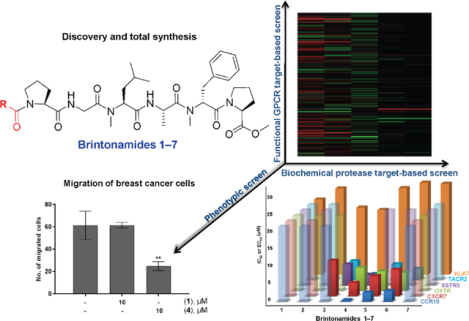
Abstract
Five novel modified linear peptides named brintonamides A‒E (1‒5) were discovered from a marine cyanobacterial sample collected from Brinton Channel, Florida Keys. The total synthesis of 1‒5 in addition to two other structurally related analogues (6 and 7) was achieved, which provided more material to allow rigorous biological evaluation and SAR studies. Compounds were subjected to cancer-focused phenotypic cell viability and migration assays and orthogonal target-based pharmacological screening platforms to identify their protease and GPCR modulatory activity profiles. The cancer related serine protease kallikrein 7 (KLK7) was inhibited to similar extents with an IC50 near 20 μM by both representative members 1 and 4, which differed in the presence or lack of the N-terminal unit. In contrast to the biochemical protease profiling study, clear SAR was observed in the functional GPCR screens, where five GPCRs in antagonist mode (CCR10, OXTR, SSTR3, TACR2) and agonist mode (CXCR7) were modulated by compounds 1‒7 to varying extents. Chemokine receptor type 10 (CCR10) was potently modulated by brintonamide D (4) with an IC50 of 0.44 μM. We performed in silico modeling to understand the structural basis underlying the differences in the antagonistic activity among brintonamides towards CCR10. Due to the significance of KLK7 and CCR10 in cancer progression and metastasis we demonstrated the ability of brintonamide D (4) at 10 μM to significantly target downstream cellular substrates of KLK7 (Dsg-2 and E-cad) in vitro, and to inhibit CCL27-induced CCR10-mediated proliferation and the migration of highly invasive breast cancer cells.
Keywords: Marine cyanobacteria, proteases, GPCR, molecular modeling, breast cancer, metastasis
Graphical Abstract
Introduction
Cancer metastasis is considered the major cause of death among cancer patients. It is a complex multistep process that often starts when cancer cells penetrate the adjacent tissues, passing the basement membrane and extracellular matrix (ECM) to enter the vascular/lymphatic circulation. Once in circulation, cancer cells travel through and eventually exit by invading the basement membrane and ECM. They adhere at the new location and ultimately proliferate and produce secondary tumors.1 Therefore, understanding the mechanisms involved in metastasis is critical for the development of treatment strategies to limit tumor progression. The metastatic process involves the regulation of many factors, where the physical barrier represented by the basement membrane and ECM surrounding the cells is considered a critical rate-limiting step for cancer invasion and metastasis.1 One mechanism by which tumor cells overcome this physical barrier is mediated by the action of proteases, which are often upregulated to degrade ECM components or digest adhesive molecules, ultimately altering the tissue cohesiveness resulting in the dissemination of tumor cells. Among the various protease families, serine proteases have been extensively studied and reported to be aberrantly expressed and associated with invasion and metastasis of several cancers.2 One example includes Kallikrein-related peptidases, consisting of 15 members of serine proteases with varying degrees of sequence homology (40‒80%).3 They are secreted as inactive zymogens and later activated by the proteolytic cleavage of their pro-signal peptide via the action of other proteases or through autoactivation.3 Among the different members of this family of proteases, kallikrein 7 (KLK7) is a chymotryptic-like serine protease, originally named stratum corneum chymotryptic enzyme.4,5 It was initially identified in human skin extracts, and functions in the shedding and desquamation of the skin via degradation of the adhesive molecules desmogleins and coreneodesmosomes.6,7 Moreover, KLK7 transcripts and protein levels have been found to be overexpressed in several cancer types including ovarian, squamous cervical, breast, and pancreatic adenocarcinomas.8–12
In addition to proteases, recent studies report the involvement of G protein-coupled receptors (GPCRs) in cancer progression.13–16 GPCRs play important roles in regulating fundamental physiological functions essential to life, such as metabolism, neurotransmission, immune responses, and homeostasis.17 However, their dysregulated activities have been linked to many diseases including diabetes,18 hypertension,19 heart failure,20 and cancer.21,22 Their vital role is reflected by the number of current GPCR drug targets, as ~34% of all FDA approved drugs act at 108 unique GPCRs.23 Several studies report the implication of GPCRs in regulating the metastatic process of various tumors, in particular GPCR linked chemokine receptors and their ligands.15,24 Chemokine receptors are overexpressed in various tumors and linked with poor prognosis. For example, the chemokine receptor type 4 (CXCR4) is one of the major chemokine receptors found to be expressed in ~23 types of cancer cells,24,25 reported to be a regulator of breast cancer and involved in lymph node metastasis.26,27 Other receptors include chemokine receptor type 10 (CCR10), which is expressed and plays a role in the metastatic cascade of adult T-cell leukemia/lymphoma (ATLL) and melanoma.24,28 In human melanoma, the expression of CCR10 enhances the invasive ability of cancer cells resulting in their dissemination to lymph nodes.24,29 Therefore, targeting such receptors might provide a potential strategy for restricting tumor metastasis.
Marine cyanobacteria are a rich source of bioactive molecules with diverse activities. In addition of being prolific producers of protease inhibitors,30–32 marine cyanobacteria often produce structurally diverse compounds that resemble endogenous ligands of GPCRs. Examples include the cyanobacterial fatty acid amides with cannabinoid receptor activity,33–38 and recently the discovery of cyanobacterial fraction targeting serotonin receptor 2C.39 Herein we describe the isolation, structure determination and synthesis of five novel modified peptides and other structurally related analogues. Also, we report the biological evaluation of their dual protease and GPCR targeting activities and subsequently their effects on breast cancer migration.
Results
Isolation and structure elucidation
Samples of intertidal cyanobacterial mats (dominated by major filament types of Oscillatoriaceae) collected from Brinton Channel, near Summerland Key, Florida were extracted with a mixture of EtOAc:MeOH (1:1) following freeze-drying. The non-polar extract was then partitioned using different solvents, yielding 1.6 g of n-BuOH fraction, which was subjected to silica gel chromatography using a gradient system of increasing polarity (i-PrOH/CH2Cl2, MeOH) to afford 10 fractions. Based on the NMR profiles of those fractions, the fraction eluted with 50% i-PrOH/CH2Cl2 showed characteristic resonances of modified peptides. Further purification of this fraction by reversed-phase HPLC resulted in the isolation of five novel compounds termed brintonamides A‒E (1‒5) (Figure 1).
Figure 1.

Cyanobacterial-derived brintonamides A‒E (1‒5) with the differences among structures 1‒5 highlighted.
The structures of those isolated compounds (1‒5) were elucidated using a combination of 1D and 2D NMR experiments (1H NMR, 13C NMR, COSY, TOCSY, HSQC, HMBC, and ROESY). The 1H and 13C NMR profiles of brintonamides A‒E (1‒5) were characteristic of modified peptides with typical chemical shifts of α-protons (δH~ 4‒5 ppm), carbonyls (δC~ 165‒175 ppm), and exchangeable protons of amides (δH~ 6‒8). Also, O-methyl (δH~ 3.73) and N-methyl (δH~ 2.76‒3.0) signals were apparent. Further analysis of 2D NMR spectra confirmed the presence of the following units in all of the isolated compounds (1‒5): O-Me-Pro, N-Me-Phe, Ala, N-Me-Leu, Gly, and Pro. However, the compounds differ in their N-terminal unit. Those units were identified to be either 2-hydroxy-3-methylpentanoic acid (Hmpa), 2-hydroxyisovaleric acid (Hiva), N,N-Me2-Phe, or cinnamic acid. The sequence of these units was determined based on correlations observed in HMBC and ROESY spectra and further confirmed by MS-MS fragmentation (Figure S1).
HRESI/APCIMS of 1 suggested a molecular formula of C39H60N6O9 (m/z 779.4321 for [M + Na]+, 757.4491 for [M + H]+). The analysis of both 1D and 2D NMR spectra (Table S1) suggested the presence of two amide NH signals (δH 7.38 and 6.75), α-protons (δH 5.65–4.02), one O-methyl singlet (δH 3.71), two N-methyl singlets (δH 2.99 and 2.84), and free OH (δH 4.73). Furthermore, examination of 2D NMR spectra allowed the identification of Hmpa as a terminal unit connected to the following moieties: O-Me-Pro, N-Me-Phe, Ala, N-Me-Leu, Gly, and Pro, making the final structure of the linear peptide.
HRESI/APCIMS of 2 suggested a molecular formula of C38H58N6O9 (m/z 765.4172 for [M + Na]+, 743.4343 for [M + H]+), indicating one less methylene compared to 1. Compound 2 exhibited similarity to 1 in terms of 1H and 13C NMR chemical shifts. Further examination of COSY, TOCSY, HSQC, HMBC, and ROESY data revealed the presence of the same units as in 1, except for Hiva instead of Hmpa at the N-terminus (Table S2).
HRESI/APCIMS of 3 exhibited molecular ion peaks at m/z 954.5311 [M + Na]+ and 932.5504 [M + H]+ suggesting a molecular formula of C50H73N7O10. Analysis of 1H NMR and edited-HSQC revealed similar chemical shifts to 1 and 2 (Table S3). The major differences were the presence of an additional N,N-dimethyl unit (δH/C 2.40, 41.3), the slightly downfield shift of the α-proton of the Hmpa moiety (δH 4.72), and an additional aromatic group. Analysis of both COSY and HMBC spectra allowed the construction of the N,N-Me2-Phe residue, which is connected to the Hmpa unit by an ester linkage. The chemical shift of the α-proton of Hmpa in 3 is indicative of an ester compared to the chemical shift of free alcohol present in 1 and 2 (δH 4.72 versus ~4.29 and 4.14, respectively).
HRESI/APCIMS of compound 4 in the positive mode exhibited a molecular ion peak at m/z of 909.4765 [M + Na]+ suggesting a molecular formula of C48H66N6O10. Analysis of 1H NMR and edited-HSQC spectra revealed chemical shifts (Table S4) very close to 3, and therefore, it is not surprising that the linear backbone of both compounds was also very similar. The major difference between 3 and 4 is the elimination of the N,N-dimethyl moiety from 4 resulting in an alkene (δH/C 6.5/117.5 and δH/C 7.7/146.1), which is connected to the aromatic ring based on correlations observed in the HMBC spectrum.
HRESI/APCIMS of compound 5 exhibited a molecular ion peak at m/z of 909.4773 [M + Na]+ suggesting a molecular formula of C48H66N6O10. Compounds 5 and 4 have the same molecular weight, and analysis of 1H NMR and edited-HSQC spectra revealed very close chemical shifts (Table S5). Examination of 2D NMR spectra confirmed the presence of the same substructures; however, analysis of 1H NMR of both compounds suggested that 4 and 5 are isomers having different configurations of the cinnamic acid moiety. In compound 4, the methine protons at the terminal unit displayed a larger coupling constant (J = 16.1 Hz) suggesting a trans configuration; whereas, the methine protons in compound 5 displayed a smaller coupling constant (J = 12.6 Hz) indicative of cis configuration.
It is important to note that some signals of brintonamides were observed as doubled signals, which are due to the presence of rotamers rather than impurities.
To assign the absolute configurations of the stereocenters present in brintonamides (1‒5), portions of 1‒5 (100 μg) were hydrolyzed using 6N HCl (110 °C, 24 h) and subjected to enantioselective HPLC-MS analysis. The analysis revealed retention times corresponding to L-Ala, L-Pro, N-Me-L-Leu, and N-Me-D-Phe, D-Hiva, and N,N-Me2-L-Phe by comparison with authentic standards. The configuration of the Hmpa unit was established by chiral HPLC analysis under different conditions using CuSO4 and was found to be (2R,3S)-Hmpa.
Total Synthesis of Brintonamides and Other Structurally Related Analogues
Because of the limited amounts of compounds 1‒5 available for biological studies through isolation, the total synthesis was pursued, which provided sufficient material for further biological investigations. Synthesis also confirmed that our structural assignments were correct, by comparison of the NMR spectra of the synthetic compounds with those of the natural products obtained through isolation. The synthesis commenced with the preparation of hexapeptide 9 by use of the standard Fmoc-based solid phase synthetic method (Scheme 1). Elongation of the peptide chain was accomplished by stepwise coupling of the required amino acids promoted by O-(7-azabenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU). After TFA-mediated cleavage of the peptide from the resin, esterification of the resulting acid 10 with tetramethylsilyldiazomethane afforded the corresponding peptide methyl ester 11. Finally, removal of the Fmoc group from 11 followed by coupling of the resulting amine with acid 12 or 13 under HATU conditions furnished brintonamides A (1) (82% yield) and B (2) (84% yield), as shown in Scheme 1 (for the detailed synthetic procedures, check the supporting information; all the synthetic samples were further purified by HPLC to give rise to samples with purity ≥ 95%).
Scheme 1.

Total synthesis of brintonamides A and B (1 and 2).
Brintonamides C‒E (3‒5) and two structurally related analogues, dehydro-brintonamide D/E (6) and dihydro-brintonamide D/E (7) were prepared from brintonamide A (1) under Yamaguchi mixed anhydride conditions (Scheme 2). Compounds 6 and 7, with different degrees of unsaturation, were designed to probe the contribution of the double bond and potential Michael acceptor in the cinnamic acid residue to activities in subsequent bioactivity studies. The esterification of acids 14‒17 with brintonamide A (1) proceeded smoothly and cleanly to furnish brintonamides D (4), E (5) and compounds 6 and 7 in good yields (61‒93%). Under identical conditions, however, brintonamide C (3) was obtained in only 39% yield.
Scheme 2.

Total synthesis of brintonamides C‒E (3‒5) and analogues 6 and 7.
Biological evaluation
In our search for new anticancer agents with activity against the highly invasive triple negative breast cancer, we initially assessed the cytotoxicity against MDA-MB-231 cells. Complementary to this phenotypic assay, we employed biochemical and functional screening strategies against druggable targets that are linked to cancer but not necessarily through a cytotoxic mechanism. We focused on proteases because marine cyanobacteria are known to produce modified peptides with propensity to inhibit various proteases, some of which play a role in breast cancer metastasis as we have shown in our prior studies.30,31,40 Additionally, as marine cyanobacteria often produce modified peptides that resemble endogenous ligands of GPCRs, and because the GPCR targeting activity of cyanobacterial secondary metabolites is largely unexplored, we carried out a functional screen against a panel of GPCRs as an orthogonal platform. Our study aimed at investigating the full potential of brintonamides in targeting invasive breast cancer.
Antiproliferative Activity of Brintonamides against Invasive Breast Cancer cells
The cytotoxic activity of brintonamides (1‒7) was evaluated against MDA-MB-231 breast cancer cells (Figure 2). Brintonamides A and B (1 and 2) did not show any cytotoxicity, while treatment with brintonamide C (3) reduced viability by 50% only at the highest concentration tested (100 μM). Brintonamides D and E (4 and 5) exhibited weak cytotoxic activity with IC50 values of 16.7 μM and 14.9 μM, respectively. It appeared that the cytotoxicity resulted from the incorporation of the Phe at the N-terminal end of the molecule, and the presence of the E-configured α,β-unsaturated carboxyl unit further enhanced the cytotoxic activity. Consequently, the synthetic analogues (6 and 7) were evaluated and were found to exhibit less potent cytotoxic activity than 4 and 5. Compound 6 resulted in 50% viability at 32 μM whereas the 7 had an IC50 of 60 μM. This preliminary SAR data suggests that role of the N-terminal residue in modulating the antiproliferative activity of these compounds (1‒7) and the presence of a hydroxy acid with free OH at the N-terminus abolishes the activity.
Figure 2.

The antiproliferative activity of brintonamides (1‒7) against MDA-MB-231 breast cancer cells. MDA-MB-231 cells were seeded (10,000 cells/well) in 96-well plates. Following 24 h incubation, the cells were treated with different concentrations of 1‒7, or solvent control for 48 h. The cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT assay). Data is presented as mean ± SD, n=3.
Protease Profiling Identifies Brintonamides as KLK7 Inhibitors
Because marine cyanobacteria are a prolific source of protease inhibitors, we thought to investigate whether brintonamides exhibit any antiproteolytic activity. To probe the activity and selectivity, brintonamides A and D (1 and 4) were profiled against a panel of 63 proteases in a dose-response format starting at 100 μM and the IC50 values were obtained (Figure 3A). Compounds 1 and 4 were selected as representative brintonamides based on their structural differences at the N-terminus. We considered proteases that were inhibited with IC50 values ~20 μM or less as potential hits. Brintonamide A (1) exhibited an inhibitory effect against the serine protease chymotrypsin and kallikrein 7 (KLK7) with IC50 values of 8.98 and 22.1 μM, respectively. Brintonamide D (4) exhibited a moderate inhibitory activity against three proteases in the low micromolar range (Table 1), including the serine proteases KLK7 and chymase, and the cysteine protease caspase 14. Despite the identification of several proteases as potential targets in the primary screen, we focused our studies on examining the inhibitory effects of brintonamides on KLK7 due to its established and growing significance in cancer metastasis.41–44 Despite the clinical significance of KLK7, there are few inhibitors reported in the literature with IC50 values in the micromolar range.45,46 From nature, a depsipeptide produced by Chondromyces bacteria was found to elicit a KLK7-specific inhibitory activity with IC50 value in the low nM range.47,48 From a clinical perspective, there are no tissue KLK7 inhibitors so far on the market. The only kallikrein inhibitor currently on the market is ecallantide, which was approved by FDA in October 2008 for the management of hereditary angioedema (HAE). Ecallantide is a potent, specific and reversible inhibitor of plasma kallikrein (Ki = 25 pM).49
Figure 3.

The protease inhibitory activity of brintonamides (1‒7). A) Profiling brintonamides A and D (1 and 4) against a panel of 63 proteases. Heat maps represent % enzyme activities at 10 μM final concentrations of 1 and 4. B) Dose response curves of the antiproteolytic activity of brintonamides 1‒7 against KLK7 as a follow up study. The dose-response is presented as % fold inhibition against solvent control (DMSO). Data is presented as mean ± SD.
Table 1.
IC50 values of brintonamides A and D (1 and 4), and positive controls against hits identified in the screen
| Protease | Kallikrein 7 | Chymase | Caspase 14 | Chymotrypsin |
|---|---|---|---|---|
| Brintonamide A (1) | 22.1 μM | 101 μM | 50.3 μM | 8.98 μM |
| Brintonamide D (4) | 18.9 μM | 23.2 μM | 10.1 μM | >100 μM |
| Gabexate mesylate | 20.4 μM | - | - | - |
| Chymostatin | - | 9.36 nM | - | 0.31 nM |
| IETD-CHO | - | - | 7.28 μM | - |
To validate KLK7 as a hit identified in the primary screen and to provide a preliminary SAR data, brintonamides A‒E (1‒5) along with the synthetic analogues (6 and 7) were tested in parallel against KLK7 in a dose-response format starting at 100 μM (Figure 3B). However, the compounds exhibited comparable IC50 values, lacking SAR against KLK7. We carried out enzyme kinetic studies, to investigate the mode of inhibition of KLK7, which revealed mixed mode of inhibition characterized by decreasing Vmax and increasing Km (Supporting Information, Figures S2). Molecular docking studies with the slightly most potent compound 4 suggested potential key interactions (Supporting Information, Figure S3). Brintonamide D (4) partially inhibited the cleavage of KLK7 downstream substrates, desmoglein-2 and E-cadherin, at 10 μM in a biochemical assay (Supporting Information, Figure S4); however, in KLK7, Dsg-2 and E-cad expressing MDA-MB-468 breast cancer cells (Supporting Information, Figure S5), compound 4 was unable to decrease Dsg-2 shedding despite inhibiting the migration of these cells similar to siRNAs targeting KLK7 (Supporting Information, Figure S6), suggesting that KLK7 is not sufficiently inhibited in a cellular context to cause the observed inhibitory phenotype on migration. The lack of cellular activity against KLK7 yet the promising inhibitory effects on migration indicated the involvement of other targets, which prompted us to apply an orthogonal functional screening platform to search for other potentially druggable targets predictive of the activity of brintonamides in breast cancer cells.
GPCR Profiling Identifies Five Additional Targets for Brintonamides
To identify relevant cellular activities, brintonamides A and D (1 and 4) were profiled against a panel of 241 GPCR targets (agonist, antagonist, and orphan) at 10 μM final concentration using cell-based functional assays (unlike the enzyme biochemical assays used for protease profiling) (Figure 4A). The screen was carried out using PathHunter β-Arrestin assay technology (Figure 4B; experimental section).
Figure 4.

GPCR profiling of brintonamides A and D (1 and 4) using cell-based functional screen. A) Heatmap showing the profiling data of brintonamides A and D (1 and 4) against the gpcrMax and orphan panels of GPCR targets. B) The GPCR assay principle based on the enzyme fragment complementation technology. C,D) Scatter plots showing the % activation or inhibition of 10 μM brintonamides A and D (1 and 4) against gpcrMax panel (agonist and antagonist modes). The hits identified in the screen with > 50% activity or inhibitions are labeled.
Although profiling brintonamide A (1) did not show any hits (Figures 4A and 4C), screening brintonamide D (4) at the same concentration (10 μM) revealed CXCR7 as the only hit in the agonist panel with 86% activity (Figure 4D). CXCR7 is a chemokine receptor type 7 which has a role in mediating tumor development, migration, and angiogenesis.50 Activating this receptor has a potential application in liver or lung fibrosis.51,52 In the antagonist panel, 4 modulated CCR10, TACR2, SSTR3, and OXTR activity with 98, 97, 77, and 76% inhibition, respectively (Figure 4D). The chemokine receptor type 10 (CCR10) is known to be expressed by melanocytes, plasma cells, and skin T cells.53 CCR10 has been reported to be involved in metastasis and T-cell mediated skin inflammation.54,55 Tachykinin receptor 2 (TACR2) is a receptor for tachykinin neuropeptide substance K (neurokinin A) that is involved in psychiatric diseases such as anxiety and depression.56 SSTR3 is a somatostatin receptor 3 which is expressed in the pancreatic islets. Somatostatin is known to suppress the production of pancreatic hormones such as insulin and therefore SSTR3 is a potential target for the management of type 2 diabetes.57,58 Oxytocin receptor (OXTR) is known to be expressed by myoepithelial cells of the mammary gland myometrium and CNS.59–62
When screened at higher concentration (20 μM), brintonamide D (4) displayed agonistic activity against CXCR7 with 91% activity. However, in the antagonist GPCR panel, several additional interactions between 4 and GPCR receptors were identified (Figure 4A). The top five hits identified were similar to those identified when screened at the lower (10 μM) final concentration but with higher % inhibitory activity, further validating these hits as targets of brintonamide D (4).
Next, to validate the hits identified in the primary screens, all brintonamides 1‒7 were tested in a dose-response manner starting at 20 μM, using an agonist and antagonist secondary screen against five targets (CXCR7, CCR10, OXTR, SSTR3, and TACR2) (Table 2).
Table 2.
IC50 or EC50 values of brintonamides (1‒7) against the GPCR hits identified in the primary screen
| Compound/GPCR target | CCR10b | CXCR7a | OXTRb | SSTR3b | TACR2b |
|---|---|---|---|---|---|
| Brintonamide A (1) | >20 | >20 | >20 | >20 | >20 |
| Brintonamide B (2) | >20 | >20 | >20 | >20 | >20 |
| Brintonamide C (3) | >20 | 10.46 | >20 | 6.11 | 5.51 |
| Brintonamide D (4) | 0.44 | 3.97 | 6.78 | 3.12 | 1.79 |
| Brintonamide E (5) | 2.71 | 5.92 | 5.76 | >20 | 4.61 |
| Compound 6 | 3.14 | 7.81 | 5.32 | >20 | 3.22 |
| Compound 7 | >20 | >20 | >20 | >20 | >20 |
EC50 agonist
IC50 antagonist (μM)
Brintonamides A and B (1 and 2) did not show any activity against the tested GPCR targets, suggesting that the N-terminus residue is important for activity and the presence of a hydroxy group abolishes the activity. Brintonamide C (3), bearing N,N-Me2-Phe residue at the N-terminus, exhibited a moderate activity against the following GPCR targets: CXCR7, SSTR3, and TACR2 with IC50 values in low micromolar range (10.4, 6.1, 5.5, respectively). However, 3 did not show any activity against CCR10 and OXTR. The regioisomeric compounds, brintonamides D and E (4 and 5), containing a cinnamic acid moiety at the N-terminus exhibited a moderate activity with IC50s in the low micromolar range. The trans isomer, brintonamide D (4), was the most potent analogue with submicromolar activity against CCR10 and was active against all five tested GPCR targets compared to the other brintonamides. The cis isomer, brintonamide E (5), exhibited comparable potency to 4 but was inactive against SSTR3, suggesting that the trans configuration is required for activity against SSTR3. The synthetic analogue 6, bearing an alkane instead of alkene, exhibited comparable potency to brintonamide E (5) and was also inactive against SSTR3. The activity of 6 suggests that the activity of brintonamides D and E (4 and 5), bearing a potential Michael acceptor, is most likely not mediated by covalent interaction with the targets. Finally, the synthetic analogue 7, containing phenylacetylene at the N-terminus, did not exhibit any activity against the tested GPCR targets indicating that the presence of an alkyne abolishes the activity.
Because CCR10 emerged as the best hit in our GPCR profiling platform, inhibited with submicromolar IC50 by brintonamide D (4), and due to its significance in cancer metastasis, we focused our studies on investigating the effects of CCR10 inhibition via brintonamides on cancer cell proliferation and migration.
Molecular Modeling of CCR10 Interactions
To gain insight into the structural basis underlying the differences in the inhibitory activity of brintonamides against CCR10, we carried out molecular docking experiments. Due to the lack of available crystal structure for CCR10, we developed and utilized a homology model from delta opioid-type receptor templates (more details in the Supporting Information).
The first visual inspection of the environment around brintonamide D (4) in the best selected model shows a highly charged binding pocket. There are 9 arginine residues (ARG 151, 238, 243, 247, 318, 322, 337, 338, and 345) in the vicinity of analogue D (4), closer than 5.0 Å to it, which accounts for 27% of the arginine content in CCR10 protein; three of them form hydrogen bonds with the ligand, including Arg247, Arg322, and Arg345, with H-bond lengths between 2.2 to 2.4 Å. The binding site is characterized by a beta-sheet which was found to be relatively stable in the same subfamilies of templates, but has movement across different families of GPCR structures. The residue Tyr227 in the middle of this beta sheet has π-π interaction with the aromatic ring on the modified side of brintonamide D (4) (Figure 5A). The perfectly parallel Tyr227 and phenyl group of 4 secures the ligand at the entrance of the binding site. This parallel stacking of aromatic rings seems to be the differentiator between the potency of the brintonamide ligands; only analogue D (4) is able to capture this key interaction, whereas the other compounds tend to engage with the other side of the binding pocket. In addition, the almost extended conformation of the ligand allows it to form three hydrogen bonds with the arginine residues in the deeper binding pocket (Figure 5A). These H-bonds are formed with arginine residues located on three adjacent helices. Several weaker H-bonds also exist with other arginine residues.
Figure 5.

The binding conformation of A) brintonamides D (4), B) Brintonamide E (5), C) Brintonamide A (1), D) Brintonamide B (2) within CCR10 model. The interactions with key amino acid residues in the binding site were shown. The compounds are displayed as stick representation.
In comparison, brintonamide E (5), which is the second most potent CCR10 antagonist, shows a completely bent conformation in a different environment (Figure 5B). While it forms two H-bonds with Arg338, it is surrounded by a more diverse set of residues ranging from Arg151, 232, 337, and 338 on one side of the ligand, to negatively charged Glu4 and 19, as well as the polar residues Gln8 and 83, Thr77, and Ser78 on the other side of the ligand. Interestingly, the two phenyl groups in compound E (5) are parallel-stacked and form π-π interactions. Analogue 6 with IC50 value of the same order as brintonamide E (5) demonstrates a bent binding pose similar to its counterpart (Figure S7A). It also captures the same two hydrogen bonds with the same residue, Arg338. For compound 7, the triple bond linker rigidifies the phenyl ring and is expected to have entropic penalty, compared to the previously discussed compounds. Due to its inflexibility at the modified side as well as its relatively long chain between the ester and phenyl groups, brintonamide 7 makes a very compact bent conformation to accommodate itself in the binding pocket, with perpendicular phenyl rings from its two sides coming together. No hydrogen bonding was observed for this analogue, presumably because of the mentioned spatial constraint (Figure S7B).
Brintonamides A and B (1 and 2) are different from other analogues in that they lack the aromatic ring at their modified side. Compound 1 occupies the same environment as analogue 4, which is surrounded by high number of arginine residues (Figure 5C). It also forms seven H-bonds with residues Arg151, Arg318, Arg322, and Arg337; nonetheless the absence of phenyl ring leads to its inability to interact with the key Tyr227 residue located on the characteristic beta sheet. This compound is in bent conformation, too (Figure 5C). Like brintonamide D (4), analogue B (2) is in extended form, while occupying the exact opposite side of the binding pocket compared to analogue D (4), is unable to form any H-bonds (Figure 5D). Finally, compound C (3) forms a hydrogen bond with Gln8 on the N-terminal part of CCR10 (Figure S7C). While the presence of aromatic ring on the modified side of this analog is supposed to potentially enable it to form π-π interactions with neighboring aromatic rings, the relatively bulky amine group next to it makes spatial hindrance, thus restricting the phenyl group to be at a small range of allowed rotamers. Therefore, it is expected to suffer from similar entropic cost as in the case of analogue 7.
Our modeling studies resulted in the development of a model derived from delta opioid-type receptor templates that is consistent with the SAR experimental data obtained for brintonamides (1−7), and enabled understanding the structural basis for the antagonistic activity of brintonamides towards CCR10, which might explain the cellular effects in breast cancer cells.
The Effects of CCR10 Inhibition on the Proliferation of Breast Cancer Cells
The role of CCR10 in promoting the proliferation of breast cancer cells was investigated using its ligand, CCL27. MDA-MB-231 cells (selected following the assessment of CCR10 expression in a panel of cancer cells; Supporting information, Figure S8) were stimulated with increasing concentrations of CCL27 under serum deprived conditions and the effect on proliferation was examined. Cell proliferation was increased in a dose-dependent manner in response to CCL27 stimulation, supporting a role of CCR10 activation in promoting the proliferation of breast cancer cells (Figure 6A). As brintonamide D (4) had no cytotoxic effects up to 10 μM (Figure 6B), we investigated the ability of our CCR10 antagonist 4 in inhibiting the CCL27 induced proliferation of MDA-MB-231 cells. Our data revealed dose-dependent inhibition of CCL27 induced proliferation in response to 4, with ~10% inhibition of proliferation at 10 μM (Figure 6C). Additionally, siRNA partial knockdown of CCR10 (50 nM, 60% knockdown) was found to reduce the proliferation by ~10%, validating the effects of CCR10 in promoting proliferation in the highly aggressive triple negative cancer cells (Figures 6D, Figure S9).
Figure 6.

The effect of targeting CCR10 on the proliferation of MDA-MB-231 cells. The effect of CCL27 (A) and brintonamides (B) on the proliferation of MDA-MB-231 cells. MDA-MB-231 cells were incubated for 48 h in the presence of different concentrations of CCL27, brintonamide A (1), or brintonamide D (4) and the effects were compared to the solvent controls. C) The effect of brintonamides on the CCL27 induced proliferation of MDA-MB-231 cells. MDA-MB-231 cells (10,000 cells/well) were pretreated with different concentrations of 1 and 4 for 2 h, followed by treatment with CCL27 (1 μg/mL) for 48 h. D) The effect of CCR10 knockdown on the proliferation of MDA-MB-231 cells. MDA-MB-231 cells were incubated in the presence of 50 nM siCCR10 (60% knockdown efficiency; see Supporting Information Figure S9A) and the effect was compared to the negative control. Data is presented as mean ± SD, n = 3. The asterisks denote significance of P < 0.05 relative to solvent control using two tailed unpaired t test (* denotes P ≤ 0.05, **** denotes P ≤ 0.0001)
The Effects of CCR10 Inhibition on the Migration of Breast Cancer Cells
To assess the effects of targeting CCR10 on metastasis, we evaluated the effects of brintonamides A and D (1 and 4) on the migration of MDA-MB-231 cells. Brintonamide D (4) inhibited the migration by 60% at 10 μM whereas brintonamide A (1) was inactive (Figure 7A). In order to link the migration inhibitory activity of 4 with the inhibition of CCR10 and potentially other GPCR hits, we carried out siRNA knockdown of the GPCR hits (Figure S9). Our data revealed ~40% inhibition of migration by siCCR10 at 50 nM, whereas knocking down the expression of other target genes had no effect on migration (Figure 7B), thus, validating the role of CCR10 in mediating cancer progression and metastasis in invasive breast cancer.
Figure 7.

The effect of CCR10 antagonists and siCCR10 on the migration of MDA-MB-231 cells. A) MDA-MB-231 cells were incubated for 48 h in the presence of 10 μM brintonamide A (1) and D (4), and the effect was compared to the solvent control. B) MDA-MB-231 cells were incubated for 72 h in the presence of 50 nM siRNA (see Supporting Information Figure S9A), and the effect was compared to the negative control. The graph represents the number of migrated cells in each treatment group. The asterisks denote significance of P < 0.05 relative to solvent control using two tailed unpaired t test (** denotes P ≤ 0.01).
Discussion
Marine cyanobacteria are producers of structurally diverse modified peptides that are not necessarily cytotoxic but possess distinct pharmacological profiles and biological functions. In particular, linear modified peptides have been widely reported, some of which contain structural features that would allow the prediction of their activity such as the aspartic protease inhibitors grassystatins A–F31,63 bearing the Leu-derived statin unit; while others, such as brintonamides A–E (1−5), lack key structural feature which relates to a particular activity, hence hindering the prediction of their potential biological activity. We carried out the first total synthesis of these compounds (1−5) in addition to other analogues (6 and 7) which enabled rigorous biological evaluation and SAR studies. To aid the identification of the biological activity of brintonamides, we applied two screening approaches. Our initial protease profiling revealed inhibitory activity against KLK7; however, due to the weak enzymatic activity and lack of SAR among brintonamides, we applied for the first time GPCR profiling as an orthogonal platform, which enabled the identification of five additional targets. The best hit identified was CCR10, which was inhibited by submicromolar concentrations of 4 (Figure 8).
Figure 8.

Summary of the SAR data of brintonamides obtained from the hits identified in the screens. The graph represents the IC50 or EC50 values of brintonamides 1‒7 against the hits identified in the target based-screens. The x-axis represents brintonamides 1‒7 while the y-axis represents IC50 or EC50 values. The GPCR targets modulated by an IC50 or EC50 values greater than 20 μM are displayed as transparent bars.
CCR10 is a chemokine receptor encoded by the CCR10 gene, and its ligands are CCL27 and CCL28, known to be overexpressed and linked to poor prognosis in melanoma, cutaneous squamous cell carcinoma, and glioma.55,64–66 The chemokine receptors play critical roles in mediating tumor progression including proliferation and metastasis.15 Studies have shown that exposure of melanoma cells overexpressing CCR10 to CCL27 protects the cells from apoptosis15 and in glioma, downregulation of CCR10 was shown to significantly impair cell growth.55 As the metastasis process resembles leukocyte trafficking, which is regulated by chemokine receptors and their ligands, chemokines may play key role in regulating tumor cell migration and metastasis. Additionally, they are involved in organ selectivity and site-specific metastasis formation.14,67,68 It was reported that chemokine expression on specific sites enables directed migration of tumor cells expressing the corresponding receptors, enhancing their adhesive and invasive properties.14,67,68 The most studied example is CXCR4 and its ligand CXCL12, known to be highly expressed in breast cancer cells.15 The metastatic pattern in breast cancer involves lymph nodes, lung, liver, and bone marrow, where CXCL12 was reported to be preferentially expressed. Hence, breast cancer cells overexpressing CXCR4 leads to targeting their migration to these organs.14 In vivo neutralization of CXCR4/CXCL12 interactions significantly reduced the metastasis of breast cancer cells to the lung and regional lymph nodes.14 Malignant melanoma, overexpressing CCR10, exhibits a similar metastatic pattern to breast cancer but with frequent metastasis to the skin where CCL27 is known to be overexpressed.14 Despite the evidence supporting the involvement of chemokine receptors in metastasis, there are few studies focusing on the molecular mechanisms and signaling pathways by which chemokine receptors, in particular CCR10, mediate cancer progression and metastasis. One study reported that CCR10/CCL27 mediates proliferation and invasion in glioma via activation of the p-AKT signaling pathway.55 Apart from melanoma and glioma, we show the expression of CCR10 in invasive breast cancer cells and that our novel CCR10 antagonist, brintonamide D (4), reduces the proliferation and migration of MDA-MB-231 cells. Our preliminary GPCR profiling data highlights the importance of the N-terminal residue in modulating the selectivity profiles (Figure 8). Therefore, we utilized our SAR experimental data along with in silico modeling to understand the structural basis underlying the antagonistic activity towards CCR10. This will aid the future design of improved inhibitors through modifications of brintonamides’ scaffold and the N-terminal residue with the possibility of tuning the activity and selectivity towards CCR10 and other GPCR targets.
Conclusions
In conclusion, we demonstrated for the first time the discovery of cyanobacterial compounds with dual protease and GPCR modulatory activities, which may have a therapeutic potential in targeting invasive breast cancer. We believe that brintonamides are starting points for the development of not only protease inhibitors but selective GPCR antagonists. Given the significance of CCR10 in cancer progression and metastasis, future studies could be directed towards the design of improved small molecule antagonists of CCR10, which could be utilized as valuable probes to understand the downstream cellular pathways mediating the antimetastatic effects in breast cancer.
Experimental section
General Experimental Procedures.
1H and 2D NMR spectra for the isolated compounds (1‒5) in CDCl3 were recorded on a Bruker 600 MHz Avance II Spectrometer, using a 5-mm cryogenic probe for indirect detection (Bruker Cryoprobe TXI). The 1H and 13C NMR spectra for the synthetic compounds (1‒7) in CDCl3 were recorded on Agilent VNMRS-600 MHz, 5-mm cold probe spectrometer. Spectra were referenced to residual solvent signals [δH/C 7.26/77.0]. HSQC experiments were optimized for 1JCH = 145 Hz and HMBC experiments were optimized for 1JCH = 7 Hz. HRESI/APCIMS data were recorded on an Agilent LC-TOF mass spectrometer equipped with an APCI/ESI multimode ion source detector in positive ion mode. ESIMS fragmentation data were obtained on an API 3200 triple quadrupole MS (Applied Biosystems) by direct injection with a syringe driver. LCMS data were obtained using API 3200 (Applied Biosystems) equipped with an HPLC system (Shimadzu). The optical rotation was measured using a Perkin-Elmer 341 polarimeter. The isolated compounds as well as the synthetic compounds were purified by HPLC and the purity was confirmed by 1H and 13C NMR, and LC-MS to be ≥ 95%.
Biological Material.
Samples of an intertidal cyanobacterial mat were collected over two years (2008–2009) from the surface of a rock seawall in Brinton Channel, near Summerland Key, Florida. The first collection was made May 4, 2008 (HL-CN2008–001), and then the sample was recollected during the week of May 21–30, 2009 (HL-CN2009–007) to obtain additional material. As is typical of many marine cyanobacterial mats, the sample contains multiple filament types. It is dominated by one filament type that is 5–7 μm wide and 8–15 μm long with square to rectangular cell shapes. The filaments have tapered ends and no heterocysts are visible. Other common filament types in the mat include Oscillatoria-like discoid cell that are 10–12 μm wide and 2 μm long with no heterocysts visible and both fine discoid filaments that are 2–3 μm wide and larger discoid filaments that are 25–30 μm wide. All filament types fit within the Oscillatoriaceae, a group of cyanobacteria currently under extensive revision. Voucher specimens are retained at the Smithsonian Marine Station at Ft. Pierce and are available for further investigation.
Extraction and Isolation.
Samples from May 2008 were freeze-dried, and the solid material was directly extracted with EtOAc-MeOH (1:1). This non-polar extract was filtered and concentrated in vacuo, then partitioned between hexanes and MeOH-H2O (80:20). The MeOH-H2O fraction was dried and further partitioned between n-BuOH and H2O. The n-BuOH-soluble fraction was dried and subjected to silica chromatography using a step gradient system of increasing i-PrOH in CH2Cl2. The material eluting with 10% i-PrOH was further purified by HPLC [column, YMC-Pack ODS-AQ, 250 × 10 mm; flow rate, 2.0 mL/min; PDA detection 200–800 nm] using a linear MeOH-H2O gradient (60–100% MeOH over 20 min, 100% MeOH for 20 min) to afford 1 (10 mg), 2 (0.9 mg), and 3 (1.3 mg) at tR 18.7 min, 17.5 min and 23.5 min, respectively.
The freeze-dried sample from May 2009 was extracted directly with the non-polar solvents EtOAc:MeOH (1:1) three times. The extract (18.6 g) was then partitioned between hexanes and 80% MeOH in H2O. The 80% aqueous MeOH fraction was evaporated and further partitioned between n-BuOH and H2O. The n-BuOH fraction (1.6 g) was concentrated in vacuo and later subjected to silica gel chromatography using a step gradient system starting the elution with CH2Cl2 followed by increasing i-PrOH in CH2Cl2, and finally with MeOH. Based on NMR-guided fractionation, The material eluting with 50% i-PrOH was selected and further purified by reversed-phase HLPC [column, YMC-Pack ODS-AQ, 250 × 10 mm; flow rate, 2.0 mL/min; PDA detection 200–800 nm] using a linear MeOH–H2O gradient (60–100% MeOH over 20 min, 100% MeOH for 20 min) to afford 1 (2.8 mg), 2 (0.4 mg), 3 (3.0 mg), 4 (0.7 mg), 5 (0.2 mg) at tR 18.7 min, 17.5 min, 23.5 min, 23.4 min, and 23.0 min, respectively.
Brintonamide A (1):
Colorless amorphous solid; [α]20D ‒37 (c 0.05, MeOH); 1H NMR, COSY, edited-HSQC, HMBC, ROESY and TOCSY (Table S1); HRESI/APCIMS m/z [M + Na]+ 779.4321 (calcd for C39H60N6O9Na, 779.4319), [M + H]+ 757.4491 (calcd for C39H61N6O9, 757.4500).
Brintonamide B (2):
Colorless amorphous solid; [α]20D ‒52 (c 0.08, MeOH); 1H NMR, COSY, edited-HSQC, HMBC, ROESY and TOCSY (Table S2); HRESI/APCIMS m/z [M + Na]+ 765.4172 (calcd for C38H58N6O9Na, 765.4163), [M + H]+ 743.4343 (calcd for C38H59N6O9, 743.4344).
Brintonamide C (3):
Colorless amorphous solid; [α]20D ‒8.3 (c 0.04, MeOH); 1H NMR, COSY, edited-HSQC, HMBC, ROESY and TOCSY (Table S3); HRESI/APCIMS m/z [M + Na]+ 954.5311 (calcd for C50H73N7O10Na, 954.5317), [M + H]+ 932.5504 (calcd for C50H74N7O10, 932.5497).
Brintonamide D (4):
Colorless amorphous solid; [α]20D ‒73 (c 0.05, MeOH); 1H NMR, COSY, edited-HSQC, HMBC, ROESY and TOCSY (Table S4); HRESI/APCIMS m/z [M + Na]+ 909.4765 (calcd for C48H66N6O10Na, 909.4765).
Brintonamide E (5):
Colorless amorphous solid; [α]20D ‒39 (c 0.05, MeOH); 1H NMR, COSY, edited-HSQC, HMBC, and ROESY (Table S5); HRESI/APCIMS m/z [M + Na]+ 909.4773 (calcd for C48H66N6O10Na, 909.4773).
Absolute Configuration
Acid Hydrolysis and Chiral Amino Acid Analysis by LC-MS.
A sample of compounds 1‒5 (100 μg each) was hydrolyzed with 6 N HCl (0.5 mL) at 110 °C for 24 h. The acid hydrolyzates were concentrated to dryness, reconstituted in 100 μL of H2O, and then analyzed by chiral HPLC [column: Chirobiotic TAG (250 × 4.6 mm), Supelco; solvent: MeOH/10 mM NH4OAc (40:60, pH 5.53); flow rate 0.5 mL/min; detection by ESIMS in positive ion mode (MRM scan)]. L-Ala, L-Pro, N-Me-L-Leu, N-Me-D-Phe, and N,N-Me2-L-Phe eluted at tR 7.5, 12.2, 13.5, 38, and 114 min, respectively. The retention times (tR , min; MRM ion pair, parent→product) of the authentic amino acids were as follows: L-Ala (7.5; 90→44), D-Ala (11.9), L-Pro (12.2; 116→70), D-Pro (33.3), N-Me-L-Leu (13.5; 146→100), N-Me-D-Leu (112), N-Me-L-Phe (22; 180.1→134.1), N-Me-D-Phe (38), N,N-Me2-L-Phe (114; 194→148), N,N-Me2-D-Phe (106). The compound-dependent MS parameters were as follows: Ala: DP 26, EP 3, CE 19, CXP 3; Pro: DP 32.4, EP 4, CE 21.8, CXP 2.8; N-Me-Leu: DP 28, EP 6, CE 17, CXP 3; N-Me-Phe: DP 29, EP 4, CE 20, CXP 3; N,N-Me2-Phe: DP 33, EP 4, CE 20, CXP 3. The source and gas-dependent MS parameters were as follows: CUR 50, CAD medium, IS 5500, TEM 750, GS1 65, and GS2 65. D-Hiva present in 2 was detected in the negative ion mode at tR 4.4 min. The retention times (tR, min; MRM ion pair, parent→product) of the authentic amino acids were as follows: L-Hiva (4.2; 117→70), D-Hiva (4.4). The compound-dependent MS parameters were as follows: DP −20, EP −4, CE −18, and CXP 1.7. The source and gas-dependent MS parameters were as follows: CUR 30, CAD medium, IS −4500, TEM 750, GS1 65, and GS2 65. The absolute configuration of the Hmpa unit in 1 was determined by chiral HPLC analysis under different conditions [column: CHIRALPAK MA (+) (50 х 4.6 mm); solvent: CH3CN/2mM CuSO4 (10:90); flow rate 1mL/min; detection by UV (254 nm)]. The retention times (tR, min) of the authentic standards were as follows: (2R,3S)-Hmpa (10.3), (2R,3R)-Hmpa (12.2), (2S,3R)-Hmpa (15), (2S,3S)-Hmpa (17.9). Under these conditions, all other units eluted at tR ˂ 8.0 min. The absolute configuration of the Hmpa unit in the hydrolyzate of brintonamide A (1) was found to be (2R,3S)-Hmpa.
Total Synthesis.
Detailed procedures for the total synthesis of brintonamides (1‒5) and analogues (6, 7) can be found in the supporting information.
Cell Culture.
MDA-MB-231, MDA-MB-468, HCT116, PANC-1 cells were propagated and maintained in Dulbecco’s modified Eagle medium (DMEM, Invitrogen), while LOX-IMVI, SNB-78 and SF-268 cells were propagated and maintained in Roswell Park Memorial Institute medium (RPMI 1640, Thermo Fisher Scientific). The media were supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT) and 1% antibiotic-antimycotic (Invitrogen) at 37 °C humidified air and 5% CO2.
Cell Viability Assay.
MDA-MB-231 cells were seeded in 96-well plates (10,000 cells/well). After 24 h incubation, the cells were treated with different concentrations of compounds (1‒7), CCL27 (376-CR/CF; R&D Systems), or solvent control (DMSO). Following 48 h of incubation, cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide according to the manufacturer’s instructions (Promega).
In the experiment evaluating the effect of CCR10 inhibition on the viability, MDA-MB-231 cells were seeded in 96-well plates (10,000 cells/well; serum free DMEM). After 24 h incubation, cells were pretreated with different concentrations of brintonamides A and D (1 and 4) for 2 h followed by treatment with CCL27 (1 μg/mL final concentration) for 2 h followed by treatment with different concentrations of brintonamides A and D (1 and 4). After 48 h incubation, cell viability was measured.
Protease Profiling and KLK7 Inhibition Assays.
The enzyme assays performed in the screen were carried out by Reaction Biology Corp. Brintonamides A and D (1 and 4) were screened in 10-dose IC50 with 3-fold serial dilution starting at 100 μM against 63 proteases in singlet. The control compounds were tested in a 10-dose IC50 with 3-fold serial dilution starting at 10 μM. To validate the hit identified from the primary screen, brintonamides A‒E (1‒5), synthetic analogues (6 and 7), and the positive control gabexate mesylate were tested against KLK7 in 10-dose IC50 with 3-fold serial dilution starting at 100 μM in duplicate. The protease activities were monitored as a time-course measurement of the increase in fluorescence signal from fluorescently-labeled peptide substrate and the initial linear portion of the slope (signal/min) was analyzed.
GPCR Profiling.
Brintonamides A (1; 10 μM) and D (4; 10 and 20 μM) were profiled against gpcrMAX panel biosensor assays (compounds were tested in the agonist and antagonist mode) and orphanMAX panel (compounds were tested in the agonist mode).The assays were performed by DiscoveRx Corporation (Fremont, CA) utilizing the PathHunter β-Arrestin enzyme fragment complementation (EFC) technology. The assay utilizes cells that are genetically engineered to overexpress the GPCR of interest as a fusion to ProLink (PK) (a small 44‒47 aa portion of β-galactosidase) and β-Arrestin as a fusion with enzyme acceptor (EA) (N-terminal deletion mutant of β-galactosidase, which is the rest of the enzyme). The removal of the 44 aa ProLink (PK) portion of β-galactosidase renders the enzyme inactive. The basic principle of the assay is based on the Enzyme Fragment Complementation (EFC) technology using β-galactosidase as the functional reporter. Briefly, GPCR activation stimulates the recruitment and binding of (EA)-tagged-β-Arrestin to the ProLink-tagged GPCR resulting in the complementation of the two enzyme fragments and restoring the activity of β-galactosidase that is capable of hydrolyzing a substrate and generating a chemiluminescent signal (Figure 4B).
For agonist determination, the sample was incubated with PathHunter cell lines (HEK293, CHO, U2OS) stably overexpressing the ProLink (PK)-tagged GPCR of interest and the Enzyme Acceptor (EA)-tagged β-Arrestin fusion proteins. The cells were seeded in a total volume of 20 μL in white 384-well plates and incubated at 37 °C for the appropriate time prior to testing to induce a response. Briefly, an intermediate dilution of sample stocks was prepared to generate 5X sample in assay buffer. To the cells, 5 μL of the 5X sample was added and further incubated at 37 °C or room temperature for 90 or 180 min.
For antagonist determination, the cells were pre-incubated with antagonist followed by agonist challenge at the EC80 concentration. To the cells 5 μL of 5X sample was added and incubated for 30 min at 37 °C or room temperature. Following the incubation period, 5 μL of 6X EC80 agonist prepared in assay buffer was added to the cells and incubated for 90 or 180 min at 37 °C or room temperature. The final assay vehicle concentration in both assay formats (agonist and antagonist) was 1%.
The assay signal detection was performed through the addition of 12.5 or 15 μL (50% v/v) of PathHunter detection reagent cocktail followed by 1 h incubation at room temperature. After the incubation period, the chemiluminescent signal was read using a PerKinElmer Envision™ instrument. The data analysis was performed using CBIS data analysis suite (ChemInnovation, CA). For the agonist mode assays, the percentage activity was calculated using the following formula: % Activity = 100% × (mean RLU of test sample ‒ mean RLU of vehicle control) / (mean MAX control ligand ‒ mean RLU of vehicle control). For antagonist mode assays, the percentage inhibition was calculated using the following formula: % Inhibition = 100% × (1 ‒ (mean RLU of test sample ‒ mean RLU of vehicle control) / (mean RLU of EC80 control ‒ mean RLU of vehicle control)).
Hit validation was performed by subjecting brintonamides A‒E (1‒5) and the synthetic analogues (6 and 7) to agonist and antagonist secondary screen utilizing five GPCR biosensor assays (Arrestin) against the following targets: CXCR7, CCR10, OXTR, SSTR3, and TACR2. The assays were performed at 10 point concentrations using 3-fold serial dilutions in duplicate.
Molecular Docking (CCR10).
Docking of brintonamides D into CCR10 requires the development of a homology model (details in the supporting information). Molecular docking of brintonamide ligands into the refined models was achieved in AutoDock VINA.69 For each model, the center of grid box was defined as the center of bound ligand, or the average of the centers of bound ligands in case the model was developed based on multiple templates. Maximum number of binding modes to generate was set as 100, with exhaustiveness of global search being set as 50. The top binding pose for each ligand was visually inspected and compared to other ligands; this was complemented by quantitative comparison of the trends in predicted binding affinities.
RNA Isolation, Reverse Transcription, and Real Time Quantitative Polymerase Chain Reaction (qPCR).
MDA-MB-231 cells (500,000 cells/well) were seeded in 6-well plates. After 24 h, RNA was isolated using RNeasy mini kit (Qiagen). cDNA synthesis was carried out using SuperScript II Reverse Transcriptase and Oligo (dT) (Invitrogen). qPCR was carried out after reverse transcription on a reaction solution (25 μL) prepared using 1 μL aliquot of cDNA, 12.5 μL of TaqMan gene expression assay mix, 1.25 μL of 20X TaqMan gene expression assay mix, and 10.25 μL of RNase-free water. The qPCR experiment was performed using ABI 7300 sequence detection system. The thermocycler program used was: 2 min at 50 °C, 10 min at 95 °C, and 15 s at 95 °C (40 cycles), and 1 min at 60 °C. KLK7 (Hs00192503_m1), CCR10 (Hs00706455_s1), and OXTR (Hs00168573_m1), SSTR3 (Hs00265633_s1), TACR2 (Hs00169052_m1) were used as target genes and GAPDH (Hs02758991_g1) was used as endogenous control.
siRNA Transfection and Assessment of Knockdown Efficiency.
MDA-MB-231 cells (300,000 cells/well) were seeded in 6-well plate. After 24 h, the cells were transfected for 48 h with 20 nM siRNA (KLK7: S11267) or 50 nM siRNA (CCR10: S194449; OXTR: S9946; SSTR3: S13503; TACR2: S13711; Neg Ctrl: AM17110; Life Technologies) using silentFect reagent according to the manufacturer’s instructions (BIO-RAD). RT-qPCR was performed to determine the extent of gene knockdown.
Transwell Migration Assay.
The effect on migration was examined using BD falcon cell culture inserts (353097) containing PET membrane with 8 μm pore size. Briefly, 5000 cells of MDA-MB-468 were suspended in complete medium (DMEM containing 10% FBS) and treated with different concentrations of brintonamide D (4) or solvent control. The cells (500 μL) were seeded into the inserts and 750 μL of complete medium containing the same concentration of 4 or solvent control was added into the other side of the membrane. The plate was incubated for 48 h (37 °C, 5% CO2). Following the incubation period, the non-migrated cells were removed using kimwipes and cotton swabs. Migrated cells were stained by crystal violet and the number was counted under the microscope. The assay was performed in triplicate.
In experiments evaluating the effects of siRNA knockdown of target genes on the migration of MDA-MB-231 cells, the cells were transfected with 20 nM siRNA (KLK7: S11267) or 50 nM siRNA (CCR10: S194449; OXTR: S9946; SSTR3: S13503; TACR2: S13711; Neg Ctrl: AM17110; Life Technologies) using silentFect reagent, seeded into the migration chambers, and incubated for 72 h.
Supplementary Material
Acknowledgment
The research was supported by the National Institutes of Health grant R01CA172310 and Shenzhen Peacock Plan (KQTD2015071714043444). We acknowledge Margaret O. James for assistance with enzyme kinetic studies.
Abbreviations
- APCI
atmospheric pressure chemical ionization
- ATLL
adult T-cell leukemia/lymphoma
- BCA
bicinchoninic acid
- DMEM
Dulbecco’s modified eagle medium
- CCR10
chemokine receptor type 10
- CCL 27
C-C motif chemokine ligand 27
- CCL28
C-C motif chemokine ligand 28
- CE
collision energy
- CUR
curtain gas
- CXCL12
C-X-C motif chemokine ligand 12
- CXCR4
chemokine receptor type 4
- CXCR7
chemokine receptor type 7
- CXP
collision cell exit potential
- DP
declustering potential
- Dsg-2
desmoglein-2, E-cad, E-cadherin
- ECM
extracellular matrix
- EP
entrance potential
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GPCR
G protein-coupled receptor
- GS1
gas 1
- GS2
gas 2
- HAE
hereditary angioedema
- HATU
O-(7-azabenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- Hiva
2-hydroxy isovaleric acid
- Hmpa
hydroxy-3-methyl pentanoic acid
- IS
ionspray voltage
- KLK7
kallikrein 7
- MRM
multiple reaction monitoring
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OXTR
oxytocin receptor
- p-AKT
phosphorylated protein kinase B
- RT-qPCR
real time quantitative polymerase chain reaction
- siRNA
small interfering RNA
- SSTR 3
somatostatin receptor 3
- TACR2
tachykinin receptor 2
- TEM
temperature
Footnotes
Supplementary Data
Includes NMR tables for brintonamides A‒E (1‒5) in CDCl3; experimental procedures for the synthesis of brintonamides A‒E (1‒5) and analogues (6, 7); results and discussions related to the KLK7 enzyme kinetic studies; effects of brintonamide D on downstream cellular substrates of KLK7 as well as the migration of breast cancer cells. Results and discussions related to CCR10 molecular modeling and expressions of CCR10 and other GPCR hits in cancer cells; experimental procedures for KLK7 experiments; relevant references; Figures S1‒S9; NMR spectra of brintonamides; HRMS of brintonamides. Molecular Formula Strings was provided as supplementary data. Coordinates for homology models were provided as supplementary data.
References
- (1).Martin Tracey A., Ye Lin, Sanders Andrew J., Lane Jane, and J. WG Cancer Invasion and Metastasis: Molecular and Cellular Perspective In adame Curie Bioscience Database [Internet]; Landes Bioscience: Austin (TX); pp 2000–2013. [Google Scholar]
- (2).Netzel-arnett S; Hooper JD; Szabo R; Madison EL; Quigley JP; Bugge H; Antalis TM Membrane Anchored Serine Proteases : A Rapidly Expanding Group of Cell Surface Proteolytic Enzymes with Potential Roles in Cancer. 2003, 22, 237–258. [DOI] [PubMed] [Google Scholar]
- (3).Yousef GM; Diamandis EP The New Human Tissue Kallikrein Gene Family: Structure, Function, and Association to Disease. Endocr. Rev 2001, 22 (February), 184–204. [DOI] [PubMed] [Google Scholar]
- (4).Proteinase ASHS Cloning , Expression , and Characterization of Stratum Corneum Chymotryptic Enzyme. 1994, 269 (30), 19420–19426. [PubMed] [Google Scholar]
- (5).Egelrud T Purification and Preliminary Characterization of Stratum Corneum Chymotryptic Enzyme: A Proteinase That May Be Involved in Desquamation. J. Invest. Dermatol 1993, 101, 200–204. [DOI] [PubMed] [Google Scholar]
- (6).Egelrud T; Lundström A A Chymotrypsin-like Proteinase That May Be Involved in Desquamation in Plantar Stratum Corneum. Arch. Dermatol. Res 1991, 283 (2), 108–112. [DOI] [PubMed] [Google Scholar]
- (7).Descargues P; Deraison C; Prost C; Fraitag S; Mazereeuw-Hautier J; D ‘alessio M; Ishida-Yamamoto A; Bodemer C; Zambruno G; Hovnanian A Corneodesmosomal Cadherins Are Preferential Targets of Stratum Corneum Trypsin- and Chymotrypsin-like Hyperactivity in Netherton Syndrome. J. Invest. Dermatol 2006, 126, 1622–1632. [DOI] [PubMed] [Google Scholar]
- (8).Santin AD; Cane’ S; Bellone S; Bignotti E; Palmieri M; De Las Casas LE; Roman JJ; Anfossi S; O’Brien T; Pecorelli S The Serine Protease Stratum Corneum Chymotryptic Enzyme (Kallikrein 7) Is Highly Overexpressed in Squamous Cervical Cancer Cells. Gynecol. Oncol 2004, 94 (2), 283–288. [DOI] [PubMed] [Google Scholar]
- (9).Shan SJC; Scorilas A; Katsaros D; Rigault de la Longrais I; Massobrio M; Diamandis EP Unfavorable Prognostic Value of Human Kallikrein 7 Quantified by ELISA in Ovarian Cancer Cytosols. Clin. Chem 2006, 52 (10), 1879–1886. [DOI] [PubMed] [Google Scholar]
- (10).Talieri M; Diamandis EP; Gourgiotis D; Mathioudaki K; Scorilas A Expression Analysis of the Human Kallikrein 7 (KLK7) in Breast Tumors: A New Potential Biomarker for Prognosis of Breast Carcinoma. Thromb. Haemost 2004, 91 (1), 180–186. [DOI] [PubMed] [Google Scholar]
- (11).Tanimoto H; Underwood LJ; Shigemasa K; Yan Yan MS; Clarke J; Parmley TH; O’Brien TJ The Stratum Corneum Chymotryptic Enzyme That Mediates Shedding and Desquamation of Skin Cells Is Highly Overexpressed in Ovarian Tumor Cells. Cancer 1999, 86 (10), 2074–2082. [PubMed] [Google Scholar]
- (12).Iakovlev V; Siegel ER; Tsao M-S; Haun RS Expression of Kallikrein-Related Peptidase 7 Predicts Poor Prognosis in Patients with Unresectable Pancreatic Ductal Adenocarcinoma. Cancer Epidemiol. Biomarkers Prev 2012, 21 (7), 1135–1142. [DOI] [PubMed] [Google Scholar]
- (13).Liu Y; An S; Ward R; Yang Y; Guo XX; Li W; Xu TR G Protein-Coupled Receptors as Promising Cancer Targets. Cancer Lett. 2016, 376 (2), 226–239. [DOI] [PubMed] [Google Scholar]
- (14).Müller a; Homey B; Soto H; Ge N; Catron D; Buchanan ME; McClanahan T; Murphy E; Yuan W; Wagner SN; Barrera JL; Mohar a; Verástegui E; Zlotnik a. Involvement of Chemokine Receptors in Breast Cancer Metastasis. Nature 2001, 410 (6824), 50–56. [DOI] [PubMed] [Google Scholar]
- (15).Sarvaiya PJ; Guo D; Ulasov IV; Gabikian P; Lesniak MS Chemokines in Tumor Progression and Metastasis. Oncotarget 2013, 4 (12), 2171–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Murakami T; Cardones AR; Hwang ST Chemokine Receptors and Melanoma Metastasis. J. Dermatol. Sci 2004, 36 (2), 71–78. [DOI] [PubMed] [Google Scholar]
- (17).Zalewska M; Siara M; Sajewicz W G Protein-Coupled Receptors: Abnormalities in Signal Transmission, Disease States and Pharmacotherapy. Acta Pol. Pharm. - Drug Res 2014, 71 (2), 229–243. [PubMed] [Google Scholar]
- (18).Hua Li J; Jain S; McMillin SM; Cui Y; Gautam D; Sakamoto W; Lu H; Jou W; McGuinness OP; Gavrilova O; Wess J A Novel Experimental Strategy to Assess the Metabolic Effects of Selective Activation of a G q -Coupled Receptor in Hepatocytes In Vivo. Endocrinology 2013, 154 (10), 3539–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ferguson SSG; Feldman RD; Taylor JA β-Adrenoceptors as Molecular Targets in the Treatment of Hypertension. Can. J. Cardiol 2014, 30, S3–S8. [DOI] [PubMed] [Google Scholar]
- (20).Zalewska M; Siara M; Sajewicz W G Protein-Coupled Receptors: Abnormalities in Signal Transmission, Disease States and Pharmacotherapy. Acta Pol. Pharm 71 (2), 229–243. [PubMed] [Google Scholar]
- (21).Liu Y; An S; Ward R; Yang Y; Guo X-X; Li W; Xu T-R G Protein-Coupled Receptors as Promising Cancer Targets. Cancer Lett. 2016, 376 (2), 226–239. [DOI] [PubMed] [Google Scholar]
- (22).Lee HJ; Wall B; Chen S G-Protein-Coupled Receptors and Melanoma. Pigment Cell Melanoma Res. 2008, 21 (4), 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hauser AS; Attwood MM; Rask-Andersen M; Schiöth HB; Gloriam DE Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov 2017, 16 (12), 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Singh S; Sadanandam A; Singh RK Chemokines in Tumor Angiogenesis and Metastasis. Cancer Metastasis Rev. 2007, 26 (3–4), 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Balkwill F Cancer and the Chemokine Network. Nat. Rev. Cancer 2004, 4 (7), 540–550. [DOI] [PubMed] [Google Scholar]
- (26).Smith MCP; Luker KE; Garbow JR; Prior JL; Jackson E; Piwnica-Worms D; Luker GD CXCR4 Regulates Growth of Both Primary and Metastatic Breast Cancer. Cancer Res. 2004, 64 (23), 8604–8612. [DOI] [PubMed] [Google Scholar]
- (27).Cabioglu N; Yazici MS; Arun B; Broglio KR; Hortobagyi GN; Price JE; Sahin A CCR7 and CXCR4 as Novel Biomarkers Predicting Axillary Lymph Node Metastasis in T1 Breast Cancer. Clin. Cancer Res 2005, 11 (16), 5686–5693. [DOI] [PubMed] [Google Scholar]
- (28).Harasawa H; Yamada Y; Hieshima K; Jin Z; Nakayama T; Yoshie O; Shimizu K; Hasegawa H; Hayashi T; Imaizumi Y; Ikeda S; Soda H; Soda H; Atogami S; Takasaki Y; Tsukasaki K; Tomonaga M; Murata K; Sugahara K; Tsuruda K; Kamihira S Survey of Chemokine Receptor Expression Reveals Frequent Co-Expression of Skin-Homing CCR4 and CCR10 in Adult T-Cell Leukemia/lymphoma. Leuk. Lymphoma 2006, 47 (10), 2163–2173. [DOI] [PubMed] [Google Scholar]
- (29).Simonetti O; Goteri G; Lucarini G; Filosa A; Pieramici T; Rubini C; Biagini G; Offidani A Potential Role of CCL27 and CCR10 Expression in Melanoma Progression and Immune Escape. Eur. J. Cancer 2006, 42 (8), 1181–1187. [DOI] [PubMed] [Google Scholar]
- (30).Al-Awadhi FH; Salvador LA; Law BK; Paul VJ; Luesch H Kempopeptin C, a Novel Marine-Derived Serine Protease Inhibitor Targeting Invasive Breast Cancer. Mar. Drugs 2017, 15 (9), 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Al-Awadhi FH; Law BK; Paul VJ; Luesch H Grassystatins D–F, Potent Aspartic Protease Inhibitors from Marine Cyanobacteria as Potential Antimetastatic Agents Targeting Invasive Breast Cancer. J. Nat. Prod 2017, 80 (11), 2969–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Al-Awadhi FH; Ratnayake R; Paul VJ; Luesch H Tasiamide F, a Potent Inhibitor of Cathepsins D and E from a Marine Cyanobacterium. Bioorg. Med. Chem 2016, 24 (15), 3276–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tan LT Bioactive Natural Products from Marine Cyanobacteria for Drug Discovery. Phytochemistry 2007, 68 (7), 954–979. [DOI] [PubMed] [Google Scholar]
- (34).Sitachitta N; Gerwick WH Grenadadiene and Grenadamide, Cyclopropyl-Containing Fatty Acid Metabolites from the Marine Cyanobacterium Lyngbya majuscula. J. Nat. Prod 1998, 61 (5), 681–684. [DOI] [PubMed] [Google Scholar]
- (35).Han B; McPhail KL; Ligresti A; Di Marzo V; Gerwick WH Semiplenamides A-G, Fatty Acid Amides from a Papua New Guinea Collection of the Marine Cyanobacterium Lyngbya semiplena. J. Nat. Prod 2003, 66 (10), 1364–1368. [DOI] [PubMed] [Google Scholar]
- (36).Montaser R; Paul VJ; Luesch H Marine Cyanobacterial Fatty Acid Amides Acting on Cannabinoid Receptors. Chembiochem 2012, 13 (18), 2676–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gutiérrez M; Pereira AR; Debonsi HM; Ligresti A; Di Marzo V; Gerwick WH Cannabinomimetic Lipid from a Marine Cyanobacterium. J. Nat. Prod 2011, 74 (10), 2313–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mevers E; Matainaho T; Allara’ M; Di Marzo V; Gerwick WH Mooreamide A: A Cannabinomimetic Lipid from the Marine Cyanobacterium Moorea bouillonii. Lipids 2014, 49 (11), 1127–1132. [DOI] [PubMed] [Google Scholar]
- (39).Lax NC; Ahmed KT; Ignatz CM; Spadafora C; Kolber BJ; Tidgewell KJ Marine Cyanobacteria-Derived Serotonin Receptor 2C Active Fraction Induces Psychoactive Behavioral Effects in Mice. Pharm. Biol 2016, 54 (11), 2723–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Al-Awadhi FH; Paul VJ; Luesch H Structural Diversity and Anticancer Activity of Marine-Derived Elastase Inhibitors: Key Features and Mechanisms Mediating the Antimetastatic Effects in Invasive Breast Cancer. ChemBiochem 2018, 19, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Emami N; Diamandis EP Utility of Kallikrein-Related Peptidases (KLKs) as Cancer Biomarkers. Clin. Chem 2008, 54 (10), 1600–1607. [DOI] [PubMed] [Google Scholar]
- (42).Obiezu CV; Diamandis EP Human Tissue Kallikrein Gene Family: Applications in Cancer. Cancer Lett. 2005, 224 (1), 1–22. [DOI] [PubMed] [Google Scholar]
- (43).Borgoño CA; Diamandis EP The Emerging Roles of Human Tissue Kallikreins in Cancer. Nat. Rev. Cancer 2004, 4 (11), 876–890. [DOI] [PubMed] [Google Scholar]
- (44).Schmitt M; Magdolen V; Yang F; Kiechle M; Bayani J; Yousef GM; Scorilas A; Diamandis EP; Dorn J Emerging Clinical Importance of the Cancer Biomarkers Kallikrein-Related Peptidases (KLK) in Female and Male Reproductive Organ Malignancies. Radiol. Oncol 2013, 47 (4), 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Freitas RF; Teixeira TSP; Barros TG; Santos JAN; Kondo MY; Juliano MA; Juliano L; Blaber M; Antunes OAC; Abrahão O; Pinheiro S; Muri EMF; Puzer L Isomannide Derivatives as New Class of Inhibitors for Human Kallikrein 7. Bioorg. Med. Chem. Lett 2012, 22 (19), 6072–6075. [DOI] [PubMed] [Google Scholar]
- (46).Oliveira JPC; Freitas RF; Melo LS de Barros TG; Santos JAN; Juliano MA; Pinheiro S; Blaber M; Juliano L; Muri EMF; Puzer L Isomannide-Based Peptidomimetics as Inhibitors for Human Tissue Kallikreins 5 and 7. ACS Med. Chem. Lett 2014, 5 (2), 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sotiropoulou G; Pampalakis G Targeting the Kallikrein-Related Peptidases for Drug Development. Trends Pharmacol. Sci 2012, 33 (12), 623–634. [DOI] [PubMed] [Google Scholar]
- (48).Krastel P Novartis Institutes for Biomedical Research Inc. Cyclic Depsipeptides. US Pat. Publ. US 2009, 156472, A1. [Google Scholar]
- (49).Farkas H; Varga L Ecallantide Is a Novel Treatment for Attacks of Hereditary Angioedema due to C1 Inhibitor Deficiency. Clin. Cosmet. Investig. Dermatol 2011, 4, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Stacer a C.; Fenner J; Cavnar SP; Xiao a; Zhao S; Chang SL; Salomonnson a; Luker KE; Luker GD Endothelial CXCR7 Regulates Breast Cancer Metastasis. Oncogene 2015, 35 (April), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cully M Lung Disease: CXCR7 Activation Overrides Lung Fibrosis. Nat. Rev. Drug Discov 2016, 15, 160. [DOI] [PubMed] [Google Scholar]
- (52).Ding B-S; Cao Z; Lis R; Nolan DJ; Guo P; Simons M; Penfold ME; Shido K; Rabbany SY; Rafii S Divergent Angiocrine Signals from Vascular Niche Balance Liver Regeneration and Fibrosis. Nature 2014, 505 (7481), 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Xiong N; Fu Y; Hu S; Xia M; Yang J CCR10 and Its Ligands in Regulation of Epithelial Immunity and Diseases. Protein Cell 2012, 3 (8), 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Daubeuf F; Jung F; Douglas GJ; Chevalier E; Frossard N Protective Effect of a Protein Epitope Mimetic CCR10 Antagonist, POL7085, in a Model of Allergic Eosinophilic Airway Inflammation. Respir. Res 2015, 16 (1), 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Chen L; Liu X; Zhang H-Y; Du W; Qin Z; Yao Y; Mao Y; Zhou L Upregulation of Chemokine Receptor CCR10 Is Essential for Glioma Proliferation, Invasion and Patient Survival. Oncotarget 2014, 5 (16), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Griebel G; Holsboer F Neuropeptide Receptor Ligands as Drugs for Psychiatric Diseases: The End of the Beginning? Nat. Rev. Drug Discov 2012, 11 (6), 462–478. [DOI] [PubMed] [Google Scholar]
- (57).Shah SK; He S; Guo L; Truong Q; Qi H; Du W; Lai Z; Liu J; Jian T; Hong Q; Dobbelaar P; Ye Z; Sherer E; Feng Z; Yu Y; Wong F; Samuel K; Madiera M; Karanam BV; Reddy VB; Mitelman S; Tong SX; Chicchi GG; Tsao KL; Trusca D; Feng Y; Wu M; Shao Q; Trujillo ME; Eiermann GJ; Li C; Pachanski M; Fernandez G; Nelson D; Bunting P; Morissette P; Volksdorf S; Kerr J; Zhang BB; Howard AD; Zhou YP; Pasternak A; Nargund RP; Hagmann WK Discovery of MK-1421, a Potent, Selective sstr3 Antagonist, as a Development Candidate for Type 2 Diabetes. ACS Med. Chem. Lett 2015, 6 (5), 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).He S; Ye Z; Truong Q; Shah S; Du W; Guo L; Dobbelaar PH; Lai Z; Liu J; Jian T; Qi H; Bakshi RK; Hong Q; Dellureficio J; Pasternak A; Feng Z; Dejesus R; Yang L; Reibarkh M; Bradley SA; Holmes MA; Ball RG; Ruck RT; Huffman MA; Wong F; Samuel K; Reddy VB; Mitelman S; Tong SX; Chicchi GG; Tsao KL; Trusca D; Wu M; Shao Q; Trujillo ME; Eiermann GJ; Li C; Zhang BB; Howard AD; Zhou YP; Nargund RP; Hagmann WK The Discovery of MK-4256, a Potent SSTR3 Antagonist as a Potential Treatment of Type 2 Diabetes. ACS Med. Chem. Lett 2012, 3 (6), 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kim SH; Bennett PR; Terzidou V Advances in the Role of Oxytocin Receptors in Human Parturition. Mol. Cell. Endocrinol 2017, 449, 56–63. [DOI] [PubMed] [Google Scholar]
- (60).Bale TL; Davis AM; Auger AP; Dorsa DM; McCarthy MM CNS Region-Specific Oxytocin Receptor Expression: Importance in Regulation of Anxiety and Sex Behavior. J. Neurosci 2001, 21 (7), 2546–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kimura T; Ito Y; Einspanier A; Tohya K; Nobunaga T; Tokugawa Y; Takemura M; Kubota Y; Ivell R; Matsuura N; Saji F; Murata Y Expression and Immunolocalization of the Oxytocin Receptor in Human Lactating and Non-Lactating Mammary Glands. Hum. Reprod 1998, 13 (9), 2645–2653. [DOI] [PubMed] [Google Scholar]
- (62).Reversi A; Cassoni P; Chini B Oxytocin Receptor Signaling in Myoepithelial and Cancer Cells. J. Mammary Gland Biol. Neoplasia 2005, 10 (3), 221–229. [DOI] [PubMed] [Google Scholar]
- (63).Kwan JC; Eksioglu EA; Liu C; Paul VJ; Luesch H Grassystatins A-C from Marine Cyanobacteria, Potent Cathepsin E Inhibitors That Reduce Antigen Presentation. J. Med. Chem 2009, 52 (18), 5732–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Kai H; Kadono T; Kakinuma T; Tomita M; Ohmatsu H; Asano Y; Tada Y; Sugaya M; Sato S CCR10 and CCL27 Are Overexpressed in Cutaneous Squamous Cell Carcinoma. Pathol. Res. Pract 2011, 207 (1), 43–48. [DOI] [PubMed] [Google Scholar]
- (65).Kühnelt-Leddihn L; Eisendle K; Müller H; Zelger B; Weinlich G Overexpression of the Chemokine Receptors CXCR4, CCR7, CCR9, and CCR10 in Human Primary Cutaneous Melanoma: A Potential Prognostic Value for CCR7 and CCR10? Arch. Dermatol. Res 2012, 304 (3), 185–193. [DOI] [PubMed] [Google Scholar]
- (66).Simonetti O; Goteri G; Lucarini G; Filosa A; Pieramici T; Rubini C; Biagini G; Offidani A Potential Role of CCL27 and CCR10 Expression in Melanoma Progression and Immune Escape. Eur. J. Cancer 2006, 42 (8), 1181–1187. [DOI] [PubMed] [Google Scholar]
- (67).Ben-Baruch A Organ Selectivity in Metastasis: Regulation by Chemokines and Their Receptors. Clin. Exp. Metastasis 2008, 25 (4), 345–356. [DOI] [PubMed] [Google Scholar]
- (68).Ben-baruch A Site-Specific Metastasis Formation. 2009, 3 (4), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Trott O; Olson AJ AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem 2010, 31 (2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.