

Abstract
Oculopharyngeal muscular dystrophy (OPMD) is a rare autosomal dominant late-onset muscular dystrophy affecting approximately 1:100 000 individuals in Europe. OPMD is mainly characterized by progressive eyelid drooping (ptosis) and dysphagia although muscles of the limbs can also be affected late in life. This muscle disease is due to a trinucleotide repeat expansion in the polyA-binding protein nuclear-1 gene. Patients express a protein with an 11–18 alanine tract that is misfolded and prone to form intranuclear inclusions, which are the hallmark of the disease. Other features of OPMD include muscle fibrosis and atrophy in affected muscles. Currently, no pharmacological treatments are available, and OPMD patients can only be referred to surgeons for cricopharyngeal myotomy or corrective surgery of extraocular muscles to ease ptosis. We recently tested a two-AAV `silence’ and `replace’ vector-based gene therapy treatment in a mouse model of OPMD. We demonstrate here that this gene therapy approach can revert already established insoluble aggregates and partially rescues the muscle from atrophy, which are both crucially important since in most cases OPMD patients already have an established disease when diagnosed. This strategy also prevents the formation of muscle fibrosis and stabilizes the muscle strength to the level of healthy muscles. Furthermore, we show here that similar results can be obtained using a single AAV vector incorporating both the `silence’ and `replace’ cassettes. These results further support the application of a gene therapy approach as a novel treatment for OPMD in humans.
Introduction
Oculopharyngeal muscular dystrophy (OPMD) is a rare autosomal dominant late-onset muscular dystrophy affecting nearly 1:100 000 people in Europe. Individuals affected by OPMD have been described in over 30 countries and mainly in few specific populations like the Bukharan Jew population and the French Canadian populations where the incidence peaks to about 1/600–1/1000 affected individuals (1–4) due to original founder effects (5). The disease primarily involves some localized muscle groups like the external ocular region (which causes eyelid drooping) and muscles of the pharyngeal area (which severely affects swallowing), but at a later stage, muscles of the limbs are also affected. Currently, there is no cure for such disease, and the only possible treatments are based on surgical interventions to correct ptosis and dysphagia (6–9). OPMD is due to an expansion of GCG triplets in the exon 1 of the poly(A)-binding protein nuclear 1 (PABPN1) gene (10). The mutated protein (expanded PABPN1 or expPABPN1) has a stretch of 11–18 alanines instead of the normal 10 that promotes the protein aggregation and the generation of insoluble intranuclear aggregates (11) where proteins (like members of the ubiquitin/proteasome pathway, RNA binding proteins and the normal PABPN1) (12–17), pre-mRNA (i.e. muscle-specific Troponin T3 pre-mRNA) (18) and polyA RNAs (14,19,20) are sequestered. Aggregates seem to be instrumental for the disease as a number of pharmacological and molecular approaches specifically targeting them ameliorate the disease in several preclinical in vivo models of OPMD (e.g. cystamine, doxycycline, trehalose, guanabenz and intrabodies) (21–25). Trehalose is currently in phase I/II clinical trial. Other promising approaches include the autologous myoblast transplantation that showed a positive outcome when considerable amounts of cells were delivered (26). We recently demonstrated the efficacy of a gene therapy treatment based on a silence and replace approach in the A17 murine model of OPMD that expresses a bovine expPABPN1 with 17 alanine residues under the control of the human skeletal actin muscle-specific promoter and presents nuclear aggregates of insoluble PABPN1 in skeletal muscles. In heterozygous mice, most of the features of human OPMD patients are recapitulated including muscle atrophy and weakness (27). For that study, two Adeno Associated Virus (AAV) vectors, the first expressing a cassette including a triple shRNA under control of RNA polymerase III promoters (AAV-shRNA3X) and the second expressing human codon-optimized PABPN1 tagged with a Myc epitope under control of a skeletal and cardiac muscle-specific promoter Spc512 (AAV-optPABPN1), were co-delivered. This treatment significantly reduced the amount of myonuclei containing PABPN1-positive insoluble intranuclear inclusions (INIs), showed significant improvements in several histopathological features (muscle regeneration, fibrosis and muscle force) and nearly completely normalized the transcriptome of A17 muscles to the ones of wild-type FvB mice (28). While this approach proved to be successful, the study was not designed to assess whether such treatment simply prevents the generation of new aggregates and the worsening of the pathology or it clears the pre-existing aggregates and reverts pathological features of OPMD instead. Furthermore, while using two AAV vectors was feasible and acceptable in the animal model as a proof of concept, the development of a single vector including both silence and replace features would be necessary to progress the gene therapy approach towards clinical stage. Here a long-term experiment in young A17 mice and a short-term experiment in older A17 mice were performed to demonstrate that the gene therapy approach effectively reverts aggregate pathology while stabilizing both fibrosis deposition and muscle strength. Furthermore, we generated a new single `dual vector’ including both shRNA3X and optPABPN1 cassettes, and we showed that it is as effective as the two-vector system in downregulating expPABPN1 and clearing aggregates. Our work represents a step further towards the design of a single-vector strategy for a clinical application of gene therapy in OPMD.
Results
`Silence and replace’ gene therapy strategy reverts the amount of insoluble aggregates containing PABPN1 to the wild-type level
A17 mice at 11 weeks of age were injected locally in tibialis anterior (TA) muscles with the combination of 2 AAV vectors (2.5 × 1010 vp of AAV-shRNA3X and 1.3 × 1011 vp of AAV-optPABPN1). Age-matched A17 and FvB mice were injected with saline as controls, and further 11-week-old A17 and FvB mice were used to assess the functional and histopathological features of TA muscles at pre-injection stage. Fifteen weeks after AAV injection (i.e. when mice were 26-week-old), muscles were collected and immunoblot for PABPN1 and Myc tag was performed. A western blot analysis detected a profound inhibition (i.e. 96% knockdown) of expPABPN1 protein expression in muscles upon co-delivery of shRNA3X and optPABPN1 (Fig. 1A). OptPABPN1-Myc was successfully expressed in muscles treated with the combination of vectors expressing shRNA3X and optPABPN1 (Fig. 1B). To assess the effects of PABPN1 knockdown on the formation of insoluble aggregates, co-immunostaining for PABPN1 and laminin was performed. 4′,6-diamidino-2-phenylindole (DAPI) was used to counterstain the nuclei. TA muscles of 11-week-old A17 mice had ~ 30% of nuclei containing INI that increased to ~ 35% in 26-week-old mice [one-way analysis of variance (ANOVA) test, P < 0.01] while muscles of FvB mice contained no INI at the corresponding ages. The injection with the combination of 2 AAVs significantly decreased the aggregates to ~ 2.5% (Fig. 1C and E). A17 muscles have higher content in collagen proteins compared with age-matched muscles of FvB mice28. The treatment with the combination of 2 AAVs significantly decreased the area covered by collagen proteins in treated muscles compared to those of 26-week-old mice and stabilized the fibrosis to the level observed in 11-week-old mice (Fig. 1D and F). These data demonstrate that a gene therapy protocol effectively down-regulates PABPN1 in vivo and clears the pre-existing aggregates from the muscles and stabilizes the collagen proteins deposition to a pre-injection stage.
Figure 1.
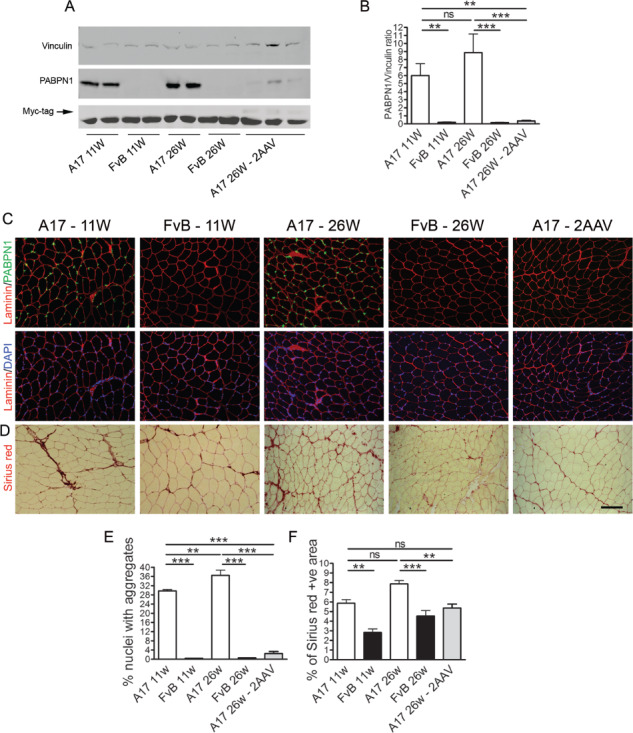
Injection of 2 AAV vectors in 11-week-old mice efficiently clears the amount of expPABPN1 aggregates, partially reverts muscle atrophy and prevents increase in fibrosis. A combination of the 2 single AAV vectors was injected in TA muscles of 11-week-old A17 or FvB mice. At 15 weeks after AAV injection, muscles were collected and analysed for PABPN1 expression and with Sirius red staining to detect the area covered by collagen I and III proteins. (A) Western blot for PABPN1 expression shows that the treatments with the combination of the two vectors efficiently knockdown the endogenous PABPN1. Western blot detecting Myc tag demonstrates that human codon-optimized PABPN1 is expressed in treated muscles. (B) Densitometric analysis of western blot for PABPN1 detection shows statistically significant reduction in protein expression after treating TA muscles with the vectors. PABPN1 levels were normalized to the relative vinculin expression. (C) Immunostaining for PABPN1 (green)/DAPI (for the nuclei, blue) to detect the percentage of nuclei containing INIs. (D) Sirius red staining to detect collagen I and III proteins in saline- and AAV-treated muscles. (E) The number of PABPN1-positive INI was significantly reduced in AAV-treated muscles compared to both 11-week-old and 26-week-old mice. Sections were pre-treated with 1 m KCl to discard all soluble PABPN1 from the tissue. (F) Morphometric evaluation of collagen I and III proteins by Sirius red staining in treated muscles showed a significant reduction in fibrosis in muscles treated with the combination of the two vectors. Scale bar, 200 μm. (One-way ANOVA test with Bonferroni post hoc test, n = 6, **P < 0.01, ***P < 0.001; ns, not significant).
Gene therapy treatment partially reverts muscle atrophy and stabilizes muscle strength
Muscles of both 11- and 26-week-old A17 mice are atrophic compared to muscles of FvB mice, and this is depicted by a difference in muscle weight (Fig. 2A). Treatment with 2 AAVs did not significantly increase the muscle weight compared to saline-injected muscles of 26-week-old mice although a clear positive trend was observed (unpaired t-test, P = 0.037; Fig. 2A). This small increase in muscle mass correlated with a significant difference in muscle strength. Indeed the injection of the 2 AAVs significantly increased the maximal tetanic force of AAV-treated TA compared to that one of saline-injected TA (one-way ANOVA test, P < 0.01) and essentially reverted the maximal force to the FvB level (one-way ANOVA test, P > 0.05; Fig. 2B and Supplementary Material, Fig. S1A and B). The normalization of the maximal force by the relative muscle weight provides a measure of the muscle strength by unit of muscle known as specific maximal force. This parameter was again improved after injection of the 2 AAVs and normalized to the pre-injection stage (Fig. 2C and Supplementary Material, Fig. 1C and D). These results show that the AAV treatment induced a partial reversion of muscle atrophy that is likely contributing to the prevention of the decrease in muscle force.
Figure 2.
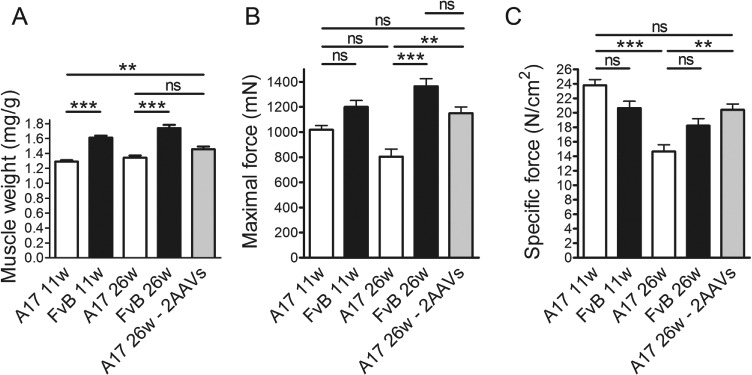
In situ TA muscle physiology of 11-week-old mice and mice after the treatment shows that the combination of 2 vectors prevented the decrease of the specific maximal force. Maximal force generated by treated TA muscles of treated mice 15 weeks after injection was measured by in situ muscle physiology. (A) TA muscle weight normalized by final bodyweight shows that the dual vector significantly increased the muscle mass compared to either age-matched saline-treated A17 muscles or muscles of 11-week-old mice. (B) Maximal force generated after 120 Hz stimulation. Co-injecting AAV-shRNA3X and AAV-optPABPN1 significantly increased the maximal force generated by treated TA muscles compared to the force observed in age-matched saline-injected muscles. (C) Normalization of maximal force by the cross-sectional area provides a measure of the muscle strength per unit of skeletal muscle called specific maximal force. Specific maximal force after 120 Hz stimulation of 26-week-old mice at the end of the treatment. The specific maximal force in 2 AAV vectors treated muscles was similar to the 1 of 11-week-old mice suggesting that the treatment prevented the decrease in muscle force. (One-way ANOVA test with Bonferroni post hoc test, n = 6, **P < 0.01, ***P < 0.001; ns, not significant).
Gene therapy treatment with a single vector clears the aggregates at advanced stage of disease
To assess if a short-term gene therapy treatment is sufficient to clear the insoluble aggregates, a short-term gene therapy study was performed in muscles when the aggregates peak in number (i.e. in 26-week-old mice). Indeed, in 11-week-old mice, the pathology and the number of aggregates are still evolving (28). The combined vectors were used at the same doses used for the previous experiment, and we analysed the muscles 8 weeks later (i.e. when mice were 34 weeks old). Furthermore, a single ‘dual’ vector presenting both the silence and replace cassettes was generated to compare its efficacy with the standard two-vector system (Fig. 3). A total of 2.5 × 1010 vp/TA were injected for the dual vector in order to have a similar expression of PABPN1 shRNAs. At the end of the treatment, saline-injected muscles presented nearly 35% of nuclei containing aggregates while in muscles of FvB no aggregates were detected. The injection with either the combination of 2 AAVs or with the dual vector significantly cleared the muscles from the presence of aggregates lowering those levels to ~ 2% and 4% of positive nuclei, respectively (P < 0.001 in both cases; Fig. 4A and B). To quantify the reduction in the expression of endogenous PABPN1 and verify that this does not affect optPABPN1 expression after dual-vector delivery, we quantified PABPN1 expression by immunoblot (Fig. 4C). PABPN1 is overexpressed in TA muscles of A17 mice compared to those of FvB wild-type mice, but the 2 gene therapy approaches reduced the amount of PABPN1 to ~ 5 and ~ 15%, respectively, compared to saline-injected A17 muscles (P < 0.001 in both cases; Fig. 4D). The different level of PABPN1 expression detected in these two treatments is likely due to the higher expression of optPABPN1 from the dual vector as confirmed by analysing the expression of Myc tag (Fig. 4E). As expected, the short time point we used did not allow to assess a change in histopathological markers of the disease. The use of either vector did not modify the percentage of centrally nucleated fibres (CNFs) and the area covered by collagen proteins (Supplementary Material, Fig. 2A–D) nor the muscle mass compared to saline-injected A17 muscles although a clear trend was observed with the dual vector (unpaired t-test, P = 0.03) (Supplementary Material, Fig. 2E). Accordingly, the maximal force generated by the muscle was not significantly modified after gene therapy treatment although, again, a positive trend was observed for the dual vector (unpaired t-test, P = 0.03) (Supplementary Material, Fig. 2F). Finally, neither treatment increased the specific maximal force (Supplementary Material, Fig. 2G). These data show that the gene therapy protocol only needs 8 weeks to clear the pre-existing aggregates. Importantly, a single vector presenting both the silence and replace cassettes has a similar potential in reducing endogenous PABPN1 and aggregates than the dual-vector system.
Figure 3.
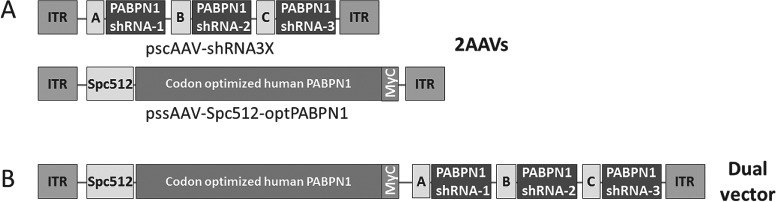
Single AAV-shRNA3X and AAV-optPABPN1 and dual AAV-shRNA3X-optPABPN1 vectors were used. (A) Three siRNAs were initially designed to bind sequences in human, mouse and bovine PABPN1. siRNAs were cloned downstream RNA polymerase III promoters and successively subcloned in self-complementary pAAV2 backbone to generate pscAAV-shRNA3X. Human PABPN1 fused with a Myc tag (optPABPN1) was sequence optimized and subcloned into single-stranded pAAV2 vector backbone downstream of Spc512 muscle-specific promoter to generate pssAAV-Spc512-optPABPN1. AAV vectors expressing shRNA3X or optPABPN1 were prepared following the standardized triple transfection protocol in HEK293T cells. (B) A single vector containing both the triple shRNA (under control of RNA polymerase III promoters) and the human codon-optimized PABPN1 (under control of Spc512 muscle-specific promoter) was prepared.
Figure 4.
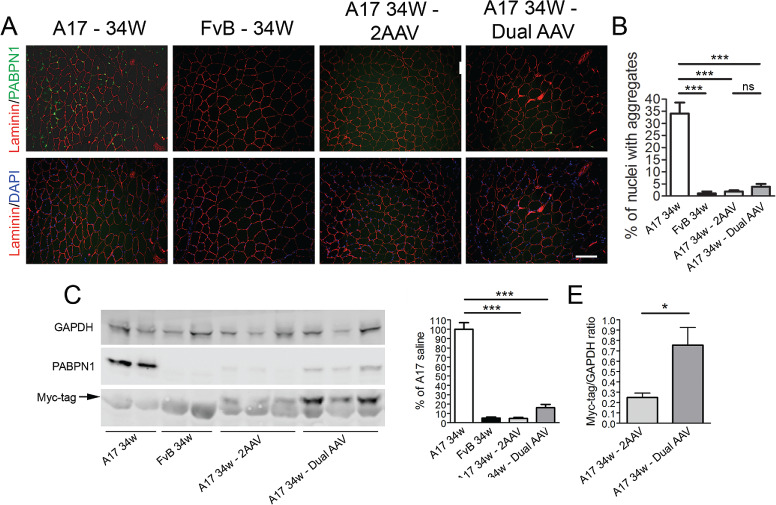
The co-injection of 2 separate AAVs or a dual bicistronic AAV in 26-week-old mice efficiently decreased the amount of insoluble aggregates containing PABPN1. A mix of the 2 single AAV vectors (2.5 × 1010 vp of scAAV-shRNA3X and 1.3 × 1011 of ssAAV-optPABPN1) or the single dual vector (2.5 × 1010 vp/TA) were injected in TA muscles of 26-week-old A17 mice and analysed at 34 weeks of age when muscles were collected and the change of histological parameters was assessed. Age-matched A17 and FvB mice were injected with saline as controls. (A) PABPN1/laminin co-immunostaining and the relative DAPI/laminin fields are shown. Sections were pre-treated with 1 m KCl to discard all soluble PABPN1 from the tissue. Scale bar, 200 μm. (B) The percentage of nuclei containing PABPN1-positive inclusions (INI) is significantly reduced in AAV-treated muscles compared to saline-injected muscles. (C) Western blot for PABPN1 expression shows that the treatments with the combination of the two vectors or the single dual vector efficiently knockdown the endogenous PABPN1. (D) Densitometric analysis of western blot for PABPN1 detection shows statistically significant reduction in protein expression compared to the level of A17 muscles when muscles are treated with the vectors. (E) Densitometric analysis of Myc tag suggests that expression of optPABPN1 is higher in samples treated with single dual vector compared with samples treated with the combination of vectors. PABPN1 and Myc levels were normalized to the relative GAPDH expression (one-way ANOVA test with Bonferroni post hoc test (for B and D) or Student’s t-test (for E), n = 6, *P < 0.05, ***P < 0.001; ns, not significant).
Discussion
Currently, there is no cure for OPMD, and the only possible treatment is based on corrective surgery to reduce ptosis and improve dysphagia (29). The use of trehalose to reduce the amount of INIs, which are considered crucial for the pathology, is currently in phase I/II clinical trial, and some preliminary results suggested beneficial effects of such treatment for the OPMD patients enrolled (30). However, this is an uncontrolled open-label trial and more objective evidences (i.e. an extension clinical trial including a placebo arm) will be necessary to really assess the beneficial effects of systemic trehalose administration. Local injection of autologous myoblasts was also studied in an open-label clinical trial. Cells were harvested from unaffected muscles of OPMD patients, and different amounts of cells were transplanted in the pharyngeal area together with a myotomy. All patients showed improvements in swallowing, and patients treated with the highest amounts of cells showed the highest benefits after the treatment (26). However, the cells are not genetically corrected, and the duration of beneficial effects for this treatment is difficult to predict. Recently, we developed a gene therapy strategy based on delivering two AAV vectors, one including triple shRNA cassette designed to knockdown endogenous PABPN1 expression and the second vector designed to express a codon-optimized human PABPN1 resistant to the RNAi degradation provided by the first AAV (27). Such treatment showed substantial reduction of aggregates and fibrosis and an improvement in muscle strength together with a near complete normalization of the muscle transcriptome to the one of normal muscles. Patients diagnosed with OPMD invariably present aggregates in muscle biopsies, and therefore, it is important to verify if the gene therapy treatment clears pre-existing aggregates or simply prevents their formation. Indeed, previous studies based on pharmacological approaches to reduce the presence of aggregates (e.g. cystamine, doxycycline, trehalose, guanabenz and intrabodies) showed that their reduction correlates to the amelioration of OPMD (21–25). To assess the effect of the gene therapy, we analysed both the amount of aggregates and the functional and histopathological features of TA muscles of 11-week-old mice (i.e. of mice age-matching the ones at the time of the injection), and we compared the results to those of muscles after the gene therapy treatment. The treatment cleared about 90–95% of aggregates present in the muscle in 8-week period suggesting that in a clinical application in human, the aggregates would potentially be quickly and almost completely reverted by this treatment. Importantly, in the previous study, we observed that PABPN1 knockdown without proper optPABPN1 replacement is detrimental for the muscle suggesting that a proper balance of PABPN1 silence and replace is needed. Here we show that the single dual vector is as effective as the two AAVs in knocking down expPABPN1 and in replacing it with optPABPN1 once administered intramuscularly. As expected, this time was too short to see any histopathological and functional improvements although a positive trend was observed for the dual vector on increasing muscle mass and muscle strength indicating that in a longer time point this vector could significantly improve these features.
Making such an effective single vector incorporating both the silence and replace features is necessary to improve the safety of the gene therapy approach, to facilitate the pathway with regulators and to reduce the cost of the vector production. Similar approaches with a single `silence and replace’ vector have been developed for a dominant form of retinitis pigmentosa (31) and for alpha-1 antitrypsin deficiency (32). Further modifications of this dual vector will be needed to obtain a construct suitable for gene therapy in human: for example, the removal of the Myc tag and the cloning of the shRNA cassette into a microRNA backbone where it could be expressed by a tissue-specific pol II polymerase promoter (33,34). This in turn would increase the safety and the specificity of the vector.
In conclusion, here we demonstrate that the gene therapy strategy reverts the aggregate in OPMD muscles while it prevents the deterioration of most of the other pathological features of this disease. Furthermore, we show that a single vector for gene therapy of OPMD providing the same beneficial effects of the dual-vector system can be generated. Once opportunely modified, this single vector may likely pave the way for the clinical translation of gene therapy for OPMD.
Materials and Methods
Preparation of single dual construct
The preparation of both the pssAAV-spc512-optPABPN1 and the pscAAV-shRNA3X has been already described (27). Briefly, three shRNA, designed to bind common sequences in human, mouse and bovine PABPN1, were cloned downstream of the U61, U69 and H1 RNA polymerase III promoters. The shRNA3X construct was cloned into a self-complementary pAAV2 backbone to generate pscAAV-shRNA3X. Human PABPN1 fused with a Myc tag (optPABPN1) and a Kozak sequence was sequence optimized by GeneArt (Waltham, US). optPABPN1 was subcloned into a single-stranded AAV2 vector backbone downstream of SPc512 muscle-specific promoter to generate pAAV-optPABPN1. The construct for the dual vector (pssAAV-spc512-optPABPN1-shRNA3X) was prepared by subcloning the triple shRNA cassette (shRNA3X) (1784 bp) in the pssAAV-PABOPT vector using restrictions enzymes NotI HF and MfeI.
Generation of adenoassociated viral vectors
scAAV2/8-shRNA3X, ssAAV2/9-optPABPN1 and the dual vector ssAAV2/9-optPABPN1-shRNA3X were prepared with the standardized double transfection protocol. Briefly, HEK293T cells were seeded in roller bottles and cultured in DMEM at 37°C and 5% CO2. Once 50% confluent, cells were transfected using PEI with the relative pAAV plasmid and either pAAV helper cap8 (pDP8) (for scAAV2/8-shRNA3X) or pAAV helper cap9 plasmids (pDP9rs) (for the other 2 vectors). Cells were cultured in roller bottles for three more days. Afterwards, cells were lysed, and recombinant pseudotyped AAV vector particles were purified by iodixanol (Sigma-Aldrich, Gillingham, UK) step-gradient ultracentrifugation. The copy number of vector genomes was quantified by quantitative polymerase chain reaction (PCR).
In vivo delivery of AAVs
A17 transgenic mice were previously described (24,28). A17 mice and wild type (WT) FvB controls were generated by crossing the heterozygous carrier A17 female mice with FvB male mice and genotyped by PCR for bovine PABPN1 4 weeks after birth. Animals were housed with food and water ad libitum in minimal disease facilities (Royal Holloway, University of London). In vivo experiments were conducted under statutory Home Office recommendation, regulatory, ethics and licencing procedures and the Animals (Scientific Procedures) Act 1986 (Project Licence 70/8271). Briefly, A17 or FvB mice were anesthetized with isoflurane and a combination of 2.5 × 1010 vp of scAAV8-shRNA3X and 1.3 × 1011 vp of ssAAV9-optPABPN1 or ssAAV2/9-optPABPN1-shRNA3X (2.5 × 1010 vp for the experiment in 26-week-old mice) were diluted in 50 μl saline and intramuscularly administered into TA muscles (n = 6 per group). Saline-injected TAs of both A17 mice and FvB mice were used as negative and healthy controls, respectively. At either 8- or 15-weeks post-injection, mice were anesthetized with Hypnorm/saline/Hypnovel 1:2:1.5, and in situ TA muscle physiology was performed. After analysis, mice were weighted and sacrificed, and TA muscles were excised from tendon to tendon, weighed, blocked in OCT and frozen in liquid nitrogen-cooled isopentane.
Immunofluorescence and histological staining
Muscle tissue sections of 10 μm thickness were cut using a Bright OTF 5000 cryostat (Bright Instruments, Huntingdon, UK), placed on coated glass slides (VWR, Lutterworth, UK) and stored at −80°C prior to use. For immunohistochemical staining, we used the following antibodies: anti-PABPN1 [rabbit monoclonal, diluted 1:100, Abcam ab75855, overnight (ON), 4°C], anti-laminin [rat monoclonal; diluted 1:800, Sigma-Aldrich, 1 h room temperature (RT)], anti-collagen VI (rabbit polyclonal, Abcam ab6588, 1:200, 1 h RT). Secondary antibodies were Alexa fluor (Life Biotechnologies, Paisley, UK) conjugated to 488 or 594 fluorochromes. PABPN1 INIs were detected by immunostaining using a previously published protocol (35). Briefly, tissue sections were fixed in paraformaldehyde 4%, and soluble proteins were removed by incubating slides with KCl buffer (1 m KCl, 30 mm HEPES, 65 mm PIPES, 10 mm EDTA, 2 mm MgCl2, pH 6.9) for 1 h at RT. Muscle sections were then blocked with 1% normal goat serum in 0.1 m phosphate-buffered saline (PBS), 0.1% Triton X-100 for 1 h and then incubated ON at 4°C with anti-PABPN1 and anti-laminin primary antibody diluted in the same buffer. After washings, sections were incubated with fluorophore-conjugated secondary antibodies. Slides were then incubated with DAPI (3 μg/ml, Sigma-Aldrich) for 5 min and finally mounted with Mowiol. For the analysis of INI content, five fields (20×) were randomly captured per each muscle, and INI were manually counted. All immunofluorescence images were captured and digitized using identical parameters of exposure, saturation and gamma-levels between specimens. Haematoxylin and eosin staining (obtained using a standard protocol) was used to calculate the percentage of CNFs. Sirius red staining was used to calculate the percentage of collagen proteins positive area in 2/3 randomly captured fields (×10 magnification). All analyses were performed using NIH ImageJ analysis software (NIH, Bethesda, Maryland, USA).
Western blot
Muscle lysates were prepared by homogenizing tissue in radioimmunoprecipitation assay buffer solution (NaCl, 0.15 m; HEPES, 0.05 m; NP-40, 1%; sodium deoxycholate, 0.5%; SDS, 0.10%; EDTA, 0.01 m) with protease inhibitor cocktail. Proteins were separated on 4–12% Bis-Tris gel (Life Technologies, Paisley, UK) and transferred onto a nitrocellulose membrane (Hybond ECL membrane; Amersham Bioscience, Little Chalfont, UK) for 1 h at 30 V constant. Membrane was blocked by incubation in 5% milk in 0.1 m PBS, 0.2% Tween-20 for 1 h at RT. Membrane was stained with anti-PABPN1 (Abcam rabbit monoclonal, ab75855, 1/10 000, ON), anti-GAPDH (Abcam mouse monoclonal, 1/2500, ON), anti-vinculin (Sigma-Aldrich, mouse monoclonal SAB4200080, 1/10 000, ON) and Myc tag (Abcam rabbit polyclonal, ab9106, 1/5000, ON). Membranes were then incubated with appropriate secondary antibodies conjugated fluorochromes (Li-cor Biosciences, Cambridge, UK; 1:10 000, 1 h RT) and the Odyssey system (Li-cor) was used to detect the signals from the membranes.
In situ muscle force measurement
Mice were anesthetized using intraperitoneal injection of a Hypnorm/Hypnovel solution, and contractile properties of TA muscles were analysed by in situ muscle electrophysiology using a protocol previously described (27). Briefly, the knee and foot were fixed with clamps, and the distal tendons of the muscles were attached to a 305B dual-mode servomotor transducer (Aurora Scientific, Aurora, Ontario, Canada) using a 4.0 braided surgical silk (Interfocus, Cambridge, UK). Mice were carefully monitored throughout the experiment to ensure that there was no reflex response to toe pinch. The sciatic nerves were proximally crushed and distally stimulated by a bipolar silver electrode using supramaximal square wave pulses of 0.02 ms duration (701A stimulator; Aurora Scientific). All data provided by the isometric transducer were recorded and analysed using a Lab-View-based DMC program (Dynamic muscle control and Data Acquisition; Aurora Scientific). All isometric measurements were made at an initial length L0 (length at which maximal tension was obtained during the tetanus). Muscle length was measured using digital calliper. Response to tetanic stimulation (pulse frequency: 10, 30, 40, 50, 80, 100, 120, 150 and 180 Hz) was recorded, and the maximal force was determined. After contractile measurements, muscles were collected, weighed to calculate the specific maximal force and frozen in isopentane cooled in liquid nitrogen and stored at −80°C. The specific force (N/cm2) was calculated by dividing Po by TA muscle cross-sectional area.
Statistical analysis
All data are presented as mean values ± standard error of the mean. Statistical analyses were performed using the one-way ANOVA with the Bonferroni post hoc analysis. A trend was considered present between two groups when a significant difference was measured using Student’s t-test. GraphPad Prism (version 4.0b; GraphPad Software, San Diego California, USA) was used for the analyses. A difference was considered to be significant at *P < 0.05, **P < 0.01 or ***P < 0.001.
Supplementary Material
Acknowledgements
P.K. held a salary from the Ministère de l'Education Nationale de la Recherche et de la Technologie (MENRT).
Conflict of interest
A patent named `Reagents for treatment of oculopharyngeal muscular dystrophy (OPMD) and use thereof’ has been filed by Benitec Biopharma. At the time of the study, D.A.S., V.S., S.H. and P.R. were employees of Benitec Biopharma. The other authors declare no conflicting financial interests.
Funding
AFM (Research Programs 15123 and 17110); Sorbonne Université; Institute of Myology; INSERM.
Author contributions
A.M., C. T. and G.D. conceived and designed the study. A.M., P.K., N.L.-N. and O.C. performed all experiments. A.M., P.K., N.L.-N., O.C. V.S.-U., S.H., P.R., D.S., C.T. and G.D. contributed to result interpretation and data analysis. A.M. wrote the manuscript with input from V.S.-U., C.T. and G.D. All authors read and approved the final manuscript.
References
- 1. Becher M.W., Morrison L., Davis L.E., Maki W.C., King M.K., Bicknell J.M., Reinert B.L., Bartolo C. and Bear D.G. (2001) Oculopharyngeal muscular dystrophy in Hispanic New Mexicans. JAMA, 286, 2437–2440. [DOI] [PubMed] [Google Scholar]
- 2. Chien Y.Y. (2012) Oculopharyngeal muscular dystrophy—an under-diagnosed disease in China? Report a China-born Chinese with PABPN1 mutation and epidemiology review of the literature. J. Formos. Med. Assoc., 111, 397–402. [DOI] [PubMed] [Google Scholar]
- 3. Cruz-Aguilar M., Guerrero-de Ferran C., Tovilla-Canales J.L., Nava-Castaneda A. and Zenteno J.C. (2017) Characterization of PABPN1 expansion mutations in a large cohort of Mexican patients with oculopharyngeal muscular dystrophy (OPMD). J. Investig. Med., 65, 705–708. [DOI] [PubMed] [Google Scholar]
- 4. Shan J., Chen B., Lin P., Li D., Luo Y., Ji K., Zheng J., Yuan Y. and Yan C. (2014) Oculopharyngeal muscular dystrophy: phenotypic and genotypic studies in a Chinese population. Neuromolecular Med., 16, 782–786. [DOI] [PubMed] [Google Scholar]
- 5. Abu-Baker A. and Rouleau G.A. (2007) Oculopharyngeal muscular dystrophy: recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies. Biochim. Biophys. Acta, 1772, 173–185. [DOI] [PubMed] [Google Scholar]
- 6. Coiffier L., Perie S., Laforet P., Eymard B. and St Guily J.L. (2006) Long-term results of cricopharyngeal myotomy in oculopharyngeal muscular dystrophy. Otolaryngol. Head Neck Surg., 135, 218–222. [DOI] [PubMed] [Google Scholar]
- 7. Gay I., Chisin R. and Elidan J. (1984) Myotomy of the cricopharyngeal muscle. A treatment for dysphagia and aspiration in neurological disorders. Rev. Laryngol. Otol. Rhinol. (Bord.), 105, 271–274. [PubMed] [Google Scholar]
- 8. Gomez-Torres A., Abrante Jimenez A., Rivas Infante E., Menoyo Bueno A., Tirado Zamora I. and Esteban Ortega F. (2012) Cricopharyngeal myotomy in the treatment of oculopharyngeal muscular dystrophy. Acta Otorrinolaringol. Esp., 63, 465–469. [DOI] [PubMed] [Google Scholar]
- 9. Perie S., Eymard B., Laccourreye L., Chaussade S., Fardeau M. and Lacau St Guily J. (1997) Dysphagia in oculopharyngeal muscular dystrophy: a series of 22 French cases. Neuromuscul. Disord., 7(Suppl. 1), S96–S99. [DOI] [PubMed] [Google Scholar]
- 10. Brais B., Bouchard J.P., Xie Y.G., Rochefort D.L., Chretien N., Tome F.M., Lafreniere R.G., Rommens J.M., Uyama E., Nohira O. et al. (1998) Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet., 18, 164–167. [DOI] [PubMed] [Google Scholar]
- 11. Tome F.M. and Fardeau M. (1980) Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol., 49, 85–87. [DOI] [PubMed] [Google Scholar]
- 12. Abu-Baker A., Messaed C., Laganiere J., Gaspar C., Brais B. and Rouleau G.A. (2003) Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum. Mol. Genet., 12, 2609–2623. [DOI] [PubMed] [Google Scholar]
- 13. Bao Y.P., Sarkar S., Uyama E. and Rubinsztein D.C. (2004) Congo red, doxycycline, and HSP70 overexpression reduce aggregate formation and cell death in cell models of oculopharyngeal muscular dystrophy. J. Med. Genet., 41, 47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calado A., Tome F.M., Brais B., Rouleau G.A., Kuhn U., Wahle E. and Carmo-Fonseca M. (2000) Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum. Mol. Genet., 9, 2321–2328. [DOI] [PubMed] [Google Scholar]
- 15. Corbeil-Girard L.P., Klein A.F., Sasseville A.M., Lavoie H., Dicaire M.J., Saint-Denis A., Page M., Duranceau A., Codere F., Bouchard J.P. et al. (2005) PABPN1 overexpression leads to upregulation of genes encoding nuclear proteins that are sequestered in oculopharyngeal muscular dystrophy nuclear inclusions. Neurobiol. Dis., 18, 551–567. [DOI] [PubMed] [Google Scholar]
- 16. Fan X., Messaed C., Dion P., Laganiere J., Brais B., Karpati G. and Rouleau G.A. (2003) HnRNP A1 and A/B interaction with PABPN1 in oculopharyngeal muscular dystrophy. Can. J. Neurol. Sci., 30, 244–251. [DOI] [PubMed] [Google Scholar]
- 17. Tavanez J.P., Bengoechea R., Berciano M.T., Lafarga M., Carmo-Fonseca M. and Enguita F.J. (2009) Hsp70 chaperones and type I PRMTs are sequestered at intranuclear inclusions caused by polyalanine expansions in PABPN1. PLoS One, 4, e6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klein P., Oloko M., Roth F., Montel V., Malerba A., Jarmin S., Gidaro T., Popplewell L., Perie S., Lacau St Guily J. et al. (2016) Nuclear poly(A)-binding protein aggregates misplace a pre-mRNA outside of SC35 speckle causing its abnormal splicing. Nucleic Acids Res., 44, 10929–10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bengoechea R., Tapia O., Casafont I., Berciano J., Lafarga M. and Berciano M.T. (2012) Nuclear speckles are involved in nuclear aggregation of PABPN1 and in the pathophysiology of oculopharyngeal muscular dystrophy. Neurobiol. Dis., 46, 118–129. [DOI] [PubMed] [Google Scholar]
- 20. Tavanez J.P., Calado P., Braga J., Lafarga M. and Carmo-Fonseca M. (2005) In vivo aggregation properties of the nuclear poly(A)-binding protein PABPN1. RNA, 11, 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chartier A., Raz V., Sterrenburg E., Verrips C.T., van der Maarel S.M. and Simonelig M. (2009) Prevention of oculopharyngeal muscular dystrophy by muscular expression of Llama single-chain intrabodies in vivo. Hum. Mol. Genet., 18, 1849–1859. [DOI] [PubMed] [Google Scholar]
- 22. Davies J.E., Rose C., Sarkar S. and Rubinsztein D.C. (2010) Cystamine suppresses polyalanine toxicity in a mouse model of oculopharyngeal muscular dystrophy. Sci. Transl. Med., 2, 34ra40. [DOI] [PubMed] [Google Scholar]
- 23. Davies J.E., Sarkar S. and Rubinsztein D.C. (2006) Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum. Mol. Genet., 15, 23–31. [DOI] [PubMed] [Google Scholar]
- 24. Davies J.E., Wang L., Garcia-Oroz L., Cook L.J., Vacher C., O'Donovan D.G. and Rubinsztein D.C. (2005) Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat. Med., 11, 672–677. [DOI] [PubMed] [Google Scholar]
- 25. Malerba A., Roth F., Harish P., Dhiab J., Lu-Nguyen N., Cappellari O., Jarmin S., Mahoudeau A., Ythier V., Laine J. et al. (2019) Pharmacological modulation of the ER stress response ameliorates oculopharyngeal muscular dystrophy. Hum. Mol. Genet., 28, 1694–1708. [DOI] [PubMed] [Google Scholar]
- 26. Perie S., Trollet C., Mouly V., Vanneaux V., Mamchaoui K., Bouazza B., Pierre Marolleau J., Laforet P., Chapon F., Eymard B. et al. (2013) Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: a phase I/IIa clinical study. Mol. Ther., 22, 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malerba A., Klein P., Bachtarzi H., Jarmin S.A., Cordova G., Ferry A., Strings V., Espinoza M.P., Mamchaoui K., Blumen S.C. et al. (2017) PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun., 8, 14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trollet C., Anvar S.Y., Venema A., Hargreaves I.P., Foster K., Vignaud A., Ferry A., Negroni E., Hourde C., Baraibar M.A. et al. (2010) Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum. Mol. Genet., 19, 2191–2207. [DOI] [PubMed] [Google Scholar]
- 29. Trollet C., Gidaro T., Klein P., Perie S., Butler-Browne G. and Lacau St Guily J. (1993 [updated 2014 Feb 20]) Oculopharyngeal Muscular Dystrophy. GeneReviews® [Internet].
- 30. Argov Z., Gliko-Kabir I., Brais B., Caraco Y. and Megiddo D. (2016) Intravenous trehalose improves dysphagia and muscle function in oculopharyngeal muscular dystrophy (OPMD): preliminary results of 24 weeks open label phase 2 trial. Neurology, 86, S28.004. [Google Scholar]
- 31. Cideciyan A.V., Sudharsan R., Dufour V.L., Massengill M.T., Iwabe S., Swider M., Lisi B., Sumaroka A., Marinho L.F., Appelbaum T. et al. (2018) Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. U. S. A., 115, E8547–E8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li C., Xiao P., Gray S.J., Weinberg M.S. and Samulski R.J. (2011) Combination therapy utilizing shRNA knockdown and an optimized resistant transgene for rescue of diseases caused by misfolded proteins. Proc. Natl. Acad. Sci. U.S.A., 108, 14258–14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McBride J.L., Boudreau R.L., Harper S.Q., Staber P.D., Monteys A.M., Martins I., Gilmore B.L., Burstein H., Peluso R.W., Polisky B. et al. (2008) Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. U.S.A., 105, 5868–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeng Y., Wagner E.J. and Cullen B.R. (2002) Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol. Cell, 9, 1327–1333. [DOI] [PubMed] [Google Scholar]
- 35. Gidaro T., Negroni E., Perie S., Mirabella M., Laine J., Lacau St Guily J., Butler-Browne G., Mouly V. and Trollet C. (2013) Atrophy, fibrosis, and increased PAX7-positive cells in pharyngeal muscles of oculopharyngeal muscular dystrophy patients. J. Neuropathol. Exp. Neurol., 72, 234–243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.