

遗传性凝血因子Ⅶ(FⅦ)缺乏症是由FⅦ基因突变引起的一种罕见的常染色体隐性遗传性出血性疾病,发病率约1∶500 000[1]。临床表现多样,轻者无明显症状,重者可发生颅内或者内脏出血。实验室检查可见凝血酶原时间(PT)延长,部分活化凝血活酶时间(APTT)及凝血酶时间(TT)正常,FⅦ活性(FⅦ∶C)降低。目前FⅦ基因突变数据库包括了279种导致遗传性FⅦ缺乏症的突变。我们联合使用直接测序法及生物信息学方法对一个遗传性FⅦ缺乏症家系进行基因分析,并探讨其发病机制。
病例和方法
1.家系资料:先证者1(Ⅱ2):男性,34岁,自幼反复鼻出血及皮肤瘀斑,无肌肉血肿及关节出血;先证者2(Ⅱ3):女性,32岁,术前体检发现PT延长,平时无明显出血倾向,无月经过多及经期延长。家系中其他成员无出血倾向,父母非近亲结婚(图1)。先证者1和先证者2的PT分别为32 s和35 s(正常参考值11~15 s),FⅦ∶C分别为2.5%和3.2%,其他凝血因子活性正常,APTT、TT和纤维蛋白原含量正常,血小板数量以及功能正常,无肝脏疾病及维生素K缺乏。本研究经山西医科大学第二医院伦理委员会批准。家系成员均知情同意。
图1. 遗传性凝血因子Ⅶ缺乏症家系图.

2.FⅦ基因功能区域PCR扩增:先证者及家系成员肘静脉取血5 ml,以3.2%枸橼酸钠1∶9抗凝。使用离心柱法DNA提取试剂盒(荷兰Fermentas公司产品)提取基因组DNA,具体操作参照试剂盒说明书。扩增引物由大连宝生物工程有限公司合成,包括整个编码区、侧翼序列及5′端启动区域,引物序列、扩增长度及退火温度参照文献[2](表1)。反应体系总体积为25 µl:基因组DNA (100 ng/µl)2 µl;PCR mixture(北京全式金公司产品)12.5 µl;引物(10 µmol/L)各1 µl。条件:95 °C预变性5 min,95 °C变性30 s,退火30 s,72 °C延伸30 s,共30个循环;72 °C延伸10 min。
表1. 凝血因子Ⅶ基因功能区域PCR扩增引物序列、片段大小及相应退火温度.
| 外显子 | 上游引物(5′→3′) | 下游引物(5′→3′) | 目的片段长度(bp) | 退火温度(°C) |
| 启动区+1 | GCATGATTGCTATGGGACAA | CTGCCCTTCCACCAAGTTTA | 491 | 58 |
| 2 | GTGGGCTGTGAGGGACAGT | GCAGGGAACACCCTCCTT | 282 | 58 |
| 3 | GTGGGCCGTGGGGCGGTCTC | GCCCCACGCGGCCTGGTTCA | 302 | 64 |
| 4+5 | TGGTGTGTCCAGTGCTTACC | CAATTTCCAACTGGGCTGAG | 425 | 58 |
| 6 | CTTCCAGGCAGAACACCACT | ATCCCACCTCACAATTGGTC | 388 | 58 |
| 7 | CTCAGAGGATGGGTGTTTCTG | TGCTAGGTGTGCTGACTTGG | 330 | 64 |
| 8 | AGGGCGAGTCATCAGAGAAA | AGTGGTACCCACCCAGCAC | 386 | 58 |
| 9A | CTTGCCCCAGAAGGAGACT | TCTCCCACCTTCCGTGACT | 452 | 58 |
| 9B | CTGGAGCTCATGGTCCTCA | TGCCCTCCTCTACCCCATTA | 441 | 58 |
3.FⅦ基因功能区域测序分析:利用ABI3730型DNA测序仪采用双脱氧链终止法对PCR凝胶回收产物进行直接测序(由北京六合华大基因有限公司完成)。所有序列使用Pairwise BLAST(www.ncbi.nlm.nih.gov/blast)与野生型FⅦ基因序列(GenBank accession NG_009262)进行比对,检测结果与FⅦ基因突变数据库(HMGD,http://www.hgmd.cf.ac.uk/ac/gene.php?gene=F7)进行比较,对于检测到的突变进行重复测序及反向测序加以验证。FⅦ基因突变核苷酸编号参照cDNA序列NM_000131命名,ATG起始密码子的A为+1。氨基酸编号参照FⅦ蛋白序列NP_000122命名,ATG起始密码子编码的Met为+1。
4.生物信息学方法:使用Clustal Omega在四种哺乳动物(人类、仓鼠、犬、猪)的FⅦ蛋白之间进行同源序列比对,进行错义突变氨基酸的保守性分析。使用PolyPhen-2(Polymorphism Phenotyping)和SIFT(Sorting Intolerant from Tolerant)两种工具分析错义突变对FⅦ蛋白结构和功能的影响。使用PyMOL软件分析错义突变FⅦ蛋白的3D结构。使用GRASP2软件分析错义突变FⅦ蛋白表面电荷的改变。
结果
1.FⅦ基因型检测结果:2例先证者FⅦ基因第5号内含子剪接供体位点出现c.430+1G>A杂合突变(图2),HMGD及PubMed数据库检索均未见此突变。同时在第9号外显子发现c.1009C>T杂合突变(图3),第337位密码子由CGC变为TGC,导致氨基酸由Arg变为Cys,为已报道突变。母亲(Ⅰ1)及另一家系成员Ⅱ1为c.430+1G>A剪接位点突变携带者,父亲(Ⅰ2)为p.Arg337Cys错义突变携带者,这3例家系成员尚有一条野生型等位基因,故无临床症状。
图2. 遗传性凝血因子Ⅶ(FⅦ)缺乏症患者FⅦ基因第5号内含子剪街位点c.430+1G>A杂合突变.
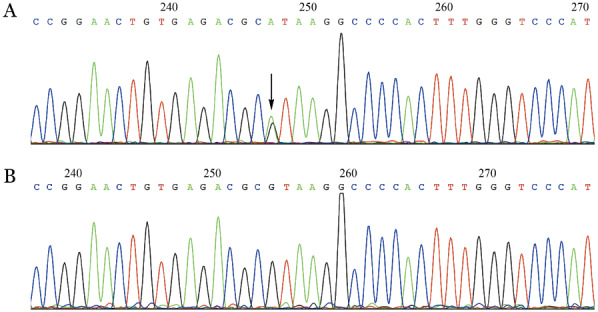
A:患者(箭头所示为突变位点);B:野生型
图3. 遗传性凝血因子Ⅶ(FⅦ)缺乏症患者FⅦ基因第9号外显子c.1009C>T错义突变.
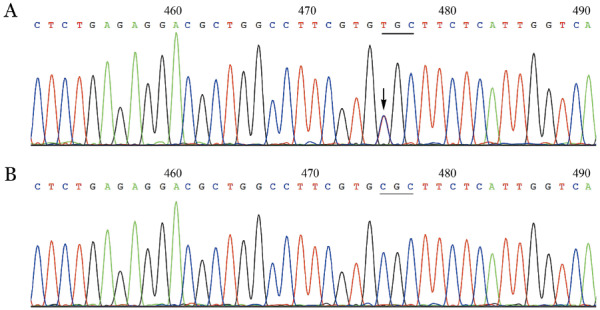
A:患者(箭头所示为突变位点,下划线表示突变所在密码子);B:野生型
2.FⅦ基因错义突变p.Arg337Cys的生物信息学分析结果:使用Clustal Omega进行氨基酸保守性分析提示FⅦ蛋白第337位氨基酸Arg在物种间高度保守,PolyPhen预测结果为“probably damaging”,积分为1,SIFT预测结果为“intolerant”。使用PyMOL软件分析p.Arg337Cys错义突变FⅦ蛋白的3D结构,未见明显变化(图4)。使用GRASP2分析p.Arg337Cys错义突变FⅦ蛋白表面电荷,提示突变前带正电荷,突变后为负电荷(图5)。
图4. 凝血因子Ⅶ(FⅦ)基因p.Arg337Cys错义突变FⅦ蛋白3D结构示意图.
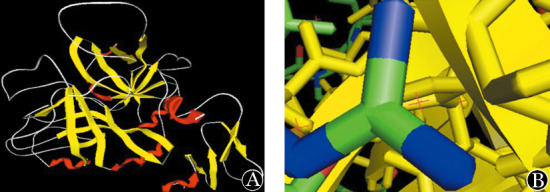
A:整体图;B:局部放大图(绿色为碳分子,蓝色为氮分子,红色为氧分子)
图5. 凝血因子Ⅶ(FⅦ)基因p.Arg337Cys错义突变FⅦ蛋白表面电荷变化示意图(蓝色代表正电荷,红色代表负电荷,白色代表中性).
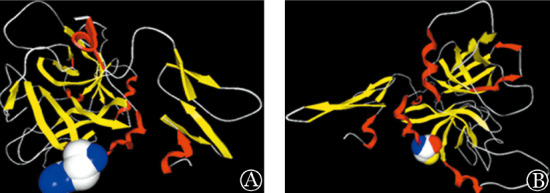
A:突变前带正电荷;B:突变后为负电荷
讨论
人类FⅦ基因位于染色体13q34,全长12 850 bp,cDNA长2 462 bp,包含9个外显子和8个内含子[3]。FⅦ是一种维生素K依赖性丝氨酸蛋白酶,含406个氨基酸,包含N末端γ-羧基谷氨酸区域、两个表皮生长因子类似区域和一个C末端的丝氨酸蛋白酶区域。血管损伤导致组织因子(TF)暴露,继而形成FⅦ/TF复合物,FⅦ转化为FⅦa后激活凝血因子Ⅸ(FⅨ)和凝血因子Ⅹ(FⅩ),启动凝血级联反应,最后形成血栓和纤维凝块[4]。FⅦ基因突变导致FⅦ缺乏引起凝血障碍。在本研究所报告的遗传性FⅦ缺乏症家系中,一种突变为新发现的剪接位点突变c.430+1G> A,为保守的剪接供体位点突变,导致移码及翻译提前终止,为肯定致病的无效突变,往往引起FⅦ重度缺乏。另一种为已报道的错义突变p.Arg337Cys,既往报道中未对该错义突变致病机制进行研究。
区别致病基因与基因多态性是遗传学研究中的难点。基因表达研究可以验证错义突变是否致病,但是FⅦ基因的表达需要复杂的翻译后修饰过程,需要高度真核细胞系统,难度较大。因为缺少这些研究,将基因多态性归为突变或者突变归为基因多态性的概率较大。目前生物信息学方法可以有效预测错义突变的致病性并分析其致病机制。首先,错义突变导致疾病的严重程度依赖突变氨基酸在蛋白序列中的位置和突变后的氨基酸。Bowen等[5]发现非保守氨基酸位点的错义突变致病可能性降低33倍,所以需要对受累的氨基酸进行不同物种间的保守性分析。PolyPhen-2和SIFT为最常见的两种分析软件,根据氨基酸替代后对蛋白质结构和(或)功能的影响判断错义突变的致病性。PolyPhen-2是全自动预测氨基酸替代后对人体蛋白质结构可能产生的影响的工具[6],预测结果包括“benign”、“possibly damaging”或“probably damaging”,同时有介于1(probably damaging)和0(benign)之间的积分。SIFT以不同物种同源蛋白或者同一物种同源基因相关蛋白之间的序列比对为基础预测氨基酸替代后的功能改变[7],结果分为“intolerant”、“potentially intolerant”、“borderline tolerant”和“tolerant”。而GRASP2用于分析氨基酸替代后蛋白表面电荷的改变[8],蛋白表面电荷改变往往影响空间构象稳定性及蛋白之间的相互作用。
本研究中,我们应用生物信息学方法对FⅦ基因p.Arg337Cys错义突变的致病性及发病机制进行了研究。氨基酸保守性分析结果显示FⅦ蛋白第337位氨基酸Arg在不同物种间高度保守,PolyPhen软件预测结果为“probably damaging”,积分为1,SIFT软件预测结果为“intolerant”,两种软件分析结果均提示该错义突变为致病基因。使用PyMOL软件分析p. Arg337Cys错义突变FⅦ蛋白的3D结构虽然未见明显变化,但是突变后氨基酸为半胱氨酸,可能形成新的二硫键导致FⅦ结构重排。另外,该位点氨基酸改变使得蛋白表面电荷由正电变为负电,电荷的改变可能影响FⅦ蛋白的空间构象,使得蛋白稳定性下降,引起FⅦ蛋白量的缺乏,同时干扰蛋白之间的相互作用影响蛋白功能,该错义突变位于FⅦ催化区,催化区为FⅦ基因的突变热点[9],故该错义突变可能直接影响了FⅦ蛋白的催化活性。
遗传性FⅦ缺乏症患者的出血表现存在较大的异质性,而且基因型和表型的关系多种多样,与FⅦ∶C的相关性较小[10]–[11]。本研究中2例先证者出血症状轻微,与杂合子患者临床表型往往较纯合子轻的报道[12]相符。这2例先证者基因型虽然相同,但临床表现存在差异,其中女性患者无症状,但男性患者有皮肤黏膜出血倾向。FⅦ基因中多种单核苷酸多态性(SNP)可以影响FⅦ∶C[10],本研究中2例先证者并未携带SNP,故除外SNP导致的表型差异,表明尚存在影响患者的表型的其他因素。
本研究我们通过直接测序法对一个遗传性FⅦ缺乏症家系进行了基因诊断,发现FⅦ基因第5号内含子剪接位点c.430+1G>A杂合突变及第9号外显子c.1009C>T错义突变。使用多种生物信息学方法确定了错义突变的致病性并探讨了发病机制,不仅有利于突变效应的理解,而且可以对家系成员进行可靠的遗传咨询。本研究我们未对新发现剪接位点突变在mRNA水平进行突变效应的研究,但是剪接位点突变引起终止密码子提前出现,往往启动无义突变介导的mRNA降解(nonsense mediated mRNA decay,NMD)途径导致mRNA量的下降,从而限制了突变效应的研究[13]。
Funding Statement
基金项目:国家自然科学基金(81270587);卫生公益性行业科研专项(201202017);山西省卫生厅科技攻关计划(20100111);山西省回国留学人员重点科研项目(2009重点7)
References
- 1.Mariani G, Bernardi F. Factor VII Deficiency[J] Semin Thromb Hemost. 2009;35(4):400–406. doi: 10.1055/s-0029-1225762. [DOI] [PubMed] [Google Scholar]
- 2.Jayandharan GR, Viswabandya A, Nair SC, et al. Molecular basis of hereditary factor VII deficiency in India: five novel mutations including a double missense mutation (Ala191Glu; Trp364Cys) in 11 unrelated patients[J] Haematologica. 2007;92(7):1002–1003. doi: 10.3324/haematol.10835. [DOI] [PubMed] [Google Scholar]
- 3.O'Hara PJ, Grant FJ, Haldeman BA, et al. Nucleotide sequence of the gene coding for human factor VII, a vitamin K-dependent protein participating in blood coagulation[J] Proc Natl Acad Sci U S A. 1987;84(15):5158–5162. doi: 10.1073/pnas.84.15.5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: initiation, maintenance, and regulation[J] Biochemistry. 1991;30(43):10363–10370. doi: 10.1021/bi00107a001. [DOI] [PubMed] [Google Scholar]
- 5.Bowen DJ, Standen GR, Mazurier C, et al. Type 2N von Willebrand disease: rapid genetic diagnosis of G2811A (R854Q), C2696T (R816W), T2701A (H817Q) and G2823T (C858F)—detection of a novel candidate type 2N mutation: C2810T(R854W)[J] Thromb Haemost. 1998;80(1):32–36. [PubMed] [Google Scholar]
- 6.Castellana S, Mazza T. Congruency in the prediction of pathogenic missense mutations: state-of-the-art web-based tools[J] Brief Bioinform. 2013;14(4):448–459. doi: 10.1093/bib/bbt013. [DOI] [PubMed] [Google Scholar]
- 7.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm[J] Nat Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 8.Petrey D, Honig B. GRASP2: visualization, surface properties, and electrostatics of macromolecular structures and sequences[J] Methods Enzymol. 2003;374:492–509. doi: 10.1016/S0076-6879(03)74021-X. [DOI] [PubMed] [Google Scholar]
- 9.Wulff K, Herrmann FH. Twenty two novel mutations of the factor VII gene in factor VII deficiency[J] Hum Mutat. 2000;15(6):489–496. doi: 10.1002/1098-1004(200006)15:6<489::AID-HUMU1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 10.Triplett DA, Brandt JT, Batard MA, et al. Hereditary factor VII deficiency: heterogeneity defined by combined functional and immunochemical analysis[J] Blood. 1985;66(6):1284–1287. [PubMed] [Google Scholar]
- 11.Cooper DN, Millar DS, Wacey A, et al. Inherited factor VII deficiency: molecular genetics and pathophysiology[J] Thromb Haemost. 1997;78(1):151–160. [PubMed] [Google Scholar]
- 12.Herrmann FH, Wulff K, Auerswald G, et al. Factor VII deficiency: clinical manifestation of 717 subjects from Europe and Latin America with mutations in the factor 7 gene[J] Haemophilia. 2009;15(1):267–280. doi: 10.1111/j.1365-2516.2008.01910.x. [DOI] [PubMed] [Google Scholar]
- 13.Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease[J] Eur J Hum Genet. 2006;14(10):1074–1081. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]