

Abstract
Thermal burn injuries are an important environmental stressor which can result in considerable morbidity and mortality. The exact mechanism by which an environmental stimulus to skin results in local and systemic effects is an area of active research. One potential mechanism to allow skin keratinocytes to disperse bioactive substances is via microvesicle particles, which are subcellular bodies released directly from cellular membranes. Our previous studies have indicated that thermal burn injury of the skin keratinocyte in vitro results in the production of the lipid mediator Platelet-activating Factor (PAF). The present studies demonstrate that thermal burn injury to keratinocytes in vitro and human skin explants ex vivo and mice in vivo generate microvesicle particles. Use of pharmacologic and genetic tools indicates that the optimal release of microvesicles is dependent upon the PAF receptor. Of note, burn injury-stimulated microvesicle particles do not carry appreciable protein cytokines, yet contain high levels of PAF. These studies describe a novel mechanism involving microvesicle particles by which a metabolically labile bioactive lipid can travel from cells in response to environmental stimuli.
Introduction
Thermal burn injury causes significant morbidity and mortality. It is estimated that more than 100,000 patients are hospitalized in the U.S. annually for severe burn injuries [1,2]. In addition to local effects, systemic effects of thermal burn injury have been described, suggesting that keratinocytes can generate bioactive agents in response to damage [3–7]. Of interest, studies have determined that thermal burn injury of keratinocytes and/or skin can generate multiple protein cytokines and lipid mediators [8–10]. Yet the exact mechanism(s) by which keratinocytes release bioactive agents are not clear.
Microvesicle particles (MVP) are defined as submicron vesicles generated from cellular membranes via exocytosis. They are irregularly shaped and range from ~100–1000 nm in size. The generation of MVP occurs in response to various stimuli, many involving intracellular calcium mobilization [11,12]. Once released into the circulation (blood and tissue), MVP can then exert functional effects on various cell types. MVP have different functions which depend on the cell of origin and stimuli. Moreover, MVP are thought to be able to target other cell types through surface membrane interactions that then allow fusion of the MVP to the target cell membrane and subsequent delivery of the intravesicular contents. The pathways by which MVP are released from cells have been studied, and are cellular- and stimulus-dependent [13]. For example, in endothelial cells, MVP release in response to agents like TNF-α are due to P38 MAPK signaling, whereas in THP-1 monocytes both Erk 1/2 (p42/44) and P38 MAPK pathways are involved [13–16].
The functions of various MVP in physiological and pathological processes depend on their carried contents (proteins, lipids and nucleic acids) [12,13]. One advantage of this arrangement is that metabolically unstable compounds inside MVP can be somewhat protected from enzymatic degradation. Studies have reported that circulating MVP are increased in inflammatory skin diseases such as psoriasis, in which many cytokines are also elevated [17–19]. In translational studies, elevated levels of MVP have been documented in vascular diseases and correlated with the severity of disease [16,18,19].
Platelet-activating factor (PAF) is the term coined by Jacques Benveniste and colleagues in the early 1970s for the substance released by activated leukocytes which induced platelet aggregation [20]. The structure of this activity was elucidated in 1979 and was found to reside in sn-1 ether linked glycerophophocholines (GPC) with a short-chained fatty acid (usually acetate) at the sn-2 position [21]. PAF is synthesized enzymatically via two separate pathways [22–24]. The remodeling pathway associated with cellular stimulation from a variety of sources, consists of phospholipase A2 (often cytosolic PLA2) followed by an acetyl-CoA-dependent acetyltransferase (LPCAT) that acetylates a lyso-PAF intermediate to form PAF. Once generated, PAF is quickly metabolized by acetylhydrolases (PAF-AH) which remove the sn-2 acetyl moiety resulting in the biologically inactive lyso-glycerophosphocholine [25]. The half-life of PAF has been estimated to be only a few minutes, due to both serum- and cell-associated PAF-AH [26,27]. Exerting its effects via a single G-protein coupled receptor (the PAFR), PAF has been found to be involved in multiple processes ranging from acute inflammatory and allergic responses to delayed immunosuppression [22,24,28,29]. PAF has been demonstrated to be produced in response to multiple environmental stressors including thermal injury [10,30,31]. The PAFR exerts its effects via multiple down-stream signaling pathways including the mitogen kinase (both ERK and P38), JNK, as well as can indirectly activate the EGFR [32].
Recent studies by our group have demonstrated that activation of the keratinocyte PAFR induces MVP release [33–35]. Of interest, UVB radiation, which also is a potent generator of PAF agonists [28–31], induces MVP in a PAFR-dependent manner [33,34]. Burn injury also generates PAF [10,36], raising the possibility that MVP could serve as effectors linking environmental stressors that act upon the skin, with the PAF system playing an intermediary role. The goal of the present studies is to assess if thermal burn injury generates MVP, what are contained in these particles, and to define the role of PAFR signaling in this process.
Materials and Methods
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless indicated otherwise. Primary murine fibroblasts from C57BL/6J mice skin and HaCaT keratinocyte-derived cell line were grown in Dulbecco’s modified Eagle’s medium (DMEM) high glucose media with 10% FCS, 6 mM L-glutamine and a 100 μg/mL mixture of penicillin and streptomycin as described [10,33,38]. PAF-R-negative KB cells were rendered PAF-R-positive (KBP) by transducing the MSCV2.1 retrovirus encoding the human leukocyte PAF-R and PAF-R-deficient (KBM) by transducing with the MSCV2.1 vector alone and grown in DMEM high glucose media with supplements as described previously [39]. Telomerase-immortalized keratinocytes (N-Terts) from neonatal foreskins and primary human keratinocytes from discarded human skin obtained as previously described [38] were grown in EpiLife medium with 10% FBS, 100 μg/mL mixture of penicillin and streptomycin and addition of Human Keratinocyte Growth Supplement (HKGS). Primary cells were experimented in passage 3–6. HaCaT cells were used between passage 70–100, and N-Terts between passages 40–60. Cell lines were regularly tested for mycoplasma. KBP and KBM cells (passage 60–80) were grown to approximately 50% confluence with small colonies and other keratinocyte derived cells were grown to approximately 80–90% confluence in 10 cm dishes, washed three times with Hanks Balanced Salt Solution (HBSS) and then incubated with pre-warmed (37°C) HBSS with 10 mg/ml fatty acid-free BSA. Thermal burn injury was performed by placement of the cell culture dish onto a 90°C water bath for various times [10,36]. In some experiments, CPAF or TPA or UVB irradiation with a Philips F20T12/UVB lamp source were employed [30,31,33,35].
Mice
Female C57BL/6 wild type mice (PAFR expressing; age 6–8 week) were purchased from The Charles River Laboratories. PAFR-KO (Ptafr−/−) mice on a C57BL/6 background, generated as previously described [40], were a kind gift of professor Takao Shimizu (Department of Biochemistry, University of Tokyo). All mice were used at approximately 7–10 weeks of age for the experiments. All mice were housed under specific pathogen-free conditions and kept on a 12-hour light/dark cycle with free access to standard animal chow and water in the animal facility at the Wright State University. All procedures were approved by the Institutional Animal Care and Use Committee of Wright State University.
Thermal burn injury in murine skin
Thermal burn injury was performed using our previously published methodology [36]. Wild-type or Ptafr−/− C57BL/6J mice were anesthetized with ketamine/xylazine (100 and 10 mg/kg, respectively) and fur removed from dorsal back skin. The dorsal skin of the mice was treated with 8 second exposure of two 1 × 1 cm stainless steel metal blocks heated to 90 °C, resulting in an ~12–15% body surface area burns. Mice were given 1 ml of normal saline i.p. immediately afterwards. At 2h post-treatment, the mice were euthanized and exsanguinated. Skin biopsies of the burned area were collected for MVP isolation. Blood was collected from heart and allowed to clot for 30 min at room temperature, followed by centrifugation at 2,000 x g for 10 min at 4 °C. Blood serum was carefully transferred into a new tube and immediately processed for MVP isolation.
Human skin explants
De-identified discarded skin was obtained from contouring surgeries (abdominoplasties and brachioplasties) [31,33,35]. Skin was washed, fat trimmed and placed in PBS warmed at 37°C. Skin blisters were generated with a Vacuubrand® vacuum pump, 100 mbar, attached to the barrel of a 20 ml syringe. Once blisters had formed (see Figure 2B), the barrels were removed, and surface of blisters were treated. As shown histologically in Figure 2B, the blisters were induced in such a fashion to separate epidermis from dermis. In the skin experiments, skin was burned with 90 °C water using a funnel for various times exactly as previously reported [36]. For other treatments, topical agents added or UVB irradiation was performed on the skin/blister roofs [35,36]. After various times, either skin biopsies were obtained or blister fluid was collected via a 25 ga syringe and weighed.
Figure 2. Thermal burn injury of human skin explants results in MVP release.
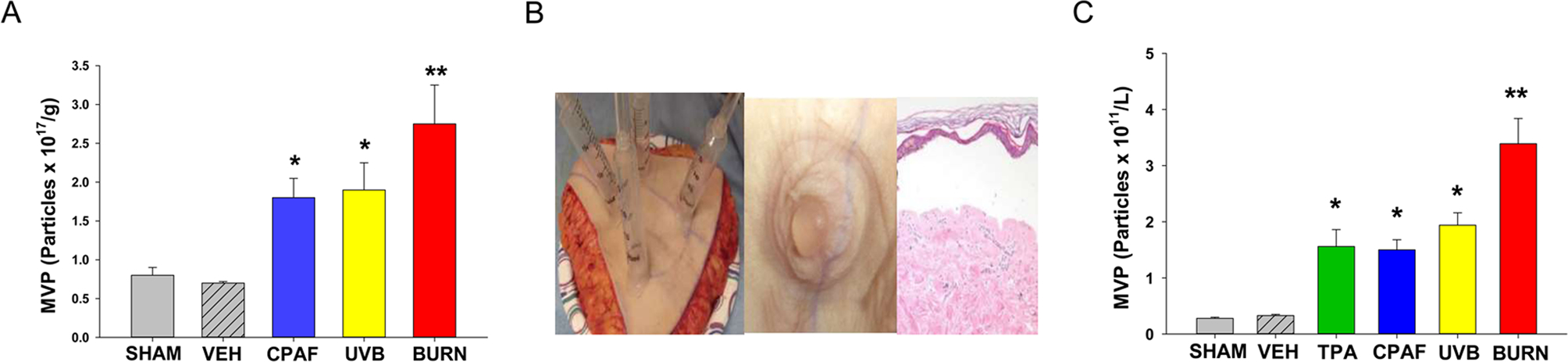
(A) Human skin tissue explants underwent treatment with a 5 sec application of aqueous 90 °C thermal burn injury, 2.5 KJ/m2 UVB, 50 μl topical CPAF (53ng), 0.1% DMSO vehicle, or no treatment (SHAM). At 4 h post treatment, 5 mm skin biopsies were obtained and weighed, then MVP were isolated and measured. (B) Suction-induced blisters were generated on human skin tissue which results in a sub-epidermal blister. (C) Blister roofs were treated with either 2.5 KJ/m2 UVB, a 5 sec thermal burn injury, topical application of 50 μl of the phorbol ester TPA (62 ng), CPAF (53 ng), 0.1% DMSO vehicle or Sham. At 4 h, blister fluid was removed and weighed, and MVP isolated and measured. The data are Mean ± SE MVP levels from at least five separate experiments. *Denotes statistically significant (P<0.05) or ** (P <0.01) changes in levels of MVP from control (sham-treated) values. Please note for A thermal burn injury generated statistically significant (P<0.05) increased MVP in comparison to CPAF but not UVB treatment. For B, thermal burn injury generated statistically significant (P<0.05) increased MVP in comparison to all other treatments.
Isolation and measurement of MVPs
Microvesicle particles were collected from culture medium as previously described with slight modifications [33,35]. In brief, cell culture medium, skin blister fluids, and mice blood serum were collected and centrifuged at 2,000 x g for 20 min to remove cells and debris. Supernatant then transferred to a new tube centrifuge at 20,000 x g for 70 min. The resulting pellet contained the isolated MVPs.
For skin biopsies, tissue was cut up finely in the microcentrifuge tube and digested in 0.5 ml of 5 mg/ml collagenase and dispase solution made in deionized filtered water shake overnight at 37°C. After overnight digestion, samples were centrifuged at 2,000 x g for 20 minutes to remove tissues, then followed with 20,000 x g centrifugation for 10 min to remove remaining tissue and subcellular component. MVP from the sample supernatants were then pelleted at 20,000 x g by centrifugation for 70 min.
The concentration of the MVP was determined by using a NanoSight NS300 instrument (NanoSight Ltd, Malvern Instruments, Malvern, UK) exactly as previously reported [33,35]. Three 30-second videos of each sample were recorded and analyzed with NTA software version 3.0 to determine the concentration and size of measured particles with corresponding standard error.
Measurement of cytokines and chemokines
HaCaT cells were treated with vehicle (0.1% ethanol), CPAF (100 nM) or burn (30 sec), after 4 hours, medium was collected for MVP isolation. MVP was re-suspended with 100 μl filtered PBS and stored in −80°C before assay. Cytokine levels were measured by Bio-Plex Pro™ Human Cytokine 27-plex Assay kit. Cytokine concentration (pg/ml) was normalized to MVP number for analysis.
Measurement of PAFR agonistic activity
The presence of PAFR agonists in lipid extracts derived from HaCaT cells was assessed by ELISA as the ability of lipid extracts to induce IL-8 release in PAFR-expressing KBP cells, but not in PAFR-deficient KBM cells, as described [31,39]. Lipid extracts were isolated either from burn treated HaCaT cells, total cell medium at various time points (using water:methanol:methylene chloride [1:1:1 v/v] exactly as reported [30]), and HaCaT induced MVP or MVP-depleted supernatant at 2 hrs. Lipids were added to PAF-R overexpressed KBP cells. The ratio of IL-8 released by treated KBP cells compared to 1 nM CPAF positive control treated KBP cells were used to determine the PAF-R agonistic activity level.
Mice ear thickness measurements
WT and Ptafr−/− mice ears were treated with: vehicle (20 μl of acetone), CPAF (20 ng), TPA, or control and burn-induced HaCaT MVP lipid extracts (from 1011 particles in 20 μl acetone). Mouse ears were measured using a PEACOCK dial thickness gauge before and 2 hours after treatment. Ear swelling levels were used as a surrogate marker for skin inflammation.
Statistics
All statistical calculations were performed using GraphPad Prism 6. All experiments were repeated at least three times in separate experiments. Statistical significance was determined using one-way or two-way ANOVA and the post-hoc Holm-Sidak method, with alpha=5%.
Results
Thermal burn injury results in increased MVP in keratinocytes.
Our previous in vitro and in vivo studies demonstrated that thermal burn injury stimulates enzymatic PAF biosynthesis [10,36]. As PAFR activation can generate MVP, our first studies tested if thermal burn injury could generate increased MVP release. For these studies, we used the human keratinocyte-derived cell line HaCaT [37]. As shown in Figure 1, burn injury resulted in the release of MVP in a concentration- and time-dependent manner in the HaCaT keratinocyte cell line. Of note, MVP were generated rapidly in response to thermal burn, with increased levels noted at 15 min post injury. Microvesicle particle levels appeared maximal at 1 h (Figure 1A). Time-response studies indicated that MVP generated in response to thermal burn injury (Burn-MVP) were elevated at similar levels from 4–8 hours post-injury, then decreased to baseline levels by 24 hours (Supplemental Figure S1). Of note, the viability of HaCaT cells following thermal burn injury at 4h was essentially 100% in response to an 8 sec injury, yet decreased to 25–30% following 30 or 45 sec burn injuries (Supplemental Figure S2). Thermal burn injury resulted in greater numbers of MVP release in HaCaT cells compared to treatment with the PAFR agonist carbamoyl-PAF (CPAF) or UVB. The amount of thermal burn injury as defined by time of exposure at 90 degrees also resulted in a concentration-dependent response (Figure 1B). Similarly, thermal burn injury resulted in MVP production in primary cultures of human keratinocytes in vitro (Figure 1C) and in immortalized N-tert keratinocytes (Supplemental Figure S3). Thermal burn injury also induced increased levels of MVP in primary cultures of murine fibroblasts indicating that other cell types residing in skin could respond to a burn injury (Figure 1D). CPAF did not result in increased MVP release in PAF-R-negative skin fibroblasts [38], yet C-2 ceramide (but not control dihydroceramide) exerted this effect. To similarly assess if burn injury can generate MVP in human skin, we treated discarded human skin tissue from body contouring surgeries (e.g., abdominoplasties [35,36]) with an aqueous burn and 4 h later did punch biopsies and tested them for MVP. As shown in Figure 2A, thermal burn injury was a potent generator of MVP, with slight but not statistically significant increased MVP in comparison to UVB radiation, but statistically significant increased MVP in comparison to topical application of the PAFR agonist CPAF in human skin explants (p<0.05). As a punch biopsy samples both epidermis and dermis, the exact source of the MVP generated and where they resided in response to thermal burn injury was not able to be determined. To assess if the MVP being generated were actually leaving the epidermis, we generated suction blisters on the skin [35,36], followed by treatment of the induced blister roofs with thermal burn injury, CPAF, TPA or UVB. As shown in Figure 2B, suction blisters result in the formation of an epidermal roof, with dermis at the base. Treatment of blister roofs with thermal burn injury, UVB, or topical application of CPAF or the phorbol ester TPA resulted in increased levels of MVP in the subepidermal blister fluid at 4 h post treatment (Figure 2C). Thermal burn injury resulted in a statistically-significant increase in blister fluid MVPs in comparison to the other stimuli employed (Figure 2C). These studies indicate that thermal burn injury induces the production and release of MVP from human skin with the likely major source being the epidermal keratinocyte.
Figure 1. Thermal burn injury generates MVP in HaCaT keratinocytes.
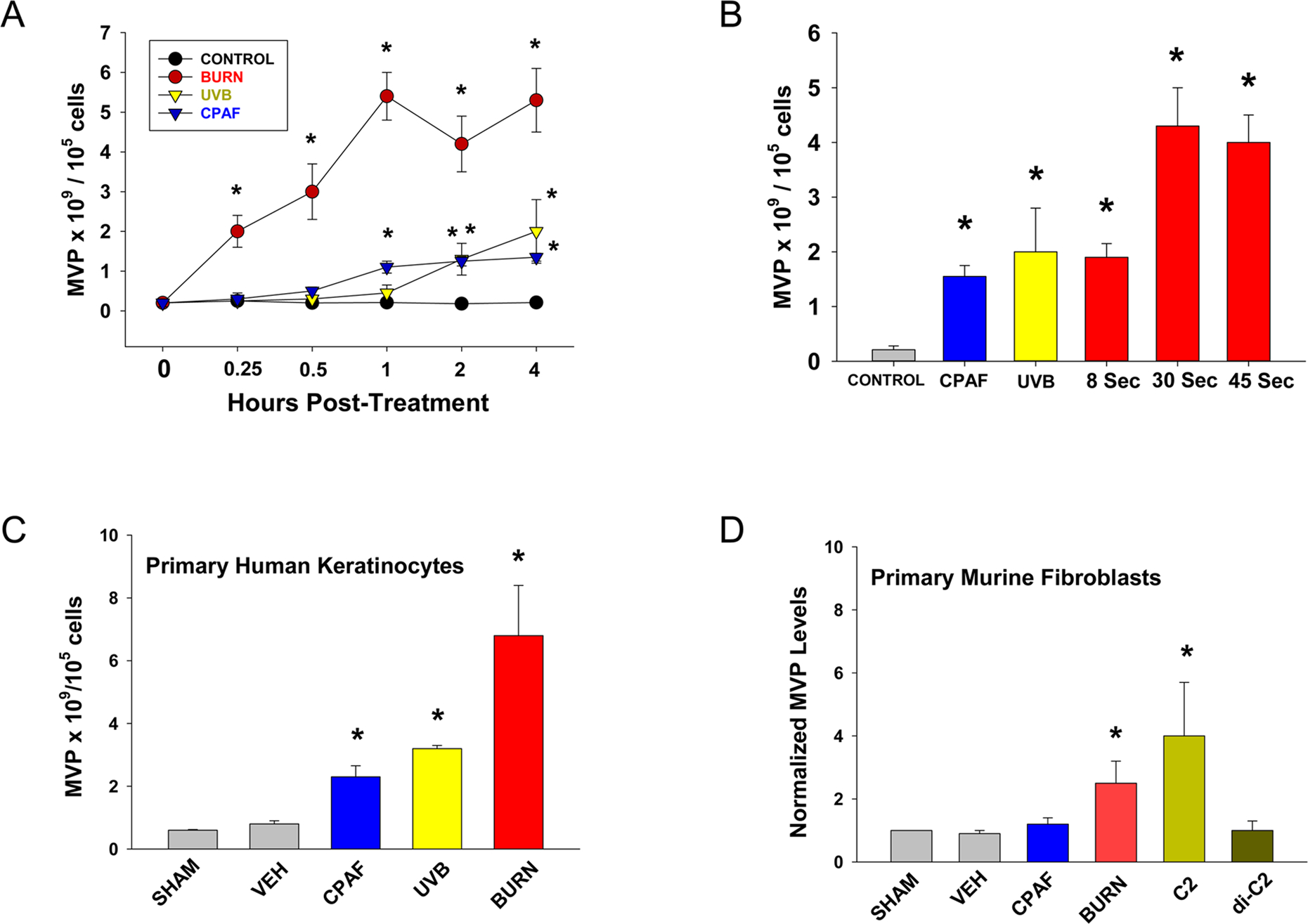
(A) Time-response studies. HaCaT cells were subjected to 90°C water bath x 30 sec, or treatment with 3.6 KJ/m2 UVB, or 100 nM CPAF, or 0.1% ethanol vehicle control. At various times, the supernatant was removed and MVP measured. (B) Concentration-response studies. HaCaT cells were treated with thermal burn injury for either 8, 30 or 45 sec, or 3.6 KJ/m2 UVB, 100 nM CPAF, or vehicle. At 4 h, the supernatant was removed and MVP measured. Primary cultures of human keratinocytes (C) or murine fibroblasts (D) were treated with CPAF, UVB (keratinocytes) or 30 sec Burn as in (A) or 2 μM c-2 ceramide or the inactive dihydroceramide (D) and 4h later MVP measured in supernatants. The data for A, B, C are the Mean ± SE MVP levels normalized to 105 cells from at least four separate experiments. The data for D are Mean ± SD MVP fold change from a representative experiment with duplicate samples which is similar to three separate experiments. *Denotes statistically significant (P<0.05) changes in MVP levels from control values. Please note for A and B, that thermal burn injury at 30 sec or 45 sec generated statistically significant (P<0.05) increased MVP in comparison to UVB/CPAF at all time points measured.
Involvement of the PAFR in MVP generation.
Given that thermal injury generates PAF [10,36], PAFR activation generates MVP [33,35], and the environmental PAF-inducing stimulus UVB generates MVP in a PAFR-dependent manner [33], we sought to define if thermal burn injury-induced MVP also involve the PAFR. Our first studies tested the ability of the PAFR antagonist WEB 2086 [41] to block thermal burn injury-induced MVP generation. As shown in Figure 3A, pretreatment of WEB 2086 blocked the increased levels of MVP generated in response to the PAFR agonist CPAF yet did not affect the PAFR-independent stimulus of the phorbol ester TPA. Pretreatment with the PAFR antagonist also blocked the MVP generation in response to burn injury. Similar findings were noted with treatment of HaCaT cells with 1 μM of the PAFR antagonist CV-6209 (data not shown). To confirm the pharmacologic studies, we tested MVP generation in cell lines with/without the PAFR. To that end, we used the PAFR-negative human epidermoid cell line KB transduced with PAFR (KBP) or control MSCV2.1 retrovirus (KBM) [39]. As shown in Figure 3B, treatment of KBM/KBP cells with CPAF or UVB resulted in MVP generation solely in the PAFR-expressing KBP cells, yet TPA generated equal levels of MVP in both cell types. Thermal burn injury generated MVP in both cell types, yet, MVP levels were much higher in KBP cells, especially in response to the increased (45 sec) burn exposure.
Figure 3. Thermal burn injury of epithelial cells induces MVP in a PAFR-dependent manner.
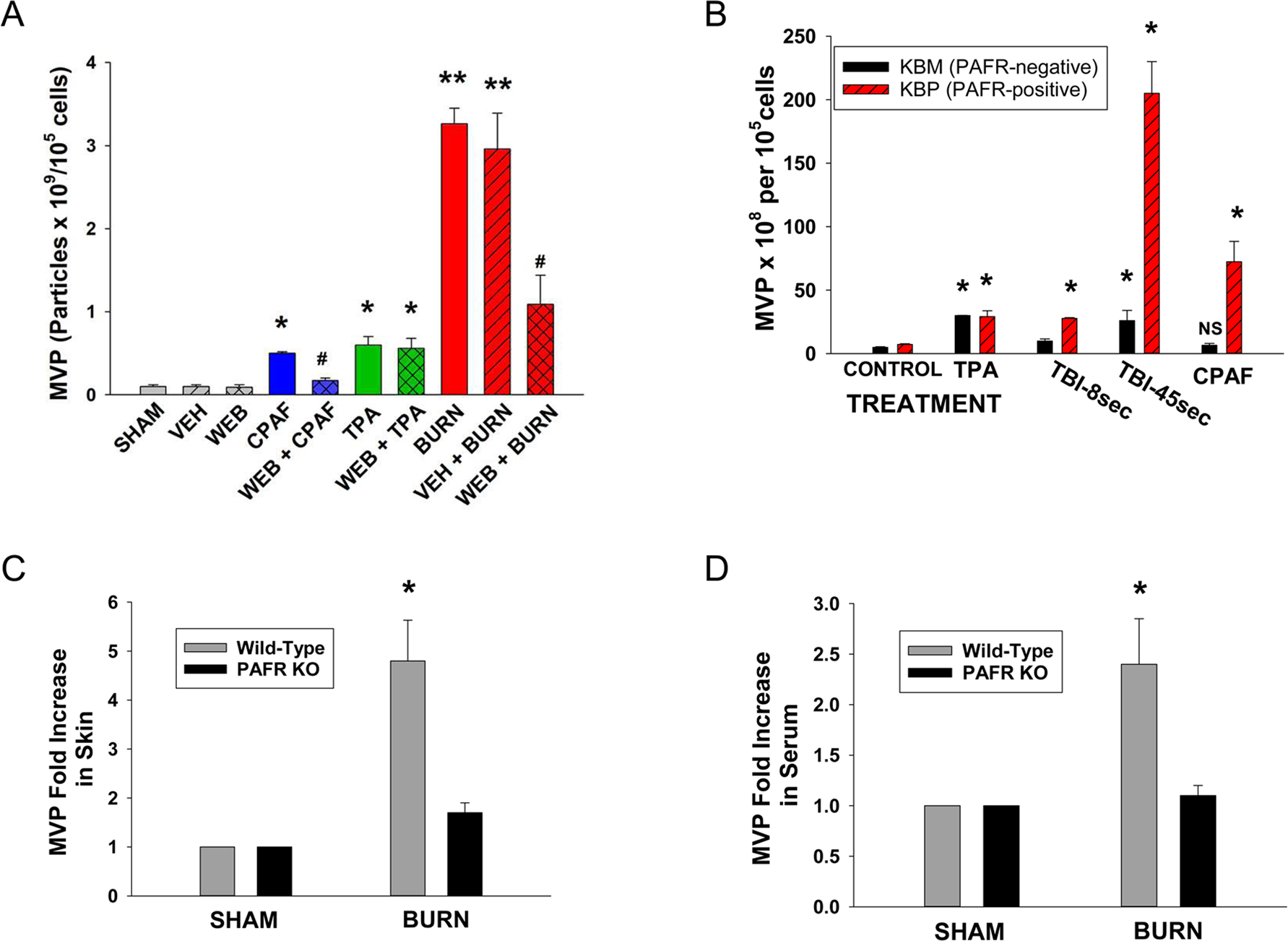
(A) HaCaT cells were treated with no treatment (sham), control (0.1% ethanol) vehicle, or preincubated with 10 μM of the PAFR antagonist WEB 2086, or 100 nM CPAF, 100 nM TPA, thermal burn injury (45 sec at 90 °C), or WEB 2086 followed 1 h later by the above agents. Two hours post-treatment, supernatants were removed and MVP quantified. (B) KBM and KBP cells were treated with control vehicle, 100 nM CPAF, 100 nM TPA, or thermal burn injury of 8 or 45 sec. Four hours later MVP were isolated from supernatants. The data are Mean ± SE MVP levels from at least four separate experiments. (C, D) Wild-type or PAFR KO mice (Ptafr−/−) mice were subjected to a thermal burn injury to approximately 15–20% body surface area, and 2 h later (C) three 5 mm punch biopsies or (D) serum was tested for MVP. The data are Mean ± SE MVP levels normalized to SHAM-treated values from at least 6–8 mice in each group. Statistically significant (*: P<0.05; **: P < 0.01) changes in levels of MVP from control (SHAM-treated) values. # Denotes statistically significant (P<0.05) differences in vehicle vs WEB 2086 treatment. N.S., not statistically significant.
The next studies sought to define if thermal burn injury can generate MVP in vivo, and the role of the PAFR in this process. Using our published protocol [36], thermal burn injury of approximately 12–15% body surface area was performed on the back skin of anesthetized wild-type or PAFR-deficient (Ptafr−/−) mice on a C57BL/6 background. At 2h post treatment, the mice were euthanized, and MVP measured in skin biopsies of injured skin and in blood. As shown in Figure 3C & 3D, increased levels of MVP were detected in both the skin and blood in wild-type mice. However, increased levels of MVP were not found in PAFR-deficient mice.
Pharmacologic strategies can also be useful adjuncts to provide mechanistic insights into complex processes. Use of pharmacologic inhibitors of second-messenger systems revealed that MVP generated in response to CPAF in HaCaT cells were blocked by preincubation with the NF-kB inhibitor ammonium pyrrolidine dithiocarbamate (PDTC) [42], MAPK/ERK kinase (MEK) inhibitor PD98,059 [32,43], JNK inhibitor SP600125 (1,9-pyrazoloanthrone) [32,44], P38 MAPK inhibitor SB203580 (4-[4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-1H-imidazol-5-yl] pyridine) [32,45], and Rho Kinase-1 (ROCK1) inhibitor Y27632 (4-[(1R)-1-aminoethyl]-N-pyridin-4-ylcyclohexane-1-carboxamide) [46]. Yet, the general caspase inhibitor Z-VAD-FMK (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone) [47,48] had no effect on PAFR-mediated MVP release, suggesting that apoptosis was not involved in this process. Of importance, Z-VAD-FMK at the concentration used (11.2 μg/ml [~24 μM]) was 2x the dose that we previously demonstrated blocks UVB-induced apoptosis in KB cells [47]. Consistent with the concept that PAFR signaling was involved in burn-mediated MVP generation, inhibitors of NF-kB, MEK, JNK and P38 also inhibited MVP release from HaCaT cells (Figure 4B) and human skin (Figure 4C). Yet, ROCK1 inhibition which effectively neutralized PAFR-mediated (Figure 4A), did not appreciably block burn-generated MVP. Altogether, these studies indicate that MVP can be generated systemically, and PAFR signaling appears to play an important role in this process.
Figure 4. Effect of pharmacologic inhibitors of various signal transduction pathways on MVP generation.
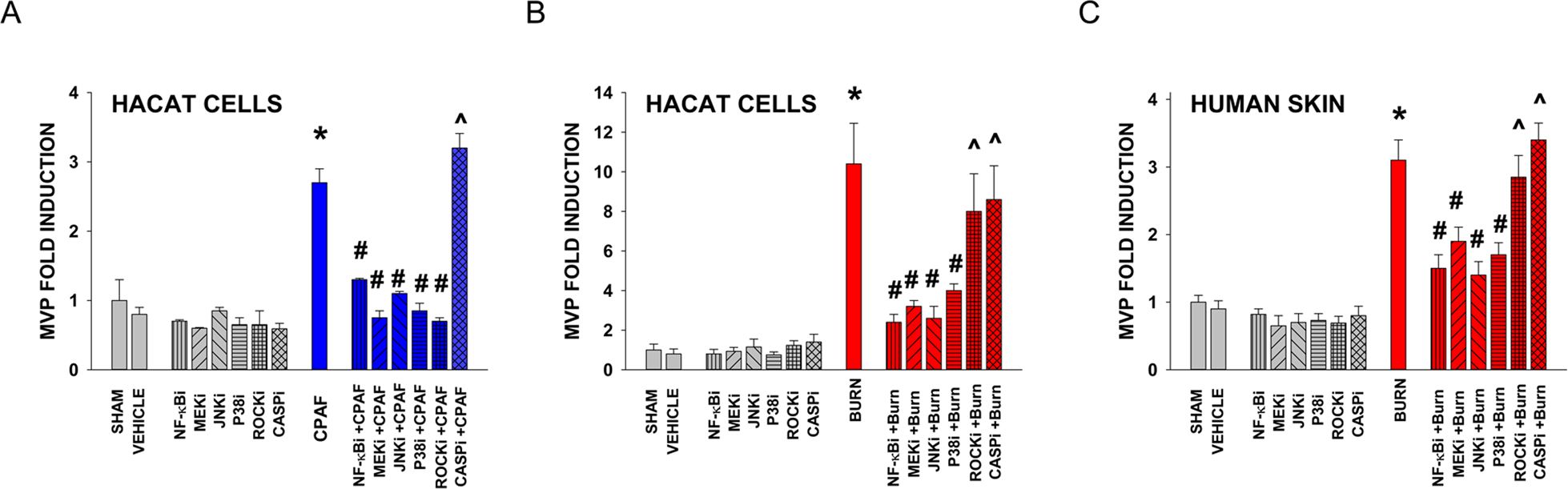
HaCaT cells were pretreated with either no treatment (SHAM), 0.1% ethanol vehicle, or the NF-κB inhibitor PDTC (1.64 μg/ml; NF-κBi) MEK inhibitor PD98,059 (2.67 μg/ml; MEKi), JNK inhibitor SP600125 (0.22 μg/ml; JNKi), P38MAPK inhibitor SB203580 (3.77 μg/ml; P38i); Rho Kinase inhibitor Y-27632 (0.32 μg/ml; ROCKi), or the general caspase inhibitor Z-VAD-FMK (11.2 μg/ml; CASPi). One hour later the cells were subjected to treatment with (A) 100 nM CPAF or (B) 45 sec thermal burn injury. The data are Mean ± SE MVP levels normalized to SHAM-treated values from at least three separate experiments. (C) Human skin explants were treated with no treatment (SHAM), 100 μl of 0.1% DMSO vehicle, or application of above amounts of various inhibitors but in 100 μl DMSO (resulting in 10x concentrations compared to in vitro studies). After one hour the tissue was treated with a 5 sec aqueous thermal burn injury (BURN). At 4 h the tissue was harvested, weighed, and MVP measured. The data are Mean ± SE MVP levels normalized to SHAM-treated values from at least three separate experiments. *Denotes statistically significant effect compared to SHAM-treated. # Denotes statistically significant differences in vehicle vs inhibitor treatment. ^ Denotes statistically significant effect compared to SHAM, but not CPAF/BURN treatment (P<0.05).
Bioactive agents in keratinocyte-derived MVP.
Microvesicles have been reported to contain a range of bioactive substances [12,13]. As thermal burn injury generates cytokines in keratinocytes [49], the next studies were designed to assess if MVP were used to carry cytokines. To that end, HaCaT cells were treated with either thermal burn injury or sham treatment. Four hours post-treatment, MVP were isolated and subjected to cytokine analysis using multiplex technology [36]. Surprisingly, cytokine levels in the MVP generated from thermal burn injury contained lower levels of all protein cytokines measured (see Figure 5 depicting a few representative cytokines and Table S-1 listing all cytokine values).
Figure 5. Burn-MVP contain only small amounts of protein cytokines.
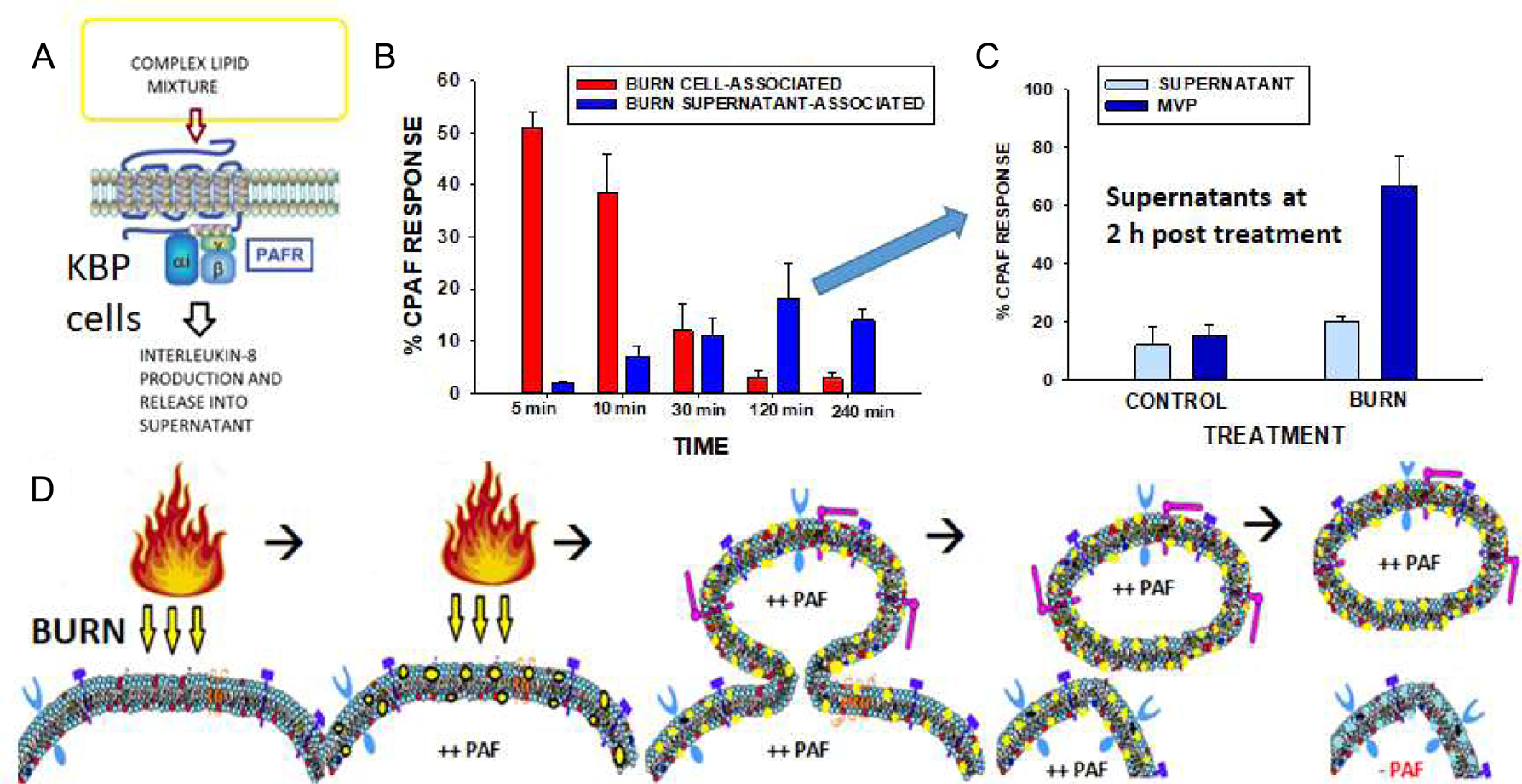
HaCaT cells were subjected to a 30 sec thermal burn injury or sham treatment and MVP isolated at 4 h. Cytokines were measured using a multiplex assay. The above depicts Mean ± SE% cytokine levels of representative ones compared to sham treatment from three separate experiments. Please see Table S1 for data of all 27 cytokines assayed. * Denotes statistically significant (P<0.01) changes in levels of cytokines in the Burn-MVP compared to untreated values.
Given that thermal burn injury generates PAF [10,36], we tested whether this glycerophosphocholine which remains in the lipid bilayer component of the cellular membrane, could be using MVP to leave the cell. Our first studies tested PAFR agonist activity in cell versus supernatant over time in response to thermal burn injury. PAFR agonist levels in the lipid extracts derived from the cell vs supernatants at various times post-burn treatment were measured by exposing them to KBM/KBP cells and measuring IL-8 release as a surrogate for PAFR activation (see Figure 6 A). As shown in Figure 6 B, at 5- and 10-min post thermal burn injury, the vast majority of PAFR agonistic activity (normalized to CPAF-induced IL-8 production in KBP cells) was cell-associated. However, by 120 min post treatment, the only appreciable PAFR agonistic activity was found in the supernatants. Of interest, even at 4 h post-treatment, significant levels of PAFR agonistic activity were found in the supernatants. To define whether the PAFR activity found in supernatants was associated with MVP, we separated the MVP from the supernatants and tested both components. As shown in Figure 6C, the PAFR agonistic activity resided in the MVP. Of importance, treatment of PAFR-negative KBM cells with lipid extracts derived from burn treated cells, supernatants or MVP did not result in IL-8 release, though positive control TPA did generate IL-8 in these cells (data not shown). The amounts of PAFR agonistic activity in MVP from thermal burn injury treated HaCaT cells at 120 min was measured in comparison to various concentrations of PAF (1-hexadecyl 2-acetyl GPC) which revealed approximately 30 ng PAF in 1011 MVP (Supplemental Figure S-4). These findings fit with the model depicted in Figure 6D that PAF being generated in response to burn injury residing in the cellular membranes then forms the MVP. The PAF lipids are preserved in the MVP, whereas acetylhydrolases in the cell remove the cellular PAF. Finally, to confirm the PAFR biological activity of MVP, we took advantage of the known ability of PAF to generate acute skin inflammation, in a process due to PAFR-dependent mast cell degranulation [50,51]. To test this, we topically treated the dorsal ears of wild-type and PAFR-deficient mice with lipid extracts from MVP derived from either untreated or thermal burn injury treated HaCaT cells, and measured ear thickness using calipers. As shown in Figure 7, lipid extracts derived from burn injury-generated MVP resulted in increased ear thickness selectively in wild-type mice. Topical treatment with CPAF also resulted in increased ear thickness in wild-type but not Ptafr−/− mice, yet topical TPA resulted in increased responses in both genotypes. These studies support the concept that MVP contain functional PAF agonists.
Figure 6. Burn-MVP contain large amounts of PAF agonistic activity.
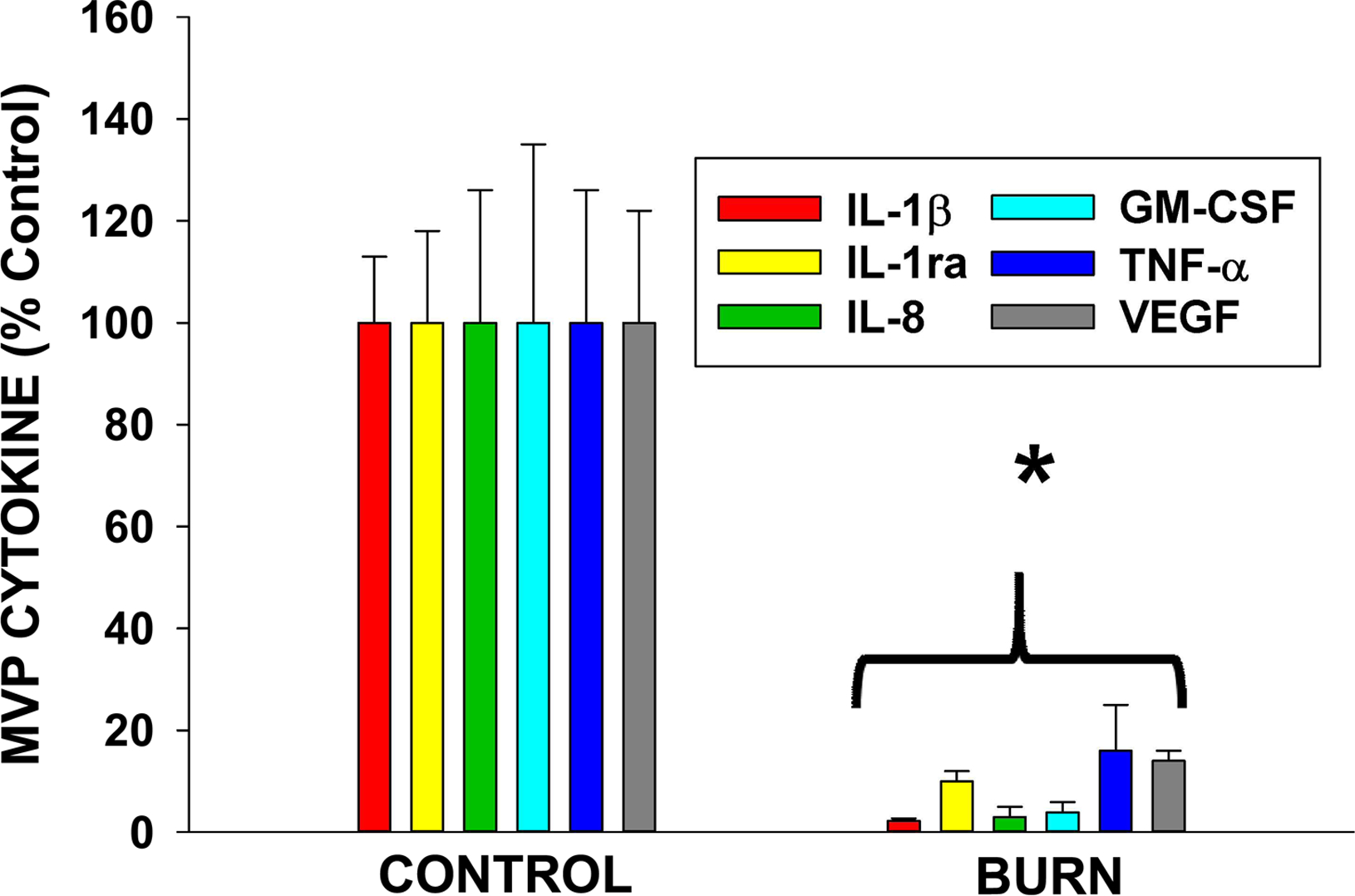
(A) Model of KBP-IL-8 assay where a complex mixture (lipid extracts) containing PAF agonists are incubated with PAFR-positive KBP cells and IL-8 released used as surrogate of PAFR activation, and compared to positive control CPAF. (B) HaCaT cells were treated with thermal burn injury (45 sec at 90°C) and at various times cells and supernatants isolated and lipids extracted, and PAFR agonistic activity assessed by measuring IL-8 release in KBP cells and comparing to 1 nM CPAF-induced IL-8 release in KBP cells. (C). Supernatants were removed at 2 h post thermal burn injury or Sham (Control) treatment, and subjected to centrifugation to remove MVP. Lipids were extracted from MVP and MVP-free supernatants and PAF-R agonistic activity measured in KBP cells and compared to 1 nM CPAF. The data are Mean ± SE % CPAF responses from three separate experiments. (D) Hypothetical model by which PAF lipids (YELLOW CIRCLES) are generated by thermal burn injury resulting in Burn-MVP containing large amounts of PAF. Note that both cell membrane and MVP contain PAF initially and over time cellular PAF-acetylhydrolases remove the cell-associated PAF, but, large amounts of labile PAF agonists remain in MVP.
Figure 7. Topical application of lipid extracts from Burn-MVP generates acute inflammation in a PAFR-dependent manner.
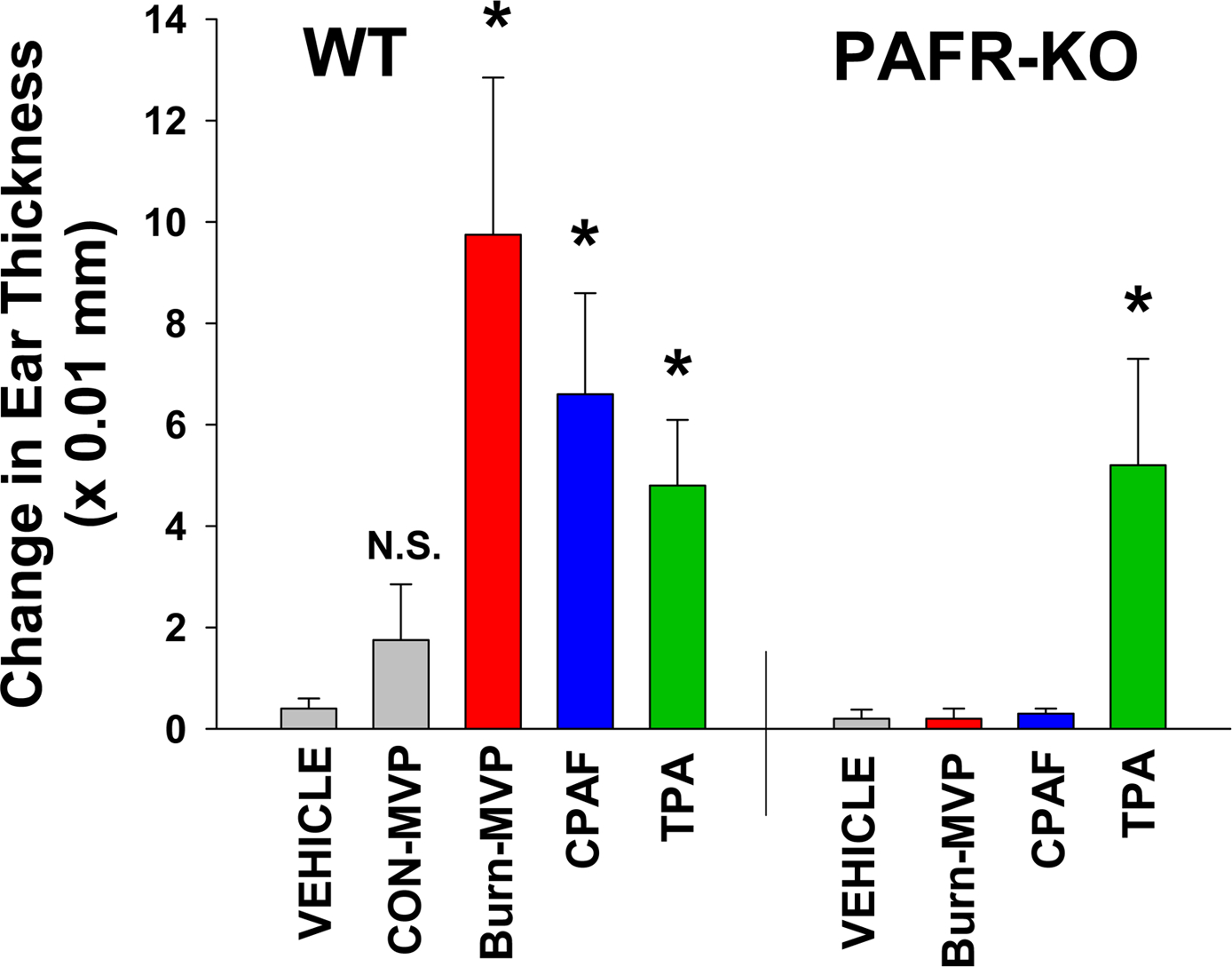
Lipid extracts were obtained from 1011 MVP derived from HaCaT cells 4 h post thermal burn injury (Burn-MVP) or untreated (CON-MVP) HaCaT cells. These lipid extracts were then applied topically to dorsal ears of wild-type or PAFR KO (Ptafr−/−) mice. Other mice were treated with topical CPAF (100 ng), TPA (1 μg), or 100% ethanol vehicle. At 2 h post-treatment, thickness of murine ears were measured and compared to pre-treatment values. The data are Mean ± SE change in ear thickness from at least six mice in each group. *Denotes statistically significant (P<0.05) changes in comparison to vehicle control.
Discussion
The keratinocyte, due to its location, commonly encounters environmental stressors, including UV radiation, thermal burn injury, and toxic chemicals. Thus, the mechanisms by which keratinocytes release bioactive substances is an important area of investigation. Though first thought to be unimportant debris, subcellular particles such as MVP have been demonstrated to play an important role in cell to cell communication, in great part through their abilities to transport bioactive compounds [9–11]. The types of bioactive agents known to be carried by MVP include cytokines and microRNAs. The potential advantage of MVP transport could be not only protection against degradation, but also the ability to traffic to specific cell types based upon cellular receptors.
The current studies demonstrate that thermal burn injury results in the formation and release of MVPs. Burn injury appears to be a potent MVP generator in comparison to other stimuli such as PAFR agonist or UVB (Figure 1). Through use of both pharmacologic inhibitors and genetic (cell lines and mice plus/minus PAFRs) approaches, optimal burn injury generated MVP appears dependent upon PAFR activation. Use of pharmacologic inhibitors of ERK, JUNK, MAPK kinase signaling pathways indicated that burn injury-generated MVP release could be affected by various signaling pathways linked to PAFR activation [32]. Yet, burn-induced MVPs are not totally dependent upon PAF as some MVP can be generated in PAFR-negative KBM as well as in murine fibroblasts (Figures 2B and 1D, respectively). These findings suggested that PAFR is not necessary for MVP generation, but, is important for optimal MVP production following a thermal burn injury.
Microvesicle particles have been reported to carry multiple bioactive agents including protein cytokines [11–13]. The current studies also indicate that MVP derived from unstimulated HaCaT cells can carry a wide range of protein cytokines. Surprisingly, MVP which are generated in response to thermal burn injury carry lower amounts of most cytokines in comparison to unstimulated MVPs (Figure 5 and Table S1). The exact mechanism(s) by which lower levels of cytokines reside in Burn-MVPs is unclear, though it is possible that this constitutes a protective response in that the host will not be subjected to high levels of cytokine-laden MVPs leaving the epidermis in response to a skin thermal burn injury. This appears to be of potential importance given our findings that thermal burn injury to mice results in increased systemic MVP. Of interest, a recent report indicates that burn injury in both murine models and in human patients resulted in increasing levels of systemic MVP, which contained high levels of IL-1beta and HMGB [52]. However, these studies examined 24 and 48 h post-injury, unlike our studies which focused on early time points (2h). The exact source of increased systemic MVP following thermal burn injury is unclear. Unfortunately, unlike other cell types, i.e., monocytes, endothelial cells and platelets, there is not a validated keratinocyte-specific marker to detect keratinocyte MVP in blood. Presumably, the increased systemic MVPs following a thermal burn injury are derived from multiple sources.
As MVP contain cellular membranes which include the lipid bilayer, it would not be surprising that these subcellular particles could also transport lipids, especially phospholipid mediators. However, little is known about bioactive lipid content of MVPs. The current studies demonstrate that not only does thermal burn injury generate MVP in keratinocytes, but these MVP contain PAF agonists. These findings fit with our previously reported findings that thermal burn injury generates PAF [10,36]. Mass spectrometric analysis of individual glycerophosphocholines (GPC) with PAF-R agonistic activity produced in response to thermal burn injury by our group has revealed that the major sn-2 acetyl GPC identified are 1-hexadecyl and 1-palmitoyl GPC species [10,36]. The current studies use a biochemical rather than a structural assay to measure PAF activity for several reasons. First, IL-8 release in KBP cells is more sensitive than the amounts needed for mass spectrometry. As shown in Supplemental Figure S5, we can routinely measure concentrations of PAF of <1 nM (~0.5ng/ml), whereas we would need 5–10 times these amounts for our mass spectrometric methodologies [10,30,36]. Second, the ability to use PAFR-negative KBM cells provides specificity to this biochemical assay. Finally, this biochemical assay measuring KBP IL-8 release measures all PAFR agonistic activity, which is important given that PAF agonists consist of multiple sn-2 acetyl GPC species [53–56]. It should be noted that there is evidence that 1-acyl PAF species which serve as weak PAF-R agonists, might serve as endogenous PAFR antagonists through PAFR desensitization [55,56]. Thus, the ability to measure all PAFR biochemical activity can provide important insights into biological activity.
The mechanism by which thermal burn injury results in the production of PAF-filled MVP is unclear. We hypothesize that PAF generated in response to the thermal burn injury is found on the cellular membranes, which then act upon the PAFR resulting in augmented MVP release. In as much as the membranes that constitute MVP contain PAF, this bioactive lipid can then travel in the formed and released MVP (see Figure 6D for model). Over time, the cell-associated PAF is degraded by PAF-acetylhydrolases. Of interest, we have tested PAF-acetylhydrolase enzymatic activity associated with HaCaT-derived MVP and fail to find any activity using our published methodology, though we can measure this activity in the cell [36] (data not shown). Areas of future study would be to test whether PAF associated with MVP are susceptible to PAF-acetylhydrolases in comparison to “free PAF”, and if PAFR agonists are more potent receptor agonists when traveling in a MVP vs free lipid in solution.
The exact role of MVP and PAF in thermal burn injury is at present unclear. In the context of skin, multiple lines of evidence suggest that the PAF system serves as an acute sensor of cutaneous damage. PAF agonists are not found in appreciable levels in normal skin but can be measured in response to multiple stressors and pathologic states including UVB, urticaria, psoriasis and blistering diseases [30,31,54,56–58]. Injection of PAF into skin of rodents or humans results in an acute inflammatory response [50]. In the context of UVB-induced sunburn responses, use of PAFR-deficient mice has demonstrated the hyperalgesia is PAF dependent [59]. Thus, it is likely that a component of the clinical manifestations thermal burn injury could involve the production of PAF, and the use of MVP as potential transporters of this bioactive lipid mediator.
In summary, the present studies indicate that keratinocytes generate MVP in response to thermal burn injury. Moreover, we provide evidence that these burn injury-induced MVP can leave the epidermis and potentially travel systemically. The optimal formation of burn injury-generated MVP involves PAFR activation. Finally, these MVP generated under pathological conditions do not contain significant protein cytokines, yet contain PAF, which could provide an important mechanism by which a metabolically labile lipid autacoid can leave the skin.
Supplementary Material
Key Points.
Thermal burn injury is a potent generator of microvesicle particles
Optimal burn-generated microvesicle particles depends upon PAF receptor signaling
Burn-generated microvesicles contain PAF lipids but not protein cytokines
Acknowledgments:
We would like to thank Mr. William C. Grunwald, Jr, manager of the Proteome Analysis Laboratory (PAL) for performing the cytokine assays and for the use of the PAL for these studies.
This research was supported in part by grants from the National Institutes of Health grant R01 HL062996 (JBT), R21 AR071110 (JCB), Veteran’s Administration Merit Award 5I01BX000853 (JBT).
References cited
- 1.Choudhry MA, and Chaudry IH. 2006. Alcohol intoxication and post-burn complications. Front. Biosci 11: 998–1005. [DOI] [PubMed] [Google Scholar]
- 2.Crowe CS, Massenburg BB, Morrison SD, Naghavi M, Pham TN, and Gibran NS. 2019. Trends of burn injury in the United Staes: 1990 to 2016. Ann. Surg 270(6):944–953. [DOI] [PubMed] [Google Scholar]
- 3.Rae L, Fidler P, and Gibran NS. 2016. The physiologic basis of burn shock and the need for aggressive fluid resuscitation. Crit. Care Clin 32: 491–505. [DOI] [PubMed] [Google Scholar]
- 4.Noel JG, Osterburg A, Wang Q, Guo X, Byrum D, Schwemberger S, Goetzman H, Caldwell CC, and Ogle CK. 2007. Thermal injury elevates the inflammatory monocyte subpopulation in multiple components. Shock. 28: 684–693. [DOI] [PubMed] [Google Scholar]
- 5.Heinrich SA, Messingham KA, Gregory MS, Colantoni A, Ferreira AM, Dipietro LA, and Kovacs EJ. 2003. Elevated monocyte chemoattractant protein-1 levels following thermal injury precede monocyte recruitment to the wound site and are controlled, in part, by tumor necrosis factor-alpha. Wound Repair Regen. 11(2):110–119. [DOI] [PubMed] [Google Scholar]
- 6.Gibran NS, Ferguson M, Heimbach DM, and Isik FF. 1997. Monocyte chemoattractant protein-1 mRNA expression in the human burn wound. J. Surg. Res 70:1–6. [DOI] [PubMed] [Google Scholar]
- 7.Piccolo MT, Wang Y, Verbrugge S, Warner RL, Sannomiya P, Piccolo NS, Piccolo MS, Hugli TE, Ward PA, and Till GO. 1999. Role of chemotactic factors in neutrophil activation after thermal injury in rats. Inflammation 23:371–385. [DOI] [PubMed] [Google Scholar]
- 8.Ou S, Liu GD, Tan Y, Zhou LS, Bai SR, Xue G, Li J, Yang Y, Cui J, Cheng JM, and Gu JW. 2015. A time course study about gene expression of post-thermal injury with DNA microarray. Int. J. Dermatol 54(7):757–764. [DOI] [PubMed] [Google Scholar]
- 9.Gragnani A, Cezillo MV, da Silva ID, de Noronha SM, Correa-Noronha SA, and Ferreira LM. 2014. Gene expression profile of cytokines and receptors of inflammation from cultured keratinocytes of burned patients. Burns 40:947–956. [DOI] [PubMed] [Google Scholar]
- 10.Alappatt C, Johnson CA, Clay KL, and Travers JB. 2000. Acute keratinocyte damage stimulates platelet-activating factor production. Arch. Dermatol. Res 292(5): 256–259. [DOI] [PubMed] [Google Scholar]
- 11.Xiao X, Ma X, Liu L, Wang J, Bi K, Liu Y, Fan R, Zhao B, Chen Y, and Bihl JC. 2015. Cellular membrane microparticles: Potential targets of combinational therapy for vascular disease. Curr. Vasc. Pharmacol 13(4):449–58. [DOI] [PubMed] [Google Scholar]
- 12.Camussi G, Deregibus MC, Bruno S, Cantaluppi V, and Biancone L. 2010. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 78(9):838–48. [DOI] [PubMed] [Google Scholar]
- 13.Norling LV, and Dalli J. 2013. Microparticles are novel effectors of immunity. Curr. Opin. Pharmacol 13:570–575. [DOI] [PubMed] [Google Scholar]
- 14.Vion A-C, Ramkhelawon B, Loyer X, Chironi G, Devue C, Loirand G, Tedgui A, Lehoux S, and Boulanger CM. 2013. Shear stress regulates endothelial microparticle release. Circ. Res 112:1323–1333. [DOI] [PubMed] [Google Scholar]
- 15.Li M, Yu D, Williams KJ, and Liu ML. 2010. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Art. Thromb. & Vas. Biol 30(9):1818–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herring JM, McMichael MA, and Smith SA. 2013. Microparticles in health and disease. J. Vet. Intern. Med 27:1020–1033. [DOI] [PubMed] [Google Scholar]
- 17.Tamagawa-Mineoka R, Katoh N, and Kishimoto S. 2010. Platelet activation in patients with psoriasis: Increased plasma levels of platelet-derived microparticles and soluble P-selectin. J. Amer. Acad. Dermatol 62:621–626. [DOI] [PubMed] [Google Scholar]
- 18.Liu M-L, Williams KJ, and Werth VP. 2016. Chapter four-Microvesicles in Autoimmune Diseases. Adv. Clin. Chem 77:125–175. [DOI] [PubMed] [Google Scholar]
- 19.Takeshita J, Mohler ER, Krishnamoorthy P, Moore J, Rogers WT, Zhang L, Gelfand JM, and Mehta NN. 2014. Endothelial cell-, platelet-, and monocyte/macrophage-derived Microparticles are elevated in psoriasis beyond cardiometabolic risk factors. J. Am. Heart Assoc 3(1):e000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benveniste J, Henson PM, and Cochrane CG. 1972. Leukocyte-dependent histamine release from rabbit platelets. The role of IgE, basophils, and a platelet-activating factor. J. Exp. Med 136(6):1356–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demopoulos CA, Pinckard RN, and Hanahan DJ. 1979. Platelet-activating factor. Evidence for 1-O-alkyl-2-acetyl-sn-glyceryl-3-phosphorylcholine as the active component (a new class of lipid chemical mediators). J. Biol. Chem 254(19): 9355–58. [PubMed] [Google Scholar]
- 22.Braquet P, Touqui L, Shen TY, and Vargaftig BB. 1987. Perspectives in platelet-activating factor research. Pharmacol. Rev 39: 97–145. [PubMed] [Google Scholar]
- 23.Nakanishi H, Shindou H, Hishikawa D, Harayama T, Ogasawara R, Suwabe A, Taguchi R, and Shimizu T. 2006. Cloning and characterization of mouse lung-type acyl-CoA:lysophosphatidylcholine acyltransferase 1 (LPCAT1). Expression in alveolar type II cells and possible involvement in surfactant production. J. Biol. Chem 281(29): 20140–47. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu T 2009. Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Ann. Rev. Pharmacol. Toxicol 49:123–150. [DOI] [PubMed] [Google Scholar]
- 25.Morimoto R, Shindou H, Oda Y, and Shimizu T. 2010. Phosphorylation of lysophosphatidylcholine acyltransferase 2 at Ser34 enhances Platelet-activating Factor production in endotoxin-stimulated macrophages. J. Biol. Chem 285: 29857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marathe GK, Pandit C, Lakshmikanth CL, Chaithra VH, Jacob SP, and D’Souza CJ. 2014. To hydrolyze or not to hydrolyze: the dilemma of platelet-activating factor acetylhydrolase. J. Lipid Res 55: 1847–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McIntyre TM, Prescott SM, and Stafforini DM. 2009. The emerging roles of PAF acetylhydrolase. J. Lipid Res 50 Suppl:S255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walterscheid JP, Ullrich SE, and Nghiem DX. 2002. Platelet-activating factor, a molecular sensor for cellular damage, activates systemic immune suppression. J. Exp. Med 195:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ullrich SE, and Byrne SN. 2012. The immunologic revolution: Photoimmunology. J. Invest. Dermatol 132: 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marathe GK, Johnson C, Billings SD, Southall MD, Pei Y, Spandau D, Murphy RC, Zimmerman GA, McIntyre TM, and Travers JB. 2005. Ultraviolet B radiation generates platelet-activating factor-like phospholipids underlying cutaneous damage. J. Biol. Chem 280(42):35448–57. [DOI] [PubMed] [Google Scholar]
- 31.Travers JB, Berry D, Yao Y, Yi Q, Konger RL, and Travers JB. 2010. Ultraviolet B radiation of human skin generates platelet-activating factor receptors agonists. Photochem. Photobiol 86(4):949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marques SA, Dy L, Southall MD, Yi Q, Smietana E, Kapur R, Marques M, Travers JB, and Spandau D. 2002. The Platelet-activating factor receptor activates the ERK MAP Kinase and induces proliferation of epidermal cells through an EGF Receptor-dependent Pathway. J. Pharmacol. Exp. Therapeut 300:1026–1035. [DOI] [PubMed] [Google Scholar]
- 33.Bihl JC, Rapp CM, Chen Y, and Travers JB. 2016. UVB generates microvesicle particle release in part due to Platelet-activating factor signaling. Photochem. Photobiol 92(3):503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Travers JB 2019. Platelet-activating Factor as an Effector for Environmental Stressors. In: Lipid Signaling in Human Diseases. Handbook of Experimental Pharmacology, eds. Cambronero JG and Frohman M PMID:31087194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fahy K, Liu L, Rapp CM, Borchers CA, Bihl J, Chen JC, Simman R, and Travers JB. 2017. UVB-generated microvesicle particles: A novel pathway by which a skin-specific stimulus could exert systemic effects. Photochem. Photobiol 93(4):937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harrison KA, Romer E, Weyerbacher J, Ocana JA, Sahu RP, Murphy RC, Kelly LE, Smith TA, Rapp CM, Borchers C, Cool DR, Li G, Simman R, and Travers JB. 2018. Enhanced Platelet-activating Factor synthesis facilitates acute and delayed effects of ethanol intoxicated thermal burn injury. J. Invest. Dermatol 138(11): 2461–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, and Fusenig NE. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell. Biol 106: 761–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Travers JB, Huff JC, Rola-Plesczynski M, Gelfand E, Morelli JG, and Murphy RC. 1995. Identification of functional platelet-activating factor receptors on human keratinocytes. J. Invest. Dermatol, 105:816–823. [DOI] [PubMed] [Google Scholar]
- 39.Pei Y, Barber LA, Murphy RC, Johnson CA, Kelly SW, Dy LC, Fertel RH, Nguyen TM, Williams DA, and Travers JB. 1998. Activation of the epidermal platelet-activating factor receptor results in cytokine and cyclooxygenase-2 biosynthesis. J. Immunol 161(4):1954–61. [PubMed] [Google Scholar]
- 40.Ishii S, Kuwaki T, Nagase T, Maki K, Tashiro F, Sunaga S, Cao WH, Kume K, Kukuchi Y, Ikuta K, Miyazaki J, Kumada M, and Shimizu T. 1998. Impaired anaphylactic responses with intact sensitivity to endotoxin in mice lacking a Platelet activating factor receptor. J. Exp. Med 187:1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Summers JB, and Albert DH. 1995. Platelet-activating factor antagonists. Adv. Pharmacol 32:67–168. [DOI] [PubMed] [Google Scholar]
- 42.Schini-Kerth V, Bara A, Mlsch A, and Busse R. 1994. Pyrrolidine dithiocarbamate selectively prevents the expression of the inducible nitric oxide synthase in the rat aorta. Eur. J. Pharmacol 265: 83–87. [DOI] [PubMed] [Google Scholar]
- 43.Vion AC, Ramkhelawon B, Loyer X, Chironi G, Devue C, Loirand G, Tedgui A, Lehoux S, and Boulanger CM. 2013. Shear stress regulates endothelial microparticle release. Circ. Res 112: 1323–1333. [DOI] [PubMed] [Google Scholar]
- 44.Li CJ, Liu Y, Chen Y, Yu D, Williams KJ, and Liu ML. 2013. Novel proteolytic microvesicles released from human macrophages after exposure to tobacco smoke. Am. J. Pathol 182: 1552–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, and Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 364: 229–233. [DOI] [PubMed] [Google Scholar]
- 46.Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, Abe K, Takeshita A, and Shimokawa H. 2005. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 91: 391–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yao Y, Wolverton JE, Zhang Q, Marathe GK, Al-Hassani M, Konger RL, and Travers JB. 2009. Ultraviolet B radiation generated Platelet-activating factor receptor agonist formation involves EGF-R-mediated reactive oxygen species. J. Immunol 182(5):2842–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW, and Cohen GM. 1996. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem. J 315: 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghahary A, and Chaffari A. Role of keratinocyte-fibroblast cross-talk in development of hypertrophic scar. 2007. Wound Repair Regen. 15: Suppl 1:S46–53. [DOI] [PubMed] [Google Scholar]
- 50.Travers J, Pei Y, Morin SM, and Hood AF. 1998. Antiinflammatory activity of the platelet-activating factor receptor antagonist A-85783. Arch. Dermatol. Res 290 (10):569–73. [DOI] [PubMed] [Google Scholar]
- 51.Sahu RP, Rezania S, Ocana JA, DaSilva-Arnold SC, Bradish JR, Richey JD, Warren SJ, Rashid B, Travers JB, and Konger RL. 2014. Topical application of a platelet-activating factor agonist suppresses phorbol ester-induced acute and chronic inflammation and has cancer chemopreventive activity in mouse skin. PLoS One 9(11):e111608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coleman LG Jr, Maile R, Jones SW, Cairns BA, and Crews ET. 2018. HMGB1/IL-1b complexes in plasma microvesicles modulate immune responses to burn injury. PLoS One 13(3):e0195335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Travers JB, Harrison KA, Johnson CA, Clay KL, Morelli JG, and Murphy RC. 1996. Platelet-activating Factor biosynthesis induced by various stimuli in human HaCaT keratinocytes. J. Invest. Dermatol 107: 88–94. [DOI] [PubMed] [Google Scholar]
- 54.Konger RL, Marathe GK, Yao Y, Zhang Q, and Travers JB. 2008. Oxidized glycerophosphocholines as biologically active mediators for ultraviolet radiation-mediated effects. Prost. Other Lipid Mediat 87(1–4):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chaithra VH, Jacob SP, Lakshmikanth CL, Sumanth MS, Abhilasha KV, Chen CH, Thyagarajan A, Sahu RP, Travers JB, McIntyre TM, Kemparaju K, and Marathe GK. 2018. Modulation of inflammatory platelet-activating factor (PAF) receptor by the acyl analog of PAF. J. Lipid Res 59 (11): 2063–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Travers JB, Murphy RC, Johnson CA, Pei Y, Morin SM, Barber LA, Clay K, Hood AF, Morelli JG, and Williams DA. 1998. Identification and pharmacological characterization of PAF and 1-acyl species found in human inflammatory blistering diseases. Prostaglandins. 56: 305–324. [DOI] [PubMed] [Google Scholar]
- 57.Grandel KE, Farr RS, Wanderer AA, Eisenstadt TC, and Wasserman SI. 1985. Association of platelet-activating factor with primary acquired cold urticaria. N. Engl. J. Med 313 (7): 405–409. [DOI] [PubMed] [Google Scholar]
- 58.Mallet AI, and Cunningham FM. 1985. Structural identification of platelet-activating factor in psoriatic scale. Biochem. Biophys. Res. Commun 126:192–196. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Q, Sitzman LA, Al-Hassani M, Cai S, Pollok KE, Travers JB, and Hingtgen CM. 2009. Involvement of Platelet-activating Factor in ultraviolet B-Induced hyperalgesia. J. Invest. Dermatol 129(1):167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.