

Abstract
Angelman syndrome (AS) is a rare neurodevelopmental disorder characterized by speech impairment, intellectual disability, ataxia, and epilepsy. AS is caused by mutations in the maternal copy of UBE3A located on chromosome 15q11-13. UBE3A codes for E6AP (E6 Associated Protein), a prominent member of the HECT (Homologous to E6AP C-Terminus) E3 ubiquitin ligase family. E6AP catalyzes the posttranslational attachment of ubiquitin via its HECT domain onto various intracellular target proteins to regulate DNA repair and cell cycle progression. The HECT domain consists of an N-lobe, required for E2~ubiquitin recruitment, while the C-lobe contains the conserved catalytic cysteine required for ubiquitin transfer. Previous genetic studies of AS patients have identified point mutations in UBE3A that result in amino acid substitutions or premature termination during translation. An AS transversion mutation (codon change from ATA to AAA) within the region of the gene that codes for the catalytic HECT domain of E6AP has been annotated (I827K), but the molecular basis for this loss of function substitution remained elusive. Here, we demonstrate that the I827K substitution destabilizes the 3D fold causing protein aggregation of the C-terminal lobe of E6AP using a combination of spectropolarimetry and nuclear magnetic resonance (NMR) spectroscopy. Our fluorescent ubiquitin activity assays with E6AP-I827K show decreased ubiquitin thiolester formation and ubiquitin discharge. Using 3D models in combination with our biochemical and biophysical results, we rationalize why the I827K disrupts E6AP-dependent ubiquitylation. This work provides new insight into the E6AP mechanism and how its malfunction can be linked to the AS phenotype.
Introduction
Angelman syndrome (AS) is a neuro-genetic disorder that effects 1 in 15,000 people characterized by symptoms such as developmental delay, speech impairment, intellectual disability, walking and balance disorders, and epilepsy [1–3]. Individuals also demonstrate a unique behavioral pattern that typically includes a happy demeanor, easily provoked laughter, short attention span, sleep disturbance, and an affinity for water [2, 4]. AS is caused by the loss of gene function of the maternal copy of UBE3A on chromosome 15. UBE3A is paternally imprinted in neurons resulting in expression of the maternal allele alone, whereas other tissues retain normal biallelic expression patterns [2, 5–8]. The Human Gene Mutation Database (HGMD) currently lists 161 different genetic mutations of UBE3A [9]. Of the genetic mechanisms that have been described as the cause of AS, an estimated 70–80% of cases contain deletions in the maternal chromosome 15q11-q13 [2, 3]. Another 10–20% of affected individuals harbor mutations in their maternally inherited UBE3A gene, 3–5% of cases are due to two paternal copies of the chromosome, and lastly 3–5% of affected individuals have the paternal imprint of the maternal chromosome leaving no functioning copy of the UBE3A gene [2, 3].
UBE3A codes for the E3 ubiquitin ligase E6-Associated Protein (E6AP). This protein is a member of the Homologous to E6AP Carboxy-Terminus (HECT) family of E3 ubiquitin ligases and plays an important role in the ubiquitylation-signaling pathway. Ubiquitylation is a post-translational modification of targeted proteins that initiates processes such as protein degradation, intracellular trafficking, and other signaling events [10, 11]. Ubiquitin is transferred onto a substrate through a cascade of three enzymes (E1, E2, E3), with the combinatorial effect of the approximately 40 E2 and over 600 E3 enzymes ultimately determining substrate specificity [12–14]. The fate of the ubiquitylated substrate is determined by the ubiquitin linkage chain type by the E2/E3 combination [10, 11, 15]. For example, K29 and K48 linkages target a substrate protein for proteasomal degradation, while K63 linkages are involved in DNA repair mechanisms and intracellular targeting [10, 11, 16]. The dysregulation of any of these components involved in the ubiquitylation process leads to a myriad of different diseases including various cancers, developmental disorders, and neurodevelopmental disorders including AS [8, 16–18].
E6AP was first identified as an E3 ubiquitin ligase through its ability to target the tumor suppressor protein p53 for degradation in conjunction with the human papilloma virus protein E6 [19, 20]. E6AP is a 100 kDa protein that catalyzes the covalent attachment of ubiquitin to its various substrates through the use of its C-terminal HECT domain (residues 518–875). The HECT domain consists of an N-terminal lobe (N-lobe) and a C-terminal lobe (C-lobe) connected by a flexible three-residue hinge [21]. A broad cleft at the interface of the two lobes contains the catalytic cysteine required for ubiquitylation [21]. E6AP selectively builds K48-polyubiquitination chains, consistent with its ability to target substrates for proteasomal degradation, and this ubiquitin chain-linkage specificity is located in the C-lobe of the E6AP HECT domain [22]. E6AP has been shown to interact with numerous cellular proteins and can regulate a number of different homeostatic cellular processes [23]. Prime examples of E6AP-regulated processes include cell cycle control through centrosomal regulation [24], DNA repair through its interaction with UV excision repair protein RAD23 homolog A (HHR23A) [25], targeting tuberin (TSC2) for proteosomal degradation [26], signal transduction by binding to multiple Src family tyrosine kinases [27], breast cell proliferation through calmodulin/Ca2+mediated proteosomal degradation of the estrogen receptor (ER) [28], and coordinating the inflammatory response in conjunction with annexin A1 [29].
While the genetic link between mutations in UBE3A and AS is well established in the literature [1–4, 6, 7], the effect that each specific AS mutation has on the translated protein product have not been fully characterized or well understood. In this study, we show that the AS I827K substitution (also described as I804K based on an alternate open reading frame start codon [30–33]) partially disrupts the overall 3D fold of the HECT C-terminal lobe of E6AP leading to its aggregation and diminished E6AP-ubiquitylation activity in vitro. We unambiguously demonstrate that the AS I827K mutation in UBE3A is a loss of function mutation. This biophysical study clarifies how the I827K substitution in the C-terminal lobe domain of E6AP contributes to the Angelman syndrome phenotype.
Methods and materials
Cloning and site-directed mutagenesis
The original DNA construct for the human HECT C-lobe of E6AP (E6APC-lobe; Uniprot Q05086, residues 761–875) was codon optimized and synthesized by ATUM (Newark, CA, USA) [34–36]. The gene was cloned into an ampicillin-resistant T7-inducible plasmid with an N-terminal His6 affinity tag followed by a TEV protease cleavage site (ENLYFQ/GS). The resulting His6-TEV-E6APC-lobe vector was subsequently used as the template to insert the I827K mutation using the SPRINP protocol [37]. The catalytic cysteine was changed to an alanine (C843A) using the same protocol. Due to subsequent precipitation issues of the I827K substituted construct and to increase solubility, the E6APC-lobe-I827K open reading frame was subcloned into an expression vector with an N-terminal His6-SUMO fusion tag using compatible 5’ BamHI and 3’ XhoI restriction sites. The HECT domain of E6AP (residues 518–875) was PCR amplified from a plasmid coding for the full-length E6AP purchased from Addgene (Plasmid #8655; Watertown, MA, USA) [27] and subcloned into the His6-SUMO vector using compatible 5’ BamHI and 3’ XhoI sites. All plasmids (pHis6-TEV-E6APC-lobe, pHis6-SUMO-E6APC-lobe-I827K, pHis6-SUMO-E6APHECT and pHis6-SUMO-E6APHECT-I827K) were isolated using the Monarch Plasmid Miniprep kit (New England Biolabs, Ipswich, MA, USA), quantified by A280 using a Nanodrop OneC UV-Vis spectrophotometer (Thermo-Fisher, Waltham, MA, USA), and verified by DNA sequencing (Macrogen, Cambridge, MA, USA).
Protein expression and purification
The pHis6-TEV-E6APC-lobe, pHis6-SUMO-E6APC-lobe-I827K, pHis6-SUMO-E6APHECT and pHis6-SUMO-E6APHECT-I827K expression plasmids were transformed into E. coli BL21 (DE3) RIL+ competent cells and grown at 37°C in Luria-Bertani media supplemented with ampicillin 100 mg/L and chloramphenicol 34 mg/L. The E6AP proteins grown for heteronuclear NMR analysis were grown in minimal M9 media (2 x 1L) supplemented with 1 g/L of 15NH4Cl and 2 g/L of 13C-glucose as the sole nitrogen and carbon sources. When the cultures reached an OD600 of 0.6–0.8, protein expression was induced with the addition of 0.5 mM IPTG for 20 hours at 16°C. The cells were harvested by centrifugation 6000 x g for 10 minutes at 4°C using a Sorvall LYNX 4000 superspeed centrifuge with a Fiberlite F10-4x1000 LEX Carbon Fiber rotor (Thermo-Fisher) and resuspended in cold wash buffer (50 mM Na2HPO4 pH 8.0, 300 mM NaCl, 10 mM imidazole) supplemented with ProBlock Gold Bacterial Protease inhibitor cocktail (GoldBio, St. Louis, MO, USA). The cells were then lysed using an Avestin EmulsiFlex-C5 Homogenizer (Avestin, Ottawa, ON, Canada) and clarified by ultracentrifugation using an Optima L-80 XP ultracentrifuge with a Ti 70.1 rotor (Beckman-Coulter) for 40 minutes at 41,000 rpm at 4°C. The clarified supernatant containing the desired His6-tagged E6AP protein was then isolated using 5 mL of HisPur Ni-NTA resin (Thermo-Fisher) and eluted with elution buffer (50 mM Na2HPO4 pH 8.0, 300 mM NaCl, 250 mM imidazole). Fractions containing E6AP protein were pooled and incubated at 25°C for one hour in the presence of TEV or SUMO protease to cleave the N-terminal His6 or His6-SUMO tag, followed by overnight dialysis at 4°C against wash buffer to remove excess imidazole. To separate the cleaved His6- or His6-SUMO tag and His6-tagged protease from the desired E6AP protein, the cleaved sample was passed through the HisPur Ni-NTA resin column a second time and the flow-through containing the desired tag-free E6AP protein was collected. The E6AP protein was then concentrated using a 10 MWCO Amicon Ultra-15 Centrifugal Filter (Millipore) to about 1 mL, and run through a Superdex-75 Gel Filtration Column using an AKTA Pure 25L Fast Performance Liquid Chromatography (FPLC) system with gel filtration buffer (50 mM HEPES, 100 mM NaCl, 1 mM DTT, pH 7.5 at 4°C) at a flow rate of 1 mL/min. Due to inherent insolubility issues and after numerous attempts, the E6APHECT and E6APC-lobe-I827K proteins were unable to be passed through the size exclusion column.
Circular dichroism spectroscopy
Circular dichroism spectroscopy was performed on the E6APC-lobe and E6APC-lobe-I827K substituted protein using a JASCO J-815 CD spectropolarimeter. The proteins were prepared by buffer exchange into low salt (10 mM Na2HPO4 pH 7.4, 30 mM NaCl) using a 10 MWCO Slide-A-Lyzer Dialysis Cassette (Thermo-Fisher). The samples were diluted to 70 μM and loaded into a quartz cuvette with a 1 mm pathlength. Wavelength scans were averaged from six trials recorded from 260 to 195 nm at 10°C using 1 nm increments and an averaging time of 1 second. The same sample was used afterwards to obtain a melting curve of the mutant monitoring changes in α-helical content at 222 nm over a temperature range of 5–90°C, temperature slope 1°C/min, data pitch 0.3, response 4 seconds, bandwidth 1 nm, sensitivity standard 100 mdeg, and voltage of 600 mV.
Ubiquitin activity assays
Ubiquitination assays were conducted with 10 μM Alexa Fluor 647 N-terminally labeled ubiquitin, 10 μM E1 activating enzyme UBE1 (Uba1), 15 μM E2 conjugating enzyme UBE2L3 (UbcH7), and E3 ligase (35 μM E6APC-lobe or E6APHECT, as well as their variants), 2 μM DTT, 20 mM ATP, 40 mM MgCl2 in 50 mM HEPES pH 7.5, 100 mM NaCl. Each reaction was incubated at 37°C in a water bath for 30 minutes. To determine the presence of ubiquitin~thioester intermediates, appropriate samples were supplemented with 10 mM DTT. Reactions were terminated by adding gel loading dye and heating at 95°C in a dry bath for one minute. The samples were then loaded onto a Bis-Tris gel at pH 6.4 and run for 1 hour at 120 V. The gels were removed from the apparatus and immediately visualized on an iBright FL1000 imaging system (Thermo-Fisher) using the fluorescent gel imaging setting for Alexa Fluor 647.
Heteronuclear NMR spectroscopy
The 1H-15N heteronuclear single quantum correlation (HSQC) spectra [38] of 15N-labeled E6APC-lobe (3.5 mM) and E6APC-lobe I827K (188 μM) were collected at 25°C in a Varian Inova 600 MHz 4-channel solution-state NMR spectrometer equipped with a 5-mm PFG triple-resonance probe housed and maintained in the Carlson School of Chemistry and Biochemistry at Clark University. The samples were prepared to 600 μL in NMR buffer (20 mM Na2HPO4 pH 7.0, 100 mM NaCl, 2 mM TCEP, 1 mM EDTA), 10% D2O. The spectra were referenced to the methyl peaks of 2 mM 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) set at 0 ppm and 2 mM imidazole was added as an internal pH indicator [39]. The backbone resonances were sequentially assigned using standard 2D and 3D experiments from the Varian Biopack including 1H-15N-HSQC, HNCACB [40], CBCA(CO)NH [41], HNCA [42–44], HN(CO)CA [44, 45], HN(CA)CO [46], and HNCO [42–44]. Side chain assignments were determined using C(CO)NH [47], H(CCO)NH [47, 48], as well as both aliphatic and aromatic [49], HCCH-TOCSY [50, 51] and 1H-15N and 1H-13C-NOESY experiments [52–54]. All data were processed using NMRPipe and NMRDraw [55] and the spectra were analyzed using NMRViewJ [56, 57]. All chemical shift assignments and experiments were deposited into the Biological Magnetic Resonance Databank (http://www.bmrb.wisc.edu) under accession code 50084.
Results and discussion
The I827K Angelman syndrome substitution destabilizes E6APC-lobe
Wild-type E6APC-lobe and E6APC-lobe-I827K Angelman syndrome substituted protein showed similar secondary structural content by circular dichroism, with minima at 208 and 222 indicating both proteins are predominantly α-helical at 10°C (Fig 1A). To test the thermal stability of wild-type E6APC-lobe and E6APC-lobe-I827K, melting curves were obtained for each protein from 5–90°C. The melting curve showed a marked decrease in thermal stability of the E6APC-lobe-I827K (Tm of 47.7°C) when compared to E6APC-lobe wild-type (Tm of 57.7°C) demonstrating that the Angelman syndrome I827K substitution appears to partially destabilize the 3D fold of the protein (Fig 1B). The Angelman mutant appears to start unfolding around 40°C, close to the physiological temperature of 37°C, whereas the E6APC-lobe wild-type begins to unfold around 55°C. These results indicate that the AS substitution has a similar global fold at cooler temperatures similar to the wild-type, whereas the melting curve suggests that the I827K AS substitution is detrimental to E6APC-lobe stability.
Fig 1. Circular dichroism spectra for E6APC-lobe (wild-type—○, I827K substitution—◆).

A) Wavelength scans of the E6APC-lobe constructs show similar secondary structure content at 10°C. B) Melting curves show the E6APC-lobe-I827K substitution has a lower the Tm (47.7°C) than wild-type E6APC-lobe (57.7°C).
I827K Angelman syndrome substitution decreases E6AP ubiquitylation
The transfer of ubiquitin through the ubiquitylation pathway onto the catalytic cysteine of the E6AP was assessed using a fluorescent ubiquitin activity assay. Using this assay, we are able to observe the sequential transfer of fluorescent ubiquitin from the E1 activating enzyme UBE1 (aka Uba1), to the E2 conjugating enzyme UBE2L3 (aka UbcH7), and finally onto the HECT E3 ubiquitin ligase. This assay allowed for the direct comparison of wild-type to the I827K Angelman substituted E6APHECT and E6APC-lobe activities. As shown in Fig 2, the progression of the ubiquitylation cascade as ubiquitin is transferred sequentially onto the E1 activating enzyme UBE1 in an ATP-dependent manner, followed by a transfer onto the E2 conjugating enzyme UBE2L3. Furthermore, the labile nature of the thiolester UBE1~ubiquitin and UBE2L3~ubiquitin complexes was demonstrated by the addition of the reducing agent DTT (Fig 2). Polyubiquitin chains formation by wild-type E6APHECT decreased in the presence of DTT. It is noteworthy that the addition of DTT did not result in a complete reduction of the E3~ubiquitin thioester bond in the E6APHECT, as was expected, possibly due to non-specific autoubiquitylation of the E6APHECT during the reaction. This is consistent with previous reports of the isolated E6APHECT being able to autoubiquitylate itself in the absence of substrate [58, 59]. Interestingly, the I827K substitution on the other hand did not result in any polyubiquitin chain formation, demonstrating that the AS substitution decreases ubiquitylation activity. With regards to the E6APC-lobe, the AS I827K mutation also showed diminished activity compared to the wild-type E6APC-lobe. Polyubiquitin chains were not formed in either of the reactions, indicating that the N-lobe is required for the efficient catalysis of chain formation, consistent with activity assays for E6APC-lobe [59] and other isolated HECT E3 ubiquitin ligase C-lobes for HUWE1, Smurf2, and UBR5 [60, 61]. Interestingly, the AS I827K substituted E6APC-lobe was still able to be monoubiquitinated, albeit to a lesser extent that wild-type E6APC-lobe. This observation could possibly be due to the structural disruption of the I827K mutation, which prevents the proper presentation of the E6APC-lobe catalytic cysteine to E2~ubiquitin complex in the absence of the E6AP N-lobe. Mutating the catalytic cysteine to an alanine (C843A) resulted in the complete loss of ubiquitylation activity, as would be expected due to the requirement of E6AP C843 in thiolester bond formation with ubiquitin.
Fig 2. E6AP I827K is a loss of function substitution.
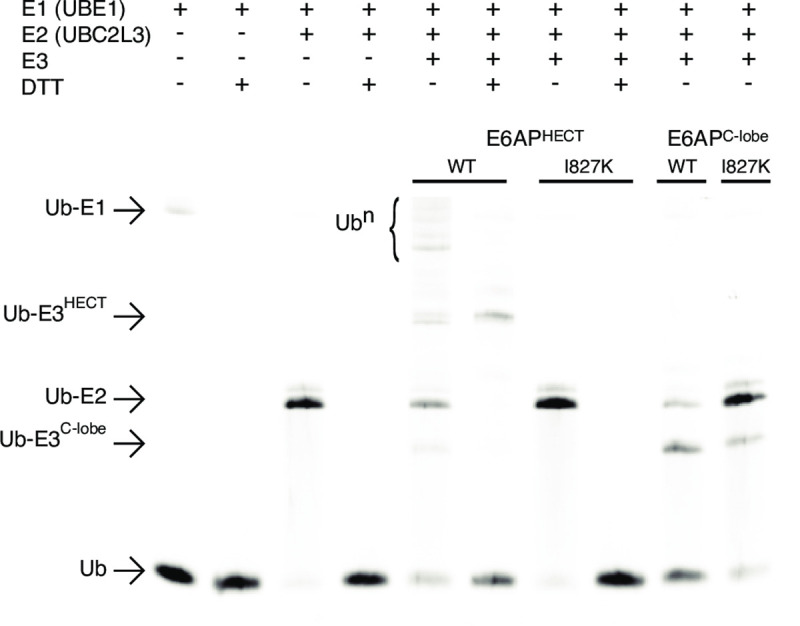
The ubiquitylation activity assay of wildtype or I827K substituted E6APHECT and E6APC-lobe consisted of a 30-minute incubation at 37°C of various combinations of 10 μM ubiquitin N-terminally tagged with Alexa Fluor 647, 10 μM E1 ubiquitin activating enzyme (UBE1), 10 μM E2 ubiquitin conjugating enzyme (UBE2L3), and 35 μM E3 ubiquitin ligase in a buffer containing 50 mM HEPES pH 7.5, 100 mM NaCl, 16 mM ATP, and 40 mM MgCl2. 10 mM DTT was added to alternate samples to reduce any thioester bonds present. The samples were run by electrophoresis a 15% Bis-Tris polyacrylamide gel and visualized on a ThermoFisher iBright FL1000 imaging system.
NMR structural analysis of E6APC-lobe
The 1H-15N heteronuclear single quantum correlation (HSQC) spectra for 15N-labeled wild-type E6APC-lobe and the E6APC-lobe-I827K substituted protein were used to analyze possible structural changes caused by the AS mutation. The wild type E6APC-lobe spectrum showed a well-folded protein, with well dispersed peaks, indicating that each amino acid was located in its own unique chemical environment (Fig 3A). In contrast, the spectrum of the AS I827K substituted E6APC-lobe showed many collapsed amide peaks that were mostly localized to the center of the spectrum (Fig 3B) indicating that the protein was partially unfolded. If the protein was completely unfolded protein would have peaks collapse to the center of the spectrum as solvent exposed peaks no longer experience shielding effects of neighboring amino acids. The disappearance of many peaks can be explained by the decreased signal/noise ratio due to the lower protein concentration as well as the line broadening effects caused by protein aggregation [62]. This is consistent with our inability to purify E6APC-lobe I827K protein after multiple attempts using gel filtration chromatography due to the protein aggregating and eluting in the void volume. Furthermore, the concentration used for I827K substituted protein in NMR was similar to the concentrations used in our activity assays and CD experiments, where we did not observe any protein precipitation issues.
Fig 3. The I827K substitution destabilizes the 3D fold of E6APC-lobe (residues 761–875).

(A) The assigned 1H-15N-HSQC spectrum of 3.5 mM human E6AP catalytic C-lobe (residues 761–875) using the one-letter amino acid code and residue number according to the human E6AP sequence. The spectrum was determined using standard 3D heteronuclear experiments, and side chain amides for asparagine and glutamine are connected with a horizontal line. (B) The E6APC-lobe I827K sample was collected under identical conditions as the wild-type except it was only 188 μM and increased transients collected due to the inherent instability of the protein. The decreased signal intensity and partial collapse amide peaks represent the aggregation of the protein caused to the I827K residue substitution. The NMR samples contained 20 mM Na2HPO4 pH 7.0, 100 mM NaCl, 2 mM TCEP, 1 mM EDTA, and 10% D2O/90% H2O, with imidazole used as an internal pH standard and DSS as the reference point. All data were collected at 25°C on a Varian Inova 600-MHz NMR spectrometer.
Based on the known E6AP crystal structure (PDB 1D5F) [21], we hypothesize that the AS I827K substitution leads to a structural disruption of the E6APC-lobe due to a hydrophobic residue being switched to a residue with a positively charged sidechain terminus that would disrupt the hydrophobic network in the core of the domain. The I827 sidechain is not solvent accessible and would not readily accommodate the polar terminus of the lysine sidechain. This is corroborated by the NOESY data that shows the I827 amino acid side chain methyl groups make numerous NOE contacts to several different surrounding hydrophobic side chains including T774, Y776, I787, F790, W791, L824, and M825 (Fig 4). The same rationale would apply to the newly published structure of the domain-swapped E6AP dimer (PDB 6TGK), as I827 is still found embedded in the monomeric hydrophobic core and is not involved in the dimerization interface [63]. This structural disruption likely induces an allosteric change that reduces the catalytic activity and leads to its subsequent aggregation. This is supported by the CD data showing the presence secondary structure at 37°C as well as our observation of aggregated protein eluted in the void volume using gel filtration chromatography. The loss of structural integrity due to the AS I827K substitution correlates with the loss of ubiquitylation activity for E6AP (Fig 2). We are confident that our NMR resonance assignments for the E6APC-lobe (residues 761–875) are correct and complete as a chemical shift index analysis, which predicts the secondary structure elements based upon chemical shift deviations of the backbone atoms (Ca, C’, Cβ, N, Ha, and NH) [64], are in good agreement with the known tertiary structure of C-terminal lobe of E6AP (Fig 5).
Fig 4. The I827 residue is an integral residue in the hydrophobic core of E6APC-lobe.

(A) The E6AP structure (PDB 1D5F) was rendered in Pymol highlighting the I827 residue (yellow with magenta methyls) surrounded by several aromatic and hydrophobic residues (cyan). (B) Representative 1H-1H-NOE strip plots for the I827 methyls showing numerous strong contacts with the surrounding hydrophobic atoms of core residues including T774, Y776, I787, F790, W791, L824, M825, and I827. The authenticity of the assignment was confirmed by reciprocal NOEs from the identified atoms.
Fig 5. Predicted secondary structural regions of E6APC-lobe.

The probability plot was made by inputting the experimentally determined resonance assignments for E6APC-lobe into the online webserver CSI 3.0 [64]. The propensity to form an α-helix or β-strand are denoted in red and blue, respectively. The position of the I827 residue is marked with a star.
This study provides a structural and biophysical rationale for the E6AP ubiquitin ligase activity loss due to the I827K substitution in AS. This is in good agreement with a previous report that showed the AS I827K substitution resulted in decreased ubiquitylation of the E6AP substrate HHR23A and instability in vivo [31]. Continued studies on the E6AP structure and function will help to understand how AS genetic mutations in UBE3A result in enzymatic insufficiency and may lead to potential treatments to alleviate the Angelman Syndrome phenotype.
Supporting information
(PDF)
Acknowledgments
The authors thank Dr. Guoxing Lin for maintaining the 600 MHz NMR spectrometer housed in the Carlson School of Chemistry and Biochemistry at Clark University.
Data Availability
All relevant data are within the manuscript. All chemical shift assignments and experiments were deposited into the Biological Magnetic Resonance Databank (http://www.bmrb.wisc.edu) under accession code 50084.
Funding Statement
This work was supported by the National Institutes of Health (R15GM126432 to D.E.S.; www.nigms.nih.gov) and start-up funds from Clark University (D.E.S.; www.clarku.edu). The funders did not play any role in the study design, data collection and analysis, decision to publish, or the preparation of the manuscript.
References
- 1.Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140(5):413–8. 10.1002/ajmg.a.31074 [DOI] [PubMed] [Google Scholar]
- 2.Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman Syndrome. Neurotherapeutics. 2015;12(3):641–50. 10.1007/s13311-015-0361-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15(1):70–3. 10.1038/ng0197-70 [DOI] [PubMed] [Google Scholar]
- 4.Bird LM. Angelman syndrome: review of clinical and molecular aspects. Appl Clin Genet. 2014;7:93–104. 10.2147/TACG.S57386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2(1):21–32. 10.1038/35047554 [DOI] [PubMed] [Google Scholar]
- 6.Buiting K, Williams C, Horsthemke B. Angelman syndrome—insights into a rare neurogenetic disorder. Nat Rev Neurol. 2016;12(10):584–93. 10.1038/nrneurol.2016.133 [DOI] [PubMed] [Google Scholar]
- 7.Khatri N, Man HY. The Autism and Angelman Syndrome Protein Ube3A/E6AP: The Gene, E3 Ligase Ubiquitination Targets and Neurobiological Functions. Front Mol Neurosci. 2019;12:109 10.3389/fnmol.2019.00109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopez SJ, Segal DJ, LaSalle JM. UBE3A: An E3 Ubiquitin Ligase With Genome-Wide Impact in Neurodevelopmental Disease. Front Mol Neurosci. 2018;11:476 10.3389/fnmol.2018.00476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21(6):577–81. 10.1002/humu.10212 [DOI] [PubMed] [Google Scholar]
- 10.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–29. 10.1146/annurev-biochem-060310-170328 [DOI] [PubMed] [Google Scholar]
- 11.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. 10.1146/annurev.biochem.67.1.425 [DOI] [PubMed] [Google Scholar]
- 12.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. 10.1146/annurev.biochem.78.101807.093809 [DOI] [PubMed] [Google Scholar]
- 13.Spratt DE, Walden H, Shaw GS. RBR E3 ubiquitin ligases: new structures, new insights, new questions. Biochem J. 2014;458(3):421–37. 10.1042/BJ20140006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lorenz S. Structural mechanisms of HECT-type ubiquitin ligases. Biol Chem. 2018;399(2):127–45. 10.1515/hsz-2017-0184 [DOI] [PubMed] [Google Scholar]
- 15.Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26(4):399–422. 10.1038/cr.2016.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rape M. Ubiquitylation at the crossroads of development and disease. Nat Rev Mol Cell Biol. 2018;19(1):59–70. 10.1038/nrm.2017.83 [DOI] [PubMed] [Google Scholar]
- 17.Wang D, Ma L, Wang B, Liu J, Wei W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017;36(4):683–702. 10.1007/s10555-017-9703-z [DOI] [PubMed] [Google Scholar]
- 18.Upadhyay A, Joshi V, Amanullah A, Mishra R, Arora N, Prasad A, et al. E3 Ubiquitin Ligases Neurobiological Mechanisms: Development to Degeneration. Front Mol Neurosci. 2017;10:151 10.3389/fnmol.2017.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129–36. 10.1016/0092-8674(90)90409-8 [DOI] [PubMed] [Google Scholar]
- 20.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10(13):4129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, et al. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286(5443):1321–6. 10.1126/science.286.5443.1321 [DOI] [PubMed] [Google Scholar]
- 22.Kim HC, Huibregtse JM. Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol Cell Biol. 2009;29(12):3307–18. 10.1128/MCB.00240-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Argiles-Castillo D, Kane EI, Zhou A, Spratt DE. HECT E3 ubiquitin ligases: emerging insights into their biological roles and disease relevance. J Cell Sci. 2020;133:jcs228072 10.1242/jcs.228072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singhmar P, Kumar A. Angelman syndrome protein UBE3A interacts with primary microcephaly protein ASPM, localizes to centrosomes and regulates chromosome segregation. PloS one. 2011;6(5):e20397 10.1371/journal.pone.0020397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar S, Talis AL, Howley PM. Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J Biol Chem. 1999;274(26):18785–92. 10.1074/jbc.274.26.18785 [DOI] [PubMed] [Google Scholar]
- 26.Zheng L, Ding H, Lu Z, Li Y, Pan Y, Ning T, et al. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells. 2008;13(3):285–94. 10.1111/j.1365-2443.2008.01162.x [DOI] [PubMed] [Google Scholar]
- 27.Oda H, Kumar S, Howley PM. Regulation of the Src family tyrosine kinase Blk through E6AP-mediated ubiquitination. Proc Natl Acad Sci U S A. 1999;96(17):9557–62. 10.1073/pnas.96.17.9557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L, Li Z, Howley PM, Sacks DB. E6AP and calmodulin reciprocally regulate estrogen receptor stability. J Biol Chem. 2006;281(4):1978–85. 10.1074/jbc.M508545200 [DOI] [PubMed] [Google Scholar]
- 29.Shimoji T, Murakami K, Sugiyama Y, Matsuda M, Inubushi S, Nasu J, et al. Identification of annexin A1 as a novel substrate for E6AP-mediated ubiquitylation. J Cell Biochem. 2009;106(6):1123–35. 10.1002/jcb.22096 [DOI] [PubMed] [Google Scholar]
- 30.Fang P, Lev-Lehman E, Tsai TF, Matsuura T, Benton CS, Sutcliffe JS, et al. The spectrum of mutations in UBE3A causing Angelman syndrome. Hum Mol Genet. 1999;8(1):129–35. 10.1093/hmg/8.1.129 [DOI] [PubMed] [Google Scholar]
- 31.Cooper EM, Hudson AW, Amos J, Wagstaff J, Howley PM. Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein. J Biol Chem. 2004;279(39):41208–17. 10.1074/jbc.M401302200 [DOI] [PubMed] [Google Scholar]
- 32.Nawaz Z, Lonard DM, Smith CL, Lev-Lehman E, Tsai SY, Tsai MJ, et al. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol Cell Biol. 1999;19(2):1182–9. 10.1128/mcb.19.2.1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russo S, Cogliati F, Viri M, Cavalleri F, Selicorni A, Turolla L, et al. Novel mutations of ubiquitin protein ligase 3A gene in Italian patients with Angelman syndrome. Hum Mutat. 2000;15(4):387. [DOI] [PubMed] [Google Scholar]
- 34.Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, et al. Design parameters to control synthetic gene expression in Escherichia coli. PLoS One. 2009;4(9):e7002 10.1371/journal.pone.0007002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welch M, Villalobos A, Gustafsson C, Minshull J. Designing genes for successful protein expression. Methods Enzymol. 2011;498:43–66. 10.1016/B978-0-12-385120-8.00003-6 [DOI] [PubMed] [Google Scholar]
- 36.Villalobos A, Ness JE, Gustafsson C, Minshull J, Govindarajan S. Gene Designer: a synthetic biology tool for constructing artificial DNA segments. BMC Bioinformatics. 2006;7:285 10.1186/1471-2105-7-285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edelheit O, Hanukoglu A, Hanukoglu I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 2009;9:61 10.1186/1472-6750-9-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kay LE, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc. 1992;114:10663–5 [Google Scholar]
- 39.Baryshnikova OK, Williams TC, Sykes BD. Internal pH indicators for biomolecular NMR. J Biomol NMR. 2008;41(1):5–7. 10.1007/s10858-008-9234-6 [DOI] [PubMed] [Google Scholar]
- 40.Grzesiek S, Bax A. An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J Magn Reson. 1992;99(1):201–7. [Google Scholar]
- 41.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc. 1992;114(16):6291–3. [Google Scholar]
- 42.Kay LE, Ikura M, Tschudin R, Bax A. Three-dimensional triple-resonance NMR Spectroscopy of isotopically enriched proteins. J Magn Reson. 1990;89(3):496–514. [DOI] [PubMed] [Google Scholar]
- 43.Muhandiram DR, Kay LE. Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J Magn Reson. 1994;103(3):203–16. [Google Scholar]
- 44.Grzesiek S, Bax A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J Magn Reson. 1992;96(2):432–40. [Google Scholar]
- 45.Bax A, Ikura M. An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the alpha-carbon of the preceding residue in uniformly 15N/13C enriched proteins. J Biomol NMR. 1991;1(1):99–104. 10.1007/BF01874573 [DOI] [PubMed] [Google Scholar]
- 46.Clubb RT, Thanabal V, Wagner G. A constant-time three-dimensional triple-resonance pulse scheme to correlate intraresidue 1HN, 15N, and 13C′ chemical shifts in 15N-13C-labelled proteins. J Magn Reson. 1992;97(1):213–7. [Google Scholar]
- 47.Grzesiek S, Anglister J, Bax A. Correlation of backbone amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J Magn Reson. 1993;101(1):114–9. [Google Scholar]
- 48.Montelione GT, Lyons BA, Emerson SD, Tashiro M. An efficient triple resonance experiment using carbon-13 isotropic mixing for determining sequence-specific resonance assignments of isotopically-enriched proteins. J Am Chem Soc. 1992;114(27):10974–5. [Google Scholar]
- 49.Vuister GW, Bax A. Resolution enhancement and spectral editing of uniformly 13C-enriched proteins by homonuclear broadband 13C decoupling. J Magn Reson. 1992;98:428–35. [Google Scholar]
- 50.Bax A, Clore GM, Gronenborn AM. 1H-1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C-enriched proteins. J Magn Reson. 1990;88(2):425–31. [Google Scholar]
- 51.Olejniczak ET, Xu RX, Fesik SW. A 4D HCCH-TOCSY experiment for assigning the side chain 1H and 13C resonances of proteins. J Biomol NMR. 1992;2(6):655–9. 10.1007/BF02192854 [DOI] [PubMed] [Google Scholar]
- 52.Marion D, Driscoll PC, Kay LE, Wingfield PT, Bax A, Gronenborn AM, et al. Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: application to interleukin 1 beta. Biochemistry. 1989;28(15):6150–6. 10.1021/bi00441a004 [DOI] [PubMed] [Google Scholar]
- 53.Marion D, Kay LE, Sparks SW, Torchia DA, Bax A. Three-dimensional heteronuclear NMR of nitrogen-15 labeled proteins. J Am Chem Soc. 1989;111(4):1515–7. [Google Scholar]
- 54.Zuiderweg ER, Fesik SW. Heteronuclear three-dimensional NMR spectroscopy of the inflammatory protein C5a. Biochemistry. 1989;28(6):2387–91. 10.1021/bi00432a008 [DOI] [PubMed] [Google Scholar]
- 55.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–93. 10.1007/BF00197809 [DOI] [PubMed] [Google Scholar]
- 56.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–52. 10.1385/1-59259-809-9:313 [DOI] [PubMed] [Google Scholar]
- 57.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4(5):603–14. 10.1007/BF00404272 [DOI] [PubMed] [Google Scholar]
- 58.Ronchi VP, Klein JM, Haas AL. E6AP/UBE3A ubiquitin ligase harbors two E2~ubiquitin binding sites. J Biol Chem. 2013;288(15):10349–60. 10.1074/jbc.M113.458059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ries LK, Sander B, Deol KK, Letzelter MA, Strieter ER, Lorenz S. Analysis of ubiquitin recognition by the HECT ligase E6AP provides insight into its linkage specificity. J Biol Chem. 2019;294(15):6113–29. 10.1074/jbc.RA118.007014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jackl M, Stollmaier C, Strohaker T, Hyz K, Maspero E, Polo S, et al. beta-Sheet Augmentation Is a Conserved Mechanism of Priming HECT E3 Ligases for Ubiquitin Ligation. J Mol Biol. 2018;430(18 Pt B):3218–33. [DOI] [PubMed] [Google Scholar]
- 61.Matta-Camacho E, Kozlov G, Menade M, Gehring K. Structure of the HECT C-lobe of the UBR5 E3 ubiquitin ligase. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68(Pt 10):1158–63. 10.1107/S1744309112036937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Redfield C. Using nuclear magnetic resonance spectroscopy to study molten globule states of proteins. Methods. 2004;34(1):121–32. 10.1016/j.ymeth.2004.03.009 [DOI] [PubMed] [Google Scholar]
- 63.Ries LK, Liess AKL, Feiler CG, Spratt DE, Lowe ED, Lorenz S. Crystal structure of the catalytic C-lobe of the HECT-type ubiquitin ligase E6 AP. Protein Sci. 2020. 10.1002/pro.3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hafsa NE, Arndt D, Wishart DS. CSI 3.0: a web server for identifying secondary and super-secondary structure in proteins using NMR chemical shifts. Nucleic Acids Res. 2015;43(W1):W370–7. 10.1093/nar/gkv494 [DOI] [PMC free article] [PubMed] [Google Scholar]