

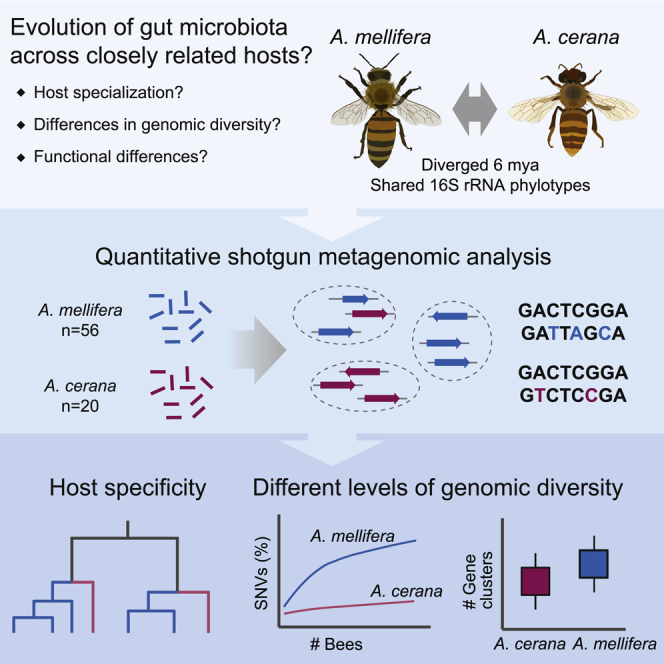
Summary
Most bacterial species encompass strains with vastly different gene content. Strain diversity in microbial communities is therefore considered to be of functional importance. Yet little is known about the extent to which related microbial communities differ in diversity at this level and which underlying mechanisms may constrain and maintain strain-level diversity. Here, we used shotgun metagenomics to characterize and compare the gut microbiota of two honey bee species, Apis mellifera and Apis cerana, which diverged about 6 mya. Although the host species are colonized largely by the same bacterial 16S rRNA phylotypes, we find that their communities are host specific when analyzed with genomic resolution. Moreover, despite their similar ecology, A. mellifera displayed a much higher diversity of strains and functional gene content in the microbiota compared to A. cerana, both per colony and per individual bee. In particular, the gene repertoire for polysaccharide degradation was massively expanded in the microbiota of A. mellifera relative to A. cerana. Bee management practices, divergent ecological adaptation, or habitat size may have contributed to the observed differences in microbiota genomic diversity of these key pollinator species. Our results illustrate that the gut microbiota of closely related animal hosts can differ vastly in genomic diversity while displaying similar levels of diversity based on the 16S rRNA gene. Such differences are likely to have consequences for gut microbiota functioning and host-symbiont interactions, highlighting the need for metagenomic studies to understand the ecology and evolution of microbial communities.
Keywords: metagenomics, gut microbiota, honey bee, Apis cerana, Apis mellifera, host associated, strain-level diversity, species, pan-genome
Graphical Abstract
Highlights
-
•
Metagenomics reveals differences in gut microbiota diversity beyond the 16S rRNA gene
-
•
Apis cerana and Apis mellifera harbor distinct species and strains in their gut
-
•
Diversity is much higher in A. mellifera per individual bee and within colonies
-
•
Major differences in functions are related to polysaccharide degradation
Bacteria have highly flexible gene content; functions of bacterial communities therefore depend on their strain-level composition. Using metagenomics, Ellegaard et al. find major differences in composition and diversity in the gut microbiota of two related honey bees, raising new questions on function and evolution of host-associated bacteria.
Introduction
Most bacteria live in complex communities, which typically encompass strains with highly variable gene content [1, 2, 3]. In host-associated bacterial communities, strain-level diversity can be substantial, despite the general assumption that genetic diversity destabilizes mutualistic interactions [4]. For example, multiple strains of a sulfur-oxidizing endosymbiont were found to co-colonize individual hosts of deep-sea mussels, presumably because they encode complementary functions [5, 6]. In contrast, strains of the human gut microbiota have been shown to segregate among individuals, resulting in host-specific genetic profiles [7, 8, 9]. However, despite the increased awareness of the existence and functional importance of strain-level diversity in host-associated bacterial communities, little is known about differences in diversity across host species or the underlying mechanisms that constrain and maintain diversity within and among hosts. This is largely due to (1) the limited ability of 16S rRNA gene amplicon sequencing to resolve strain-level diversity [10, 11, 12], (2) the technical challenges associated with the deep sequencing and analysis of bacterial genomes from complex natural communities [13], and (3) the difficulty to generate comparable datasets across host organisms.
Unlike most animals, eusocial corbiculate bees (honey bees, stingless bees, and bumble bees) have been shown to harbor a relatively simple, yet specialized, gut microbiota with a highly conserved taxonomic composition, consisting of up to 10 phylotypes, as based on 16S rRNA gene analyses (with phylotypes having >97% sequence identity in the 16S rRNA gene) [14, 15]. Some of these phylotypes are common across a wide range of social bees, suggesting they were acquired around the time when eusociality evolved in the bees [14]. Several studies based on genomic data have demonstrated an impressive amount of genetic flexibility within 16S rRNA phylotypes of the honey bee gut microbiota [7, 16, 17, 18, 19, 20, 21], indicating that major functional differences are encoded at the genomic level, as has also been shown to be the case for other bacteria [3, 22, 23]. Given these characteristics, the gut microbiota of social bees represents an emerging model for studies on genomic diversity and evolution of host-associated bacterial communities [7, 14, 15, 24].
Despite extensive horizontal gene transfer in bacteria [3], recent studies have shown that bacteria exist as discrete populations, which can be identified based on metagenomic read recruitment [25, 26, 27, 28] or gANI (genomic average nucleotide identity) [11, 29, 30]. Bacteria belonging to the same population generally have more than 95% gANI with each other, whereas bacteria from different populations typically have less than 90% gANI [11, 30]. These populations have therefore also been referred to as “sequence-discrete populations” (SDPs) or even “species” [11, 27, 30], although the latter term continues to be controversial for bacteria. For the western honeybee, Apis mellifera, a recent metagenomic study identified 1–4 SDPs per 16S rRNA phylotype in the gut microbiota [7]. It is therefore possible for bee species with similar phylotype-level gut microbiota composition to display major differences, due to changes occurring at the SDP or strain level (Figure 1A). Indeed, two previous studies based on amplicon sequencing data have provided evidence that the extent of diversity can differ between different species of social bees [14, 31]. However, comparative community-wide analyses, based on data with genome-level resolution, are lacking.
Figure 1.

Analysis of Community Composition and Diversity in Bacterial Communities
(A) Schematic phylogeny illustrating three levels of diversity, which can be analyzed for bacterial communities: phylotypes; sequence-discrete populations (SDPs); and strains. While amplicon sequencing of the 16S rRNA gene can be used to characterize the phylotype-level composition, analysis of SDPs or strains requires either genomic or metagenomic data [7, 11, 30]. SDPs have recently been proposed to represent bacterial species [11, 30], and even closely related strains can differ widely in gene content [3]. Therefore, bacterial communities that appear similar when analyzed using the 16S rRNA gene can potentially display major differences in composition, diversity, and gene content (as also illustrated by the Venn diagram).
(B) Relative abundance of phylotypes in A. mellifera and A. cerana for metagenomic samples analyzed in the current study. Most of the phylotypes are shared between both hosts. Shared phylotypes are indicated in blue and green colors and highlighted by black outline. Relative abundances correspond to approximately 90% of the host-filtered reads in all samples.
See also Figure S1.
In the current study, we perform a comparative metagenomic analysis of the gut microbiota of two closely related species of honey bees, Apis mellifera and Apis cerana. Based on molecular data, their last common ancestor has been dated to approximately 6 mya [32, 33, 34, 35], and previous 16S rRNA-based studies have shown that they are colonized mostly by the same 16S rRNA phylotypes [14]. With the exception of A. mellifera, all extant species of honey bees (genus Apis) are confined to Asia, pointing toward an Asian origin of the Apis genus [33]. Based on molecular analysis, A. mellifera expanded into its native range (Africa, Europe, and Western Asia) approximately 300,000 years ago [32]. However, A. mellifera was recently re-introduced to Asia by humans [36, 37], thereby bringing not only the bees but also their associated bacterial communities into close proximity, potentially resulting in a homogenization of their gut microbiota.
In order to compare the composition, diversity, and evolution of the gut microbiota of A. mellifera and A. cerana at the strain level, we analyzed shotgun metagenomes of individual bees using a common DNA extraction protocol and comparable sequencing depth. We find that each host species harbors a highly distinct bacterial community, composed of different SDPs and strains, with occasional transfers among sympatric bees. Quantitative analysis revealed that the gut microbiota diversity of A. mellifera is much higher than for A. cerana, resulting in a larger metabolic potential at both the individual and colony level. These results represent the first comparative genome-wide analysis of strain-level diversity between related host-associated microbial communities, raising new questions regarding underlying mechanisms and functional consequences.
Results
Metagenomic Data Reveal that the Gut Microbiota of A. mellifera and A. cerana Are Distinct
A total of 40 shotgun metagenome samples were collected from individual bees, with 20 bees per host species (A. mellifera and A. cerana). Bees were collected from inside the colonies, targeting nurse bees (see STAR Methods). Two colonies were sampled for each host species, with all colonies originating from different apiaries, no more than 100 km apart, close to Tsukuba (Japan). We also included 36 previously published metagenomes from individual bees sampled from two colonies from Switzerland. In order to analyze the community composition across samples, we first established a genomic database of isolated strains (Data S1) representative of both hosts. Eleven new genomes isolated from A. cerana were sequenced (see STAR Methods) and added to a previously established, non-redundant honey bee gut microbiota database [7], together with previously published genomes isolated from more distantly related bee species. Approximately 90% of the host-filtered reads mapped to the new database, regardless of the host affiliation of the samples, indicating that the database is highly and equally representative of the gut microbiota of both host species (Figure S1).
To make an initial broad comparison of the gut microbiota composition, the relative abundance of all phylotypes in the database was quantified, based on mapped read coverage to single-copy core genes. Overall, the phylotype-level composition was consistent with previous studies employing amplicon sequencing of the 16S rRNA gene [14] (Figure 1B). The five phylotypes that have been proposed to constitute the core microbiota of corbiculate bees [14] were found to colonize both host species, although the relative abundance profiles were distinct between the hosts (Figure 1B). Other phylotypes associated with A. mellifera (Bartonella apis, Frischella perrara, and Commensalibacter sp.) were not detected in any of the A. cerana samples (Figure 1B), consistent with a very low prevalence in this host [14]. Conversely, although Apibacter was not detected in any of the A. mellifera samples, it was prevalent among the A. cerana samples (Figure 1B).
To determine whether the five core phylotypes colonizing both host species are distinct at the SDP level (Figure 1A), candidate SDPs were inferred from isolate genomes in the database, based on core genome phylogenies (Figures 2A–2C, 2G, and 2H) and pairwise gANIs (Data S2). Subsequent metagenomic validation (Figure S2) confirmed three new SDPs within the Gilliamella phylotype (Figure 2A) and one new SDP within the Lactobacillus Firm5 phylotype (Figure 2B), all of which were represented exclusively by A. cerana-derived isolates. A new SDP was also confirmed for Snodgrassella (Figure 2C), based on isolates from A. cerana, A. andreniformis, and A. florea, suggesting that this SDP may be shared among other species of honey bees than A. mellifera. In contrast, for the two remaining core phylotypes Lactobacillus Firm4 and Bifidobacterium, no new candidate SDPs were inferred (Figures 2G and 2H), because the A. cerana-derived genome isolates had gANI values of up to 95% and 90% to the A. mellifera isolates, thus falling within the range of gANI values observed among the A. mellifera isolates (Data S2).
Figure 2.

The Gut Microbiota of A. mellifera and A. cerana Are Composed of Divergent SDPs and Strains
(A–C, G, and H) Core genome phylogenies of the five core phylotypes (A) Gilliamella; (B) Lactobacillus Firm5; (C) Snodgrassella; (G) Lactobacillus Firm4; and (H) Bifidobacterium; colonizing both A. mellifera and A. cerana. For (A)–(C) and (H), the trees were rooted with isolates derived from bumble bees, while for (G), the tree was rooted with the genome isolate from the Lactobacillus Firm4-2 SDP. Confirmed SDPs are indicated by the labels of the clades, with the SDPs identified in the current study highlighted with bold fonts. Genomes are highlighted with blue shades for isolates from A. mellifera and red shades for isolates from A. cerana. Gray shades indicate isolates from other honey bee species. Bars correspond to 0.1 substitutions per site.
(D–F) Barplots displaying relative abundance of the confirmed SDPs for the phylotypes (D) Gilliamella; (E) Lactobacillus Firm5; and (F) Bifidobacterium, across metagenomes from Japan.
(I and J) Principal coordinate analysis plots based on the pairwise fractions of shared SNVs (Jaccard distance) for the SDPs (I) "Firm4-1" and (J) "Bifido-1". Dots represent individual samples, color-coded by host and colony origin, as indicated by the legend. CH, Switzerland; JP, Japan.
(K) Number of host-specific and mixed sequence clusters generated from all metagenomic ORFs at different clustering thresholds. Metagenome assemblies were generated separately for each sample, using 20 million host-filtered paired-end reads per sample. Number of metagenomic samples included is as follows: n = 48 for A. mellifera and n = 20 for A. cerana.
See also Figures S2 and S3 and Data S1, S2, and S3.
To further validate the host specificity of the novel SDPs, we quantified the relative abundance of each SDP across the metagenomic samples, including samples previously collected in Switzerland [7]. All SDPs found to be host specific in the genomic database displayed a clear host preference across the metagenomic samples, but a small number of transfers were nevertheless detected among the Japanese samples within the Lactobacillus Firm5 and Gilliamella phylotypes (Figures 2D–2F and S3). These results therefore indicate that honey bees do get exposed to non-native SDPs, at least in Japan, occasionally resulting in colonization.
For the two SDPs represented by genome isolates from both hosts (Figures 2G and 2H), rooting of the core genome phylogenies with isolates from bumble bees (Figure 2G) or the most closely related SDP (Figure 2H) suggested that the A. cerana-derived isolates diverged prior to the A. mellifera-derived isolates, as would be expected if a more recent host specialization had occurred. Therefore, to determine whether these SDPs differ between the host species at the strain level (Figure 1A), the fraction of shared single-nucleotide variants was calculated for all sample pairs and visualized using principal coordinate analysis (Figures 2I and 2J). Remarkably, the samples were found to cluster strongly by host, indicating that each host species is colonized by a distinct population of strains. In contrast, there was no clustering by country or colony affiliation (Figures 2I and 2J). For other community members, clustering by country or colony was observed for only a subset of SDPs (Data S3), indicating that strains are not necessarily geographically specialized.
Finally, to obtain a database-independent estimate of the host specificity of the gut microbiota of A. mellifera and A. cerana, we de novo assembled the metagenomes (using 20 million host-filtered paired-end reads per sample) and compared the gene contents by clustering all predicted ORFs (open reading frames) by sequence identity. Clustering was done using a range of thresholds (80%–95% nucleotide identity), resulting in 101.581–185.585 clusters. Regardless of clustering threshold, only a very low number of clusters contained sequences from both hosts (Figure 2K), further corroborating the small overlap of the gut microbiota between the two host species.
In conclusion, although the gut microbiota of A. mellifera and A. cerana is colonized largely by the same phylotypes (Figure 1B), metagenomic analysis clearly demonstrates that the communities are distinct, being composed of divergent SDPs and strains.
The Diversity of the Gut Microbiota Is Higher in A. mellifera Compared to A. cerana
As shown in Figure 2K, the clustering analysis of the metagenomic ORFs resulted in a much larger number of clusters for A. mellifera compared to A. cerana. This difference may in part be explained by A. mellifera housing a bacterial community composed of more SDPs. Although 90% of the host-filtered reads mapped to the database for both hosts (Figure S1), these mapped reads represent 12 SDPs for the five core phylotypes in A. mellifera (plus up to 4 non-core members) but only 7 in A. cerana (plus Apibacter sp. and occasionally Lactobacillus kunkeei; Figures 2 and S3). Indeed, the number of gene clusters and the total genome assembly size per bee were both approximately twice as large for A. mellifera when using comparable subsets of host-filtered reads (Figures 3A and 3B). However, the sharp drop in the number of clusters from 95% to 90% sequence identity observed only for A. mellifera (Figure 2K) indicates that strain-level diversity is also a contributing factor.
Figure 3.

Major Differences in Strain-Level Diversity in the Gut Microbiota of A. mellifera and A. cerana
(A) Number of sequence clusters per sample for each host, based on metagenomic ORFs from assemblies generated with 20 million paired-end host-filtered reads. For each boxplot, the center line displays the median, and the boxes correspond to the 25th and 75th percentiles; all data points are shown (n = 48 for A. mellifera; n = 20 for A. cerana).
(B) Total length of metagenome assemblies generated from different amounts of host-filtered reads. Each line represents different subsets of reads derived from the same sample. Samples from the two Swiss colonies are shown in the same plot in different colors. CH, Switzerland; JP, Japan.
(C) Fraction of single-nucleotide variants (SNVs) within core genes in each sample, for SDPs corresponding to the core phylotypes, plus Apibacter sp. SDP labels correspond to Figure 1.
(D and E) Cumulative number of sequence clusters for each host, relative to the number of samples, shown (D) per host and (E) per colony.
(F and G) Cumulative fractions of SNVs within core genes relative to the number of samples for the SDPs (F) “Bifido-1” and (G) “Firm4-1” (Figures 2G and 2H). Blue-green shades represent different A. mellifera colonies, whereas the samples for the two A. cerana colonies were pooled due to the smaller number of samples.
See also Figure S4.
Previous analysis of strain-level diversity in A. mellifera showed that strains segregate among individuals within colonies [7], in contrast to the SDPs, which mostly co-exist (Figures 2D–2F and S3). Therefore, sequence clusters occurring only in a subset of bees are likely to represent strain-level diversity. To determine how the sequence clusters distribute across bees, we plotted the number of clusters relative to the number of samples, using the 95% nucleotide sequence identity threshold. From the cumulative curves (Figure 3D), it is evident that the gene content harbored within individual bees represent a minor fraction of the total gene content present across hosts. Moreover, the number of sequence clusters increased more rapidly with sample size for A. mellifera as compared to A. cerana and did not appear to have reached saturation with the current sampling size. This difference was not related to diversity between colonies or countries for A. mellifera (Figure 3E). Rather, the gene content of the gut microbiota is highly variable among bees, also within colonies, and much more so for A. mellifera compared to A. cerana (Figures 3D and 3E). Taken together with the consistent taxonomic profile among bees (Figures 2D–2F and S3), these results indicate that a major fraction of the variation in gene content is related to strain-level diversity.
To quantify the extent of strain-level diversity, the fraction of single-nucleotide variants (SNVs) within core genes was determined for all SDPs (615–1,046 genes per SDP, with the same positions being evaluated for all profiled samples for each SDP; see STAR Methods). At the level of individual bees, only two of the SDPs colonizing A. cerana were found to harbor more than 2% SNVs in any of the samples (“Firm5-7” and “Bifido-1”; Figure 3C). In contrast, nearly all the SDPs colonizing A. mellifera had more than 2% SNVs per bee in a major fraction of the samples (Figure 3C). For example, “Bifido-1”, an SDP shared between both bee species, had on average 9.6% SNVs per individual bee in A. mellifera, while in A. cerana the average percentage SNVs was as low as 0.8%.
To quantify diversity at the colony level, we again generated cumulative curves of SNVs as a function of the number of analyzed bees (Figures 3F, 3G, and S4). Interestingly, the two SDPs occurring in both hosts, “Bifido-1” and “Firm4-1” (Figures 2G and 2H), displayed a clear difference in strain-level diversity between the host species (Figures 3F and 3G). This difference was not explained by the choice of reference genome, because the pattern persisted after swapping the reference genome with an isolate from the alternate host (Figure S4).
Based on these results, we conclude that strain-level diversity in A. mellifera is substantially higher than in A. cerana, both in individual bees and within colonies.
The Gut Microbiota of A. mellifera Encodes More Diverse Enzymes for Polysaccharide Degradation
The observed differences in SDP composition and strain-level diversity raise the question whether the gut microbiota is functionally distinct between the two host species. To address this question, we annotated metagenomic ORFs using the COG and CAZyme databases. Although the number of ORFs per sample was approximately twice as high for A. mellifera compared to A. cerana, the relative COG profiles were indistinguishable among hosts (Figures 4A and S5). Consistent with previous studies [20], carbohydrate metabolism and transport (COG category “G”) was abundant across the metagenomic samples of both host species. In order to identify possible differences within this important functional category, metagenomic ORFs encoding glycoside hydrolases (GHs) and polysaccharide lyases (PLs) were annotated with dbCAN2 [38]. Notably, when calculating the mean number of ORFs annotated per CAZyme family for each host species, a linear correlation was observed (Figure 4B), indicating that the relative abundance of each GH/PL family is highly similar. However, samples from A. mellifera harbored approximately twice as many genes per family, as evidenced from the slope of the correlation (Figure 4B).
Figure 4.

Functional Comparison of the Gut Microbiota of A. mellifera and A. cerana
All ORFs were obtained from assemblies generated with 20 million paired-end host-filtered reads per sample.
(A) Relative abundance of COG annotations according to general functional COG categories, across metagenomes from Japan.
(B) Mean number of ORFs assigned to each CAZyme family, calculated separately for each host (n = 48 for A. mellifera; n = 20 for A. cerana). As shown by the blue regression line, there was a linear correlation between the counts, indicating that the CAZyme families display similar relative abundance patterns in both hosts. But the total number of ORFs assigned to each CAzyme family is larger in A. mellifera samples as compared to A. cerana samples, as indicated by the deviation from the black line.
(C) Number of sequence clusters within each CAZyme family (clustered at 50% amino acid sequence identity), as estimated across metagenomes from Japan (n = 20 for A. mellifera; n = 20 for A. cerana). Colors indicate the subsets of clusters specific to each host and clusters containing ORFs derived from both hosts. Only families with at least two clusters are shown.
(D) SDP affiliation for ORF sequences annotated as GH/PL CAZyme families, as estimated from blast hits against the honey bee gut microbiota database). Results are only shown for close hits (>95% amino acid sequence identity). The upper panel includes all ORFs passing this threshold; the lower panel includes the subset of these occurring within host-specific clusters (C). SDP labels correspond to Figure 1. Ap, Apibacter sp.; Fp, Frischella perrara; Co, Commensalibacter sp.; Lk, Lactobacillus kunkeei.
See also Figure S5.
Although enzyme substrate specificities are known to be variable within CAZyme families, the substrate specificity of most sequences in the CAZyme database still await experimental characterization [39, 40]. Therefore, to estimate the potential for carbohydrate metabolism within the gut microbiota of each host species, the sequences of each GH/PL family were clustered separately, using only the Japanese samples (in order to have the same sampling depth for each host). Remarkably, even with a highly conservative clustering threshold (50% amino acid sequence identity), an average of 10.4 clusters per family was generated, indicative of very high diversity among genes annotated to the same family. A total of 52 out of 62 GH/PL families contained host-specific clusters (Figure 4C). However, only 17 families contained clusters specific to A. cerana, whereas all 52 contained clusters specific to A. mellifera (Figure 4C). Moreover, the mean number of host-specific clusters per family was 6.0 for A. mellifera but only 3.1 for A. cerana. Taken together, these results therefore indicate that both host species may harbor specialized functions for polysaccharide degradation, with a higher versatility for A. mellifera as compared to A. cerana.
In order to gain further insights into the origin of the GH/PL families, the sequences were blasted against the honey bee gut microbiota database. Overall, 97% of the sequences had significant hits to the database (e-value < 10e−05; >80% query coverage), with 79% having a close hit (>95% amino acid identity). Among the sequences with close hits, the vast majority of hits were to genomes of the “Bifido-1” SDP, followed by other SDPs of the Lactobacillus Firm4 and Firm5 phylotypes (Figure 4D, upper panel). For the host-specific GH/PL clusters, only 52% of the sequences falling within A. mellifera-specific clusters had a close hit to the database, whereas 67% of the A. cerana-specific clusters had close hits. Thus, although the current database contains fewer genome isolates from A. cerana compared to A. mellifera, it is more representative of A. cerana in terms of GH/PL families, likely as a consequence of GH/PL genes being at least partly associated with strain-level diversity. Among the sequences corresponding to host-specific GH/PL clusters, the majority of blast hits were once again to “Bifido-1” (Figure 4D, lower panel). Strikingly, for both of the SDPs shared between the host species (“Bifido-1” and “Firm4-1”), almost all host-specific sequences came from A. mellifera. In contrast, for the clusters specific to A. cerana, most of the hits were to Apibacter sp. and “Firm5-7”, suggesting that diversity occurring at the phylotype- and SDP-level contribute more to functional specialization than strain-level diversity in A. cerana.
In conclusion, despite the similarity in the general functional profiles, A. mellifera harbors a much more diverse repertoire of GH/PL families, with many more host-specific GH/PL clusters compared to A. cerana. Moreover, the majority of GH/PL sequences were associated with the “Bifido-1” SDP, which is much more diverse in A. mellifera compared to A. cerana, suggesting that strain-level diversity in A. mellifera is a major contributor to functional differences between the two host species.
A. mellifera and A. cerana Differ in Bacterial Community Size within Individual Bees
According to neutral theory, diversity is expected to correlate with population size, where smaller populations are subject to increased genetic drift [41, 42, 43, 44]. The strong segregation of strains among bees within colonies suggests that the bacterial populations found within individual bees are temporally isolated from each other. Therefore, if the census population size of the gut microbiota within individuals is smaller for A. cerana than for A. mellifera, this could potentially explain the observed differences in strain-level diversity. Based on wet-weight, the hindgut, where most of the bacteria reside, was not significantly different between the hosts (Figure 5A). However, the bacterial community size, as estimated from quantitative real-time PCR with universal 16S rRNA primers (normalized to the copy number of the host gene actin) was still found to be significantly larger for A. mellifera compared to A. cerana (p < 0.001; Mann-Whitney U test; Figure 5B).
Figure 5.

Possible Factors Explaining the Difference in Gut Microbiota Diversity between A. mellifera and A. cerana
(A and B) Comparison across hosts for (A) wet weight of the hindgut (n = 18 for A. mellifera; n = 16 for A. cerana) and (B) bacterial community size (estimated with qPCR, targeting the 16S rRNA gene and normalized by copy number of the host gene actin; n = 20 for A. mellifera; n = 18 for A. cerana). For each boxplot, the center line displays the median, and the boxes correspond to the 25th and 75th percentiles; all data points are shown. Statistical significance was calculated using a Mann-Whitney U test (ns, not significant; ∗∗∗p < 0.001).
(C) Schematic illustration of three possible factors explaining differences in diversity. “Human influence”: transportation and mixing of A. mellifera colonies and genotypes around the world by beekeepers results in mixing of strains from different geographic origins and thereby increasing strain-level diversity in A. mellifera. “Dietary specialization”: A. mellifera may have a more generalist diet (here illustrated by pollen grain diversity) as compared to A. cerana and thereby be able to sustain a more diverse community. “Species-area relationship”: although previously applied to species-level diversity in animals, this concept may also apply to strain-level diversity in bacteria. For the honey bee gut microbiota, spatial differences are applicable at three levels: the size of the bacterial community within individual bees; the size of honey bee colonies; and the size of the geographic range.
Discussion
In the current study, we carried out a community-wide metagenomic characterization of the gut microbiota of two closely related honey bee species, A. mellifera and A. cerana. From this analysis, three key results emerged. First, we found that the gut bacterial communities of the two host species were highly divergent, consisting of different SDPs and strains, despite having a very similar phylotype-level composition. Second, the two host species displayed major differences in the magnitude of strain-level diversity within their bacterial communities. And third, the gut microbiota of A. mellifera harbored a much larger repertoire of enzymes related to polysaccharide breakdown. Thus, in the time since their last common ancestor, approximately 6 mya [32, 35], the gut bacterial communities of A. mellifera and A. cerana have undergone substantial changes in composition, genomic diversity, and functionality, with likely consequences for the interaction with their hosts.
Based on amplicon sequencing of the 16S rRNA gene, multiple studies have shown that gut microbiota composition is influenced by host phylogeny [45, 46], with the overall observation that closely related host species harbor more similar gut bacterial communities at the phylotype level than more distant ones [47]. However, the slow evolutionary rate of the 16S rRNA gene [10] does not permit evolutionary analysis of phylotypes that are shared across related hosts. Therefore, there are currently little data providing insights into the evolution of the gut microbiota for closely related animal hosts. Targeting the fast-evolving gyrA gene for three bacterial families colonizing hominids, Moeller et al. [48] found evidence of co-diversification in the Bacteroidaceae and Bifidobacteriaceae, but not the Lachnospiraceae. Similarly, for honey bees and bumble bees, amplicon sequencing of the minD gene uncovered both host-specific and more generalist clades within the core phylotype Snodgrassella [31]. Consistently, comparative genome analyses of bacterial isolates have also uncovered several examples of host-specific lineages [16, 17, 18, 19]. However, the current study is the first to report community-wide patterns of host specialization using metagenomic data.
Interestingly, although we found evidence of host specialization for all of the five core phylotypes colonizing both hosts, the extent of divergence differed widely among them. For three of the phylotypes (Lactobacillus Firm5, Gilliamella, and Snodgrassella), each host was found to be colonized by different SDPs, i.e., discrete bacterial populations that are sufficiently divergent to be classified as different species (having less than 90% gANI to each other) [30]. In contrast, the host specialization of the Bifidobacterium and Lactobacillus Firm4 phylotypes only became evident when analyzed with strain-level resolution, consistent with the comparatively short branch lengths observed for the core genome phylogenies. Assuming a 16S rRNA gene divergence rate of about 1% per 50 Ma [49, 50], it seems unlikely that any of the SDPs could have emerged within the 6 Ma separating A. mellifera and A. cerana. More likely, the current SDP composition represents a selection of pre-existing SDPs, with secondarily evolved traits resulting in the observed host preference. For example, we found that all SDPs within the Lactobacillus Firm5 phylotype contributed to highly divergent host-specific glycoside hydrolases, which could potentially allow for dietary specialization and host adaptation. In contrast, the comparatively little sequence divergence between the host-specialized strains of Lactobacillus Firm4 and the Bifidobacteria could match a time span of 6 Ma and thus be a product of co-diversification. However, a larger genomic dataset from multiple host species will be needed to properly test this hypothesis.
Remarkably, we also found that the two host species differed substantially in terms of the extent of genomic diversity in their gut microbiota. In comparison, a previous study employing amplicon sequencing of the 16S rRNA gene found only a subtle trend toward lower diversity in A. cerana compared to A. mellifera, even when using a highly elevated sequence identity clustering threshold of 99.5% [14]. Because the 20 metagenomic samples of A. cerana came from only two colonies in Japan, it remains to be confirmed whether our findings also apply to other subspecies of A. cerana outside of Japan. However, given that many studies have shown a lack of concordance between the 16S rRNA gene and genome-level divergence in bacteria [10, 12, 22, 23, 25], also in the honey bee gut microbiota [21], these results rather seem to highlight that genome-wide data are needed for quantification of strain-level diversity, especially at short evolutionary timescales. This is also supported by amplicon sequencing of the minD gene, which revealed marked differences in strain-level diversity for the phylotype Snodgrassella between bumble bees and honey bees, indicating that the 16S rRNA gene strongly underestimates diversity in the honey bee gut microbiota [31].
However, despite a rapid increase in metagenomic studies during the last decade, quantification of strain-level diversity from metagenomic data is still technically challenging and quantitative comparisons across hosts are therefore rare. Differences in sampling, DNA extraction, and sequencing depth can have a strong impact on both composition and diversity, making cross-comparisons between samples and studies particularly difficult [5, 51, 52]. In the current study, sequencing and sampling biases were limited by using a common sampling and DNA extraction protocol and a comparable sequencing depth. Because of the simple taxonomic composition of the honey bee gut microbiota, we obtained high and comparable mapping and de novo metagenome assembly efficiencies in both hosts. Furthermore, normalization by sampling and sequencing depth was applied in all analyses. Taken together, we are therefore confident that the observed community-wide differences in strain-level diversity are not due to technical biases.
Several factors could potentially have contributed to the observed difference in strain-level diversity (Figure 5C). First, although A. cerana has long been used for honey production in Asia, A. mellifera differs from all other extant honey bee species by having been extensively transported around the globe by humans for hundreds of years [37]. Thus, it is possible that humans have contributed to a mixing of locally adapted strains, thereby increasing diversity within colonies (Figure 5C, “human influence”). As of yet, large-scale studies on the geographic distribution of strains are still lacking, and previous studies have reported mixed results [14, 31]. Likewise, in the current study, ordination plots based on shared SNVs clustered by country for only a subset of SDPs (Data S3). Future studies on wild honey bee populations should provide further insights into this question. Interestingly, if the high strain-level diversity in A. mellifera is caused by human interference, it raises the possibility that the gut microbiota of A. mellifera is sub-optimal for local conditions, potentially resulting in reduced colony fitness.
However, based on molecular data, A. mellifera has had a large and varied geographic range long before human interference [32, 33]. Moreover, despite the occurrence of colony collapse disorder in managed colonies around the world [53, 54, 55], A. mellifera has been found to successfully establish feral colonies even outside its native range (i.e., the New World), indicative of a remarkable ability to survive under highly varied conditions [56]. Thus, it is possible that A. mellifera is more of a generalist than A. cerana, for example, by having a wider foraging range, and therefore is able to maintain a more varied bacterial community and larger repertoire of sugar breakdown functions (Figure 5C, “dietary specialization”). Indeed, diet has been shown to have an impact on diversity in the gut microbiota in multiple studies, with, for example, a reduction of diversity reported for the human gut microbiota as a consequence of westernized diet [57, 58]. As of yet, large-scale systematic studies on the foraging preferences of A. cerana and A. mellifera have not been conducted, but they appear to have largely overlapping foraging ranges in Asia [59, 60]. Further studies are therefore needed to determine whether differences in strain-level diversity are related to dietary differences.
Finally, it is also possible that the difference in strain-level diversity between A. mellifera and A. cerana is driven by neutral processes (Figure 5C, “species-area relationship”). Specifically, the species-area relationship posits a positive correlation between habitat size and diversity and is widely held to constitute one of the few laws in ecology [61, 62, 63]. However, it is still debated whether this relationship also applies to bacteria [64, 65, 66]. The gut microbiota represents an attractive model system for testing the hypothesis, because bacterial populations in this case can be easily delineated, due to the host association. Indeed, it has previously been proposed that differences in gut microbiota diversity in different bee species could be explained by the species-area relationship, because 16S rRNA gene diversity was found to correlate with both colony size and bacterial population size within individual bees when compared among honey bees, bumble bees, and stingless bees [14, 31]. Interestingly, our results uncovered much more dramatic differences in diversity at the strain level compared to the species level, raising the question of whether the species-area relationship must be adapted to include strain-level diversity in bacterial communities. In the case of A. mellifera and A. cerana, which are very similar in terms of colony cycle and behavior, spatial differences exist at multiple levels, possibly explaining the differences observed in strain-level diversity (Figure 5C, species-area relationship). First, we found that the size of the bacterial communities within individuals was significantly larger for A. mellifera compared to A. cerana, a trend that was also observed in the previous study comparing diversity across bees [14]. Second, A. mellifera is known to form larger colonies than A. cerana [34]. And third, the native range of A. mellifera is also larger compared to A. cerana [37, 67, 68]. Our analysis clearly shows that the diversity within individual bees is much lower than the diversity found within their colonies. Thus, it seems plausible that competition among strains is alleviated when strains colonize different hosts. If so, it also follows that larger colonies should be able to support more strain-level diversity, by providing more colonization opportunities for strains that would otherwise compete. However, given that the diversity was also consistently higher for individual bees of A. mellifera as compared to A. cerana, the species-area relationship may be equally applicable at this level. In contrast, the inclusion of multiple colonies had very little impact on diversity estimates, suggesting that host geographic range is of less importance for maintenance of strain-level diversity in honeybees.
In conclusion, the results of the current study brought several fundamental questions regarding the evolution and maintenance of diversity in host-associated bacterial communities to the foreground. Although the term “diversity” has an inherently positive connotation, it is not obvious whether diversity in host-associated bacterial communities should be beneficial and, if so, in what sense [69]. For example, high strain-level diversity in the gut microbiota of A. mellifera may provide more metabolic flexibility, facilitating foraging on more diverse pollen sources and thereby faster adaptation to changing environmental conditions. On the other hand, high strain-level diversity could also lead to increased competition within the gut microbiota, with resources being diverted toward inter-bacterial warfare rather than host-symbiont mutualistic interactions. It is also possible that a less diverse gut microbiota consisting of strains adapted to local conditions would be more beneficial than a more diverse one. These possibilities can be experimentally tested in honey bees, thereby providing novel insights into the functional relevance of strain-level diversity in host-associated bacterial communities and for honey bee health.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Apis mellifera | AIST (National institute of advanced sciences), Japan | N/A |
| Apis mellifera | University of Tokyo, Japan | N/A |
| Apis cerana | Chiba (local beekeeper), Japan | N/A |
| Apis cerana | Hodogaya (local beekeeper), Japan | N/A |
| Apis cerana | NIES (National intitute for environmental studies), Japan | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tryptic Soy Agar | BD Difco | 236950 |
| Columbia blood agar base | Oxoid | CM0331 |
| Sheep blood defibrinated | Oxoid | SR0051E |
| MRS agar (DE MAN, ROGOSA, SHARPE) | Oxoid | CM0361 |
| BHI agar (Brain Heart Infusion) | BD Difco | 11708872 |
| Glucose | Sigma | G7021 |
| Fructose | Sigma | F2543 |
| L-cysteine | Fluka | 30120 |
| Bacto Tryptone | BD Difco | 211705 |
| Yeast extract | Oxoid | LP0021 |
| Cellobiose | Sigma | 22160 |
| Vitamin K | Sigma | V3501 |
| FeSO4 | Sigma | F8633 |
| CaCl2 | Sigma | 746495 |
| MgSO4 | Sigma | 63140 |
| NaHCO3 | Sigma | S6297 |
| NaCl | Acros | 207790010 |
| Hematin | Sigma | H-3281 |
| Histidine | Sigma | 53319 |
| Na2HPO4 | Applichem | A2530 |
| NaH2PO4 | Sigma | 71504 |
| Ethanol | Wako | Cat#057-00451 |
| NaCl | Wako | Cat#191-01665 |
| KCl | Wako | Cat#163-03545 |
| Na2HPO4 | Wako | Cat#194-02875 |
| KH2PO4 | Wako | Cat#169-04245 |
| UltraPure™ DNase/RNase-free distilled water | Invitrogen | Cat#10977023 |
| 1.0mm glass beads | TOMY | Cat#GB-10 |
| 0.1mm zirconia/silica beads | TOMY | Cat#ZSB-01 |
| Ethylenediamine-N,N,N’,N’-tetraacetic acid, disodium salt, dihydrate | DOJINDO | Cat#N001 |
| Proteinase K | Wako | Cat#160-14001 |
| Proteinase K | TaKaRa | Cat#U0506A |
| 2-Mercaptoethanol | Wako | Cat#133-06864 |
| Trizma® base Primary Standard and Buffer, ≥ 99.9% (titration), crystalline | Sigma-Aldrich | Cat#T1503 |
| HCl | Wako | Cat#080-01066 |
| Hexadecyltrimethylammonium Bromide (synonym of CTAB) | Wako | Cat#036-021-02 |
| Phenol/Chloroform/Isoamyl alcohol (25:24:1) | nacalai-tesque | Cat#25970-14 |
| Phenol/Chloroform/Isoamyl alcohol (25:24:1) | NIPPON GENE | Cat#311-90151 |
| Chloroform | Wako | Cat#038-02606 |
| NaOAc | NIPPON GENE | Cat#316-90081 |
| Glycogen | Wako | Cat#079-00832 |
| RNase A (17,500U) | QIAGEN | Cat#19101 |
| T-Vector pMD-20 | TaKaRa | Cat#3270 |
| TB Green premix Ex TaqII (Tli RNaseH Plus) | TaKaRa | Cat#RR820S |
| ROX reference dye II | TaKaRa | Cat#RR820S |
| Critical Commercial Assays | ||
| NexteraXT DNA Library Preparation kit (96 samples) | Illumina | Cat#FC-131-1096 |
| Deposited Data | ||
| Raw sequence reds for metagenomes have been deposited on the NCBI Sequence Read Archive | This paper | PRJNA598094 |
| Code used for bioinformatic analysis | This paper | http://doi.org/10.5281/zenodo.3747314 |
| Oligonucleotides | ||
| Primers for V3-V4 region of 16S rRNA | [70] | N/A |
| Primers for actin gene, A. mellifera | See STAR Methods section | N/A |
| Primers for actin gene, A. cerana | See STAR Methods section | N/A |
| Software and Algorithms | ||
| QuantStudio Design & Analysis Software v1.4 (Firmware version 1.3.0) | Applied Biosystems | N/A |
| EZR | [71] | http://www.jichi.ac.jp/saitama-sct/SaitamaHP.files/statmed.html |
| Perl v.5.26.1 | [72] | https://www.perl.org |
| BioPerl v.1.7.5 | [73] | https://bioperl.org |
| Bash v. 4.4 23 | https://www.gnu.org/software/bash | https://www.gnu.org/software/bash |
| R version 3.5.0 | R Development Core Team, 2008 | https://www.r-project.org |
| BLAST+ version 2.2.31 | [74] | https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+ |
| FastQC v.0.11.4 | [75] | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| Trimmomatic 0.35 | [76] | http://www.usadellab.org/cms/?page=trimmomatic |
| fastANI v.1.3 | [30] | https://github.com/ParBLiSS/FastANI |
| Orthofinder v.2.3.5 | [77] | https://github.com/davidemms/OrthoFinder |
| mafft v.7.312 | [78] | https://mafft.cbrc.jp/alignment/software/ |
| RAxML v.8.1.24 | [79] | https://github.com/stamatak/standard-RAxML |
| FigTree | [80] | https://github.com/rambaut/figtree/releases |
| SPAdes v.3.10.1 | [81] | http://cab.spbu.ru/software/spades/ |
| bwa v.0.7.15-r1142-dirty | [82] | https://sourceforge.net/projects/bio-bwa |
| Samtools v.1.9 | [83] | https://github.com/samtools/samtools |
| Picard tools v.2.7.1 | [84] | https://broadinstitute.github.io/picard/ |
| Prodigal v.2.6.3 | [85] | https://github.com/hyattpd/Prodigal |
| Freebayes v.1.0.2 | [86] | https://github.com/ekg/freebayes |
| GNU parallel 20180422 | [87] | http://www.gnu.org/software/parallel |
| Vcflib v.41 | [88] | https://github.com/vcflib/vcflib |
| CD-HIT v.4.7 | [89] | https://github.com/weizhongli/cdhit |
| eggnog v.5.0 | [90] | http://eggnogdb.embl.de/#/app/home |
| eggNOG-mapper v. 1.0.3 | [91] | https://github.com/eggnogdb/eggnog-mapper |
| HMMER 3.1b2 | http:/hmmer.org | http:/hmmer.org |
| dbCan2 v.8 | [38] | http://bcb.unl.edu/dbCAN2/ |
| Other | ||
| Tweezer No. 5-Dumoxel BIOLOGIE | Dumont | N/A |
| Micro SmashTM MS-100 bead-beater | TOMY | N/A |
| QuantStudio 3 Real-Time PCR Instrument (96-well, 0.2mL Block) | Applied Biosystems | Cat#A28132 |
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philipp Engel (philipp.engel@unil.ch)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The accession number for raw sequence data reported in this paper is NCBI BioProject: PRJNA598094. Accession numbers for genomes included in the genomic database are provided in Data S1. All databases used for analysis, files corresponding to metagenomic assemblies, and scripts generated for analysis, have been deposited on zenodo at: https://doi.org/10.5281/zenodo.3747314.
Experimental Model and Subject Details
Sample acquisition
Dissection of hindguts from bees
For dissection of bees, the cuticle of the abdomen was peeled off and inner tissues were exposed, using tweezers (UV irradiated for 10 min). Furthermore, midgut and Malpighian tubules were removed, in order to obtain the hindgut section only (pylorus to rectum).
Metagenomic samples
A total of 40 metagenomic samples were collected from individual bees, with 20 samples from A. cerana japonica and A. mellifera, respectively. For each host species, 10 bees were collected from each of two colonies, all estimated to be nurse bees based on behavior (A. mellifera only) and the presence of a well-developed hypopharyngeal gland (both host species). The bees were soaked in ethanol and stored at −20°C until dissection. All sampled colonies were from different apiaries (“AIST,” “UT,” “Chiba,” “Kanagawa”) located less than 100km apart, close to Tsukuba, Japan, in September 2017. Further metadata, including GPS coordinates, is available for each sample via accession numbers in NCBI BioSample: SAMN13698777-SAMN13698816.
Bacterial isolates
Eleven new bacterial isolates were obtained from two A. cerana nurse bees, collected from Kanagawa (September 2017) and NIES (November 2017), respectively. The nurse bee from Kanagawa came from the same colony as the metagenomic samples.
qPCR (bacterial load)
In order to estimate the size of the bacterial communities colonizing the gut of A. mellifera and A. cerana, 8-10 honeybees were collected from each of two colonies, for each host. For A. mellifera, samples were collected in May 2019, from the same apiaries as the metagenomic samples (but corresponding to different colonies). For A. cerana, samples were collected in September 2017 from Kanagawa (same colony as for metagenomic samples) and in October 2017 from NIES (different apiary). Dissected hindguts were placed in bead-beating tubes (2 mL) with 364 μL ultra-pure DNase/RNase-free ddH2O and zirconia/silica beads (0.1mm), and stored at −80°C until DNA extraction.
Gut weights
In order to estimate the wet-weight of the hindguts for each host, additional honey bees were collected from AIST (A. mellifera, n = 18, sampled in August 2019), and NIES (A. cerana, n = 16, sampled in September 2019). Dissected guts were weighed on the day of collection, without freezing.
Method Details
Metagenomic DNA extraction and sequencing
Bacterial enrichment
The metagenomic samples were enriched for bacterial cells, as also described previously [7]. Dissected hindguts were placed in bead-beating tubes with 1 mL of PBS (136.9mM NaCl, 2.7mM KCl, 8.1mM Na2HPO4, 1.5mM KH2PO4, pH 7.5), and kept on ice for the duration of the dissection. Gut tissue was homogenized with a bead-beater (MicroSmash MS-100, TOMY) using glass-beads (1mm, TOMY) for 30 s at 5,500rpm. A series of centrifugation and filtration steps were then carried out to enrich for bacterial cells in the sample relative to host cells/tissue/pollen, all in PBS at room temperature. First, the homogenate was centrifuged at 2,500rpm, 5 min, to remove debris, and the supernatant was collected into new eppendorf tubes. The samples were then centrifuged at 9,000rpm, 15 min, to pellet bacterial cells. The supernatant was removed, and the bacterial pellets were re-suspended in 800 μL PBS. The suspension was again centrifuged at 2,500rpm, 5 min, to remove additional host-cells/debris. Finally, the sample was passed through a 10 μm filter (Merck), to remove large particles (pollen, remaining debris), and centrifuged at 10,000rpm for 15 min to pellet bacterial cells.
DNA extraction
DNA was extracted from the enriched bacterial pellet using a CTAB-based DNA extraction protocol. For each sample, the bacterial pellet was re-suspended in 485.3 μL CTAB lysis buffer (100mM Tris-HCl, pH 8, 1.4M NaCl, 20mM EDTA pH 8.0, 2% w/v CTAB) with 1.3 μL 2-mercaptoethanol and 12 μL proteinase K (22 mg/ml, TaKaRa). The samples were transferred to bead-beating tubes with zirconia/silica beads (0.1mm, TOMY), and homogenized on a bead-beater (MicroSmash MS-100, TOMY) for two times 90 s, at 5,500rpm. Samples were incubated at 56°C over-night. 5 μL RNase A (10 mg/mL) was added to each sample, followed by incubation at 37°C for 1 h. Finally, the DNA was extracted with PCI (phenol/chloroform/isoamyl alcohol 25:24:1)(nacalai-tesque), washed with chloroform and precipitated with 1/10 vol NaOAc (3M, pH 5.2) and 2.5 vol 99.5% ethanol, with 4 μL glycogen (20 mg/ml) added as a DNA carrier. Glycogen was purified through PCI treatment 4 times and precipitated using 1/10 vol NaOAc (3M, pH 5.2) and 2.5 vol ethanol. After air-drying, the weight of the pellet was measured, and the pellet was re-suspended in ddH2O, to obtain 20 mg/ml glycogen solution. DNA pellets were re-suspended in 20 μl ddH2O, and stored at −20°C until library preparation.
Sequencing and quality filtering
Samples were prepared for metagenomic sequencing using the NexteraXT library kit, and sequenced on an Illumina HiSeq2500 instrument (paired-end, 2 × 100bp). The quality of the raw data was checked with fastQC [75], and the reads were subsequently trimmed with Trimmomatic [76] using the settings: LEADING:28, TRAILING:28 MINLEN:60, and trimming for the Nextera adaptor. The same filtering was also applied to 36 previously published metagenomes [7], corresponding to age-controlled bees (Day 10 and Day 22/24), collected from the apiary at the University of Lausanne, Switzerland [7].
Quantification of bacterial loads
Total DNA extraction
DNA was extracted using a CTAB-based protocol [92]. For each sample, 364 μL of CTAB lysis buffer (200mM Tris-HCl pH 8.0, 2.8M NaCl, and 40mM EDTA pH 8.0, 4% CTAB (w/v)), 2 μL of 2-mercaptoethanol, and 20 μL of 20 mg/mL proteinase K (Wako) was added. Bead-beating was done twice for 90 s, at 3,500rpm (MicroSmash MS-100, TOMY), with 1 min rest on ice in between. 1 μL of 10 mg/mL RNase A was added, and the tubes were incubated overnight at 55°C. Next, 750 μL PCI (phenol/chloroform/isoamyl alcohol)(NIPPON GENE) was added, the samples were mixed by shaking, placed on ice for 2 min, and centrifuged at 13,300rpm at 4°C for 30 min. DNA was precipitated with ethanol, washed, air-dried and dissolved in 50 μL ultra-pure DNAase/RNase-free ddH2O.
qPCR assay
Bacterial loads were estimated with quantitative real-time PCR, targeting the V3-V4 region of 16S rRNA gene with the following primers: 5′-ACTCCTACGGGAGGCAGCAGT-3′ (forward) and 5′-ATTACCGCGGCTGCTGGC-3′ (reverse) [70]. Normalization was done relative to the actin gene of the host, using the following primers for A. mellifera: 5′-TGCCAACACTGTCCTTTCTG-3′ (forward) and 5′-AGAATTGACCCACCAATCCA-3′ (reverse). For A. cerana, the reverse primer was 5′-AGAATTGATCCACCAATCCA-3′. Standards were prepared as also described in [93]. The target sequence was cloned into plasmid vector pMD-20 (TaKaRa). The insertion of the target sequence was confirmed by sequencing, and the insert was amplified by PCR and purified. The copy number of the PCR product was calculated, serially diluted and used as standard. qPCR reactions were performed in triplicates in a total volume of 10 μL, containing 5 μL of 2 x TB Green premix Ex TaqII, 0.2 μL ROX reference dye II, 0.2 μM of each primer and 1 μL of 100x-diluted extracted DNA, on a QuantStudio 3 instrument (Applied Biosystems). The thermal cycling conditions were as follows: denaturation at 95°C for 30 s, followed by 40 amplification cycles at 95°C for 5 s, and 60°C for 1 min (actin) or 30 s (16S rRNA).
qPCR data analysis
The data was analyzed using QuantStudio Design & Analysis Software v1.4 (Firmware version 1.3.0) (Applied Biosystems) and Excel (Microsoft).
Isolation of strains from A. cerana
To obtain bacterial isolates from A. cerana, the hindgut was dissected as described above and homogenized in PBS (136.9mM NaCl, 2.7mM KCl, 8.1mM Na2HPO4, 1.5mM KH2PO4, pH 7.5) with a bead-beater (MicroSmash MS-100, TOMY) using glass-beads (1mm, TOMY) for 30 s at 5,500rpm. The homogenized hindgut tissue was plated in different dilutions on the following media used for cultivation of the honey bee gut microbiota [94]: modified tryptone glucose yeast extract agar (0.2% Bacto tryptone, 0.1% Bacto yeast extract, 2.2 mM D-glucose, 3.2mM L-cysteine, 2.9mM cellobiose, 5.8mM vitamin K, 1.4μM FeSO4, 72.1 μM CaCl2, 0.08mM MgSO4, 4.8mM NaHCO3, 1.36mM NaCl, 1.8μM Hematin in 0.2mM Histidine, 1.25% Agar adjusted to pH 7.2 with potassium phosphate buffer), De Man, Rogosa and Sharpe agar supplemented with 2% w/v fructose and 0.2% w/v L-cysteine-HCl, tryptic soy agar, brain heart infusion agar, and Columbia agar base supplemented with 5% (v/v) defibrinated sheep blood. The specific medium on which each isolate was obtained is provided in Data S1. Details on sequencing and assembly of genomes is accessible via their IMG accession numbers (Data S1).
Bioinformatic analyses
Custom scripts
All the following computational tasks were done with custom scripts written in perl [72], bash or R unless otherwise indicated, and have been deposited on zenodo (https://doi.org/10.5281/zenodo.3747314), with README.txt files explaining their usage.
Establishment of genomic database
A previously published honey bee gut microbiota genomic database [7] was updated to include recently published genomes isolated from A. mellifera, plus genomes isolated from other social bee species. Moreover, the genomes of 11 new isolates of A. cerana were sequenced with PacBio to increase the database representation for this host species for the current study. Pairwise genomic average nucleotide identities (gANI) were calculated with fastANI [30] for all isolates of the same phylotype, and used to streamline the database for redundancy. Genomes isolated from A. mellifera or A. cerana were required to have a maximum of 98.5% gANI to other genomes within the database, whereas genomes isolated from other species were streamlined at circa 95% gANI, in both cases prioritizing the most complete genome assemblies. Metadata and accession numbers for all genomes included in the database are provided in Data S1.
Metagenomic assemblies
To filter off host-derived reads, the reads of each metagenomic sample were first mapped against a database containing the genomes of A. cerana (PRJNA235974) and A. mellifera (PRJNA471592, version Amel_Hav3.1), using bwa mem [82] with default settings. Bam-files containing unmapped reads were generated with samtools [83] (flag -f 4), and paired reads were extracted from the bam-files with Picard tools [84]. Additionally, subsets of the host-filtered reads were generated in increments of 10 million read pairs for each sample. Metagenomic assemblies were generated independently for each sample and each read subset, using SPAdes [81] with default settings for metagenomic assembly. The resulting contigs were filtered to have a minimum length of 500bp and a minimum kmer coverage of 1 (parsed from the contig fasta header). To check for eventual differences in assembly efficiency related to community complexity, the reads of each subset were mapped back to the corresponding assembly with bwa mem, and the number of mapped reads was counted with samtools (samtools view, flags: -F 4, -c).
Identification and validation of SDPs
Candidate SDPs were identified within the non-redundant honey bee gut microbiota genomic database based on core genome phylogenies and pairwise genomic average nucleotide identities (gANI) (Data S2). Orthologous gene families were estimated separately for each phylotype, for all genomes included in the genomic database (Data S1), using Orthofinder [77]. For the phylogenies, sequences of single-copy core gene families were aligned at the amino acid level with mafft [78] (option–auto), back-translated into codon-aligned nucleotide alignments, and trimmed by removing positions represented by less than 50% of the sequences, using BioPerl [73]. The alignments were merged into a single alignment, from which phylogenies were inferred with RAxML [79], with 100 bootstrap replicates, using the GTRCAT model. Since genomes isolated from bumble bees fell into separate well-supported clades, as also observed in previous studies [16], the phylogenies were rooted with these isolates when possible. For phylotype Lactobacillus Firm4, no isolates are available for other bee species, the tree was therefore rooted using the “Firm4-2” SDP. After inspection of the phylogenies and ANI tables, candidate SDPs were identified as forming discrete clades with 100% bootstrap support, and having a minimum pairwise ANI of 89% within clusters, as also described previously [7].
Validation was done separately for each candidate SDP. First, a filtered set of core gene families was generated for each phylotype according to two criteria: i) core gene families containing sequences shorter than 300bp were removed, ii) core gene families for which the sequence alignment identity was larger than 95% for any pair of sequences belonging to different SDPs were removed. These filtered core gene families were then used for SDP validation, as illustrated schematically in Figures S2A–S2D. In step 1, amino acid alignments of the core gene sequences were generated and back-translated to nucleotide alignments (as for the core genome phylogenies), for all the genomes associated with the candidate SDP (Figure S2B, illustrated by colored arrows and lines in the alignments). Furthermore, the core gene sequences were used as queries in a blastn search against a database containing all ORFs (predicted with Prodigal [85]) on all metagenomic assemblies (generated using all host-filtered reads). Sequences of metagenomic ORFs were extracted from the blast file for hits where the ORF length was at least 50% of the query length and the blast alignment identity was above 70%. In step 2, the sequence of each recruited metagenomic ORF was added individually to the corresponding core gene alignment, using mafft [78] with the option –addfragment (Figure S2B, metagenomic ORFs illustrated by gray arrows and lines in the alignments), and their maximum percentage identity within the alignment was recorded (using Bioperl). Additionally, the recruited metagenomic ORFs were blasted against the gut microbiota genomic database, and their closest SDP was recorded (based on blast hit percentage identity).
For validation, the percentage identities calculated for the recruited ORFs were plotted as density distributions. ORFs with a best hit to the SDP being evaluated were assigned to the first density distribution, while ORFs with a best hit to other SDPs were assigned to the second density distribution (Figures S2C and S2D, shown in color and gray respectively). An SDP was considered confirmed if the two distributions were non-overlapping (Figure S2C), indicating that the metagenomic ORFs recruited to the SDP were discrete relative to related candidate SDPs contained within the database.
Community profiling
For each sample, all quality-filtered paired-end reads were mapped against the honey bee gut microbiota genomic database, using bwa mem with default settings, and the resulting bam-files were subsequently filtered to retain mapped reads, using a minimum alignment length of 50bp as filtering threshold. The unmapped reads were also extracted from the bam-files, using samtools (samtools view, flag -f4) and picard tools, and mapped to the host genomic database to calculate the fraction of host-derived reads per sample.
The relative abundance of community members within samples was quantified based on mapped read coverage to the filtered single-copy core gene families generated for the SDP validation, using the approach described previously [7], with some minor modifications. First, the mapped read coverage of each core gene was obtained from the bamfiles using samtools (samtools bedcov), and divided by the gene length (resulting in mean mapped read coverage per bp, per gene). Next, the mean coverages were summed per core gene family, per SDP. Finally, the summed coverages were plotted relative to their position on an SDP reference genome, and the abundance was taken as the coverage at the terminus of replication, estimated using a fitted segmented regression line (R package “segmented” [95]). For generating plots on phylotype abundances, the terminus coverages estimated per SDP were summed for all SDPs associated with the phylotype.
SNV profiling
SNVs occurring within core genes were profiled using a reduced genomic database, containing one representative genome per SDP. For the two SDPs represented by isolates derived from both A. cerana and A. mellifera (“Bifido-1” and “Firm4-1”), the pipeline was repeated using a database containing a reference genome isolated from the alternate host species, in order to check the impact of reference genome choice on SNV quantification. The reads were mapped against the reduced database (using bwa mem), and the bam-files were filtered by both alignment length (min 50bp) and edit distance (max 5). Candidate SNVs were predicted with freebayes [86] and GNU parallel [87], using the following options: “-C 5–pooled-continuous–min-alternative-fraction 0.1–min-coverage 10–no-indels–no-mnps–no-complex.” Thus, complex variants were not predicted, and rare SNVs were excluded, in order to avoid inflation of diversity estimates in samples where SDPs had very high coverage.
The VCF file was subset for SNVs occurring within core genes, and filtered, using a combination of custom perl scripts and vcflib [88]. For each SDP, only samples with at least 20x terminus coverage were profiled, in order to avoid underestimating diversity in samples where SDPs had low coverage. Furthermore, core gene families were excluded, if they had less than 10x mean coverage in any of the profiled samples. Finally, candidate SNVs were excluded if freebayes reported “missing data” for more than 10% of the profiled samples (indicative of localized low coverage in variable regions within genes). Thereby, the same core gene positions were evaluated in all profiled samples for each SDP. Only SNVs remaining polymorphic across samples after all filtering steps were included in the downstream analysis.
To quantify diversity within SDPs, the fraction of variable sites among all profiled sites was calculated for each SDP, both per sample and across the full dataset. Furthermore, the cumulative increase in the fraction of variable sites relative to the number of bees sampled per host/colony was calculated, using 10 random sampling orders per SDP. To visualize the distribution of SNVs across samples, and identify eventual patterns related to host species, country or colony affiliation, a Jaccard distance matrix was generated based on shared polymorphic sites. Specifically, an SNV was considered to be shared between two samples if it occurred with an intra-sample relative abundance of at least 10% in both samples. The jaccard distance was calculated as the fraction of non-shared polymorphic sites divided by the number of sites profiled. The resulting matrix was visualized with a principal coordinate analysis (R package: ape [96]) (Data S3).
Gene content diversity in metagenomes
To compare the total genetic diversity in the gut microbiota among individuals, the length of all the metagenomic assemblies corresponding to increments of 10 Million host-filtered paired-end reads was first calculated. Since assembly length increases with sequencing depth (due to increased coverage on rare community members or strains), the downstream quantitative analysis was based on assemblies using 20 Million paired-end host-filtered reads per sample. ORFs were predicted with prodigal [85] (option –meta), and were filtered for ORFs shorter than 300bp or flagged as partial by Prodigal (partial flag 11, 01 or 10), in order to minimize the impact of spurious annotations. Sample affiliations were added to the fasta headers of the filtered ORFs, after which the sequences were concatenated.
To quantify gene content diversity within and across samples and hosts, the filtered ORFs were clustered with CD-HIT [89], using a range of thresholds (80%–95% nucleotide identity), with hierarchical clustering as recommended in the CD-HIT manual. For each threshold, the number of shared and host-specific clusters were counted from the cluster file, taking advantage of the host-affiliation contained within the headers of the ORFs. Likewise, the number of clusters in which each sample was represented was counted, in order to quantify the number of clusters per sample. Finally, to estimate the segregation of sequence clusters among individuals, the cumulative increase in the number of clusters relative to the number of individuals per host/colony was calculated, using 10 random sampling orders.
Functional characterization of metagenomes
As for the quantification of gene content diversity, the functional analysis was based on the filtered ORFs generated from assemblies of 20 Million paired-end host-filtered reads. Amino acid sequences were annotated using the eggNOG database v.5 [90] with the eggnog-mapper (version 1.0.3) [91], from which COG category annotations were extracted and counted. Polysaccharide lyases and glycoside hydrolases were annotated using the dbCan2 database [38]. The database was queried with hmmsearch, and the results were filtered by e-value (max e-value 1e-05 for alignments longer than 80 amino acids, otherwise 1e-03), and HMM coverage (min fraction 0.3). To investigate whether the gut microbiota of the two host species have a similar representation of CAZyme families, the mean number of ORFs per sample annotated to each family was calculated, for each host.
To quantify and compare the diversity within CAZyme families, ORF sequences annotated as either glycoside hydrolases (family “GH”) or Polysaccharide lyases (family “PL”) were clustered separately for each CAZyme family. For this analysis, only the Japanese samples were used, in order to have the same sampling depth per host. The clustering was done with CD-HIT, with a conservative amino acid identity threshold of 50% (and word-size of 3 as recommended in the CD-HIT manual). Furthermore, the ORFs were blasted (blastp) against the honey bee gut microbiota database, in order to estimate which SDPs contribute most to these enzyme families (threshold of significance: e-value < 10e-05 and query-coverage > 80%, with “close hits” corresponding to the subset of these having an amino acid percentage identity > 95%).
Quantification and Statistical Analysis
The data shown in Figure 5A was obtained from individual honey bees (n = 18 for A. mellifera, n = 16 for A. cerana), collected from one apiary per host species. The data shown in Figure 5B was obtained from individual honey bees (n = 20 for A. mellifera, n = 18 for A. cerana), collected from two apiaries per host species. For both data-sets, statistical significance was calculated using a Mann-Whitney U test [71] (with p < 0.05 as significance threshold).
Acknowledgments
We would like to thank Yoshiko Sakamoto, Shohei Kanari, Nao Tsuyuki, Yoshihiro Saito, Hiroki Kohno, and Takeo Kubo for their help during the sampling of honey bees. We also thank Kohei Fukuda for the technical assistance of molecular work. P.E. is supported by the European Research Council ERC-StG “MicroBeeOme” (714804) and the Swiss National Science Foundation (SNFS) project grant 31003A_179487. R.M. and P.E. are supported by the Human Frontier Science Program (HFSP) Young Investigator grant RGY0077/2016. R.M. is supported by Japan Science and Technology Agency ERATO (JPMJER1502).
Author Contributions
P.E., K.M.E., and R.M. designed the study. S.S. performed the experimental work, including sample collection, DNA extraction, and qPCR. K.M.E. developed the bioinformatic pipeline and analyzed the data. P.E. contributed to the data analysis. K.M.E. and P.E. wrote the first draft of the manuscript. R.M. contributed to the editing of the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: June 11, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cub.2020.04.070.
Contributor Information
Kirsten M. Ellegaard, Email: kirsten.ellegaard@unil.ch.
Philipp Engel, Email: philipp.engel@unil.ch.
Supplemental Information
For each genome, the accession numbers for both NCBI and IMG are provided (if available). Annotation files generated by IMG were used for analyses when possible. Genomes and genes are represented by their locus-tags in the database, and throughout all analysis files deposited on zenodo. SDP names are given only when metagenomic validation has been performed. For isolates published in the current study, the culture medium used for isolation is indicated. (A) Bifidobacterium spp., (B) Lactobacillus spp., phylotype “Firm5,” (C) Lactobacillus spp., phylotype “Firm4,” (D) Gilliamella spp., (E) Snodgrassella spp., (F) Apibacter spp., (G) Frischella perrrara, (H) Bartonella apis, (I) Bombella spp., (J) Commensalibacter spp., (K) Lactobacillus spp.
Genome identifiers correspond to locus-tags, see Data S1 for associated metadata. (A) Bifidobacterium spp., (B) Lactobacillus spp., phylotype “Firm5,” (C) Lactobacillus spp., phylotype “Firm4,” (D) Gilliamella spp., (E) Snodgrassella spp., (F) Apibacter spp., (G) Frischella perrrara, (H) Bartonella apis, (I) Bombella spp., (J) Commensalibacter spp., (K) Lactobacillus spp.
The distance matrix was based on the pairwise fractions of shared SNVs (jaccard distance), among all pairs of samples with sufficient coverage of SNV profiling (minimum 20x terminus coverage). Blue dots represent A. cerana samples, green dots represent A. mellifera samples from Japan, and red dots represent A. mellifera samples from Switzerland, with different shades indicating colony affiliation.
References
- 1.Kashtan N., Roggensack S.E., Rodrigue S., Thompson J.W., Biller S.J., Coe A., Ding H., Marttinen P., Malmstrom R.R., Stocker R. Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science. 2014;344:416–420. doi: 10.1126/science.1248575. [DOI] [PubMed] [Google Scholar]
- 2.Cordero O.X., Polz M.F. Explaining microbial genomic diversity in light of evolutionary ecology. Nat. Rev. Microbiol. 2014;12:263–273. doi: 10.1038/nrmicro3218. [DOI] [PubMed] [Google Scholar]
- 3.Brockhurst M.A., Harrison E., Hall J.P.J., Richards T., McNally A., MacLean C. The ecology and evolution of pangenomes. Curr. Biol. 2019;29:R1094–R1103. doi: 10.1016/j.cub.2019.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Mitri S., Foster K.R. The genotypic view of social interactions in microbial communities. Annu. Rev. Genet. 2013;47:247–273. doi: 10.1146/annurev-genet-111212-133307. [DOI] [PubMed] [Google Scholar]
- 5.Ansorge R., Romano S., Sayavedra L., Porras M.A.G., Kupczok A., Tegetmeyer H.E., Dubilier N., Petersen J. Functional diversity enables multiple symbiont strains to coexist in deep-sea mussels. Nat. Microbiol. 2019;4:2487–2497. doi: 10.1038/s41564-019-0572-9. [DOI] [PubMed] [Google Scholar]
- 6.Romero Picazo D., Dagan T., Ansorge R., Petersen J.M., Dubilier N., Kupczok A. Horizontally transmitted symbiont populations in deep-sea mussels are genetically isolated. ISME J. 2019;13:2954–2968. doi: 10.1038/s41396-019-0475-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellegaard K.M., Engel P. Genomic diversity landscape of the honey bee gut microbiota. Nat. Commun. 2019;10:446. doi: 10.1038/s41467-019-08303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu A., Sunagawa S., Mende D.R., Bork P. Inter-individual differences in the gene content of human gut bacterial species. Genome Biol. 2015;16:82. doi: 10.1186/s13059-015-0646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenblum S., Carr R., Borenstein E. Extensive strain-level copy-number variation across human gut microbiome species. Cell. 2015;160:583–594. doi: 10.1016/j.cell.2014.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim M., Oh H.S., Park S.C., Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014;64:346–351. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 11.Olm M.R., Crits-Christoph A., Diamond S., Lavy A., Matheus Carnevali P.B., Banfield J.F. Consistent metagenome-derived metrics verify and delineate bacterial species boundaries. mSystems. 2020;5 doi: 10.1128/mSystems.00731-19. e00731-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez-R L.M., Castro J.C., Kyrpides N.C., Cole J.R., Tiedje J.M., Konstantinidis K.T. How much do rRNA gene surveys underestimate extant bacterial diversity? Appl. Environ. Microbiol. 2018;84 doi: 10.1128/AEM.00014-18. e00014-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sczyrba A., Hofmann P., Belmann P., Koslicki D., Janssen S., Dröge J., Gregor I., Majda S., Fiedler J., Dahms E. Critical assessment of metagenome interpretation-a benchmark of metagenomics software. Nat. Methods. 2017;14:1063–1071. doi: 10.1038/nmeth.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwong W.K., Medina L.A., Koch H., Sing K.W., Soh E.J.Y., Ascher J.S., Jaffé R., Moran N.A. Dynamic microbiome evolution in social bees. Sci. Adv. 2017;3:e1600513. doi: 10.1126/sciadv.1600513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwong W.K., Moran N.A. Gut microbial communities of social bees. Nat. Rev. Microbiol. 2016;14:374–384. doi: 10.1038/nrmicro.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steele M.I., Kwong W.K., Whiteley M., Moran N.A. Diversification of type VI secretion system toxins reveals ancient antagonism among bee gut microbes. MBio. 2017;8 doi: 10.1128/mBio.01630-17. e01630-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwong W.K., Engel P., Koch H., Moran N.A. Genomics and host specialization of honey bee and bumble bee gut symbionts. Proc. Natl. Acad. Sci. USA. 2014;111:11509–11514. doi: 10.1073/pnas.1405838111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng H., Nishida A., Kwong W.K., Koch H., Engel P., Steele M.I., Moran N.A. Metabolism of toxic sugars by strains of the bee gut symbiont Gilliamella apicola. MBio. 2016;7 doi: 10.1128/mBio.01326-16. e01326-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellegaard K.M., Tamarit D., Javelind E., Olofsson T.C., Andersson S.G., Vásquez A. Extensive intra-phylotype diversity in lactobacilli and bifidobacteria from the honeybee gut. BMC Genomics. 2015;16:284. doi: 10.1186/s12864-015-1476-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engel P., Martinson V.G., Moran N.A. Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. USA. 2012;109:11002–11007. doi: 10.1073/pnas.1202970109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel P., Stepanauskas R., Moran N.A. Hidden diversity in honey bee gut symbionts detected by single-cell genomics. PLoS Genet. 2014;10:e1004596. doi: 10.1371/journal.pgen.1004596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welch R.A., Burland V., Plunkett G., 3rd, Redford P., Roesch P., Rasko D., Buckles E.L., Liou S.R., Boutin A., Hackett J. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biller S.J., Berube P.M., Lindell D., Chisholm S.W. Prochlorococcus: the structure and function of collective diversity. Nat. Rev. Microbiol. 2015;13:13–27. doi: 10.1038/nrmicro3378. [DOI] [PubMed] [Google Scholar]
- 24.Ellegaard K.M., Engel P. Beyond 16S rRNA community profiling: intra-species diversity in the gut microbiota. Front. Microbiol. 2016;7:1475. doi: 10.3389/fmicb.2016.01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia S.L., Stevens S.L.R., Crary B., Martinez-Garcia M., Stepanauskas R., Woyke T., Tringe S.G., Andersson S.G.E., Bertilsson S., Malmstrom R.R., McMahon K.D. Contrasting patterns of genome-level diversity across distinct co-occurring bacterial populations. ISME J. 2018;12:742–755. doi: 10.1038/s41396-017-0001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rusch D.B., Halpern A.L., Sutton G., Heidelberg K.B., Williamson S., Yooseph S., Wu D., Eisen J.A., Hoffman J.M., Remington K. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caro-Quintero A., Konstantinidis K.T. Bacterial species may exist, metagenomics reveal. Environ. Microbiol. 2012;14:347–355. doi: 10.1111/j.1462-2920.2011.02668.x. [DOI] [PubMed] [Google Scholar]
- 28.Bendall M.L., Stevens S.L., Chan L.K., Malfatti S., Schwientek P., Tremblay J., Schackwitz W., Martin J., Pati A., Bushnell B. Genome-wide selective sweeps and gene-specific sweeps in natural bacterial populations. ISME J. 2016;10:1589–1601. doi: 10.1038/ismej.2015.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goris J., Konstantinidis K.T., Klappenbach J.A., Coenye T., Vandamme P., Tiedje J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 30.Jain C., Rodriguez-R L.M., Phillippy A.M., Konstantinidis K.T., Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Powell E., Ratnayeke N., Moran N.A. Strain diversity and host specificity in a specialized gut symbiont of honeybees and bumblebees. Mol. Ecol. 2016;25:4461–4471. doi: 10.1111/mec.13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallberg A., Han F., Wellhagen G., Dahle B., Kawata M., Haddad N., Simões Z.L., Allsopp M.H., Kandemir I., De la Rúa P. A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat. Genet. 2014;46:1081–1088. doi: 10.1038/ng.3077. [DOI] [PubMed] [Google Scholar]
- 33.Han F., Wallberg A., Webster M.T. From where did the western honeybee (Apis mellifera) originate? Ecol. Evol. 2012;2:1949–1957. doi: 10.1002/ece3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park D., Jung J.W., Choi B.S., Jayakodi M., Lee J., Lim J., Yu Y., Choi Y.S., Lee M.L., Park Y. Uncovering the novel characteristics of Asian honey bee, Apis cerana, by whole genome sequencing. BMC Genomics. 2015;16:1. doi: 10.1186/1471-2164-16-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C., Wang H., Liu Z., Chen X., Tang J., Meng F., Shi W. Population genomics provide insights into the evolution and adaptation of the eastern honey bee (Apis cerana) Mol. Biol. Evol. 2018;35:2260–2271. doi: 10.1093/molbev/msy130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takamura N., Tokuda Y., Nakano S., Horikiri M. Chikusan Hattatsu Shi, M.o.A.a.F. Livestock Industry Bureau. Chuo Kouron Jigyo; 1966. Yoho no Seisei; pp. 1313–1334. [Google Scholar]
- 37.Requier F., Garnery L., Kohl P.L., Njovu H.K., Pirk C.W.W., Crewe R.M., Steffan-Dewenter I. The conservation of native honey bees is crucial. Trends Ecol. Evol. 2019;34:789–798. doi: 10.1016/j.tree.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., Busk P.K., Xu Y., Yin Y. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46(W1):W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helbert W., Poulet L., Drouillard S., Mathieu S., Loiodice M., Couturier M., Lombard V., Terrapon N., Turchetto J., Vincentelli R., Henrissat B. Discovery of novel carbohydrate-active enzymes through the rational exploration of the protein sequences space. Proc. Natl. Acad. Sci. USA. 2019;116:6063–6068. doi: 10.1073/pnas.1815791116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lombard V., Golaconda Ramulu H., Drula E., Coutinho P.M., Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimura M. Cambridge University; 1983. The Neutral Theory of Molecular Evolution. [Google Scholar]
- 42.Charlesworth B. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009;10:195–205. doi: 10.1038/nrg2526. [DOI] [PubMed] [Google Scholar]
- 43.Leffler E.M., Bullaughey K., Matute D.R., Meyer W.K., Ségurel L., Venkat A., Andolfatto P., Przeworski M. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biol. 2012;10:e1001388. doi: 10.1371/journal.pbio.1001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bobay L.M., Ochman H. Factors driving effective population size and pan-genome evolution in bacteria. BMC Evol. Biol. 2018;18:153. doi: 10.1186/s12862-018-1272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishida A.H., Ochman H. Rates of gut microbiome divergence in mammals. Mol. Ecol. 2018;27:1884–1897. doi: 10.1111/mec.14473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ley R.E., Hamady M., Lozupone C., Turnbaugh P.J., Ramey R.R., Bircher J.S., Schlegel M.L., Tucker T.A., Schrenzel M.D., Knight R., Gordon J.I. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Groussin M., Mazel F., Sanders J.G., Smillie C.S., Lavergne S., Thuiller W., Alm E.J. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 2017;8:14319. doi: 10.1038/ncomms14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moeller A.H., Caro-Quintero A., Mjungu D., Georgiev A.V., Lonsdorf E.V., Muller M.N., Pusey A.E., Peeters M., Hahn B.H., Ochman H. Cospeciation of gut microbiota with hominids. Science. 2016;353:380–382. doi: 10.1126/science.aaf3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ochman H., Elwyn S., Moran N.A. Calibrating bacterial evolution. Proc. Natl. Acad. Sci. USA. 1999;96:12638–12643. doi: 10.1073/pnas.96.22.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moran N.A., Munson M.A., Baumann P., Ishikawa H. A molecular clock in endosymbiotic bacteria is calibrated using the insect hosts. Proc. R. Soc. B. 1993;253:167–171. [Google Scholar]
- 51.Reese A.T., Dunn R.R. Drivers of microbiome biodiversity: a review of general rules, feces, and ignorance. MBio. 2018;9 doi: 10.1128/mBio.01294-18. e01294-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schloissnig S., Arumugam M., Sunagawa S., Mitreva M., Tap J., Zhu A., Waller A., Mende D.R., Kultima J.R., Martin J. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox-Foster D.L., Conlan S., Holmes E.C., Palacios G., Evans J.D., Moran N.A., Quan P.L., Briese T., Hornig M., Geiser D.M. A metagenomic survey of microbes in honey bee colony collapse disorder. Science. 2007;318:283–287. doi: 10.1126/science.1146498. [DOI] [PubMed] [Google Scholar]
- 54.Stokstad E. Entomology. The case of the empty hives. Science. 2007;316:970–972. doi: 10.1126/science.316.5827.970. [DOI] [PubMed] [Google Scholar]
- 55.Oldroyd B.P. What’s killing American honey bees? PLoS Biol. 2007;5:e168. doi: 10.1371/journal.pbio.0050168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seeley T. Princeton University; 2019. The Lives of Bees: The Untold Story of the Honey Bee in the Wild. [Google Scholar]
- 57.Sonnenburg E.D., Smits S.A., Tikhonov M., Higginbottom S.K., Wingreen N.S., Sonnenburg J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529:212–215. doi: 10.1038/nature16504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fragiadakis G.K., Smits S.A., Sonnenburg E.D., Van Treuren W., Reid G., Knight R., Manjurano A., Changalucha J., Dominguez-Bello M.G., Leach J., Sonnenburg J.L. Links between environment, diet, and the hunter-gatherer microbiome. Gut Microbes. 2019;10:216–227. doi: 10.1080/19490976.2018.1494103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tatsuno M., Osawa N. Flower visitation patterns of the coexisting honey bees Apis cerana japonica and Apis mellifera (Hymenoptera: Apidae) Entomol. Sci. 2016;19:255–267. [Google Scholar]
- 60.Suryanarayana M.C., Mohana Rao G., Singh T. Studies on pollen sources for Apis cerana Fabr and Apis mellifera L bees at Muzaffarpur, Bihar, India. Apidologie. 1992;23:33–46. [Google Scholar]
- 61.Rosenzweig M. Cambridge University; 1995. Species Diversity in Space and Time. [Google Scholar]
- 62.Arrhenius O. Species and area. J. Ecol. 1921;9:95–99. [Google Scholar]
- 63.Gleason H.A. On the relation between species and area. Ecology. 1922;3:158–162. [Google Scholar]
- 64.Logue J.B., Langenheder S., Andersson A.F., Bertilsson S., Drakare S., Lanzén A., Lindström E.S. Freshwater bacterioplankton richness in oligotrophic lakes depends on nutrient availability rather than on species-area relationships. ISME J. 2012;6:1127–1136. doi: 10.1038/ismej.2011.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bell T., Ager D., Song J.I., Newman J.A., Thompson I.P., Lilley A.K., van der Gast C.J. Larger islands house more bacterial taxa. Science. 2005;308:1884. doi: 10.1126/science.1111318. [DOI] [PubMed] [Google Scholar]
- 66.Koskella B., Hall L.J., Metcalf C.J.E. The microbiome beyond the horizon of ecological and evolutionary theory. Nat. Ecol. Evol. 2017;1:1606–1615. doi: 10.1038/s41559-017-0340-2. [DOI] [PubMed] [Google Scholar]
- 67.Koetz A.H. Ecology, behaviour and control of Apis cerana with a focus on relevance to the Australian incursion. Insects. 2013;4:558–592. doi: 10.3390/insects4040558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruttner F. Springer; 1988. Biogeography and Taxonomy of Honeybees. [Google Scholar]
- 69.Shade A. Diversity is the question, not the answer. ISME J. 2017;11:1–6. doi: 10.1038/ismej.2016.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gareau M.G., Wine E., Rodrigues D.M., Cho J.H., Whary M.T., Philpott D.J., Macqueen G., Sherman P.M. Bacterial infection causes stress-induced memory dysfunction in mice. Gut. 2011;60:307–317. doi: 10.1136/gut.2009.202515. [DOI] [PubMed] [Google Scholar]
- 71.Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48:452–458. doi: 10.1038/bmt.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Christiansen T., Orwant J., Wall L., Foy B. Fourth Edition. O’Reilly Media; 2012. Programming Perl. [Google Scholar]
- 73.Stajich J.E., Block D., Boulez K., Brenner S.E., Chervitz S.A., Dagdigian C., Fuellen G., Gilbert J.G., Korf I., Lapp H. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andrews S. Babraham Bioinformatics; 2010. FastQC: A Quality Control Tool for High Throughput Sequence Data. [Google Scholar]
- 76.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Emms D.M., Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Katoh K., Misawa K., Kuma K., Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 80.Rambaut A. Institute of Evolutionary Biology, University of Edinburgh; 2010. FigTree v1.4.4. [Google Scholar]
- 81.Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Broad Intitute . Broad Institute; 2019. Picard Tools. [Google Scholar]
- 85.Hyatt D., Chen G.L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. arXiv, 2012 https://arxiv.org/abs/1207.3907 arXiv:1207.3907v2. [Google Scholar]
- 87.Tange O. SCL; 2018. GNU Parallel 2018. [Google Scholar]
- 88.Garrison E. 2016. Vcflib, a simple C++ library for parsing and manipulating VCF files. [Google Scholar]
- 89.Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huerta-Cepas J., Szklarczyk D., Heller D., Hernández-Plaza A., Forslund S.K., Cook H., Mende D.R., Letunic I., Rattei T., Jensen L.J. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huerta-Cepas J., Forslund K., Coelho L.P., Szklarczyk D., Jensen L.J., von Mering C., Bork P. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017;34:2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Powell J.E., Martinson V.G., Urban-Mead K., Moran N.A. Routes of acquisition of the gut microbiota of the honey bee Apis mellifera. Appl. Environ. Microbiol. 2014;80:7378–7387. doi: 10.1128/AEM.01861-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kešnerová L., Mars R.A.T., Ellegaard K.M., Troilo M., Sauer U., Engel P. Disentangling metabolic functions of bacteria in the honey bee gut. PLoS Biol. 2017;15:e2003467. doi: 10.1371/journal.pbio.2003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Engel P., James R.R., Koga R., Kwong W.K., McFrederick Q.S., Moran N.A. Standard methods for research on Apis mellifera gut symbionts. J. Apic. Res. 2013;52:1–24. [Google Scholar]
- 95.Muggeo V.M.R. Interval estimation for the breakpoint in segmented regression: a smoothed score-based approach. Aust. N. Z. J. Stat. 2017;59:311–322. [Google Scholar]
- 96.Paradis E., Schliep K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For each genome, the accession numbers for both NCBI and IMG are provided (if available). Annotation files generated by IMG were used for analyses when possible. Genomes and genes are represented by their locus-tags in the database, and throughout all analysis files deposited on zenodo. SDP names are given only when metagenomic validation has been performed. For isolates published in the current study, the culture medium used for isolation is indicated. (A) Bifidobacterium spp., (B) Lactobacillus spp., phylotype “Firm5,” (C) Lactobacillus spp., phylotype “Firm4,” (D) Gilliamella spp., (E) Snodgrassella spp., (F) Apibacter spp., (G) Frischella perrrara, (H) Bartonella apis, (I) Bombella spp., (J) Commensalibacter spp., (K) Lactobacillus spp.
Genome identifiers correspond to locus-tags, see Data S1 for associated metadata. (A) Bifidobacterium spp., (B) Lactobacillus spp., phylotype “Firm5,” (C) Lactobacillus spp., phylotype “Firm4,” (D) Gilliamella spp., (E) Snodgrassella spp., (F) Apibacter spp., (G) Frischella perrrara, (H) Bartonella apis, (I) Bombella spp., (J) Commensalibacter spp., (K) Lactobacillus spp.
The distance matrix was based on the pairwise fractions of shared SNVs (jaccard distance), among all pairs of samples with sufficient coverage of SNV profiling (minimum 20x terminus coverage). Blue dots represent A. cerana samples, green dots represent A. mellifera samples from Japan, and red dots represent A. mellifera samples from Switzerland, with different shades indicating colony affiliation.
Data Availability Statement
The accession number for raw sequence data reported in this paper is NCBI BioProject: PRJNA598094. Accession numbers for genomes included in the genomic database are provided in Data S1. All databases used for analysis, files corresponding to metagenomic assemblies, and scripts generated for analysis, have been deposited on zenodo at: https://doi.org/10.5281/zenodo.3747314.