

Abstract
The pentameric γ-aminobutyric acid type A receptors (GABAARs) are the major inhibitory ligand-gated ion channels in the central nervous system. They mediate diverse physiological functions, mutations in them are associated with mental disorders and they are the target of many drugs such as general anesthetics, anxiolytics and anti–convulsants. The five subunits of synaptic GABAARs are arranged around a central pore in the order β-α-β-α-γ. In the outer third of the transmembrane domain (TMD) drugs may bind to five homologous intersubunit binding sites. Etomidate binds between the pair of β – α subunit interfaces (designated as β+/α−) and R–mTFD-MPAB binds to an α+/β− and an γ+/β− subunit interface (a β− selective ligand). Ligands that bind selectively to other homologous sites have not been characterized. We have synthesized a novel photolabel, (2,6-diisopropyl-4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)methanol or pTFD-di-iPr-BnOH). It is a potent general anesthetic that positively modulates agonist and benzodiazepine binding. It enhances GABA–induced currents, shifting the GABA concentration-response curve to lower concentrations. Photolabeling–protection studies show that it has negligible affinity for the etomidate sites and high affinity for only one of the two R–mTFD-MPAB sites. Exploratory site–directed mutagenesis studies confirm the latter conclusions and hint that pTFD-di-iPr-BnOH may bind between the α+/β− and α+/γ− subunits in the TMD, making it an α+ ligand. The latter α+/γ− site has not previously been implicated in ligand binding. Thus, pTFD-di-iPr-BnOH is a promising new photolabel that may open up a new pharmacology for synaptic GABAARs.
Keywords: GABA-A Receptor, Transmembrane domain, Positive allosteric modulator, General Anesthetics Binding Sites, Photolabeling, Diazirine
Graphical abstract
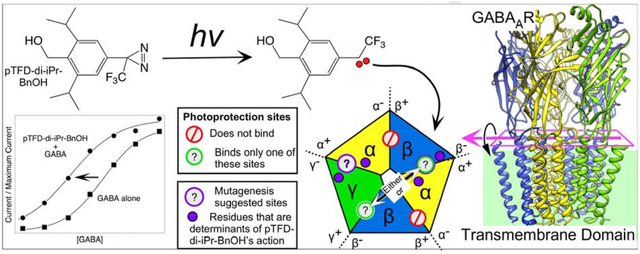
1. INTRODUCTION
The family of γ-aminobutyric acid type-A receptors (GABAARs) are the major inhibitory ligand-gated ion channels in the central nervous system. They are located in both synaptic and extrasynaptic neuronal membranes, respectively generating phasic and tonic currents. GABAAR activity affects many neurophysiological phenomena, including consciousness, learning and memory, sedation, anxiety, epilepsy, and mood [1]. GABAAR mutations are associated with epilepsy, depression, Down’s syndrome, schizophrenia and autism [2–4]. They are also targets for many neuromodulatory drugs such as general anesthetics, anticonvulsants and anxiolytics [5, 6].
GABAAR are members of the pentameric ligand-gated ion channel superfamily, composed of five homologous subunits arranged pseudo–symmetrically around a central chloride conducting pore (Figure 1). Mammals possess genes for 19 such homologous subunits and about two dozen receptor isoforms have been identified. Typical synaptic receptors consist of α, β, and γ subunits arranged anti–clockwise in the order α–γ–β–α–β viewed from the extracellular space [7, 8]. The various isoforms occupy distinct locations in the CNS where they may mediate distinct behaviors and pharmacological effects [9, 10]. This rich complexity has spurred researchers to identify isoform–selective modulators that may target specific CNS functions (reviewed in [5]).
Figure 1. The structure of the human full length α1β3γ2L GABAAR showing the main amino acid residues mentioned in this manuscript.
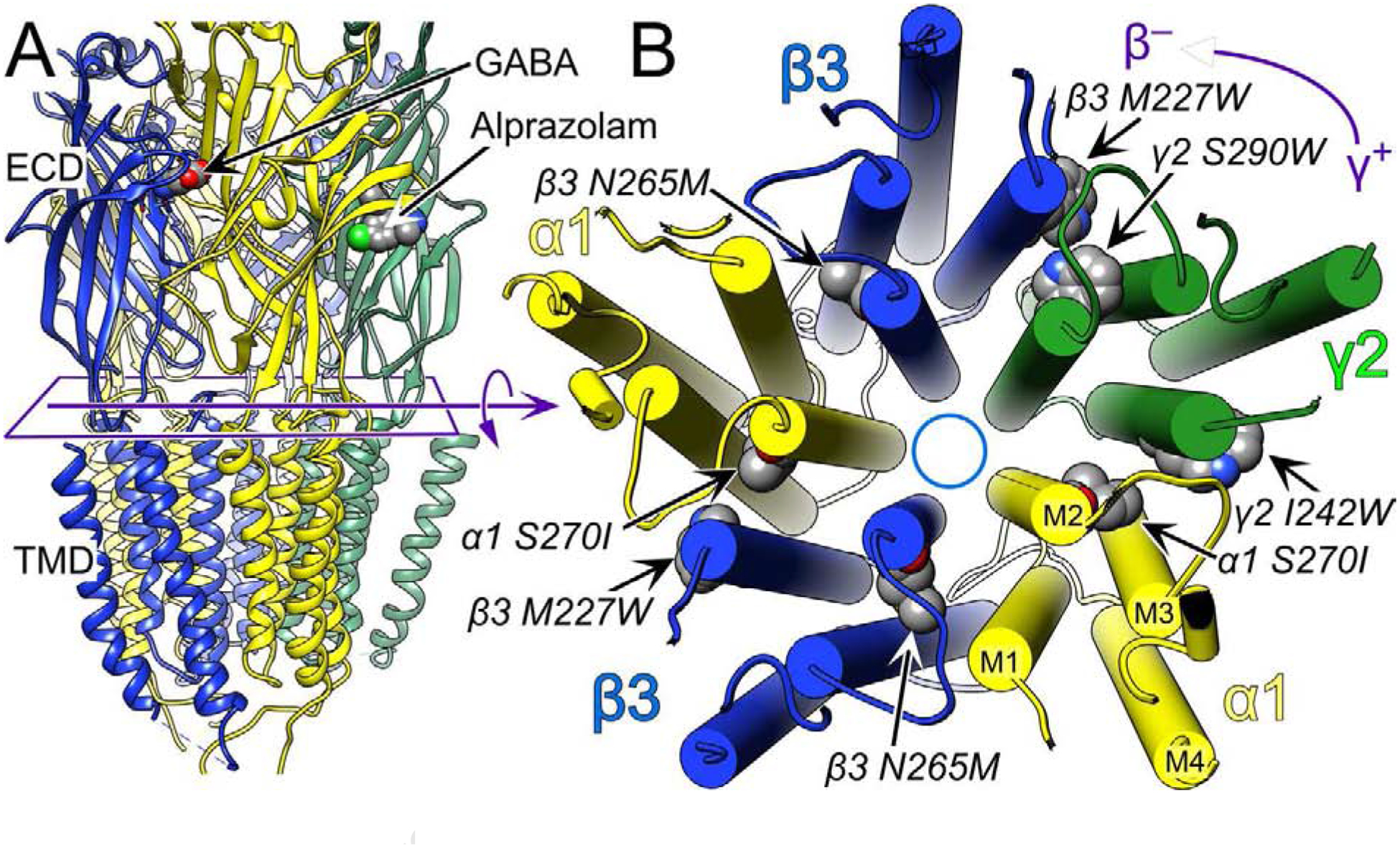
Panel A shows a side view of the extracellular domain (ECD) and transmembrane domain (TMD) depicted in ribbon mode. The intracellular domain is unstructured and therefore not shown. Panel B shows a cross section of the TMD viewed from the extracellular side with cylindrical helices. The subunits are labeled and color coded as indicated. The convention is to refer to the subunits in counter clockwise order with the plus and minus side of the subunit interface defined as indicated in the top right corner, which defines the γ+/β− subunit interface. In the ECD, GABA binds to the two β+/α− interfaces and alprazolam, a benzodiazepine, binds in the single α+/γ− interface. The structure shown is from the Protein Data Base, 6HUO.pdb, which is the α1β3γ2L GABAAR with both GABA and Alprazolam bound [7]. The figure was created using UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311 [39].
In typical synaptic receptors, GABA binds to a pair of nearly identical sites in the extracellular domain (ECD) between the β and α subunits (β+/α− sites). Benzodiazepines act at the single homologous α+/γ− site (Figure 1A; [7]). Derivatives that target specific α isotypes at α(1–6)+/γ− ECD subunit interfaces have been developed and have proved useful together with knock–in mice in implicating distinct α isotypes in the sedative, amnestic, and anxiolytic effects of classical benzodiazepines [1, 11].
In the transmembrane domain (TMD), where anesthetics and their derivatives bind, selective targeting of allosteric inter-subunit sites also occurs. Thus, etomidate and neurosteroid hypnotics target the two β+/α− interfaces at adjacent sites located near the extracellular and intracellular ends of the TMD, respectively, whereas the mephobarbital derivative R–mTFD-MPAB (5-allyl-5-(3-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)pyrimidine-2,4,6(1H,3H,5H)-trione) selectively targets both the γ+/β− and the α+/β− outer TMD interfaces (reviewed in [12]). Photolabeling protection experiments using [3H]azietomidate and [3H] R–mTFD-MPAB have been used to classify the selectivity of drugs for the outer TMD interfaces [13]. Notably, propofol occupies all four TMD sites with little selectivity [14], whereas another propofol analog, 4-benzoyl-propofol, has high selectivity between these sites, only having high affinity for one of the two R–mTFD-MPAB interfaces [15]. Meanwhile, no drug has been found that binds in the fifth homologous site in the α+/γ− outer TMD interface [16].
Thus, characterization of the pharmacology of the TMD as a drug target remains far from complete [17]. We and others have explored photolabels related to propofol, but this has proved difficult because the simplest derivatives are either relatively unstable, have limited efficacy as GABAAR modulators or have limits as photolabels [18–20]. Such chemical instability may be related to the presence of the phenolic hydroxyl. We previously used a 4-(trifluoromethyl)-diazirine derivative of benzyl alcohol to characterize an alkanol–binding cleft on luciferase [21]. Although that photolabel has low potency, we have now synthesized a new propofol derivative, pTFD-di-iPr-BnOH, that is both a potent and an efficacious GABAAR modulator. Preliminary experiments indicate it displays novel selectivity for the outer TMD subunit interfaces on the GABAAR. It is also an effective and stable photolabel whose photoincorporation into GABAARs is enhanced by GABA.
2. RESULTS
2.1. Synthesis of pTFD-di-iPr-BnOH.
The photoprobe 8 was synthesized as described below. Bromoaniline 1 was diazotized and subjected to a Sandmeyer reaction with cuprous cyanide to yield the substituted benzonitrile 2. The subsequent reduction of the cyano group with DIBAL and hydrolysis of the imine provided the corresponding substituted benzaldehyde 3. Aldehyde 3 was reduced with sodium borohydride into an alcohol 4 and protected with tert-butyldimethyl silyl chloride into the silyl ether 5. Compound 5 was lithiated with n-BuLi and acylated using ethyl trifluoroacetate into the corresponding substituted trifluoroacetophenone 6. The conversion of 6 into the aziridine was performed using an earlier described sequence [22] including oximation, formation of oxime tosylate, reaction with ammonia and the oxidation of the incipient diaziridine to afford the corresponding diazirine 7 with satisfactory yield. Finally, the silyl group protection was removed by the treatment with TBAF to provide the photoprobe 8.
Synthesis of the tritiated 8 was carried out by the sequence of periodinane oxidation of alcohol 8 into a corresponding aldehyde 9 and its reduction with NaB3H4 by Vitrax Corporation into tritiated alcohol 3H-8 with specific radioactivity of 15 Ci/mmol.
2.2. Anesthetic potency.
The fraction of tadpoles showing loss of righting reflexes (LoRR) increased with the concentration of Di-iPr-BnOH ((2,6-diisopropylphenyl)benzyl alcohol) between 3 – 100 μM, and analysis yielded an EC50 of 16 ± 1.4 μM; number of animals, N = 30. Further adding a 4-trifluoromethydiazirine group to (2,6-diisopropylphenyl)benzyl alcohol gave pTFD-di-iPr-BnOH, which produced LoRR in tadpoles with an EC50 of 2.5 ± 0.6 μM (N = 73; concentration range 0.5 – 10 μM), a 6–fold increase in potency. pTFD-di-iPr-BnOH had a low therapeutic index in tadpoles with an LC50 of 3.6 ± 0.6 μM. At 2, 3, and 10 μΜ, the fraction of tadpoles that did not recover from anesthesia and were therefore excluded from the analysis for anesthesia was 4/7, 6/9, and 7/10, respectively.
2.3. Modulation of [3H]muscimol binding.
We next examined the ability of the benzyl alcohol derivatives to modulate the binding of [3H]muscimol to α1β3γ2L receptors in native HEK cell plasma membranes. The base structure, benzyl alcohol (BnOH) caused no modulation up to 50 mM. However, adding either two isopropyl groups or a p-trifluorodiazirine group to BnOH conferred potency (Table 1) with the di-isopropyl addition being particularly effective with an EC50 of 40 ± 6 μM (mean ± standard deviation). Adding a pTFD group to Di-iPr-BnOH caused a further 6 fold increase in potency, whereas adding the two 4,6-isopropyl groups to pTFD-BnOH increased potency nearly 100–fold to 7.4 ± 0.7 μM (Figure 2A), a value that was both comparable to that of propofol and of sufficient potency to encourage its development as a photolabel.
Table 1.
The pharmacological properties of the benzyl alcohol derivatives studied compared to propofol.
| Structure | Abbreviated name | LoRR in Tadpoles EC50 ± SD, μM (# of animals) | Enhancement of [3H]muscimol binding in α1β3γ2L GABAARs EC50 ± SD, μM (N) | Protection against photoincorporation in the β subunit of α1β3γ2L GABAARs in the presence of GABA | ||
|---|---|---|---|---|---|---|
| [3H]azietomidate IC50, μM) (N, R2) | [3H]pTFD-di-iPr-BnOH IC50, μM) (N, R2) | [3H]R-mTFD-MPAB IC50, μM (N, R2) | ||||
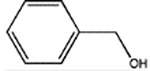 |
BnOH | 2,000 ± 210a | >50,000 | ND | ND | ND |
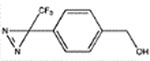 |
pTFD-BnOH | 28 ± 7b (30) | 700 ± 81 (3) | 305 ± 33 (5, 0.92) | ND | 465 ± 5 (5, 0.87) |
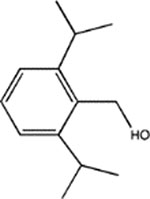 |
Di-iPr-BnOH | 16 ± 1 (30) | 40 ± 6 (2) | 92 ± 6 (5, 0.97) | ND | 243 ± 23 (5, 0.91) |
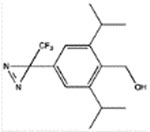 |
pTFD-di-iPr-BnOH | 2.5 ± 0.6 (73) | 7.4 ± 0.7 (6) | 460 ± 70 (4, 0.59) | 18 ± 2 (4, 0.94) | 26 ± 5 Bns = 42 ± 3% (4, 0.87) |
 |
Propofol | 0.63 ± 0.09c | 5.4 ± 0.9 (2) | 8 ± 1d | 32 ± 6 (4, 0.87) | 44 ± 4d |
Figure 2. pTFD-di-iPr-BnOH is a positive allosteric modulator of α1β3γ2L GABAARs.
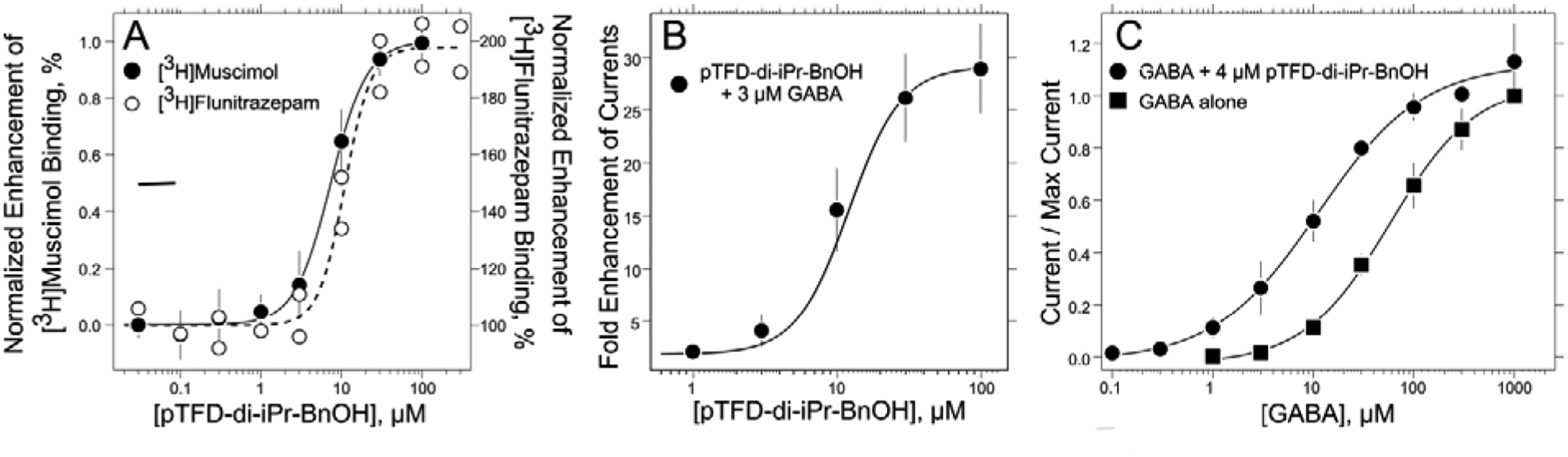
A. pTFD-di-iPr-BnOH enhances the specific binding of both [3H]muscimol and [3H]flunitrazepam to α1β3γ2L receptors in HEK293 cell membranes as described in Methods. The [3H]muscimol data points and their standard deviation are calculated from six separate experiments, forty four individual points, each of whose maximum modulation was normalized to one; the EC50 = 7.4 ± 0.6 μM, nH = 1.9 ± 0.3. The eighteen [3H]flunitrazepam data points are individual points from two independent experiments; EC50 = 11 ± 1 μM, maximum enhancement = 198 ± 4% and nH = 2.7 ± 1.0. B. pTFD-di-iPr-BnOH enhances 3 μM GABA (~EC02) currents in α1β3γ2L GABAARs expressed in oocytes. The displayed points are the means and SD of five oocytes; EC50 = 10 ± 1 μM, maximum enhancement = 30 ± 1–fold and nH = 2.0 ± 0.4 (n = 33). C. The concentration-response curves for GABA–stimulated currents normalized to the current at 1 mM GABA are plotted in the absence and presence of 4 μM pTFD-di-iPr-BnOH. For GABA alone the EC50 = 58 ± 5 μM, nH = 1.0 ± 0.1 (n = 33). For GABA at fixed pTFD-di-iPr-BnOH the EC50 = 11 ± 2 μM, the nH = 0.8 ± 0.1 (n = 40). All curves are nonlinear least squares fits of the individual data points to equations 1 or 2. Means and SD are shown for display purposes.
In α1β3 GABAARs, pTFD-di-iPr-BnOH’s EC50 for modulating [3H]muscimol binding was slightly lower than in α1β3γ2 receptors at 2.9 ± 0.7 μM, showing that the switch from a γ2 subunit to a β3 subunit in the pentamer does not attenuate its action. We compared modulation of [3H]muscimol binding by pTFD-di-iPr-BnOH with that by propofol, a ligand that binds in both the etomidate and R–mTFD-MPAB binding sites. Substituted cysteine modification protection experiments show that β M286C is protected from modification by propofol whereas α A291C is not [23]. We found that in α1β3(M286C), propofol’s EC50 was dramatically increased from 7.0 ± 1.7 in WT to >500 μM in the mutant, while leaving that of pTFD-di-iPr-BnOH unchanged at 3.1 ± 0.4 μM, an observation consistent with pTFD-di-iPr-BnOH not binding in the β+/α− interfaces. In α1(A291C)β3 receptors that contain a mutation on α1 M3 in the α+/β− interface, the EC50s of pTFD-di-iPr-BnOH and propofol were only modestly increased to 13 ± 3 and 32 ± 10 μM respectively. In all these experiments two independent concentration-response curves were determined and the combined 18 points fitted to Equation 1 (see Materials and Methods).
We also asked if pTFD-di-iPr-BnOH binds in the benzodiazepine site in the α+/γ− interface in the extracellular domain. To the contrary, pTFD-di-iPr-BnOH enhanced [3H]flunitrazepam binding over the concentration range 0.03 to 300 μM with no inhibition evident at the highest concentration (Figure 2A). This effect was characterized by an EC50 of 11 ± 1 μM, a Hill coefficient, nH, of 2.7 ± 1.0 and maximum modulation of 198 ± 4.4 %, comparable to modulation of [3H]muscimol binding (EC50 = 7.4 ± 0.6 μM, nH = 1.9 ± 0.27 and maximal modulation of 260 ± 46%). Similar Hill coefficients were observed in [3H]muscimol binding experiments using α1β3 receptors (nH = 2.3 ± 0.76).
2.4. Enhancement of GABAA receptor currents by pTFD-di-iPr-BnOH.
Synaptic type α1β3γ2L receptors were expressed in Xenopus oocytes and receptor activation was measured using two-microelectrode voltage–clamp electrophysiology. Currents elicited by a low concentration of GABA (3 μM ~EC02) were enhanced in a concentration–dependent manner (Figure 2B) when GABA was co-applied with pTFD-di-iPr-BnOH, plateauing at 30 to 100 μM. The concentration-response curve with the combined data from 5 oocytes was fitted to equation (2) (see Materials and Methods) to yield a maximum enhancement of 30 ± 1 fold, an EC50 of 10 ± 1 μM and a Hill coefficient of 2.0 ± 0.4. No surge currents were seen with pTFD-di-iPr-BnOH concentrations up to 100 μM.
2.5. Leftward shift of the GABA concentration-response curve.
In the presence of a fixed hypnotic concentration of pTFD-di-iPr-BnOH (4 μM), EC50s derived from GABA concentration-response curves in α1β3γ2L receptors were reduced about 5–fold, from 58 ± 5 μM to 11 ± 2 μM (Figure 2C). At 1 mM GABA, pTFD-di-iPr-BnOH enhanced maximal GABA-activated currents by 1.13 ± 0.07 fold (n = 4). Thus, pTFD-di-iPr-BnOH’s is a potent positive allosteric modulator, and its action resembles that of other potent intravenous general anesthetics.
At high concentrations, most general anesthetics that modulate GABAA receptors also directly activate receptors in the absence of GABA. In contrast, application of 100 μM pTFD-di-iPr-BnOH without GABA to oocytes expressing α1β3γ2L GABAA receptors resulted in no detectable current greater than the background noise (mean ± SD = 0.008 ± 0.013 pA), while 1 mM GABA elicited currents from every oocyte tested (3.2 ± 1.5 μA; n = 5).
2.6. Effect of mutations in previously characterized general anesthetic binding pockets.
To ascertain if interfacial transmembrane sites on GABAA receptors mediate modulation by pTFD-di-iPr-BnOH, we introduced mutations that are known to reduce the actions of etomidate or R–mTFD-MPAB (Figure 3A–F). Mutations were first introduced on the positive side of all three subunits at the homologous M2–15´ positions (α1 S270I, β3 N265M, and γ2 S280W). Additionally, mutations were introduced on the negative side of β3 and γ2 subunits at the M1–11´ position (numbered from the conserved arginine at the start of M1; β3 M227W and γ2 I242W) to differentiate effects mediated by α+/β− and α+/γ− interfaces. Enhancement of currents activated with subsaturating GABA concentrations in α1β3γ2L receptors containing each of the mutations above was compared to that in wild type receptors in the presence of etomidate (5 μM), R–mTFD-MPAB (8 μM) and pTFD-di-iPr-BnOH (10 μM).
Figure 3. Anesthetic compounds produce variable gating enhancement patterns in α1β3γ2L GABAA receptors harboring single-point mutations.
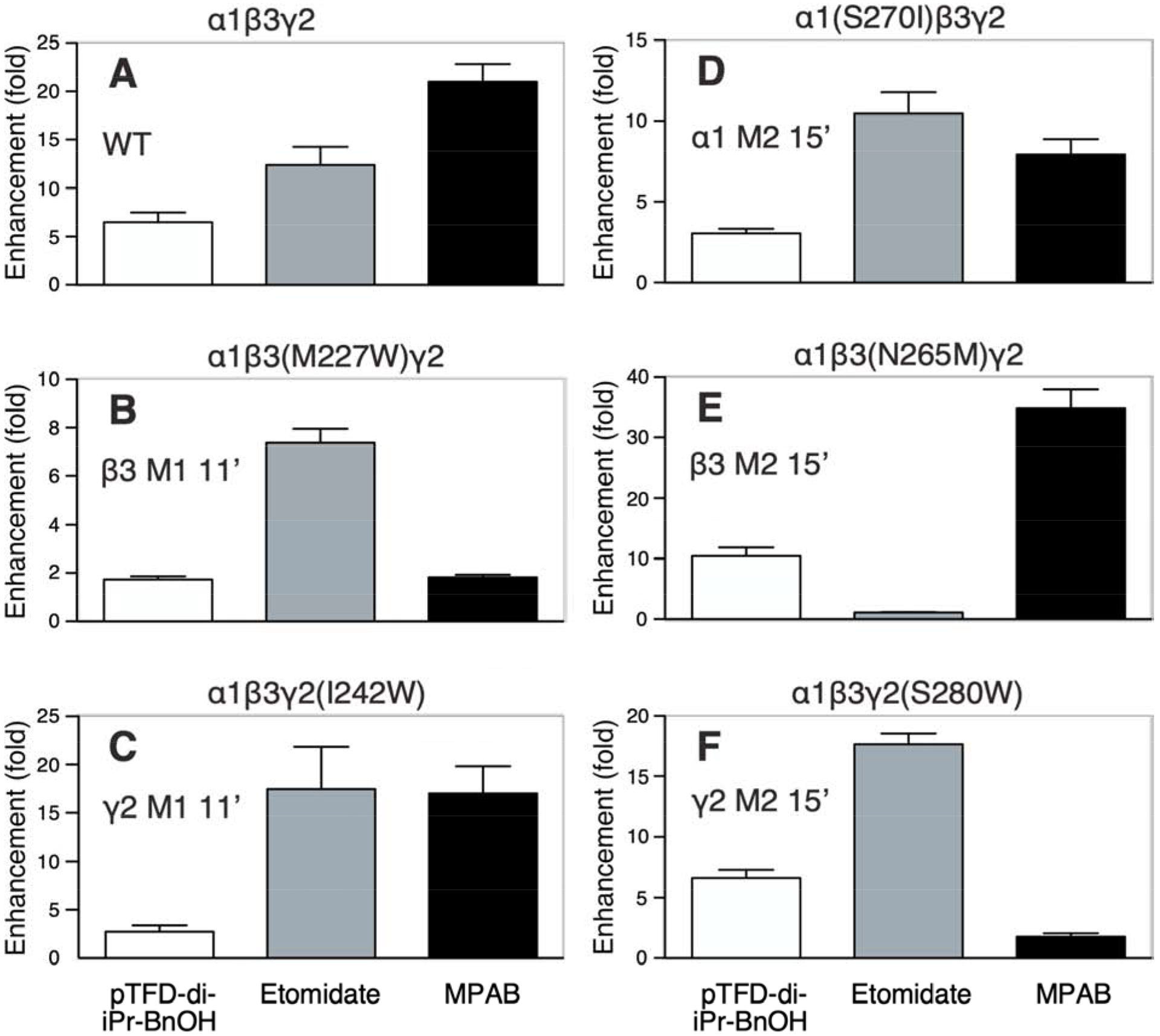
Bars represent mean ± SEM results from oocyte electrophysiology experiments (n = 8 to 11 for each drug-receptor condition) measuring mean (IGABA+Drug/IGABA, where IGABA+Drug is the current elicited with GABA combined with anesthetic drug). Results for each of six receptors (wild-type and five mutants) is displayed in separate panels labeled A to F and with the expressed receptor subunit mixture. GABA concentrations used elicited 2 to 5% of maximal GABA-elicited currents. Anesthetic drug conditions were 10 μM pTFD-di-iPr-BnOH (white), 5 μM etomidate (gray); and 8 μM R–mTFD-MPAB (black).
The anesthetic enhancement results for wild-type receptors is shown in Figure 3A. In the etomidate sites, the M2–15´ mutation β3 N265M (Figure 3E) eliminated etomidate enhancement, consistent with prior reports [24, 25], whereas modulation by both R–mTFD-MPAB and pTFD-di-iPr-BnOH was greater than that in wild-type. In the two nonidentical R–mTFD-MPAB sites, M2–15´ mutations α1S270I (Figure 3D) and γ2 S280W (Figure 3F) both significantly reduced R–mTFD-MPAB modulation, as expected [26]. In α1(S270I)β3γ2L receptors, etomidate enhancement was comparable to that in wild-type, whereas enhancement by pTFD-di-iPr-BnOH was reduced more than 50%. However, in α1β3γ2(S280W) receptors, enhancement by etomidate and pTFD-di-iPr-BnOH was maintained or increased.
To test whether mutations on the negative side of the α+/β− and α+/γ− interfaces were effective, we introduced β− and γ− mutations at M1–11´. In α1β3(M227W)γ2L receptors (Figure 3B), modulation by R–mTFD-MPAB and pTFD-di-iPr-BnOH was much lower than in wild-type, but etomidate’s action was unaffected. Modulation of α1β3γ2(I242W) receptors by both etomidate and R–mTFD-MPAB was similar to wild-type, but that of pTFD-di-iPr-BnOH was strongly attenuated (Figure 3C). These actions of etomidate and R–mTFD-MPAB on the β3 M227W and γ2 I242W mutations agree with a prior report [16]. Thus, we can conclude that both β− and γ− mutations at M1–11´ strongly reduce pTFD-di-iPr-BnOH’s enhancing action of GABA–induced currents.
To more readily compare the pattern of mutant effects on different drugs, the enhancement ratios with each drug were normalized to its mean wild-type value (Figure 4) and the results analyzed using two-way ANOVA, which revealed highly significant effects of mutant [F(5, 130) = 26.2; P < 0.0001] and drug [F(2,130) = 7.15; P = 0.0008] and factor interaction [F(10,130) = 33.2; P < 0.0001]. Normalized enhancement ratio patterns across the three M2–15´ mutations for etomidate and R–mTFD-MPAB are very similar to recently reported allosteric shifts for the same drugs and mutants [26]. We infer adjacent drug binding if a mutation reduces normalized enhancement with a significance value of P < 0.01 adjusted for 5 wild-type vs. mutant comparisons per drug (i.e. P < 0.01 = 0.05/5). The analysis of the M2–15´ mutations is consistent with their established selectivity [12], with etomidate selectively binding in the two β+/α− sites, while R–mTFD-MPAB binds only in the α+/β− and γ+/β− sites. The analysis also indicates that pTFD-di-iPr-BnOH binds near α1S270, which contributes to the α+/β− and α+/γ− transmembrane anesthetic binding sites, but not elsewhere.
Figure 4. Drug-dependent normalized enhancement patterns in α1β3γ2L GABAA receptors with mutations in the M1 or M2 transmembrane domain helices.
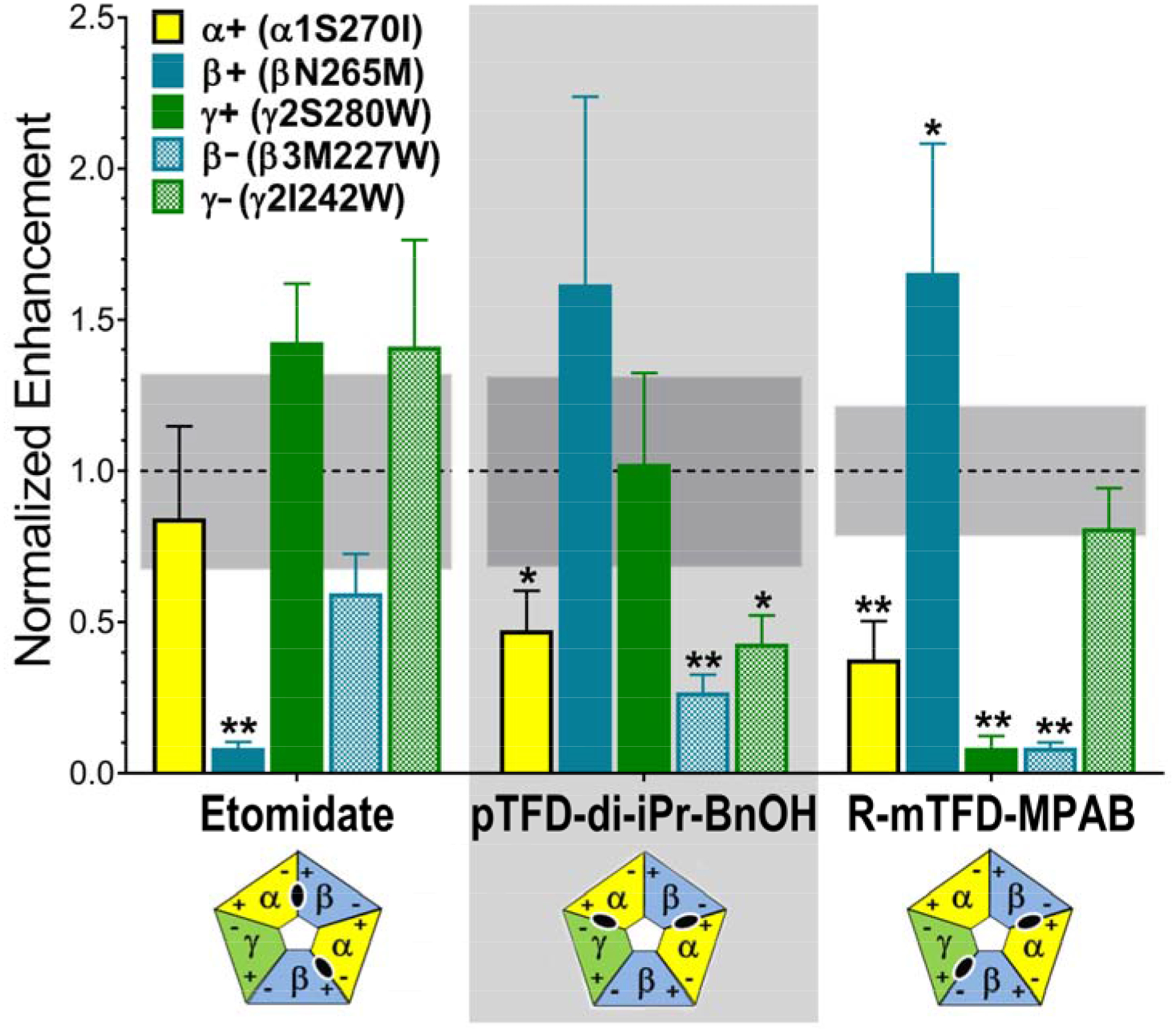
Drug enhancement ratios from experiments summarized in Figure 3 were normalized to average drug-specific enhancement ratios in wild-type receptors (see Figure 3, panel A). Normalized ratios are displayed as bars (mean ± SD), colored by mutation (M2 15’ mutants: α1 S270I = yellow; β3 N265M = solid blue; γ2 S280W = solid green. M1 11’ mutants: β3 M227W = checkered blue; γ2 I242W = checkered green). Combined mean ± SD values for normalized wild-type enhancement were: for etomidate, 1.0 ± 0.32 (n =9); for pTFD-di-iPr-BnOH, 1.0 ± 0.30 (n = 9), and for R-mTFD-MPAB, 1.0 ± 0.17 (n =8). The 95% confidence intervals of the wild type mean is shown as a gray band. Data were analyzed using two-way ANOVA and P-values (mutant vs. wild-type) were calculated with Student’s t-tests (* P < 0.01; ** P < 0.001). A significance threshold of P < 0.01 was based on the Bonferroni correction for 5 mutant comparisons to each wild-type data set for a given drug. Results where significant reductions in drug enhancement relative to wild-type are found indicate the following drug-residue contacts: etomidate binds near β3 N265; pTFD-di-iPr-BnOH binds near α1 S270, β3 M227 and γ2 I242; and R–mTFD-MPAB binds near α1 S270, γ2 S280; and β3 M227. The diagrams below each drug name depict the established subunit arrangement for αβγ GABAA receptors with subunits colored α =yellow; β = blue; and γ = green, and with + and − faces labeled. The inferred transmembrane inter-subunit sites occupied by each drug are shown in the corresponding diagrams as black ovals.
The β− mutation β3 M1 M227W, which abuts both α+/β− and γ+/β− interfacial sites where R–mTFD-MPAB binds, reduces modulation by both R–mTFD-MPAB and pTFD-di-iPr-BnOH. Notably, the γ− mutation, γ2 M1 I242W, significantly weakened modulation by pTFD-di-iPr-BnOH, but had no effect on modulation by either etomidate or R–mTFD-MPAB. These results are consistent with pTFD-di-iPr-BnOH binding at both the α+/β− and the α+/γ− intersubunit TMD sites. Until now there has been no evidence that anesthetics can bind in the latter interface [12, 16], making pTFD-di-iPr-BnOH a possible first ligand to do so. Thus it could prove to be a useful new addition to the photolabel arsenal for characterizing sites.
2.7. Inhibition of [3H]azietomidate and [3H]R–mTFD-MPAB photolabeling.
In order to conduct photolabeling experiments, it is necessary to purify and reconstitute the GABAARs into asolectin:CHAPS (200 μM:5 mM) micelles. Under these conditions, pTFD-di-iPr-BnOH still modulated [3H]muscimol binding strongly (216 ± 6% in micelles vs. 273 ± 14% in native membranes). The EC50 in micelles was about double that in native membranes (Table 1) at 13 ± 2 μM (3 independent concentration-response curves and 22 separate points between 1 and 300 μM pTFD-di-iPr-BnOH).
Competition of photolabeling was used to determine the relative potencies of the benzyl alcohol anesthetics as inhibitors of photolabeling by [3H]azietomidate in the α subunit (primarily into α1 M1 M236 [27]) and by [3H]R–mTFD-MPAB in the β subunit (primarily into β3 M1 M227 [13]) (Figures 5A & B respectively). GABAARs were photolabeled in the presence of GABA, and after photolabeling, subunits were resolved by SDS-PAGE, and 3H incorporation was determined by liquid scintillation counting. None of the benzyl alcohols inhibited [3H]azietomidate photolabeling in the α subunit at concentrations comparable to those that modulate GABAAR function. Di-iPr-BnOH was 3–4-fold more potent than either pTFD-BnOH or pTFD-di-iPr-BnOH, but more than 10-fold less potent than propofol (Table 1). In contrast, inhibition of [3H]R–mTFD-MPAB photoincorporation in the β subunit by pTFD-di-iPr-BnOH was 10-fold more potent than di-iPr-BnOH or pTFD-BnOH, but comparable in potency to propofol (Table 1). However, even at the highest concentrations, pTFD-di-iPr-BnOH only inhibited [3H]R–mTFD-MPAB photolabeling of the β subunit by 60% in contrast to the ~90 % inhibition seen in the presence of excess R–mTFD-MPAB (60 μM) itself (Figure 5B). Such partial inhibition indicates that pTFD-di-iPr-BnOH binds with high affinity to only one of R–mTFD-MPAB’s two β− intersubunit anesthetic sites.
Figure 5. Inhibition by benzyl alcohols of [3H]azietomidate (A) and [3H]R–mTFD-MPAB (B) photolabeling of α1β3γ2 GABAARs.
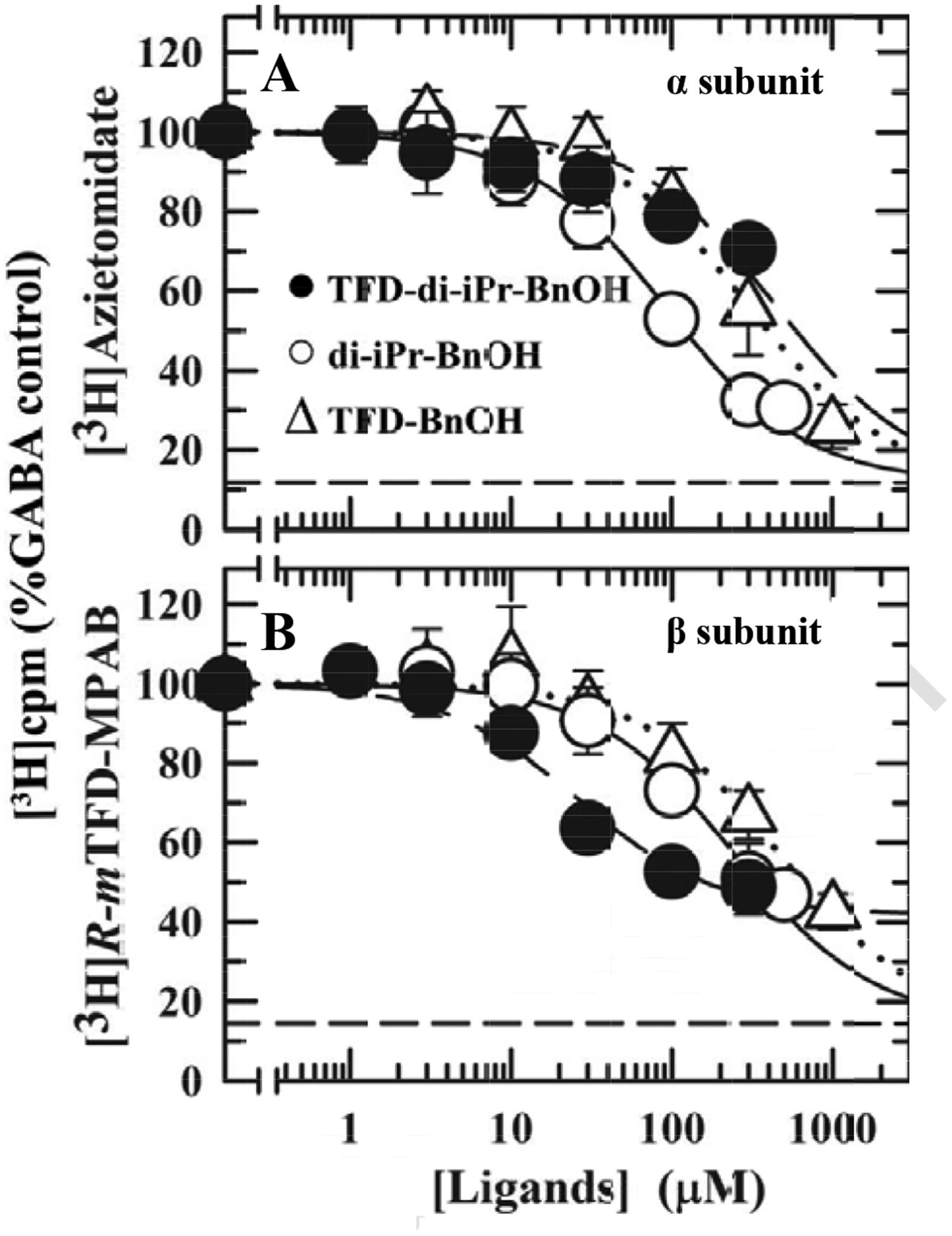
GABAARs were photolabeled in the presence of 300 μM GABA and the indicated concentrations of pTFD-di-iPr-BnOH (Filled circles), di-iPr-BnOH (Open circles) or pTFD-BnOH (Open triangles). After photolabeling, GABAAR subunits were isolated by SDS-PAGE, and covalent incorporation of 3H cpm was determined by liquid scintillation of excised gel bands containing the α ([3H]azietomidate) or β ([3H] R–mTFD-MPAB) subunit. Non-specific photolabeling (Bns), indicated by the dashed lines, was determined in the presence of 300 μM etomidate (Bns = 13 ± 3%, N = 6) or 60 μM R–mTFD-MPAB (Bns = 13 ± 4%, N = 6). For each independent experiment, data were normalized to the control condition, and the plotted data are the means (± SD) from the pooled independent experiments. The pooled data from the independent experiments were fit to Eq. 3 with IC50 as a variable parameter and Bns fixed and also, for pTFD-di-iPr-BnOH inhibition of [3H]R–mTFD-MPAB photolabeling, with IC50 and Bns variable. Based upon the extra-sum of the squares principle (F test, α = 0.05) the concentration dependence of pTFD-di-iPr-BnOH inhibition of [3H]R–mTFD-MPAB photolabeling was better fit (P<0.0001, F(Dfn,Dfd) = 44.1 (1,26) with variable Bns (IC50 = 26 ± 5 μM, Bns = 42 ± 3%) than with fixed Bns (IC50 = 73 ± 10 μM, Bns = 13%). Parameters for the fits and the number of independent experiments (N) are tabulated in Table 1.
2.8. [3H]pTFD-di-iPr-BnOH photolabeling of α1β3γ2L GABAARs.
We first compared photolabeling of GABAARs by [3H]pTFD-di-iPr-BnOH in the resting and desensitized states, that is in the presence of either the inverse agonist bicuculline or the agonist GABA. GABAAR subunits were separated by SDS-PAGE, and covalent incorporation of 3H into receptor subunits was characterized by fluorography (Figure 6A) and by liquid scintillation counting (Figure 6B). Based upon fluorography, 3H was incorporated primarily into the β subunit, and photolabeling was strongly enhanced in the presence of GABA compared to bicuculline, showing that an allosteric site had been photolabeled.
Figure 6. Pharmacological specificity of [3H]pTFD-di-iPr-BnOH photolabeling of α1β3γ2 GABAARs. A & B.
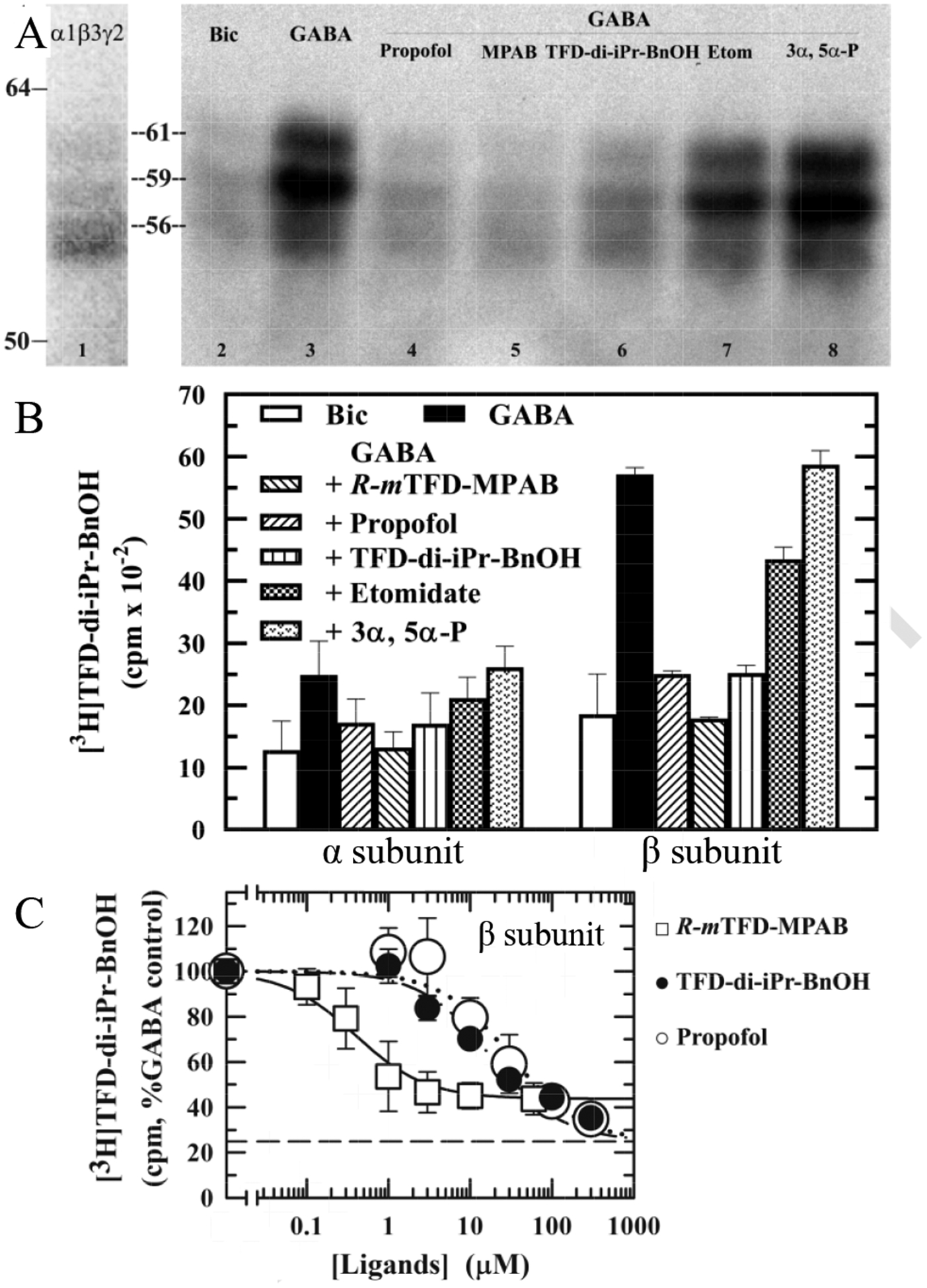
Aliquots of GABAARs were photolabeled with 1.5 μM [3H]pTFD-di-iPr-BnOH in the presence of 100 μM bicuculline (Bic; resting state receptors) or 300 μM GABA (desensitized receptors) in the absence of other drugs and in the presence of 100 μM propofol, 60 μM R–mTFD-MPAB (MPAB), 100 μM pTFD-di-iPr-BnOH, 300 μM etomidate (Etom), or 30 μM allopregnanolone (3,5-P). After photolabeling, receptor subunits were resolved by SDS-PAGE and 3H incorporation was determined by fluorography (A, except that lane 1 is a representative Coomassie Blue stained gel lane) or by liquid scintillation counting of excised subunits (B). Indicated on the left are the mobilities of the molecular mass markers and the calculated mobilities of the GABAAR subunit bands (α1, 56 kDa; β3, 59/61 kDa; with the γ2 subunit distributed diffusely in the three bands). B. In parallel with the fluorogram, 3H incorporation in the excised α (56 kDa) and β (59/61 kDa) subunit gel bands was determined by liquid scintillation counting. The means ± 1/2 range are plotted from 2 gels. C. The concentration dependence of inhibition of β subunit photolabeling by pTFD-di-iPr-BnOH (closed circles), propofol (open circles) or R–mTFD-MPAB (open squares). Non-specific photolabeling (Bns) was determined in the presence of 300 μM etomidate and 60 μM R-mTFD-MPAB. For inhibition by pTFD-di-iPr-BnOH and propofol, which was determined for [3H]pTFD-di-iPr-BnOH at 2 μM, Bns was 25 ± 3%, (N = 4, dashed line). For R–mTFD-MPAB inhibition, determined at 3 μM [3H]pTFD-di-iPr-BnOH, Bns was 42 ± 8% (N = 5). For each independent experiment, data were normalized to the control condition, and the plotted data are the means (± SD) from the pooled independent experiments. The pooled data from the independent experiments were fit to Eq. 3 (see Materials and Methods) with IC50 as a variable parameter. Parameters for the fits and the number of independent experiments (N) are tabulated in Table 1.
We next examined the pharmacology of the pTFD-di-iPr-BnOH sites. In the presence of GABA, photoincorporation of [3H]pTFD-di-iPr-BnOH into the β subunit was reduced to levels similar to those in the presence of bicuculline by non-radioactive pTFD-di-iPr-BnOH, propofol or R–mTFD-MPAB, whereas etomidate and allopregnanolone (3α, 5α–P) caused little inhibition of photolabeling (Figure 6A). When 3H photoincorporation was quantified by liquid scintillation counting (Figure 6B), R–mTFD-MPAB (60 μM) inhibited total β subunit photolabeling by 70%, reducing it to the level seen in the presence of bicuculline, whereas 100 μM propofol and pTFD-di-iPr-BnOH reduced the photolabeling by 55%. Although a similar pattern of photoincorporation is seen in the α subunit, the pharmacologically specific photolabeling was 3–fold less than that in the β subunit rendering quantification less secure.
Based upon the radiochemical specific activity of [3H]pTFD-di-iPr-BnOH (15 Ci/mmol) and the amount of GABAAR loaded on the gel (4 pmol), the 3,900 cpm of R-mTFD-MPAB-inhibitable β subunit photolabeling indicated specific photolabeling of ~6 % of GABAAR β subunits, a photolabeling efficiency similar to that seen for [3H]R–mTFD-MPAB [22].
We determined the concentration dependence of inhibition of [3H]pTFD-di-iPr-BnOH photoincorporation in the presence of GABA only in the GABAAR’s β subunit for reasons enunciated above (Figure 6C). At higher concentrations (300 μM) than used in Figure 6B, pTFD-di-iPr-BnOH and propofol now each inhibited photolabeling to the same extent as the background labeling, which was determined with a combination of 60 μM R–mTFD-MPAB and 300 μM etomidate and is denoted by the horizontal dashed line. The IC50s for pTFD-di-iPr-BnOH and propofol were18 ± 2 and 32 ± 6 μM respectively, and similar in each case to their values for inhibition of [3H]R–mTFD-MPAB photolabeling (Table 1). R–mTFD-MPAB also inhibited [3H]pTFD-di-iPr-BnOH photoincorporation with an IC50 value of 0.4 ± 0.1 μM, similar to its affinity in the presence of GABA for its reported binding sites at the two β− subunit interfaces [13].
3. DISCUSSION
3.1. Does pTFD-di-iPr-BnOH bind in novel sites?
In the search for new allosteric sites in the TMD of the GABAAR, we have synthesized a novel photoactivatable benzyl alcohol agent. It is a potent general anesthetic and a positive allosteric modulator of GABAARs. After tritiation, it photo-incorporated in both the α and the β subunits and that photoincorporation was positively modulated in the presence of GABA (Figure 6). For technical reasons, we cannot rule out photoincorporation in the γ subunit.
Three lines of evidence support the assertion that pTFD-di-iPr-BnOH does not bind in the well–characterized etomidate site in the two β+/α− subunit interfaces of the transmembrane domain. First, pTFD-di-iPr-BnOH only protected against photolabeling by [3H]azietomidate weakly and at high concentrations (Figure 5A). Second, site–directed mutagenesis of a residue in the β+/α− interface (β3 M3 M286C) attenuated propofol’s action on [3H]muscimol binding but not that of pTFD-di-iPr-BnOH. Third, a mutation in β3 M2 15’ (N265M), which strongly attenuated etomidate’s ability to enhance currents elicited by subsaturating concentrations of GABA [28], had no effect on pTFD-di-iPr-BnOH’s action (Figure 3 & 4).
Interaction with the two R–mTFD-MPAB sites, one in the γ+/β− and the other in the α+/β− interface, was more complex. Although, pTFD-di-iPr-BnOH protected against photoincorporation by [3H]R–mTFD-MPAB with high affinity, it only protected against half of the photolabeling (Figure 5B). Furthermore, R–mTFD-MPAB inhibited [3H]pTFD-di-iPr-BnOH photoincorporation into the β subunit to the same extent as non-radioactive pTFD-di-iPr-BnOH (Figure 6B and C). Taken together, these two results imply that pTFD-di-iPr-BnOH only interacts with one of the two R–mTFD-MPAB sites, and that this site accounts for the pharmacologically specific [3H]pTFD-di-iPr-BnOH photolabeling in the β subunit. In the α subunit a similar pharmacological pattern was observed, but photoincorporation was at less than 25% the efficiency of that in the β subunit. Thus, direct identification of the amino acids photolabeled in the α subunit will be necessary to determine whether or not they contribute to an R–mTFD-MPAB site. Our photolabeling results left two unresolved questions. First, which of the two R–mTFD-MPAB sites does pTFD-di-iPr-BnOH interact with and, second, is there an additional pTFD-di-iPr-BnOH binding site?
Because it was particularly important to assess whether pTFD-di-iPr-BnOH was a photolabel that would reveal new sites, and therefore if it was worth pursuing in further detail, we conducted a rapid survey using site–directed mutagenesis. Although mutagenesis studies have well known limitations, there is a sufficient body of experience that, when used carefully, they can point towards potential binding sites [16, 26]. We assumed that a robust criterion for identifying a binding site was that a drug’s action was affected by adjacent mutations on both the positive and the negative side of a given subunit interface. Applying this criterion, our observations suggest, but do not prove, that pTFD-di-iPr-BnOH binds in the α+/β− and not the γ+/β− interface. Thus, on the one hand, mutations on each side of the α+/β− interface (α1 M2 S270I and β3 M1 M227W respectively, see Figure1 for location of all residues mentioned here) both attenuate pTFD-di-iPr-BnOH and R–mTFD-MPAB’s enhancing actions on GABA currents (Figures 3B & D; Figure 4). On the other hand, a mutation in the γ+/β− interface (γ2 M2 15’ S280W) that attenuated R–mTFD-MPAB’s enhancing action was ineffective on pTFD-di-iPr-BnOH’s action (Figure 3F; Figure 4).
Next, we tested the hypothesis that the second site is in the remaining homologous interface, namely the α+/γ− interface, which has formerly been termed the orphan (or “undruggable”) site because photolabeling and substituted cysteine modification protection experiments have failed to find ligands that binds in it [12, 16]. We placed a mutation on the negative side of this interface (γ2 M1 I242W, which is homologous to β3 M1 M227W), and found that it attenuated only the action of pTFD-di-iPr-BnOH and certainly not that of R–mTFD-MPAB (Figure 3C; Figure 4). Considering that the α1 M2 15’ mutation on the other side of this interface also attenuated pTFD-di-iPr-BnOH’s action (Figure 3D; Figure 4), our observations point to the second pTFD-di-iPr-BnOH site being in the α+/γ− interface. Definitively answering these questions will take much more detailed and time consuming experiments using photolabeling and sequencing in purified reconstituted receptors and substituted cysteine modification protection experiments in intact cells
If it is confirmed that pTFD-di-iPr-BnOH binds in the α+/γ− interface, it would be the first photolabel to bind in this, so–called, orphan interface. In that case a third type of action and pharmacology in the transmembrane domain is added to those previously established. First, pTFD-di-iPr-BnOH would be the first agent that binds in the pair of α+ interfaces, whereas R–mTFD-MPAB binds in the two β− interfaces and etomidate in the two β+ interfaces. Thus, pTFDdi-iPr-BnOH promises to be an important new tool to aid the development of further subunit–interface selective pharmacological agents.
3.2. In vivo potency of benzyl alcohols and their sites of action
BnOH and p-TFD-BnOH caused loss of righting reflexes in tadpoles at some 20-times lower concentrations than they modulated [3H]muscimol binding to GABAARs in HEK cell membranes (Table 1), suggesting that they may have other sites of action most likely on glutamate receptors [29]. On the other hand, all the ligands with 2,6-isopropyl groups, had comparable general anesthetic and GABAAR modulatory potencies. This is consistent with the isopropyl group conferring a strong association with the latter receptors, a conclusion that is backed by other studies [30, 31].
3.3. Structure activity relationships of benzyl alcohols acting on GABAARs
BnOH had no action on GABAARs but addition of either a pTFD or two ortho isopropyl groups conferred potency, with the latter substitution being some twenty fold more effective than the former, again consistent with a special role for the isopropyl group. When these two substitutions were combined the net increase in potency over BnOH was greater than one thousand fold.
The photolabeling–protection data summarized in Table 1 make it possible to study the structure–activity relationship in each of the homologous interfaces in the transmembrane domain. At the etomidate site in the β+/α− interface, these studies showed that any substitution on BnOH’s aromatic ring conferred potency, but the most notable finding was that the addition of two ortho isopropyl groups to pTFD-BnOH did not enhance potency, whereas in the absence of the p-TFD substitution potency was moderately good. On the other hand, at the R–mTFD-MPAB site neither of the above substitutions in BnOH produced a high affinity interaction, but addition of a pTFD group to di-iPr-BnOH increased potency some tenfold, whereas the same addition at the etomidate sites decreased potency some fivefold. This site was also slightly less sensitive to the replacement of propofol’s phenolic hydroxyl with a –CH2–OH group, which reduced potency fivefold, half as much as at the etomidate site.
The overall effect of the differing apparent affinities for the etomidate and R–mTFD-MPAB sites is that: (1) pTFD-BnOH binds unselectively to both sites with low affinity; (2) di-iPr-BnOH favors the etomidate site 2–3-fold and has moderate affinity for it, and (3) pTFD-di-iPr-BnOH has high affinity for one of the R–mTFD-MPAB sites but low affinity for the etomidate sites. Thus, it is the combination of the two substitutions that confers binding selectivity for one of the two R–mTFD-MPAB sites without having much influence at the etomidate sites.
The role of the pTFD- group in conferring selectivity to the R–mTFD-MPAB sites may be compared to that in a series of 4-substituted propofols in a recent study [15]. Propofol itself (4-H) and 4-Cl-propofol favored the etomidate site by 5.5- and 4-fold respectively, but the following larger substitutions favored the R–mTFD-MPAB sites: Me–CO– by 1.5-fold; Ph–C(OH)– by 5.3 fold, and t-Bu– by 8-fold.
Unlike pTFD-di-iPr-BnOH, none of the above compounds distinguished between the two R–mTFD-MPAB sites but it has been reported that 4-benzoylpropofol does so [15]. Its low affinity R–mTFD-MPAB site was favored over the etomidate sites by only 3-fold, whereas its high affinity R–mTFD-MPAB site was favored over the etomidate site by 200-fold, about ten-fold more than pTFD-di-iPr-BnOH, suggesting that the rigidity of this derivative of propofol confers an advantage.
4. CONCLUSIONS
The aim of this study was to devise a new anesthetic photolabel that would identify novel binding sites on GABAA receptors. Our findings show that pTFD-di-iPr-BnOH fulfills this goal. It is a positive allosteric modulator of synaptic GABAARs that at hypnotic concentrations does not bind to the etomidate sites in two β+/α− interfaces, but does interact with one of the two R–mTFD-MPAB α+/β− and γ+/β− sites and with another site. This additional site is most likely in the homologous α+/γ− interface, for which there is currently no known ligand. Thus, pTFD-di-iPr-BnOH promises to be a useful new tool for characterizing the pharmacology of the five homologous intersubunit drug binding sites in the outer third of the transmembrane domain of GABAARs. It will complement the two well–established tools, azietomidate and R–mTFD-MPAB.
5. MATERIALS AND METHODS
5.1. Materials.
4-Bromo-2,6-diisopropylaniline and anhydrous grade solvents used in synthesis were from Aldrich, and were not further dried or purified. Common chemicals, etomidate, asolectin, FLAG peptide and polyethyleneimine were from Sigma. Buffer chemicals, CHAPS and DDM were from Fisher–Anatrace. pTFD-BnOH was obtained from TCI America. R–mTFD-MPAB, [3H]R–mTFD-MPAB (38 Ci/mmol, 26 μM in ethanol, and [3H]azietomidate (19 Ci/mmol, 53 μM in ethanol) were synthesized and tritiated previously [22, 32]. [3H]Muscimol and [3H]flunitrazepam were from Perkin Elmer (Cat. # NET 574 250UC and NET 567250UC respectively).
The human GABAARs used for the biochemical and photolabeling studies described herein and designated as α1β3γ2L or α1β3 had the composition N-FLAG–α1β3γ2L–C–(GGS)3GK–1D4 or N-FLAG–α1β3 respectively and were expressed in tetracycline-inducible HEK293 cells as previously described [33, 34]. They were used as native membranes or after solubilization and purification on a FLAG antibody column, from which they were eluted in micelles of 200 μM asolectin and 5 mM CHAPS with 100 μg/mL (~100 μM) FLAG peptide. Membranes and reconstituted receptors were stored at (–80°) until needed. In electrophysiological studies, the human subunits lacked the purification tags (see below).
5.2. Analytical Chemistry.
Analytical Chemistry.
The methods used were as previously described [35]. 1H, 13C and 19F NMR spectra were recorded on a Bruker AVANCE spectrometer at 400 MHz, 100 MHz and 376 MHz, respectively, unless otherwise noted. The NMR chemical shifts were referenced indirectly to TMS for 1H and 13C, and to CFCl3 for 19F NMR. High resolution mass spectrometry was performed with a Q-TOF-2TM (Micromass). TLC was performed using Merck 60 F254 silica gel plates. Purity of the final compounds was assessed by HPLC analysis with a Synergy Hydro-RP column (4 μm, 4.60 × 150 mm) using a methanol and methanol-water gradient running from 1% methanol to 99% methanol over 32 min, followed by isocratic elution. Elution was monitored by UV at 254 nm. These HPLC analyses indicated purity > 96%.
4-Bromo-2,6-diisopropylbenzonitrile (2).
Hydrochloric acid (37%, 1.5 mL) was added at room temperature to a stirred suspension of 4-bromo-2,6-diisopropylaniline (1) (1.57 g, 6.13 mmol) in H2O (5 mL), and the mixture was allowed to stir at room temperature for 30 min. A solution of sodium nitrite (0.31 g, 4.49 mmol) in H2O (1 mL) was added in a dropwise manner at 0°C (ice-bath), and the mixture was allowed to stir at 0°C for 30 min. The mixture was neutralized by saturated aqueous solution of NaHCO3 and added in portions at 70°C to a stirred solution of CuCN (0.43 g, 4.80 mmol) and KCN (0.65 g, 9.98 mmol) in H2O (1 mL). The reaction mixture was allowed to stir at 70°C for 30 min, cooled to room temperature, and extracted with toluene (15 mL). The organic layer was washed with water (5 mL), brine (5 mL), dried with Na2SO4, and evaporated to dryness under reduced pressure to yield a crude product which was purified by flash column chromatography (silica gel, hexane-ethyl acetate, 97:3) to afford 0.72 g (44%) of 4-bromo-2,6-diisopropylbenzonitrile (2) as a brown solid. 1H NMR (CDCl3): δ 1.32 (d, J = 6.8 Hz, 12H, CH3), 3.35–3.45 (m, 2H, CH), 7.37 (s, 2H, ArH). 13C NMR (CDCl3): δ 154.66, 128.33, 126.84, 116.46, 110.24, 32.61, 23.15.
4-Bromo-2,6-diisopropylbenzaldehyde (3).
Diisobutylaluminum hydride (1M in toluene, 6.5 mL) was added in a dropwise manner at 0°C (ice-bath) to a stirred solution of 4-bromo-2,6-diisopropylbenzonitrile (2) (0.734 g, 2.76 mmol) in CH2Cl2 (1 mL) under an argon atmosphere. The ice bath was removed, and the reaction mixture was allowed to stir at room temperature overnight, cooled to 0°C (ice-bath), and quenched by the addition of HCl (1.5M, 5 mL). The reaction mixture was heated at reflux for 30 min, cooled to room temperature, and extracted with CH2Cl2 (3 × 5 mL). The combined organic portion was washed with brine (5 mL), dried with Na2SO4, and concentrated to dryness under reduced pressure to yield a crude product which was purified by flash column chromatography (silica gel, hexane-ethyl acetate, 97:3) to afford 0.631 g (85%) of 4-bromo-2,6-diisopropylbenzaldehyde (3) as a brown oil. 1H NMR (CDCl3): δ 1.28 (d, J = 6.8 Hz, 12H, CH3), 3.48–3.58 (m, 2H, CH), 7.40 (s, 2H, ArH), 10.64 (s, 1H, CHO). Compound 3 was previously synthesized from 5-bromo-2-iodo-1,3-diisopropylbenzene through a Bouveault reaction [36]; however, the reported route was not followed to avoid lithiation of the aromatic bromo group by n-butyllithium used during the reaction.
(4-Bromo-2,6-diisopropylphenyl)methanol (4).
A solution of NaBH4 (0.243 g, 6.42 mmol) in EtOH (5 mL) was added to 4-bromo-2,6-diisopropylbenzaldehyde (3) (0.631 g, 2.34 mmol) under an argon atmosphere, and the reaction mixture was allowed to stir at room temperature for 3 h, cooled to 0°C (ice-bath), and quenched by the addition of HCl (1.5M, 5 mL). The solvent was evaporated under reduced pressure. Water (10 mL) was added, and the mixture was extracted with ethyl acetate (3 × 5 mL). The combined organic portion was washed with brine (5 mL), dried with Na2SO4, and concentrated to dryness under reduced pressure to yield a crude product which was purified by flash column chromatography (silica gel, hexane-ethyl acetate, 60:40) to afford 0.496 g (78%) of 4-bromo-2,6-diisopropylbenzyl alcohol (4) as a yellowish white solid. 1H NMR (CDCl3): δ 1.28 (d, J = 6.7 Hz, 12H, CH3), 3.33–3.40 (m, 2H, CH), 4.77 (s, 2H, CH2OH), 7.31 (s, 2H, ArH). 13C NMR (CDCl3): δ 150.59, 132.96, 126.55, 123.22, 57.03, 29.37, 24.34.
((4-Bromo-2,6-diisopropylbenzyl)oxy)(tert-butyl)dimethylsilane (5).
A solution of 4-bromo-2,6-diisopropylbenzyl alcohol (4) (0.496 g, 1.83 mmol), imidazole (0.200 g, 2.94 mmol), and tert-butyldimethylsilyl chloride (0.400 g, 2.65 mmol) in CH2Cl2 (5 mL) was allowed to stir at room temperature under argon atmosphere for 40 h. 4-Dimethylaminopyridine (0.400 g, 3.27 mmol) was added, and the reaction mixture was stirred at room temperature for 24 h, heated at reflux for 22 h, cooled to room temperature, washed with water (5 mL), brine (5 mL), dried with Na2SO4, and concentrated to dryness under reduced pressure to yield a crude product which was purified by flash column chromatography (silica gel, hexane-ethyl acetate, 97:3) to afford 0.570 g (86%) of ((4-bromo-2,6-diisopropylbenzyl)oxy)(tert-butyl)dimethylsilane (5) as a colorless oil. 1H NMR (CDCl3): δ 0.15 (s, 6H, CH3), 0.93 (s, 9H, CH3), 1.26 (d, J = 6.8 Hz, 12H, CH3), 3.28–3.38 (m, 2H, CH), 4.72 (s, 2H, CH2O), 7.28 (s, 2H, ArH). 13C NMR (CDCl3): δ 150.51, 133.20, 126.19, 122.58, 57.15, 29.26, 25.89, 24.22, 18.28, −5.35.
1-(4-(((tert-Butyldimethylsilyl)oxy)methyl)-3,5-diisopropylphenyl)-2,2,2-trifluoroethan-1-one (6).
n-Butyllithium (1.6M in hexane, 1.6 mL) was added in a dropwise manner over 10 min at −78°C into a stirred solution of ((4-bromo-2,6-diisopropylbenzyl)oxy)(tert-butyl)dimethylsilane (5) (0.476 g, 1.24 mmol) in THF (5 mL) under an argon atmosphere, and the reaction mixture was allowed to stir at −78°C for 2 h. A solution of ethyl trifluoroacetate (0.358 g, 2.52 mmol) in THF (1.5 mL) was added in a dropwise manner over 10 min at −78°C, the reaction mixture was allowed to stir at −78°C for 1 h and quenched by the addition of saturated aqueous solution of NaHCO3 (5 mL). Water (5 mL) was added, and the mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic portion was washed with brine (2 × 10 mL), dried with Na2SO4, and concentrated to dryness under reduced pressure to yield a crude product which was purified by flash column chromatography (silica gel, hexane-ethyl acetate, 95:5) to afford 0.330 g (66%) of 1-(4-(((tert-butyldimethylsilyl)oxy)methyl)-3,5-diisopropylphenyl)-2,2,2-trifluoroethan-1-one (6) as a pale yellow oil. 1H NMR (CDCl3): δ 0.17 (s, 6H, CH3), 0.94 (s, 9H, CH3), 1.31 (d, J = 6.9 Hz, 12H, CH3), 3.36–3.47 (m, 2H, CH), 4.81 (s, 2H, CH2O), 7.88 (s, 2H, ArH). 13C NMR (CDCl3): δ 180.60 (q, J = 34.3 Hz), 149.33, 142.17, 129.55, 124.92, 116.87 (q, J = 289.5 Hz), 57.21, 29.33, 25.82, 24.15, 18.26, −5.41. 19F NMR (CDCl3): δ −71.02.
3-(4-(((tert-Butyldimethylsilyl)oxy)methyl)-3,5-diisopropylphenyl)-3-(trifluoromethyl)-3H-diazirine (7).
Pyridine (99.8%, 1.5 mL) was added at room temperature to a stirred solution of the substituted acetophenone 6 (0.330 g, 0.82 mmol) in EtOH (3 mL). Solid hydroxylamine hydrochloride (0.103 g, 1.48 mmol) was added, and the reaction mixture was heated at 80°C for 4 h and cooled to room temperature. Water (30 mL) was added, and the mixture was extracted with ethyl acetate (3 × 30 mL). The combined organic portion was washed with brine (3 × 30 mL), dried with Na2SO4, and concentrated to dryness under reduced pressure to yield a crude product which was purified by flash column chromatography (silica gel; CH2Cl2) to afford 0.267 g of an oxime as a white solid which was subjected to further steps. Tosyl chloride (0.206 g. 1.08 mmol) was added at 0°C (ice-bath) to a stirred solution of oxime (0.267 g, 0.64 mmol), N,N-diisopropylethylamine (0.151 g, 1.17 mmol), and 4-dimethylaminopyridine (0.011 g, 0.09 mmol) in CH2Cl2 (5 mL). The ice bath was removed, and the reaction mixture was allowed to stir at room temperature overnight. Water (20 mL) was added, and the mixture was extracted with CH2Cl2 (3 × 30 mL). The combined organic portion was washed with brine (25 mL), dried with Na2SO4, and concentrated under reduced pressure to yield a crude product which was purified by flash column chromatography (silica gel, hexane-ethyl acetate, 50:50) to afford the corresponding tosylate (0.359 g) as a colorless oil. Ammonia solution (7M in methanol, 3 mL) was added at −78°C into a stirred solution of the tosylate (0.359 g, 0.63 mmol) in THF (4 mL) under argon. The reaction mixture was stirred at room temperature for 24 h and concentrated under reduced pressure. Dichloromethane (5 mL) was added, and the mixture was filtered, and the collected solid was washed with CH2Cl2. The filtrate was concentrated under reduced pressure to yield the corresponding diaziridine (0.250 g) as an orange oil. Triethylamine (99%, 0.25 mL) was added to a stirred solution of diaziridine (0.250 g, 0.60 mmol) in methanol (5 mL). Iodine powder was added in portions until the color of iodine disappeared. The reaction mixture was allowed to stir at room temperature for 1 h. An aqueous solution of sodium thiosulfate solution (10% in H2O) was added in a dropwise manner until the color of iodine was discharged. The solvent was evaporated under reduced pressure. Water (10 mL) was added, and the mixture was extracted with CH2Cl2 (3 × 15 mL). The combined organic portion was washed with brine (5 mL), dried with Na2SO4, and concentrated to dryness under reduced pressure to yield a crude product which was purified by flash column chromatography (silica gel, hexane-ethyl acetate, 96:4) to afford diazirine 7 (0.177 g, 52%) as a colorless oil.
1H NMR (CDCl3): δ 0.15 (s, 6H, CH3), 0.93 (s, 9H, CH3), 1.25 (d, J = 6.8 Hz, 12H, CH3), 3.31–3.41 (m, 2H, CH), 4.74 (s, 2H, CH2O), 6.95 (s, 2H, ArH). 13C NMR (CDCl3): δ 148.95, 135.85, 128.62, 123.65, 120.97, 57.12, 29.23, 25.88, 24.18, 18.30, −5.38. 19F NMR (CDCl3): δ −65.04.
3-(4-(((tert-Butyldimethylsilyl)oxy)methyl)-3,5-diisopropylphenyl)-3-(trifluoromethyl)-3H-diazirine (8).
Tetrabutylammonium fluoride solution (1M in THF, 0.73 mL) was added to a stirred solution of silyldiazirine 7 (0.177 g, 0.43 mmol) in THF (4 mL), and the reaction mixture was allowed to stir at room temperature for 1 h. The mixture was concentrated under reduced pressure and a crude product which was purified by flash column chromatography (silica gel, hexane-ethyl acetate, 94:6) to afford 0.093 g (73%) of desilylated alcohol 8 as a white solid. 1H NMR (CDCl3): δ 1.28 (d, J = 6.8 Hz, 12H, CH3), 3.35–3.45 (m, 2H, CH), 4.81 (d, J = 4.6 Hz, 2H, CH2OH), 6.99 (s, 2H, ArH). 13C NMR (CDCl3): δ 149.10, 135.60, 129.24, 123.58, 121.31, 120.86, 56.96, 29.37, 24.30. 19F NMR (CDCl3): δ −65.09.
2,6-Diisopropyl-4-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzaldehyde (9).
Dess-Martin periodinane (0.3 M in CH2Cl2, 0.5 mL) was added to a stirred solution of alcohol 11 (0.026 g, 0.09 mmol) in CH2Cl2 (1.5 mL), and the reaction mixture was allowed to stir at room temperature for 1 h and diluted with ethyl ether (12 mL). A solution of Na2S2O3 (0.5 M in saturated aqueous NaHCO3, 3 mL) was added. The mixture was stirred at room temperature until a clear biphasic solution was obtained. The organic portion was washed with saturated aqueous solution of NaHCO3 (5 mL), brine (5 mL), dried with Na2SO4, and concentrated to dryness under reduced pressure to yield 0.019 g (75%) of aldehyde 9 as a pale yellow oil. 1H NMR (CDCl3): δ 1.27 (d, J = 6.8 Hz, 12H, CH3), 3.44–3.51 (m, 2H, CH), 7.04 (s, 2H, ArH), 10.68 (s, 1H, CHO). 13C NMR (CDCl3): δ 195.14, 150.12, 134.11, 132.72, 121.46, 121.02, 120.90, 29.00, 23.90. 19F NMR (CDCl3): δ −64.87.
Tritiation procedure for alcohol 8.
A solution of NaBH4 (0.003 g, 0.079 mmol) in EtOH (1 mL) was added to aldehyde 9 (0.004 g, 0.013 mmol) under argon atmosphere, and the reaction mixture was allowed to stir at room temperature for 4 h, quenched by the careful addition of HCl (1M, 2.5 mL), and extracted with ethyl ether (6 mL). The organic portion was dried with Na2SO4, and concentrated under reduced pressure to yield a crude product which was purified by flash column chromatography (silica gel, hexane – ethyl acetate, 97:3 v/v) to afford alcohol 8 (0.003 g, 100%). Aldehyde 9 undergoes rapid addition of water to form a hydrate which is resistant to reduction by sodium borohydride. To ensure successful tritiation, aldehyde 9 was store under strictly anhydrous conditions. The analogous procedure was carried out by Vitrax Co. using NaB3H4 to provide tritiated alcohol with specific radioactivity of 15 Ci/mmol.
5.3. Anesthetic properties.
Studies with tadpoles were conducted according to animal protocols pre-approved by the Massachusetts General Hospital (MGH) Subcommittee on Research Animal Care (Protocols #2006N000124 & 2015N000012) with the approval of the MGH Institutional Animal Care and Use Committee. Early pre-limb bud stage Xenopus laevis tadpoles (2 – 2.5 cm in length) were obtained from Xenopus One (Ann Arbor, MI) and were housed in the MGH Center for Comparative Medicine Facilities until needed. Loss of Righting Reflexes (LoRR) assays were conducted as previously described [37]. Briefly, tadpoles were immersed in anesthetic solution until their response stabilized (30–60 minutes). LoRR was assessed by inverting a tadpole with a bent Pasteur pipette. Those that righted themselves within 5 s, were scored zero, the remainder were scored one. Finally, animals were allowed to recover in dechlorinated tap water overnight. Those not recovering were eliminated from the analysis. After completion, tadpoles were euthanized with 5% tricaine or a lethal concentration of pentobarbital. Data for individual animals were fitted to a logistic equation by nonlinear least squares using Igor Pro (Wavemetrics, OR).
5.4. Allosteric modulation of agonist binding to GABAA receptors
To 100 μg of membrane protein was added 500 μL of 2 nM (final concentration) [3H]muscimol and appropriate modulator stock solution and brought up to 2mL with assay buffer (10mM phosphate buffer, 200 mM KCl, and 1 mM EDTA; pH 7.4). This was sufficient for four filtrations. After incubation for 10 minutes, 500 μL aliquots were filtered on GF/B glass fiber filters (Whatman, Cat. #1821–025) that had been pretreated for ≥ 1 h in 0.5% w/v poly(ethyleneimine). Filters were then washed twice under vacuum with 5 mL of cold assay buffer, removed and dried under a lamp for 60–80 minutes. Next, they were equilibrated in Liquiscint (Atlanta, GA Cat. # LS-121) and counted overnight (Tri-Carb 1900, Liquid Scintillation analyzer, Perkin–Elmer/Packard, Waltham, MA). Each independent concentration-response curve had 9–10 points and the number of repetitions of each curve is given in Table 1.
Data was fit by nonlinear least squares (Igor, Wavemetrics, OR) to an equation of the form:
| Eqn. 1 |
where y is the measured quantity, [3H]muscimol or [3H]flunitrazepam binding in this instance, Max and Min are the maximum and minimum measured quantity, x is the variable concentration of agent applied, EC50 is the half effect concentration and nH is the Hill coefficient.
5.5. Electrophysiology of GABAA receptors.
Oocytes were harvested from Xenopus laevis frogs using a surgical procedure that was approved by the Institutional Animal Care and Use Committee (IACUC) of Massachusetts General Hospital (Protocol #2010N000002). Frogs were housed in a veterinarian supervised facility, and every effort was made to reduce the stress and minimize the number of animals used. GABAA receptor coding DNA sequences for wild type α1, β3 and γ2L as well as for mutant α1 S270I, β3 M227W, β3 N265M, γ2L I242W and γ2L S280W subunits were cloned into pCDNA3.1 plasmids. Capped mRNAs were synthesized from the DNA plasmids, as previously described [16, 26]. Oocytes were injected with 0.5–1 ng total mRNA mixtures (1:1:5 ratio for αβγ), and electrophysiological recordings were performed about 24 hr after injection. Two-electrode voltage-clamp electrophysiological studies on recombinant GABAA receptors were carried out at room temperature, as previously described [26]. Oocytes were transferred to a custom-made low volume chamber, perfused with ND96 solutions (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.6). The resistance of electrodes filled with 3 M KCl was < 2 MΩ. Oocytes were voltage clamped at −50 mV (OC-725C, Warner Instruments, Hamden, CT, USA), and whole-cell currents were filtered at 1 kHz, and digitized at 2 kHz (Digidata 1550B, Molecular Devices, Sunnyvale, CA, USA). Gravity-fed delivery of GABA or pTFD-di-iPr-BnOH solutions alone or in combination was controlled with computer-actuated valves (VC-8, ALA Scientific Instruments, Farmingdale, NY, USA) and a micro-manifold (VM-8, ALA Scientific Instruments, Farmingdale, NY, USA). Drug solutions were applied for 5–30 s, followed by washout in ND96 for up to 10 min before subsequent drug exposure.
5.6. Analysis of wild type electrophysiological data.
Whole-cell currents were analyzed offline using Clampfit 10.6 (Molecular Devices). Data are reported as mean ± SD unless otherwise stated. Normalized concentration-response data were fitted using a nonlinear logistic equation with a variable slope:
| Eqn. 2 |
in which EC50 is the GABA concentration that elicits 50% of maximal response and nH is the Hill coefficient (Figure 2C).
Anesthetic current enhancement (fold-enhancement) was calculated for individual cells by dividing the peak current evoked by co-application of the anesthetic with a low concentration of GABA (EC2–5) by the peak current evoked by the same low concentration of GABA alone (Figure 2C). One-way ANOVA with post hoc Tukey’s test was used to compare the fold of current enhancement among different modulators for a wild type or mutant receptor
5.7. Analysis of mutant receptor modulation data.
Drug enhancement of receptor activation by low GABA (EC02 to EC05) was tested in oocytes using three drug conditions (5 μM etomidate, 10 μM pTFD-di-iPr-BnOH, and 8 μM R-mTFD-MPAB) in wild-type α1β3γ2L receptors and five receptors with the following mutations: α1 S270I, β3 N265M, γ2 S280W, β3 M227W, and γ2L I242W. Enhancement ratios were calculated as in the paragraph above. For a given drug-receptor combination (n = 8 to 11 oocytes per condition) were combined to calculate mean ± SEM for display (Figure 3).
Recorded peak currents were corrected for baseline, and enhancement ratios for individual oocytes were calculated as (Mean IGABA+Drug / Mean IGABA).
Wild-type and mutant enhancement ratio results from individual oocytes were normalized to the wild-type mean enhancement ratio for the appropriate drug condition: etomidate, pTFD-di-iPr-BnOH, or R-mTFD-MPAB (IGABA+Drug / Mean IGABA). The resulting data set of normalized enhancement results was used to calculate the upper and lower 95% confidence intervals for the wild-type means for each drug (Figure 4). Normalized enhancement results for all three drugs and all six receptor types were summarized for display as mean ± SD and analyzed using two-way ANOVA, testing the significance (P < 0.05 threshold) of mutant (5 comparisons to wild-type using Dunnett’s pos–hoc test), drug, and mutant-drug factor interaction effects. Within data subsets for each drug, P-values were calculated using two-tailed Student’s t-tests comparing each mutant to wild-type, and significance was inferred at P < 0.01, based on the Bonferroni correction with 5 comparisons to each wild-type data set. All statistical calculations were performed in Graphpad Prism v7.05 (Graphpad software, San Diego, CA, USA). Drug-residue contact was inferred if a mutation significantly reduced that drug’s modulation relative to wild-type.
5.8. Photolabeling of α1β3γ2L and α1β3 GABAARs.
Aliquots of purified α1β3γ2L GABAAR in elution buffer were used for analytical scale photolabeling (50 – 70 μL per gel lane, ~2 – 4 pmol [3H]muscimol binding sites) to determine the interactions of the novel benzyl alcohol derivatives with the [3H]azietomidate and [3H]R–m-TFD-MPAB binding sites and to characterize the pharmacological specificity of [3H]TFD-di-iPr-BnOH photolabeling. Receptors were photolabeled as described [15] with [3H]azietomidate (2 μM, 2 Ci per aliquot) or [3H]R–mTFD-MPAB (2 μM, 3 μCi per aliquot) in the presence of 300 μM GABA and varying concentrations of the benzyl alcohols. After irradiation at 365 nm for 30 min, GABAAR subunits were resolved by SDS-PAGE [13], and gels were stained for protein using GelCode Blue Safe Protein Stain (ThermoFisher Scientific). The three stained bands enriched in GABAAR α (56 kDa) and β (59/61 kDa) subunits were excised separately, and 3H incorporation in each band was determined by liquid scintillation counting. The GABAAR γ2 subunit is distributed diffusely in this region of the gel and not identifiable as a distinct stained band [13]. For [3H]azietomidate and [3H]R–mTFD-MPAB, non-specific photolabeling was determined in the presence of 300 μM etomidate or 60 μM non-radioactive R–mTFD-MPAB, respectively. The same experimental protocol was used to photolabel GABAARs with [3H]pTFD-di-iPr-BnOH (2 – 3 μM, ~2 μCi per aliquot), with 3H photoincorporation determined by fluorography using Amplify (GE Healthcare) as well as by liquid scintillation counting. Stock solutions of drugs were prepared at 60 mM in ethanol, with the exception of GABA and bicuculline methochloride that were prepared at 60 mM in distilled water. During photolabeling, ethanol was present at 0.25% (v/v) in all samples.
For [3H[azietomidate and [3H] R–mTFD-MPAB, parameters for the concentration dependence of inhibition were determined from the 3H incorporation in the α (56 kDa) and β (59/61 kDa) subunit gel bands, respectively, that reflects photolabeling of α1 Met-236 and β3 Met-227 [13]. For [3H] pTFD-di-iPr-BnOH, inhibition parameters were determined for the β subunit gel bands, which contained >75% of inhibitable photolabeling (see Results ). The concentration dependence of inhibition was fit to Equation 3.
| Eqn. 3 |
where B(x) is the 3H cpm incorporated in a subunit at an inhibitor concentration of x; B0 is the 3H incorporation in the absence of inhibitor, IC50 is the total inhibitor concentration reducing specific 3H incorporation by 50%, and Bns is the non-specific 3H incorporation in the presence of 300 μM etomidate for [3H]azietomidate, 60 μM R–mTFD-MPAB for [3H] R–mTFD-MPAB, or 300 μM etomidate and 60 μM R–mTFD-MPAB for [3H]pTFD-di-iPr-BnOH. The data plotted in individual figures are the means (±SD) from N independent experiments. To combine data from multiple independent experiments, 3H incorporation at each inhibitor concentration was normalized to that in the absence of inhibitor (as %), and the full data set was fit using Sigma Plot (v11.0, Systat Software) either with IC50 as an adjustable parameter or, when noted, with IC50 and Bns as adjustable parameters.
Supplementary Material
Scheme 1. Synthesis of propofol analog 8.
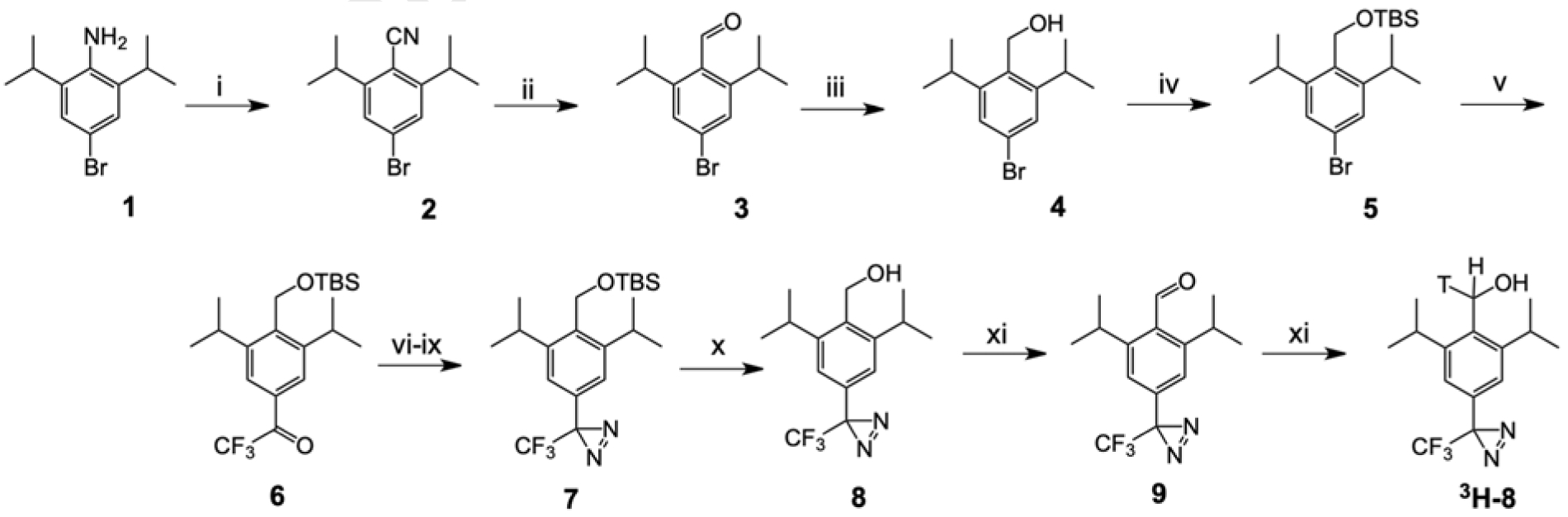
i: (a) HCl/NaNO2, 0°C; (b) CuCN-KCN, 70°C; ii: DIBAL-H; iii: NaBH4; iv: (a) t-butyldimethylsilyl chloride – 4-dimethylaminopyridine, 40°C; v: (a) n-BuLi, −78°C; (b) ethyl trifluoroacetate, −78°C; vi: hydroxylamine-HCl, 80°C; vii: tosyl chloride, 4-dimethylaminopyridine, diisopropylethylamine; viii: NH3/methanol; ix: I2, triethylamine; x: tetra-n-butylammonium fluoride,; xi: Dess-Martin periodinane; xii: NaB3H4.
Highlights.
pTFD-di-iPr-BnOH is a potent general anesthetic and enhances GABA’s actions
It photo-incorporates into intersubunit sites in the GABA(A)R’s transmembrane domain
Photo-protection shows it binds at either the γ+/β− or the α+/β− subunit interface
Similarly, it does not bind in the β+/α− subunit interfaces (etomidate sites)
Site–directed mutagenesis suggests the α+/γ− interface is another site
ACKNOWELEDGEMENTS
This research was supported by a grant from the National Institute for General Medicine to K.W.M. (GM 58448) and by the Department of Anesthesia, Critical Care & Pain Medicine, Massachusetts General Hospital. We thank Ervin Pejo for help with some tadpole experiments.
Abbreviations
- BnOH
benzyl alcohol
- di-iPr-BnOH
(2,6-diisopropylphenyl)methanol
- ECD
extracellular domain
- nH
Hill coefficient
- pTFD-BnOH
(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)methanol
- pTFD-di-iPr-BnOH
(2,6-diisopropyl-4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)methanol
- R–mTFD-MPAB
(5-allyl-1-methyl-5-(3-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)pyrimidine-2,4,6(1H,3H,5H)-trione)
- TBAF
tetra-n-butylammonium fluoride
- TMD
transmembrane domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF COMPETING INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Engin E, Benham RS, Rudolph U, An Emerging Circuit Pharmacology of GABAA Receptors, Trends Pharmacol Sci, 39 (2018) 710–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hernandez CC, Macdonald RL, A structural look at GABAA receptor mutations linked to epilepsy syndromes, Brain Research, 1714 (2019) 234–247. [DOI] [PubMed] [Google Scholar]
- [3].Rudolph U, Mohler H, GABAA receptor subtypes: Therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism, Annu Rev Pharmacol Toxicol, 54 (2014) 483–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Whissell PD, Lecker I, Wang DS, Yu J, Orser BA, Altered expression of deltaGABAA receptors in health and disease, Neuropharmacology, 88 (2015) 24–35. [DOI] [PubMed] [Google Scholar]
- [5].Sieghart W, Savic MM, International Union of Basic and Clinical Pharmacology. CVI: GABAA Receptor Subtype- and Function-selective Ligands: Key Issues in Translation to Humans, Pharmacoligal Reviews, 70 (2018) 836–878. [DOI] [PubMed] [Google Scholar]
- [6].Olsen RW, Allosteric ligands and their binding sites define gamma-aminobutyric acid (GABA) type A receptor subtypes, Advances in Pharmacology, 73 (2015) 167–202. [DOI] [PubMed] [Google Scholar]
- [7].Masiulis S, Desai R, Uchanski T, Serna Martin I, Laverty D, Karia D, Malinauskas T, Zivanov J, Pardon E, Kotecha A, Steyaert J, Miller KW, Aricescu AR, GABAA receptor signalling mechanisms revealed by structural pharmacology, Nature, 565 (2019) 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Laverty D, Desai R, Uchanski T, Masiulis S, Stec WJ, Malinauskas T, Zivanov J, Pardon E, Steyaert J, Miller KW, Aricescu AR, Cryo-EM structure of the human alpha1beta3gamma2 GABAA receptor in a lipid bilayer, Nature, 565 (2019) 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Olsen RW, Sieghart W, GABAA receptors: subtypes provide diversity of function and pharmacology, Neuropharmacology, 56 (2009) 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Olsen RW, Sieghart W, International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update, Pharmacological Reviews, 60 (2008) 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Crestani F, Assandri R, Tauber M, Martin JR, Rudolph U, Contribution of the alpha1-GABA(A) receptor subtype to the pharmacological actions of benzodiazepine site inverse agonists, Neuropharmacology, 43 (2002) 679–684. [DOI] [PubMed] [Google Scholar]
- [12].Forman SA, Miller KW, Mapping General Anesthetic Sites in Heteromeric gamma-Aminobutyric Acid Type A Receptors Reveals a Potential For Targeting Receptor Subtypes, Anesth Analg, 123 (2016) 1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW, Cohen JB, Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human alpha1beta3gamma2 gamma-aminobutyric acid type A (GABAA) receptor, The Journal of biological chemistry, 288 (2013) 19343–19357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jayakar SS, Zhou X, Chiara DC, Dostalova Z, Savechenkov PY, Bruzik KS, Dailey WP, Miller KW, Eckenhoff RG, Cohen JB, Multiple propofol-binding sites in a gamma-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog, J Biol Chem, 289 (2014) 27456–27468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jayakar SS, Zhou X, Chiara DC, Jarava-Barrera C, Savechenkov PY, Bruzik KS, Tortosa M, Miller KW, Cohen JB, Identifying Drugs that Bind Selectively to Intersubunit General Anesthetic Sites in the alpha1beta3gamma2 GABAAR Transmembrane Domain, Mol Pharmacol, 95 (2019) 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nourmahnad A, Stern AT, Hotta M, Stewart DS, Ziemba AM, Szabo A, Forman SA, Tryptophan and Cysteine Mutations in M1 Helices of alpha1beta3gamma2L gamma-Aminobutyric Acid Type A Receptors Indicate Distinct Intersubunit Sites for Four Intravenous Anesthetics and One Orphan Site, Anesthesiology, 125 (2016) 1144–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Iorio MT, Vogel FD, Koniuszewski F, Scholze P, Rehman S, Simeone X, Schnurch M, Mihovilovic MD, Ernst M, GABAA Receptor Ligands Often Interact with Binding Sites in the Transmembrane Domain and in the Extracellular Domain-Can the Promiscuity Code Be Cracked?, Int J Mol Sci, 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yip GM, Chen ZW, Edge CJ, Smith EH, Dickinson R, Hohenester E, Townsend RR, Fuchs K, Sieghart W, Evers AS, Franks NP, A propofol binding site on mammalian GABAA receptors identified by photolabeling, Nat Chem Biol, 9 (2013) 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hall MA, Xi J, Lor C, Dai S, Pearce R, Dailey WP, Eckenhoff RG, m-Azipropofol (AziPm) a photoactive analogue of the intravenous general anesthetic propofol, J Med Chem, 53 (2010) 5667–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Stewart DS, Savechenkov PY, Dostalova Z, Chiara DC, Ge R, Raines DE, Cohen JB, Forman SA, Bruzik KS, Miller KW, p-(4-Azipentyl)propofol: a potent photoreactive general anesthetic derivative of propofol, Journal of Medicinal Chemistry, 54 (2011) 8124–8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shanmugasundararaj S, Lehle S, Yamodo HI, Husain SS, Tseng C, Nguyen K, Addona GH, Miller KW, The location and nature of general anesthetic binding sites on the active conformation of firefly luciferase; a time resolved photolabeling study, PLoS One, 7 (2012) e29854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Savechenkov PY, Zhang X, Chiara DC, Stewart DS, Ge R, Zhou X, Raines DE, Cohen JB, Forman SA, Miller KW, Bruzik KS, Allyl m-trifluoromethyldiazirine mephobarbital: an unusually potent enantioselective and photoreactive barbiturate general anesthetic, Journal of Medicinal Chemistry, 55 (2012) 6554–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bali M, Akabas MH, Defining the propofol binding site location on the GABAA receptor, Molecular Pharmacology, 65 (2004) 68–76. [DOI] [PubMed] [Google Scholar]
- [24].Desai R, Ruesch D, Forman SA, Gamma-Amino Butyric Acid Type A Receptor Mutations at beta2N265 Alter Etomidate Efficacy While Preserving Basal and Agonist-dependent Activity, Anesthesiology, 111 (2009) 774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Siegwart R, Krahenbuhl K, Lambert S, Rudolph U, Mutational analysis of molecular requirements for the actions of general anaesthetics at the gamma-aminobutyric acidA receptor subtype, alpha1beta2gamma2, BMC pharmacology, 3 (2003) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Szabo A, Nourmahnad A, Halpin E, Forman SA, Monod-Wyman-Changeux Allosteric Shift Analysis in Mutant alpha1beta3gamma2L GABAA Receptors Indicates Selectivity and Crosstalk among Intersubunit Transmembrane Anesthetic Sites, Mol Pharmacol, 95 (2019) 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chiara DC, Dostalova Z, Jayakar SS, Zhou X, Miller KW, Cohen JB, Mapping general anesthetic binding site(s) in human alpha1beta3 gamma-aminobutyric acid type A receptors with [(3)H]TDBzl-etomidate, a photoreactive etomidate analogue, Biochemistry, 51 (2012) 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ, The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid, Proc Natl Acad Sci U S A, 94 (1997) 11031–11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Colton CA, Colton JS, Depression of glutamate-mediated synaptic transmission by benzyl alcohol, Can J Physiol Pharmacol, 55 (1977) 917–922. [DOI] [PubMed] [Google Scholar]
- [30].James R, Glen JB, Synthesis, biological evaluation, and preliminary structure-activity considerations of a series of alkylphenols as intravenous anesthetic agents, J Med Chem, 23 (1980) 1350–1357. [DOI] [PubMed] [Google Scholar]
- [31].Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL, General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility, Journal of Pharmacology & Experimental Therapeutics, 297 (2001) 338–351. [PubMed] [Google Scholar]
- [32].Husain SS, Ziebell MR, Ruesch D, Hong F, Arevalo E, Kosterlitz JA, Olsen RW, Forman SA, Cohen JB, Miller KW, 2-(3-Methyl-3H-diaziren-3-yl)ethyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate: a derivative of the stereoselective general anesthetic etomidate for photolabeling ligand-gated ion channels, J Med Chem, 46 (2003) 1257–1265. [DOI] [PubMed] [Google Scholar]
- [33].Dostalova Z, Liu A, Zhou X, Farmer SL, Krenzel ES, Arevalo E, Desai R, Feinberg-Zadek PL, Davies PA, Yamodo IH, Forman SA, Miller KW, High-level expression and purification of Cys-loop ligand-gated ion channels in a tetracycline-inducible stable mammalian cell line: GABAA and serotonin receptors, Protein Science, 19 (2010) 1728–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dostalova Z, Zhou X, Liu A, Zhang X, Zhang Y, Desai R, Forman SA, Miller KW, Human alpha1beta3gamma2L gamma-aminobutyric acid type A receptors: High-level production and purification in a functional state, Protein Science, 23 (2014) 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wu B, Jayakar SS, Zhou X, Titterton K, Chiara DC, Szabo AL, Savechenkov PY, Kent DE, Cohen JB, Forman SA, Miller KW, Bruzik KS, Inhibitable photolabeling by neurosteroid diazirine analog in the beta3-Subunit of human hetereopentameric type A GABA receptors, Eur J Med Chem, 162 (2019) 810–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Knowles DB, Lin C, MacKenzie PB, Tsai J, Walters R, Beers SA, Brown CS, Yeager WH, Barron E, Blue Phosphorescent Iridium Complexes and Light-Emitting Devices Using Them, in: WO 2008156879, 2008. [Google Scholar]
- [37].Kent DE, Savechenkov PY, Bruzik KS, Miller KW, Binding site location on GABAA receptors determines whether mixtures of intravenous general anaesthetics interact synergistically or additively in vivo, Br J Pharmacol, 176 (2019) 4760–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kita Y, Bennett LJ, Miller KW, The partial molar volumes of anesthetics in lipid bilayers, Biochim.Biophys.Acta, 647 (1981) 130–139. [DOI] [PubMed] [Google Scholar]
- [39].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis, J Comput Chem, 25 (2004) 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.