

PM specificity of Pr55Gag membrane binding is mediated through the interaction of PI(4,5)P2 with the matrix (MA) basic residues. It was shown that overexpression of a PI(4,5)P2-depleting enzyme strongly impaired PM localization of Pr55Gag. However, cellular factors that control PI(4,5)P2 production required for Pr55Gag-PM targeting have not yet been characterized. In this study, by individually inhibiting PIP5K1 isoforms, we elucidated a correlation between PI(4,5)P2 metabolism pathways mediated by PIP5K1 isoforms and the targeting of Pr55Gag to the PM of TZM-bl HeLa cells. Confocal microscopy analyses of cells depleted from PIP5K1α and PIP5K1γ show a rerouting of Pr55Gag to various intracellular compartments. Notably, Pr55Gag is degraded by the proteasome and/or by the lysosomes in PIP5K1α-depleted cells, while Pr55Gag is targeted to endosomal vesicles in PIP5K1γ-depleted cells. Thus, our results highlight, for the first time, the roles of PIP5K1α and PIP5K1γ as determinants of Pr55Gag targeting to the PM.
KEYWORDS: HIV-1, PIP5K1, Pr55Gag
ABSTRACT
HIV-1 assembly occurs principally at the plasma membrane (PM) of infected cells. Gag polyprotein precursors (Pr55Gag) are targeted to the PM, and their binding is mediated by the interaction of myristoylated matrix domain and a PM-specific phosphoinositide, the phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2]. The major synthesis pathway of PI(4,5)P2 involves the activity of phosphatidylinositol-4-phosphate 5-kinase family type 1 composed of three isoforms (PIP5K1α, PIP5K1β, and PIP5K1γ). To examine whether the activity of a specific PIP5K1 isoform determines proper Pr55Gag localization at the PM, we compared the cellular behavior of Pr55Gag in the context of PIP5K1 inhibition using siRNAs that individually targeted each of the three isoforms in TZM-bl HeLa cells. We found that downregulation of PIP5K1α and PIP5K1γ strongly impaired the targeting of Pr55Gag to the PM with a rerouting of the polyprotein within intracellular compartments. The efficiency of Pr55Gag release was thus impaired through the silencing of these two isoforms, while PIP5K1β is dispensable for Pr55Gag targeting to the PM. The PM mistargeting due to the silencing of PIP5K1α leads to Pr55Gag hydrolysis through lysosome and proteasome pathways, while the silencing of PIP5K1γ leads to Pr55Gag accumulation in late endosomes. Our findings demonstrated that, within the PIP5K1 family, only the PI(4,5)P2 pools produced by PIP5K1α and PIP5K1γ are involved in the Pr55Gag PM targeting process.
IMPORTANCE PM specificity of Pr55Gag membrane binding is mediated through the interaction of PI(4,5)P2 with the matrix (MA) basic residues. It was shown that overexpression of a PI(4,5)P2-depleting enzyme strongly impaired PM localization of Pr55Gag. However, cellular factors that control PI(4,5)P2 production required for Pr55Gag-PM targeting have not yet been characterized. In this study, by individually inhibiting PIP5K1 isoforms, we elucidated a correlation between PI(4,5)P2 metabolism pathways mediated by PIP5K1 isoforms and the targeting of Pr55Gag to the PM of TZM-bl HeLa cells. Confocal microscopy analyses of cells depleted from PIP5K1α and PIP5K1γ show a rerouting of Pr55Gag to various intracellular compartments. Notably, Pr55Gag is degraded by the proteasome and/or by the lysosomes in PIP5K1α-depleted cells, while Pr55Gag is targeted to endosomal vesicles in PIP5K1γ-depleted cells. Thus, our results highlight, for the first time, the roles of PIP5K1α and PIP5K1γ as determinants of Pr55Gag targeting to the PM.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) assembly, budding, and release involve a highly orchestrated series of interactions between the viral RNA, viral proteins, and host factors of the plasma membrane (PM). These late steps of the viral replication cycle are coordinated by the Gag precursor (Pr55Gag) and are initiated by the targeting of Pr55Gag complexes to the inner leaflet of the PM (1). Pr55Gag contains three major structural domains—the matrix (MA), capsid (CA), and nucleocapsid (NC) domains—together with two spacer peptides (sp1 and sp2) and an unstructured C-terminal p6 peptide (2). The N-terminal MA domain targets Pr55Gag to the PM via two specific motifs, an N-terminal myristoyl moiety and a highly basic region (HBR) spanning residues 17 to 31 (3–5). Pr55Gag targeting to the PM is dependent on the binding of MA HBR to phosphatidylinositol-(4,5)-biphosphate [PI(4,5)P2] (6–10). The electrostatic interactions of Pr55Gag with PI(4,5)P2 at the PM, together with the initiation of Pr55Gag multimerization, expose the N-terminal myristoyl moiety outside its hydrophobic pocket, thereby reinforcing the Pr55Gag-PM association driven by the HBR (10–13).
The principal mechanism of PI(4,5)P2 production is the phosphorylation of phosphatidylinositol-4-monophosphate [PI(4)P] at the D5 position of the inositol ring by phosphatidylinositol-4-phosphate 5-kinase family type 1 (PIP5K1). The PIP5K1s family includes three isoforms (PIP5K1α, PIP5K1β, and PIP5K1γ) with unique tissue distributions, present in different subcellular compartments and generating functionally different specialized pools of PI(4,5)P2 (14). PIP5K1α has been detected in different subcellular compartments, including the Golgi compartment, PM, and nuclei, in which it produces the PI(4,5)P2 involved in reorganization of the actin cytoskeleton, membrane ruffle formation, and pre-mRNA splicing (14–18). PIP5K1α activity, which is controlled by Arf6, is also crucial for trafficking through the Arf6 PM-endosomal recycling pathway (19). PIP5K1β is localized to vesicular structures in the perinuclear region in which the PI(4,5)P2 pool may be involved in actin dynamics and endocytosis (14, 18). Finally, the PIP5K1γ isoform has six splice variants, named PIP5K1γ-v1 to PIP5K1γ-v6 (18, 63, 64). PIP5K1γ proteins are preferentially localized to the PM, cell-cell junctions, or focal adhesions (18, 20–22). They are involved in various signaling processes, including the production of the PI(4,5)P2 pool used to generate inositol-3-phosphate (InsP3), which is involved in Ca2+ efflux and secretion (23).
PI(4,5)P2 was shown to be essential for the targeting of Pr55Gag to the PM in studies using phosphoinositide-5-phosphatase IV, which depletes the cell of PI(4,5)P2, causing the retargeting of Pr55Gag to late endosomes (6). More recently, the manipulation of PI(4,5)P2 levels was shown to prevent the correct targeting of Pr55Gag to the PM and to cause the loss of preassembled Pr55Gag lattice from the PM (12). However, both these approaches deplete all PI(4,5)P2 pools, regardless of the implication of PIP5Ks isoforms in PI(4,5)P2 metabolism. We investigated the role of PI(4,5)P2 in the targeting of Pr55Gag to the PM inner leaflet in more detail by focusing on the cellular activity of PIP5Ks type 1 to study the effect of PI(4,5)P2 metabolism on the intracellular distribution of Pr55Gag.
To address this question, we transfected TZM-bl HeLa cells with siRNAs targeting the various PIP5K1 isoforms (PIP5K1α, PIP5K1β, and PIP5K1γ). The decrease in total cellular PI(4,5)P2 through PIP5K1 silencing was assessed by ultra-high-pressure liquid chromatography coupled with high-resolution mass spectrometry (UHPLC-HRMS-MS). This method made a semiquantitative analysis of the distribution of PI(4,5)P2 molecular species possible. PIP5K1 silencing led to a decrease in total cellular PI(4,5)P2 levels, and this effect was particularly strong for PIP5K1α silencing. We then showed that the silencing of PIP5K1α or PIP5K1γ decreased the accumulation of Pr55Gag at the PM, while the silencing of PIP5K1β had no effect. The PM mistargeting due to the silencing of PIP5K1α led to Pr55Gag hydrolysis through lysosome and proteasome pathways, while the silencing of PIP5K1γ led to Pr55Gag accumulation in late endosomes. The disruption of these two pathways of PI(4,5)P2 metabolism was thus found to alter the extracellular release of Pr55Gag.
RESULTS
The silencing of PIP5K1 isoforms decreases PI(4,5)P2 production.
For identification of the PIP5K1 isoform involved in targeting Pr55Gag to the PM, we individually knocked down the expression of PIP5K1α, PIP5K1β, and PIP5K1γ. We therefore transfected TZM-bl HeLa cells with siRNA mixtures or with untargeted siRNA as a control, and the absolute number of copies of targeted mRNA was determined by reverse transcriptase quantitative PCR (RT-qPCR) 72 h posttransfection. The basal level of PIP5K1α mRNA was approximately seven times higher than those of PIP5K1β and PIP5K1γ (Fig. 1A, dark gray bars). PIP5K1 silencing decreased the amounts of the PIP5K1α, PIP5K1β, and PIP5K1γ mRNAs by 80% ± 12%, 70% ± 7%, and 76% ± 10%, respectively (Fig. 1A, dashed bars). Consistent with this decrease, the levels of the corresponding protein isoforms were about 70% to 80% lower than those in control cells (Fig. 1B). An additional faint band, migrating slightly slower than the PIP5K1, was observed in Western blotting using the PIP5K1α and PIP5K1β antibodies, which may correspond to an unspecific binding of the antibodies (24). Note that the antibodies used here for the Western blot analysis were found inefficient for immunofluorescence staining. The effect of each PIP5K1 isoform RNA knockdown on their PIPK5K1 RNA counterparts was also checked (Fig. 1C). Only the PIP5KIα mRNA was shown to be affected significantly, albeit weakly, in the context of siRNAs targeting PIP5K1β or PIP5K1γ (Fig. 1C, gray and green bars). However, PIP5K1β or γ mRNAs represent only ∼15% of the overall PIP5K1 RNAs (Fig. 1A). Therefore, we assumed that the effect of PIP5K1α RNA knockdown on Pr55Gag subcellular localization was mostly linked to this isoform enzymatic activity.
FIG 1.

The silencing of PIP5K1 isoforms decreases PI(4,5)P2 production. TZM-bl HeLa cells were transfected with either a nontargeting control siRNA or with siRNAs targeting PIP5K1α, PIP5K1β, or PIP5K1γ. Data were collected 72 h after siRNA transfection. (A) The silencing of each PIP5K1 isoform was assessed by RT-qPCR measurements of the corresponding mRNA expression on the basis of comparison with the nontargeting control siRNA. The absolute number of PIP5K1 mRNA copies was determined by comparison with a range of standards, with normalization against HPRT1 mRNA as an internal control. (B) The knockdown of PIP5K1 proteins was assessed by Western blotting, with HPRT1 proteins used to verify protein loading between lanes. (C) The impact of silencing a single PIP5K1 isoform on the two others was assessed by RT-qPCR on the basis of comparison with the nontargeting control siRNA, as described previously for panel A. (D) The effect of each PIP5K1 isoform silencing on total cellular PI(4,5)P2 level was determined by UHPLC-HRMS-MS. For panels A, C, and D, the data shown are expressed as the mean ± the standard error of the mean (SEM) from three independent experiments performed in triplicate. The P values shown were obtained with Mann-Whitney tests for panels A, C, and D. Significant results are indicated (****, P < 0.0001; ***, 0.0001 < P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; ns, not significant).
We next investigated the impact of PIP5K1 isoform silencing on PI(4,5)P2 production by extracting lipids from siRNA-transfected cells. The concentrations of PI(4,5)P2 and its various fatty acyl species were determined by UHPLC-HRMS-MS. The addition of a known amount of PI(4,5)P2 internal standard (C16:0/C16:0) before lipid extraction made it possible to estimate the amount of PI(4,5)P2 total pool. The knockdown of PIP5K1α decreased the amount of PI(4,5)P2 by 70% ± 9% (Fig. 1D, blue bar) relative to the amount in experiments with nontargeting control siRNA (Fig. 1D, black bar), whereas the silencing of PIP5K1β and PIP5K1γ decreased the amount of cellular PI(4,5)P2 by 45% ± 8% and 44% ± 12%, respectively (Fig. 1D, gray and green bars). Taking into account the weak proportion of PIP5K1β and PIP5K1γ RNAs (Fig. 1A), this important decrease of PI(4,5)P2 production was unexpected. Thus, the metabolic flux rate of each PIP5K1 isoform does not seem to be directly correlated with its concentration, as previously shown for other metabolic functions (25). Taken together, the larger amount of RNA found for the PIP5K1α isoform and the larger drop of PI(4,5)P2 observed after its silencing suggest that this isoform produces most of the total cellular PI(4,5)P2.
The knockdown of PIP5K1 isoforms has various effects on the broad range of PI(4,5)P2 species.
We then investigated the impact of the silencing of the various PIP5K1 isoforms on the distribution of various fatty acyl PI(4,5)P2 species. By UHPLC-HRMS-MS, the most sensitive tools to analyze this minor lipids family, we were able to identify 13 molecular species of PI(4,5)P2 (Fig. 2). The response ratio of these molecular species ranged from 0.4 for the minority species 20:4/16:0 to 11.5 for the majority species 20:2/18:0 (Fig. 2, black bars). The depletion of an individual isoform significantly altered the production of all the species identified, with the exception of one of the minority species (C18:0/C16:0) identified in this cell type. The knockdown of PIP5K1α resulted in a sharp decrease in PI(4,5)P2 levels, particularly for molecular species bearing the C18:0 acyl chain and an unsaturated fatty acyl chain (Fig. 2, blue bars). Indeed, the levels of the C20:1/C18:0, C18:1/C18:0, C22:2/C18:0, and C20:2/C18:0 species were reduced by 6.5-, 5-, 4.5-, and 3.5-fold, respectively (Fig. 2; compare the blue bars with the black bars). In contrast, the silencing of PIP5K1β (gray bars) and PIP5K1γ (green bars) had a lesser impact on the production of PI(4,5)P2 molecular species. For example, the knockdown of these two isoforms had no significant effect on the production of the three species bearing the C20:4 fatty acyl chain. In addition, PIP5K1β depletion did not alter the basal expression of C18:1/C18:1 species (Fig. 2). Moreover, the production of the three species bearing the C18:1 fatty acyl chain was more strongly affected by the knockdown of PIP5K1α than by the knockdown of PIP5K1β and PIP5K1γ. The distribution of PI(4,5)P2 molecular species was similar in HeLa cells and TZM-bl HeLa-modified cells (data not shown). Our findings show that PIP5K1α activity is involved in the metabolism of a large range of PI(4,5)P2 species, whereas the activities of PIP5K1β and PIP5K1γ have a more restricted involvement in this metabolism, consistent with the finding that PIP5K1α is responsible for most PI(4,5)P2 production (Fig. 1D). Thus, PIP5K1α is the principal producer of PI(4,5)P2 in the context of TZM-bl HeLa cells.
FIG 2.

The knockdown of PIP5K1 isoforms has various effects on the distribution of PI(4,5)P2 species. Lipids were extracted from the lysate of TZM-bl HeLa cells 3 days after transfection with either a nontargeting control siRNA or with siRNAs targeting PIP5K1α, PIP5K1β, or PIP5K1γ. The effect of PIP5K1 isoform silencing on the distribution of PI(4,5)P2 molecular species was assessed by UHPLC-HRMS-MS. The histogram shows the response ratio of each PI(4,5)P2 molecular species area peak from the lysate of cells transfected with the nontargeting control siRNA (black bars) or an siRNA targeting PIP5K1α, PIP5K1β, or PIP5K1γ (blue, gray, and green bars, respectively). The data shown are the means and SEM of three independent experiments conducted in triplicate. Mann-Whitney tests were performed to assess the significance of differences. Significant results are indicated (****, P < 0.0001; ***, 0.0001 < P < 0.001; **, 0.001 < P < 0.01; *, 0.01 < P < 0.05; ns, not significant). Stars indicate a significant decrease in the amount of a particular molecular species relative to the basal amount present in control cells. The stars above the black lines indicate significant differences in amounts of molecular species between siRNA-PIP5K1α-transfected cells and the cells transfected with other siRNAs.
The knockdown of PIP5K1α and PIP5K1γ impairs HIV-1 Pr55Gag targeting to the plasma membrane.
We investigated whether the silencing of each PIP5K1 isoform and the resulting PI(4,5)P2 depletion led to HIV-1 Pr55Gag mislocalization. We transfected TZM-bl HeLa cells with siRNAs and then with a mixture of plasmids encoding Pr55Gag and Pr55Gag incorporating enhanced green fluorescent protein (Pr55Gag-eGFP) for an additional 24 h. The cells were then stained with the dSQ12S probe to delineate the PM (26) and analyzed by confocal microscopy. As shown in the top row of Fig. 3A, Pr55Gag was partially distributed at the PM, while the dSQ12S probe was uniformly delineating this PM. As a consequence, a partial colocalization of Pr55Gag and the dSQ12S probe was observed with yellow staining (Fig. 3A, row a). Punctate staining corresponding to the accumulation of Pr55Gag was also observed in the cytoplasm. The overlap between the two fluorescent reporters was calculated by the Squassh method, with double labeling evaluated by calculating Pearson’s correlation coefficients (R) across the whole image (27). Consistent with the yellow staining of the merged image (Fig. 3A, row a, Merge), the Pr55Gag staining overlapped with staining for the dSQ12S probe with R ∼0.5 (Fig. 3B, black dots). In sharp contrast, the depletion of PIP5K1α and PIP5K1γ had a major impact on the distribution of Pr55Gag, with an accumulation of the viral protein in intracellular compartments (Fig. 3A, rows b and d). This accumulation resulted in a loss of colocalization of the two reporters with 0.05<R > 0.1 (Fig. 3B, blue and green dots). Conversely, the silencing of PIP5K1β had no effect on the cellular distribution of Pr55Gag, with cells displaying the same R coefficient as cells transfected with the nontargeting control siRNA (Fig. 3A, row c, and Fig. 3B, gray dots). Similar results for the silencing of the various PIP5K1 isoforms were obtained when Pr55Gag was expressed together with Pr55Gag translated from the pNL4.3 proviral construct, suggesting that the role of PIP5K1 in Pr55Gag targeting is identical when the full-length HIV-1NL4.3 provirus genome is transfected (Fig. 3C and D). Overall, these results provide the first evidence of a role for the metabolism of PI(4,5)P2 mediated by PIP5K1α and PIP5K1γ in the accumulation of Pr55Gag at the PM.
FIG 3.

The knockdown of PIP5K1α and PIP5K1γ impairs HIV-1 Pr55Gag localization to the plasma membrane. TZM-bl HeLa cells, transfected with either a nontargeting control siRNA or with siRNAs targeting each PIP5K1 isoform, were then transfected with plasmids expressing Pr55Gag/Pr55Gag-eGFP or with a replication-competent HIV provirus plasmid pNL4.3 and a Pr55Gag-eGFP-expressing plasmid. One day after transfection, the PM of the cells were stained with the dSQ12S probe, and living cells were imaged by confocal microscopy. (A) Effect of PIP5K1 isoform silencing on the distribution of Pr55Gag in transfected cells. Representative z-stack images (green for Pr55Gag, red for the dSQ12S probe, and yellow for the merged channels) for cells transfected with nontargeting control siRNA or siRNA targeting PIP5K1α, PIP5K1β, or PIP5K1γ (a, b, c, and d, respectively) are shown. (B) The colocalization of Pr55Gag with the PM probe (dSQ12S) was quantified with ImageJ software. Each dot represents the average of R for 10 z-stack images of a cell. Horizontal black bars stand for the mean value. (C) Effect of PIP5K1 isoforms silencing on the distribution of Pr55Gag produced by the replication-competent HIV-1NL4.3 in transfected cells. Representative z-stack images (green for Pr55Gag, red for the dSQ12S probe, and yellow for the merged channels) from cells transfected with nontargeting control siRNA or siRNAs targeting PIP5K1α, PIP5K1β, or PIP5K1γ (a, b, c, and d, respectively) are shown. (D) The colocalization of Pr55Gag produced by the replication-competent HIV-1NL4.3 with the PM probe dSQ12S was quantified with ImageJ software. Each dot represents the average of R for 10 z-stack images of a cell. Horizontal black bars stand for the mean value. Mann-Whitney tests were performed to assess the significance of differences. Significant results are indicated (****, P < 0.0001; ns, not significant).
The silencing of PIP5K1α and PIP5K1γ has different effects on the retargeting of HIV-1 Pr55Gag to endosomal pathways.
We then sought to identify the subcellular compartments to which Pr55Gag was redirected in cells depleted of PIP5K1α or PIP5K1γ. Among vesicular markers, Rab GTPases are widely used to distinguish early or late endosomes and lysosomes (28). Rab 5 and Rab 7A are proteins associated with early and late endosomal compartments, respectively (29). Thus, we transfected siRNA-treated cells with plasmids encoding Pr55Gag/Pr55Gag-eGFP together with a plasmid encoding mCherry-tagged versions of Rab5 (Fig. 4A) or Rab7A (Fig. 4C). In order to make a comprehensive figure, only images corresponding to the overlay of green and red channels (merged images) were presented in Fig. 4. In cells expressing mCherry-Rab5, we found that Pr55Gag did not accumulate in early endosomes, whether the cells were treated with the nontargeting control siRNA or depleted from each PIP5K1 isoform (Fig. 4A and B). Likewise, in cells expressing mCherry-Rab7A, we found that Pr55Gag did not accumulate in late endosomes in cells transfected with the nontargeting control siRNA or depleted of PIP5K1β (Fig. 4C, images a and c, and Fig. 4D, black and gray dots). Conversely, following the depletion of PIP5K1α or PIP5K1γ, much less Pr55Gag was detected at the PM (Fig. 3A and C, rows b and d), with accumulation of internal yellowish vesicles in the cytoplasm (Fig. 4C, images b and d). Quantification of the colocalization between Pr55Gag and Rab7A following the depletion of PIP5K1α and PIP5K1γ showed double staining for Pr55Gag and mCherry-Rab7A with R ∼0.25 (Fig. 4D, blue and green dots). These observations strongly suggest that knocking down the expression of PIP5K1α and PIP5K1γ led to the specific accumulation of the Pr55Gag precursor in late endosomes, whereas the knockdown of PIP5K1β had no impact on Pr55Gag distribution.
FIG 4.

The silencing of PIP5K1α and PIP5K1γ has different effects on the retargeting of HIV-1 Pr55Gag to endosomal pathways. (A) Effect of the silencing of PIP5K1 isoforms on the rerouting of Pr55Gag to early endosomes. TZM-bl HeLa cells, transfected with either a nontargeting control siRNA or with siRNAs targeting each PIP5K1 isoform, were then transfected with plasmids encoding Pr55Gag/Pr55Gag-eGFP and mCherry-Rab5. Z-stack confocal images were acquired from living cells as previously described. Representative yellow merged images (green for Pr55Gag and red for mCherry-Rab5) are shown for cells transfected with nontargeting control siRNA or siRNA targeting PIP5K1α, PIP5K1β, or PIP5K1γ (a, b, c, and d, respectively). (B) The colocalization of Pr55Gag-eGFP with mCherry-Rab5 was quantified with ImageJ software. Each dot represents the average of R for 10 z-stack images of a representative cell. Horizontal black bars stand for the mean value. (C) Effect of the silencing of PIP5K1 isoforms on the rerouting of Pr55Gag to late endosomes. TZM-bl HeLa cells, transfected with either a nontargeting control siRNA or with siRNAs targeting each PIP5K1 isoform, were then transfected with plasmids encoding Pr55Gag/Pr55Gag-eGFP and mCherry-Rab7A. Z-stack confocal images were acquired from living cells as previously described. Representative yellow merged images (green for Pr55Gag and red for mCherry-Rab7A) are shown for cells transfected with nontargeting control siRNA or siRNA targeting PIP5K1α, PIP5K1β, or PIP5K1γ (a, b, c, and d, respectively). (D) The colocalization of Pr55Gag-eGFP with mCherry-Rab7A was quantified with ImageJ software. Each dot represents the average of R for 10 z-stack images of a representative cell. Horizontal black bars stand for the mean value. (E) Effect of PIP5K1 isoform silencing on the relocalization of Pr55Gag to acidic vesicles. TZM-bl HeLa cells, transfected with either a nontargeting control siRNA or with siRNAs targeting each PIP5K1 isoform, were cotransfected with Pr55Gag- and Pr55Gag-mCherry-expressing plasmids. Lysosomes were stained in green with the LysoTracker probe 24 h after DNA transfection. Acquisitions of z-stack confocal images were performed as previously described, and the colors of Pr55Gag-mCherry and the green LysoTracker probe were inversed to prevent confusion. Representative yellow merged images (green for Pr55Gag and red for the LysoTracker probe) are shown for cells transfected with nontargeting control siRNA or siRNA targeting PIP5K1α, PIP5K1β, or PIP5K1γ (a, b, c, and d, respectively). (F) The colocalization of Pr55Gag-mCherry with the green LysoTracker probe was quantified with ImageJ software. Each dot represents the average of R for 10 z-stack images of a representative cell. Horizontal black bars stand for the mean value. Mann-Whitney tests were performed to assess the significance of differences. Significant results are indicated (****, P < 0.0001; ***, 0.0001 < P < 0.001; ns, not significant).
We then investigated the endosomal pathways followed by the late endosomes containing Pr55Gag, using the LysoTracker probe, which stains lysosome vesicles. In cells treated with the nontargeting control siRNA or depleted of the PIP5K1β protein, a weak, if any, colocalization between Pr55Gag and the LysoTracker was observed (Fig. 4E, images a and c), giving rise to R < 0.1 (Fig. 4F, black and gray dots). Intriguingly, the same absence of colocalization between Pr55Gag and LysoTracker staining was observed in cells depleted of PIP5K1γ (Fig. 4E, image d, and Fig. 4F, green dots). In contrast, the depletion of PIP5K1α induced double staining for Pr55Gag and the LysoTracker probe (R ∼0.29) (Fig. 4F, blue dots), as shown by the yellow staining in PIP5K1α-depleted cells (Fig. 4E, image b). These data indicate that Pr55Gag follows the lysosomal pathway and that late endosomes containing Pr55Gag fuse with the acidic vesicles only after depletion of the PIP5K1α isoform but not after depletion of the PIP5K1γ isoform.
Effects of PIP5K1α and PIP5K1γ silencing on Pr55Gag release.
We investigated the role of each PIP5K1 isoform on Pr55Gag release by quantifying the level of Pr55Gag in cell lysate and supernatant of PIP5K1-depleted cells (Fig. 5). PIP5K1α-depleted cells had much lower levels of Pr55Gag in the cytoplasm than control cells (Fig. 5A, red rectangle in upper panel), whereas Pr55Gag levels were similar to those in control cells for both PIP5K1β- and PIP5K1γ-depleted cells (Fig. 5A, top panel). The levels of Pr55Gag in the supernatant were lower than those recorded for control cells by a factor of 3 for PIP5K1α-depleted cells and 1.5 for PIP5K1γ-depleted cells, whereas levels of Pr55Gag in the supernatant were unaffected in PIP5K1β-depleted cells (Fig. 5B, white bars). Thus, the treatment of cells by siRNA against PIP5K1α decreased the amount of cytoplasmic Pr55Gag and its release, while the treatment of cells by siRNA against PIP5K1γ decreased only the release of Pr55Gag. Then, the efficiency of the Pr55Gag release was calculated by dividing the amount of Pr55Gag expression in the supernatant by the total amount of Pr55Gag expressed in both the supernatant and the cell lysate. In PIP5K1α- and PIP5K1β-depleted cells, Pr55Gag release efficiency was similar to that recorded for nontargeting control siRNA-treated cells (Fig. 5C, second and third white bars). In contrast, PIP5K1γ depletion decreased Pr55Gag release efficiency by one-third (Fig. 5C, fourth white bar).
FIG 5.
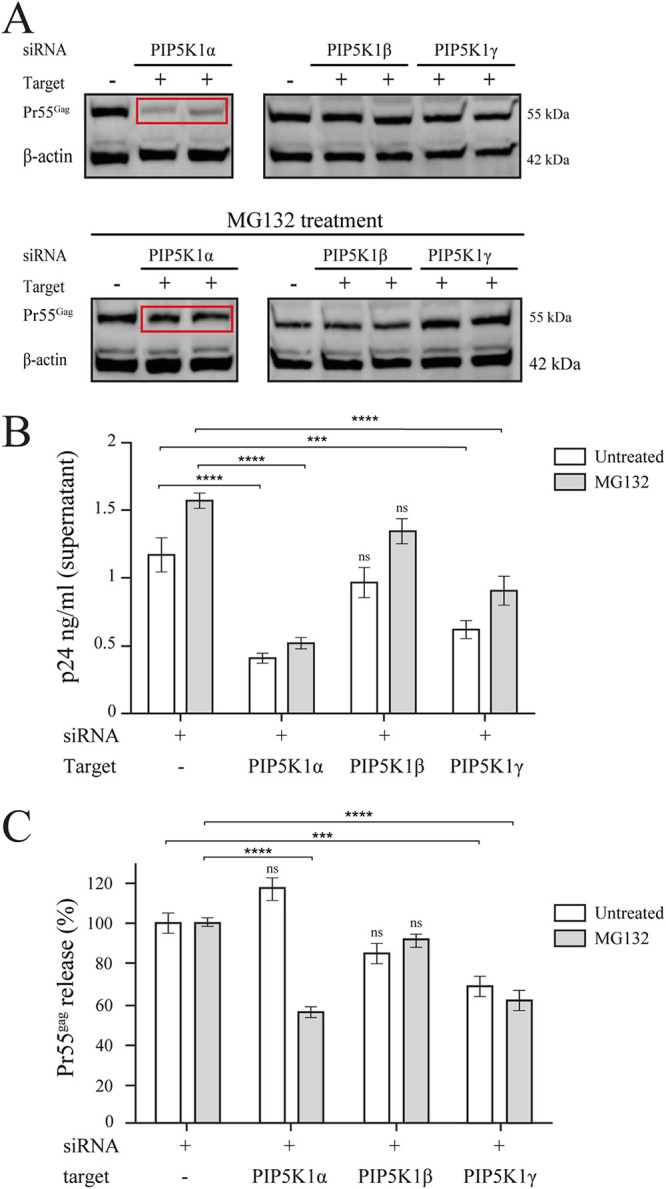
Silencing of PIP5K1α and PIP5K1γ impairs Pr55Gag targeting to the PM and release into the cell supernatant. TZM-bl HeLa cells, transfected with siRNA targeting the PIP5K1 isoforms, were then transfected with a Pr55Gag-encoding plasmid. One day posttransfection, cells were treated with 10 μM MG132. (A) The level of intracellular Pr55Gag expression was analyzed by Western blotting 48 h posttransfection. The β-actin protein was used to normalize protein loading between lanes. The bands in the top part of each panel correspond to the Pr55Gag in siRNA-transfected cells without (top) or with MG132 (bottom) treatment, and the results shown are representative of three independent experiments (each in duplicate). Chemiluminescence analysis was performed with ImageJ software to assess Pr55Gag levels. The red box highlights Pr55Gag level in PIP5K1α-depleted cells. (B) Pr55Gag release in the supernatant was assessed using an anti-p24 ELISA 48 h posttransfection. The histogram shows the mean and SEM of p24 concentration (ng/ml) in the supernatants of cells left untreated (white bars) or treated with MG132 (gray bars) in three independent experiments. (C) Pr55Gag release efficiency was calculated as described in Materials and Methods in three independent experiments. This histogram shows the Pr55Gag release efficiency of transfected TZM-bl HeLa cells left untreated (white bars) or treated with MG132 (gray bars), expressed as percentage compared to control cells transfected with the nontargeting siRNA. Mann-Whitney tests were performed to assess the significance of differences. Significant results are indicated (****, P < 0.0001; ***, 0.0001 < P < 0.001; ns, not significant).
To overcome Pr55Gag hydrolysis, these experiments were repeated in the presence of MG132 proteasome inhibitor (Fig. 5A, bottom panel). The addition of MG132 to PIP5K1α-depleted cells restored the amount of cytoplasmic Pr55Gag almost to control levels (Fig. 5A, red rectangle in bottom panel). A similar result was obtained when the cells were treated with the lysosomal inhibitor bafilomycin A1 (data not shown) (30). These observations confirmed that the depletion of PIP5K1α causes Pr55Gag hydrolysis. Surprisingly, this increase in the amount of Pr55Gag in the cytoplasm did not result in an increase in the amount of Pr55Gag in the supernatant, as the amount of p24 in the supernatant was similar for cells with and without MG132 treatment (Fig. 5B, compare second white and gray bars). The efficiency of the Pr55Gag release was, therefore, almost halved (Fig. 5C, second gray bar). Thus, a wild-type level of Pr55Gag in the cells treated with PIP5K1α siRNA and MG132 is not sufficient to counteract the absence of PI(4,5)P2 in the membrane, emphasizing the essential role of this recognition during viral budding. Conversely, the amounts of Pr55Gag present in the cytoplasm (Fig. 5A) and supernatant of PIP5K1γ-depleted cells (Fig. 5B) were similar in the presence and absence of MG132 treatment. Pr55Gag release efficiency thus remained low in the presence of the proteasome inhibitor (Fig. 5C, fourth gray bar). This result, together with the lack of Pr55Gag and LysoTracker colocalization (Fig. 4), indicates that an absence of PIP5K1γ-mediated PI(4,5)P2 production does not seem to induce Pr55Gag hydrolysis.
Overall, these results show that PIP5K1β is dispensable for Pr55Gag release, whereas PIP5K1α and PIP5K1γ are both involved in the Pr55Gag release. In addition to decreasing release efficiency, our results indicate that the depletion of PIP5K1α has a critical effect on cytoplasmic Pr55Gag levels.
DISCUSSION
The inner leaflet of the PM, which is enriched in PI(4,5)P2, is targeted by Pr55Gag at late stages of the replication cycle. In this study, we determined the role of each PIP5K1 isoform in the cellular localization of Pr55Gag and its accumulation at the assembly site in TZM-bl HeLa cells, a well-characterized model for studies of the HIV-1 replication cycle. Using a semiquantitative UHPLC-HRMS-MS approach, we showed that the knockdown of PIP5K1 isoforms decreased PI(4,5)P2 levels to various extents (Fig. 1D and Fig. 2). The siRNA-based approach used here did not completely eliminate PI(4,5)P2 from the cells, probably due to the well-described redundancy of production pathways (18, 31, 32). Consistent with the data shown in Fig. 1A, indicating that PIP5K1α is the principal isoform produced (in terms of the amount of mRNA), we showed that this isoform is also the principal isoform underlying total PI(4,5)P2 production in TZM-bl HeLa cells (Fig. 1D). We identified 13 molecular species of PI(4,5)P2 with different fatty acyl chain compositions in TZM-bl HeLa cells. The principal phosphoinositide species are known to carry C20:4/C18:0 acyl chains (33–35), but our findings show that C20:2/C18:0 is the major PI(4,5)P2 species in TZM-bl HeLa cells, followed by C18:1/C18:0 and C18:1/C18:1 (Fig. 2). Similar results have been reported for several transformed cell lines, and the low abundance of the C20:4/C18:0 species is thought to be due to the culture medium used (36–39). Our results suggest that molecular species bearing C20:1/C18:0, C18:1/C18:0, C22:2/C18:0, and C20:2/C18:0 could be the principal species generated by PIP5K1α activity, suggesting the specificity of PIP5K1 isoforms for certain PI(4,5)P2 species, consistent with previous in vitro studies in which PI(4)P was used as a substrate (40, 41).
The sequential depletion of PIP5K1 isoforms performed here revealed, for the first time, the importance of the isoforms PIP5K1α and PIP5K1γ and the corresponding PI(4,5)P2 metabolism for the cellular trafficking of Pr55Gag. Based on the effects of the depletion of PIP5K1α and PIP5K1γ, a relationship between PI(4,5)P2 metabolism and Pr55Gag localization was established. Pr55Gag expression in TZM-bl HeLa cells leads to the accumulation of the protein mainly at the PM, although there is also some cytoplasmic labeling (Fig. 3A and C, row a). The depletion of PIP5K1α results in a much lower level of Pr55Gag accumulation at the PM (Fig. 3A and C, row b). This strong reduction may result from the decrease of PI(4,5)P2 level at the membrane, consistent with the known importance of PI(4,5)P2 concentration for the PM targeting of Pr55Gag (6, 12). Consequently, Pr55Gag is degraded by the proteasome (Fig. 5A) and/or by the lysosomes (Fig. 4E) in PIP5K1α-depleted cells. The MG132-mediated inhibition of Pr55Gag proteolysis restores cytoplasmic Pr55Gag level (Fig. 5A, bottom panel) but not Pr55Gag release efficiency (Fig. 5C, second gray bar). Interestingly, the depletion of Arf6, an activator of PIP5K1α, also resulted in a lower expression of Pr55Gag in HeLa cells (42). These findings indicate that PIKP5Kα inhibition results in a lower level of intracellular Pr55Gag and a significantly lower extracellular release of Pr55Gag. Another study did not find any decrease in HIV particle production in cells for which Arf6 was downregulated by siRNA (43). However, mammary epithelial MCF-7 cells were used to produce the HIV particles, which could explain the discrepancy in results. In our study, the ratio between the Pr55Gag expression in the supernatant and the total amount of Pr55Gag expression (supernatant and lysate of PIP5K1α-depleted cells) is similar to that obtained with the nontargeting siRNA (Fig. 5C, compare first and second white bars). This suggests that the residual level of P(4,5)P2 generated by the PIP5K1β and PIP5K1γ isoforms or by other pathways is sufficient for the targeting of at least some Pr55Gag at the PM (31, 32). Moreover, the PIP5K1 isoforms have been reported to homo- or heterodimerize (44), and we cannot exclude a potential role of PIP5K1α/PIP5K1γ heterodimers in rescuing this residual level of PM-PI(4,5)P2. This heterodimerization of the PIP5K1 isoforms and their specific cellular distribution may also explain why almost 50% of cellular PI(4,5)P2 were conserved in cells treated with siRNAs targeting PIP5K1β or PIP5K1γ mRNAs (Fig. 1D). In conclusion, the silencing of PIP5K1α, a protein located principally at the PM, where it controls membrane events and actin dynamics (14, 17, 18, 45), decreases the PI(4,5)P2 concentration, in turn strongly reducing the release of Pr55Gag from the cell.
The impact of the PI(4,5)P2 generated by PIP5K1γ on the intracellular fate of Pr55Gag seems to involve another mechanism. In PIP5K1γ-depleted cells, much less Pr55Gag accumulates at the PM than in control cells (Fig. 3A and C, row d), but Pr55Gag is mostly targeted to intracellular vesicles corresponding to late endosomes (Fig. 4C, image d). This retargeting of Pr55Gag to late endosomes seems to be similar to that observed in PIP5K1α-depleted cells (Fig. 4C, image b). However, there are two key differences between PIP5K1α and PIP5K1γ. On one hand, Pr55Gag is not localized in lysosomes when the PIP5K1γ level is decreased (Fig. 4E, image d), and, on the other hand, the level of intracellular Pr55Gag is not affected by PIP5K1γ silencing (Fig. 5A). Nevertheless, despite this cytoplasmic accumulation of Pr55Gag, lower levels of PM targeting result in lower levels of Pr55Gag release (Fig. 5C). The accumulation of Pr55Gag in late endosomes might have also favored an alternative trafficking pathway to the PM, which, however, does not rescue the release defect observed (46, 47). Thus, the depletion of PIP5K1γ has a profound impact on the cellular behavior of Pr55Gag despite the moderate contribution of this isoform to PI(4,5)P2 production (Fig. 1D and Fig. 2). Furthermore, given that the PIP5K1α isoform, which is considered to be the principal producer of PI(4,5)P2, is still active in PIP5K1γ-depleted cells, the observed defects seem to be related to local and specific pools of PI(4,5)P2 dependent on PIP5K1γ activity rather than total PI(4,5)P2 levels. Interestingly, PIP5K1γ has been reported to display lipid kinase behavior in focal adhesion compartments (21, 48), and a previous study showed that an intracellular compartment referred to as a “focal adhesion-like structure” was essential for HIV assembly and budding in monocyte-derived macrophages (49). Thus, since depriving cells of PIP5K1γ disrupts the focal adhesion plaque (50), we can speculate that it may, in turn, decrease Pr55Gag release efficiency (Fig. 5).
In summary, we have identified PIP5K1α and PIP5K1γ as new determinants required for targeting the Pr55Gag to the PM in TZM-bl HeLa cells. We demonstrated the mistargeting of Pr55Gag from the PM to internal cytoplasmic compartments when PIP5K1α and PIP5K1γ are silenced. Further investigations are underway to determine whether these PIP5K1 isoforms act similarly in other cell models such as CD4 T cells or macrophages where the molecular determinants directing Pr55Gag assembly to virus-containing compartments may be different (51, 52).
MATERIALS AND METHODS
Plasmids.
Constructs encoding PIP5K1α and PIP5K1β were kindly provided by Yungfeng Feng (53). Plasmids encoding GFP-PIP5K1γ90 (22300 and 22299) (54) and mCherry-labeled Rab5 and Rab7A (49201 [55] and 61804 [56]) were purchased from Addgene. The construction of human codon-optimized plasmids encoding Pr55Gag, Pr55Gag-eGFP, and Pr55Gag-mCherry has been described elsewhere (57, 58). The HIV-1 proviral DNA (pNL4-3) (57, 58) was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID.
Antibodies.
The anti-PIP5K1α (C17; catalog no. sc-11774), anti-HPRT (FL-218; catalog no. sc-20975), and anti-β-actin antibodies (I19; catalog no. sc-1616) were purchased from Santa Cruz Biotechnology. The anti-PIP5K1β (catalog no. ab154818), anti-PIP5K1γ (catalog no. ab109192), and anti-HIV1 p24 (catalog no. ab53841) antibodies were purchased from Abcam. The horseradish peroxidase (HRP)-conjugated donkey anti-goat (catalog no. 6420-05) and donkey anti-rabbit (catalog no. 6440-05) secondary antibodies were purchased from Southern Biotech, and the rabbit anti-goat secondary antibody (catalog no. ab6741) was obtained from Abcam.
siRNA and siRNA transfection.
All ON-TARGETplus RNA oligonucleotides and nontargeting control siRNAs (catalog no. D-001810-01-05) were obtained from Thermo Scientific Dharmacon. The siRNA target sequences are as follows: PIP5K1α, 5′-AAAUCAGUGAGGGCUCGCCUA-3′ and 5′-UUGAAAGGUGCCAUCCAGUUAGGC-3′; PIP5K1β, 5′-UGUUGGGAAUUCAUUUCCUGGA-3′ and 5′-CAGCAAAGGGUUACCUUCCAGUUCA-3′; and PIP5K1γ, 5′-AUCAUCAAGACCGUCAUGCAC-3′ and 5′-GCCACCUUCUUUCGAAGAA-3′ (59–61). For siRNA transfection, 2.5 × 105 TZM-bl HeLa cells were cultured in a 6-well plate and transfected with 100 pmol of a pool of two siRNA sequences per isoform, with Lipofectamine RNAiMAX used according to the reverse protocol of the manufacturer (Invitrogen). Flow cytometry analyses showed that around 80% of cells were transfected when a fluorescently labeled siRNA (GLO siRNA; Invitrogen) was used to check the transfection efficiency (data not shown).
Cell culture, transfection, and Pr55Gag release assays.
TZM-bl HeLa cells (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID) were maintained in high-glucose pyruvate-supplemented Dulbecco modified Eagle medium (DMEM), with 10% fetal calf serum, 25 mM HEPES, and gentamicin (50 μg/ml).
For live-cell imaging, 2 × 105 TZM-bl HeLa cells were cultured in a 35-mm imaging dish (Ibidi) and transfected with siRNA as described above. After 2 days, cells were transfected with 0.8 μg of plasmid encoding wild-type Pr55Gag and 0.2 μg of plasmid encoding Pr55Gag-eGFP, with or without plasmids encoding mCherry-Rab5 or mCherry-Rab7A (ratio of 1:0.25:0.075) in the presence of jetPEI (Ozyme) according to the manufacturer’s instructions. The Pr55Gag assembly obtained with the mixture of plasmids encoding wild-type Pr55Gag and Pr55Gag-eGFP, mixed in a ratio of 1:0.25, has been shown to resemble the wild-type virus in terms of Pr55Gag localization and particle morphology (57, 58, 62). The cells were mixed with 1 ml of phenol-free DMEM imaging medium 24 h after DNA transfection. For PM staining, cells were washed twice with phenol-free Opti-MEM medium and incubated with 1 ml of phenol-free medium containing a 15-nM dSQ12S probe (a gift from Andrey Klymchenko, Strasbourg). The incubation time was kept to 30 min to minimize the entry of the dSQ12S probe and the staining of intracellular membranes (26). Alternatively, the Pr55Gag-eGFP-encoding plasmid was replaced with a plasmid encoding Pr55Gag-mCherry in the transfection mix, and LysoTracker Green DND-26 (catalog no. 7526; Invitrogen) was used to stain acidic vesicles. The colors were then inverted with ImageJ to facilitate interpretation of the images obtained.
To quantify the efficiency of Pr55Gag release, Pr55Gag expression levels were measured in the cell supernatants by enzyme-linked immunosorbent assay (ELISA) (Innotest HIV Antigen mAb; Fujirebio) and in the cell lysates by Western blotting (see below). The cells were treated or not with the proteasome inhibitor MG132 (Sigma-Aldrich). MG132 was used at a final concentration of 10 μM and incubated for 6 h before cell lysis. The amounts of Pr55Gag found in the supernatant and the lysate of cells transfected with the nontargeting control siRNA were given the arbitrary value of 1. The levels of Pr55Gag in the supernatants and in the lysates of PIP5K1-depleted cells were expressed relative to this value of 1. Pr55Gag release efficiency was then calculated by dividing the amount of Pr55Gag found in the supernatant by the total amount of Pr55Gag (supernatant and lysate) and expressed as percentage compared to control cells transfected with nontargeting siRNA.
Microscopy.
Live-cell imaging was performed at 37°C with 5% CO2 and a Leica TCS SP8 g-STED 3× inverted confocal microscope equipped with a 63 × 1.4-numerical aperture (NA) oil immersion objective (HCX PL APO 63×/1.40 Oil CS) (Leica). The analysis of the entire cell was performed by a z-stack acquisition. We obtained enhanced green fluorescent protein (eGFP) images by scanning the cells with a 488-nm laser line and a 500- to 555-nm band-pass filter for emission. The PM was detected after incubating the cells with the dSQ12S probe for 30 min with excitation at 632 nm and the use of a 640- to 750-nm band-pass filter for emission. For the mCherry images, a 558-nm laser line was used with a 605- to 730-nm band-pass filter.
SDS-PAGE and Western blot analysis.
Proteins were isolated with the NucleoSpinRNA/protein kit (Macherey-Nagel), and the protein pellet was then solubilized in protein solubilizing buffer containing Tris(2-carboxyethyl)phosphine (PSB-TCEP) in accordance with the manufacturer’s protocol. The total amount of protein was determined with a DS-11 spectrophotometer (DeNovix). Proteins were then denatured by heating and analyzed by SDS-PAGE in 8% to 16% polyacrylamide gels (Eurogentec). The protein bands obtained were transferred onto membranes, which were then probed with primary antibodies followed by the appropriate HRP-conjugated secondary antibodies diluted in blocking solution (50 mM Tris-buffered saline [TBS], 0.05% Tween, and 5% milk powder). Membranes were incubated with the Pierce ECL substrate (Thermo Scientific), and the signals were acquired with the ImageQuant LAS 500 system and analyzed with ImageQuant TL 8.1 (GE Healthcare) or ImageJ software.
RT-qPCR.
Total RNA was isolated with the NucleoSpinRNA/protein kit (Macherey-Nagel) according to the manufacturer’s instructions. We subjected 1 μg of total RNA to reverse transcription with the SuperScript III first-strand synthesis system (Invitrogen). The synthesized cDNA was then used for qPCR with the LightCycler 480 SYBR green I Master system (Roche) and the following primers: PIP5K1α, forward, GAACGGTTCCAGCGCTTCAT, and reverse, GTCTCTCCAACTAGAGGTGA; PIP5K1β, forward, CCAGGAATGGAAGGATGAGA, and reverse, AATTGTGGTTGCCAAGGAAG; PIP5K1γ, forward, GCTACTTCCGGGAGCTCTTT, and reverse, CGTAGAAGAGGGAGCCACTG; and hypoxanthine phosphoribosyltransferase 1 (HPRT1), forward, TGACCTTGATTTATTTTGCATAC, and reverse, CGAGCAAGACGTTCAGTCCT. The data were analyzed with LightCycler 480 software (Roche) and normalized to that of HPRT1 mRNA. Absolute quantification was achieved by comparing the cycle threshold (CT) values of our samples to those of a range of standards.
Lipid extraction and LC-MS analysis.
For PI(4,5)P2 quantification, we transfected 5 × 105 TZM-bl HeLa cells with siRNA as described above. Seventy-two hours posttransfection, cells were scraped into 1 ml of 1 M HCl at 4°C and centrifuged (15,000 × g for 5 min at 4°C). Pellets were resuspended in a mixture of methanol, CHCl3, and 1 M HCl (52.6, 26.2, and 2.6%, respectively) in water, with PI(4)P diC16 and PI(4,5)P2-diC16 (tebu-bio) added as internal standards. After addition of CHCl3 and 2 M HCl (80 and 20%, respectively) and centrifugation (1,500 × g for 5 min at room temperature), the organic phase was collected. The solvent was then evaporated with a SpeedVac at 35°C, and the residues were resuspended in 500 μl of methanol. A derivatization step was performed by adding 50 μl of CH2N2 followed by incubation at room temperature for 30 min. This operation was done twice. Finally, the reaction was stopped by adding 10 μl of acetic acid. The reaction mixture was evaporated, and the residues were resuspended in 100 μl of CH3CN/H2O (10%/90%). Twenty-five microliters of each sample were then injected for liquid chromatography-mass spectrometry (LC-MS) analysis using a UHPLC Dionex Ultimate 3000 (Waters Acquity UPLC BEH300 C4 column; 45°C) with 0.1% formic acid in water as phase A and 0.1% formic acid in CH3CN as phase B at a flow rate of 0.26 ml/min. The gradient phase was as follows: 100% of A at 0 to 5 min, 80% to 55% of A at 5 to 8 min, 55% to 30% of A at 8 to 25 min, 30% to 100% of A at 25 to 30 min, 100% of A at 30 to 32.5 min, and 0% of A at 32.5 to 37 min. LC was coupled to a Q-Exactive mass spectrometer (Thermo Scientific) equipped with a heated electrospray ionization source (HESI). The HESI parameters in positive polarity were as follows: sheath gas flow rate, 35; auxiliary gas flow rate, 10; sweep gas flow rate, 1; spray voltage, 3.50 kV; capillary temperature, 350°C; S-lens RF level, 60.0; and heater temperature, 250°C. The full-scan acquisition parameters were as follows: resolution, 70,000; automatic gain control (AGC) target, 100 ms; and scan range, 850 to 1,300 m/z. The parameters of the top 10 data-dependent MS-MS were as follows: resolution, 35,000; isolation window, 1.0 m/z; collision energy, 20; AGC target, 1e6; and max IT, 200 ms. Identification of each lipid was done based on the retention time associated with high-resolution full MS parent ion and confirmed by high-resolution MS-MS acquisition. The distinction between phosphatidylinositol phosphate (PIP) and phosphatidylinositol bisphosphate (PIP2) was realized based on neutral loss of each lipid family. Chain lengths were determined based on the mass fragment obtained during MS-MS experiments. MS and MS-MS of each PIP and PIP2 are available upon request. For each identified lipid, chromatographic peak areas were obtained using Excalibur processing setup (Thermo Fisher). Internal standards peak areas were used to normalize the amounts of each lipid. The PI(4)P diC16 internal standard was used for normalized PIP lipids and PI(4,5)P2 diC16 for PIP2 lipids. This normalization step was done by dividing signal intensity by that of the appropriate internal standard. Semiquantitative results are thus obtained and enable comparisons between samples.
Statistical and image analysis.
All our data were analyzed in nonparametric Mann-Whitney tests. The distribution of Pr55Gag-eGFP and its colocalization with the dSQ12S probe and mCherry-Rab7A were quantitatively analyzed from the two-color confocal images obtained with the Squassh plugin in ImageJ software (27). The Squassh method allows image segmentation and quantification of the distribution, size, and intensity of transmitted signals. Therefore, the software delineates the fluorescent object in each channel and calculates their overlap. The software then provides the Pearson correlation coefficient accounting for the colocalization of the different markers under each condition. We followed, step-by-step, the protocol previously described by Rizk et al. (27). The software generates the R script R_analysis. R was used to perform one-way analysis of variance (ANOVA) for the statistical analysis of differences between data sets. The distribution of Pr55Gag-mCherry and its colocalization with the LysoTracker Green probe were evaluated by the same method.
ACKNOWLEDGMENTS
This study was financially mass supported by a grant from the Region Centre-Val de Loire (CalViC project number 201500103990).
REFERENCES
- 1.Hermida-Matsumoto L, Resh MD. 2000. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J Virol 74:8670–8679. doi: 10.1128/JVI.74.18.8670-8679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freed EO. 2015. HIV-1 assembly, release and maturation. Nat Rev Microbiol 13:484–496. doi: 10.1038/nrmicro3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryant M, Ratner L. 1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci U S A 87:523–527. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gottlinger HG, Sodroski JG, Haseltine WA. 1989. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A 86:5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou W, Parent LJ, Wills JW, Resh MD. 1994. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J Virol 68:2556–2569. doi: 10.1128/JVI.68.4.2556-2569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A 101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shkriabai N, Datta SAK, Zhao Z, Hess S, Rein A, Kvaratskhelia M. 2006. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry 45:4077–4083. doi: 10.1021/bi052308e. [DOI] [PubMed] [Google Scholar]
- 8.Chukkapalli V, Hogue IB, Boyko V, Hu W-S, Ono A. 2008. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient Gag membrane binding. J Virol 82:2405–2417. doi: 10.1128/JVI.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan R, Uchil PD, Jin J, Shui G, Ott DE, Mothes W, Wenk MR. 2008. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J Virol 82:11228–11238. doi: 10.1128/JVI.00981-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A 103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercredi PY, Bucca N, Loeliger B, Gaines CR, Mehta M, Bhargava P, Tedbury PR, Charlier L, Floquet N, Muriaux D, Favard C, Sanders CR, Freed EO, Marchant J, Summers MF. 2016. Structural and molecular determinants of membrane binding by the HIV-1 matrix protein. J Mol Biol 428:1637–1655. doi: 10.1016/j.jmb.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mücksch F, Laketa V, Müller B, Schultz C, Kräusslich H-G. 2017. Synchronized HIV assembly by tunable PIP2 changes reveals PIP2 requirement for stable Gag anchoring. Elife 6:1–26. doi: 10.7554/eLife.25287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. 2004. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A 101:517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doughman RL, Firestone AJ, Anderson RA. 2003. Phosphatidylinositol phosphate kinases put PI4,5P 2 in its place. J Membr Biol 194:77–89. doi: 10.1007/s00232-003-2027-7. [DOI] [PubMed] [Google Scholar]
- 15.Mellman DL, Gonzales ML, Song C, Barlow CA, Wang P, Kendziorski C, Anderson RA. 2008. A PtdIns4,5P2-regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature 451:1013–1017. doi: 10.1038/nature06666. [DOI] [PubMed] [Google Scholar]
- 16.Boronenkov IV, Loijens JC, Umeda M, Anderson RA. 1998. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol Biol Cell 9:3547–3560. doi: 10.1091/mbc.9.12.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, Kawamoto K, Nakayama K, Morris AJ, Frohman MA, Kanaho Y. 1999. Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99:521–532. doi: 10.1016/S0092-8674(00)81540-8. [DOI] [PubMed] [Google Scholar]
- 18.Balla T. 2013. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93:1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown FD, Rozelle AL, Yin HL, Balla T, Donaldson JG. 2001. Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J Cell Biol 154:1007–1017. doi: 10.1083/jcb.200103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oude Weernink PA, Schmidt M, Jakobs KH. 2004. Regulation and cellular roles of phosphoinositide 5-kinases. Eur J Pharmacol 500:87–99. doi: 10.1016/j.ejphar.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Ling K, Doughman RL, Firestone AJ, Bunce MW, Anderson RA. 2002. Type Iγ phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature 420:89–93. doi: 10.1038/nature01082. [DOI] [PubMed] [Google Scholar]
- 22.Akiyama C, Shinozaki-Narikawa N, Kitazawa T, Hamakubo T, Kodama T, Shibasaki Y. 2005. Phosphatidylinositol-4-phosphate 5-kinase gamma is associated with cell-cell junction in A431 epithelial cells. Cell Biol Int 29:514–520. doi: 10.1016/j.cellbi.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 23.Wang YJ, Li WH, Wang J, Xu K, Dong P, Luo X, Yin HL. 2004. Critical role of PIP5KIγ87 in InsP3-mediated Ca2+ signaling. J Cell Biol 167:1005–1010. doi: 10.1083/jcb.200408008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volpicelli-Daley LA, Lucast L, Gong L-W, Liu L, Sasaki J, Sasaki T, Abrams CS, Kanaho Y, De Camilli P. 2010. Phosphatidylinositol-4-phosphate 5-kinases and phosphatidylinositol 4,5-bisphosphate synthesis in the brain. J Biol Chem 285:28708–28714. doi: 10.1074/jbc.M110.132191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoppe A. 2012. What mRNA abundances can tell us about metabolism. Metabolites 2:614–631. doi: 10.3390/metabo2030614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karpenko IA, Collot M, Richert L, Valencia C, Villa P, Mély Y, Hibert M, Bonnet D, Klymchenko AS. 2015. Fluorogenic squaraine dimers with polarity-sensitive folding as bright far-red probes for background-free bioimaging. J Am Chem Soc 137:405–412. doi: 10.1021/ja5111267. [DOI] [PubMed] [Google Scholar]
- 27.Rizk A, Paul G, Incardona P, Bugarski M, Mansouri M, Niemann A, Ziegler U, Berger P, Sbalzarini IF. 2014. Segmentation and quantification of subcellular structures in fluorescence microscopy images using Squassh. Nat Protoc 9:586–596. doi: 10.1038/nprot.2014.037. [DOI] [PubMed] [Google Scholar]
- 28.Zerial M, Parton R, Chavrier P, Frank R. 1992. Localization of Rab family members in animal cells. Methods Enzymol 219:398–407. doi: 10.1016/0076-6879(92)19039-9. [DOI] [PubMed] [Google Scholar]
- 29.Wandinger-Ness A, Zerial M. 2014. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb Perspect Biol 6:a022616. doi: 10.1101/cshperspect.a022616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ochiai H, Sakai S, Hirabayashi T, Shimizu Y, Terasawa K. 1995. Inhibitory effect of bafilomycin A1, a specific inhibitor of vacuolar-type proton pump, on the growth of influenza A and B viruses in MDCK cells. Antiviral Res 27:425–430. doi: 10.1016/0166-3542(95)00040-S. [DOI] [PubMed] [Google Scholar]
- 31.Vicinanza M, D'Angelo G, Di Campli A, De Matteis MA. 2008. Function and dysfunction of the PI system in membrane trafficking. EMBO J 27:2457–2470. doi: 10.1038/emboj.2008.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burke JE. 2018. Structural basis for regulation of phosphoinositide kinases and their involvement in human disease. Mol Cell 71:653–673. doi: 10.1016/j.molcel.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Pettitt TR, Dove SK, Lubben A, Calaminus SDJ, Wakelam M. 2006. Analysis of intact phosphoinositides in biological samples. J Lipid Res 47:1588–1596. doi: 10.1194/jlr.D600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Milne SB, Ivanova PT, DeCamp D, Hsueh RC, Brown HA. 2005. A targeted mass spectrometric analysis of phosphatidylinositol phosphate species. J Lipid Res 46:1796–1802. doi: 10.1194/jlr.D500010-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.D'Souza K, Epand RM. 2014. Enrichment of phosphatidylinositols with specific acyl chains. Biochim Biophys Acta Biomembr 1838:1501–1508. doi: 10.1016/j.bbamem.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 36.Ogiso H, Taguchi R. 2008. Reversed-phase LC/MS method for polyphosphoinositide analyses: changes in molecular species levels during epidermal growth factor activation in A431 cells. Anal Chem 80:9226–9232. doi: 10.1021/ac801451p. [DOI] [PubMed] [Google Scholar]
- 37.Clark J, Anderson KE, Juvin V, Smith TS, Karpe F, Wakelam MJO, Stephens LR, Hawkins PT. 2011. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat Methods 8:267–272. doi: 10.1038/nmeth.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rouzer CA, Ivanova PT, Byrne MO, Milne SB, Marnett LJ, Brown HA. 2006. Lipid profiling reveals arachidonate deficiency in RAW264.7 cells: structural and functional implications. Biochemistry 45:14795–14808. doi: 10.1021/bi061723j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Traynor-Kaplan A, Kruse M, Dickson EJ, Dai G, Vivas O, Yu H, Whittington D, Hille B. 2017. Fatty-acyl chain profiles of cellular phosphoinositides. Biochim Biophys Acta Mol Cell Biol Lipids 1862:513–522. doi: 10.1016/j.bbalip.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shulga YV, Anderson RA, Topham MK, Epand RM. 2012. Phosphatidylinositol-4-phosphate 5-kinase isoforms exhibit acyl chain selectivity for both substrate and lipid activator. J Biol Chem 287:35953–35963. doi: 10.1074/jbc.M112.370155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shulga YV, Topham MK, Epand RM. 2011. Study of arachidonoyl specificity in two enzymes of the PI cycle. J Mol Biol 409:101–112. doi: 10.1016/j.jmb.2011.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joshi A, Garg H, Nagashima K, Bonifacino JS, Freed EO. 2008. GGA and Arf proteins modulate retrovirus assembly and release. Mol Cell 30:227–238. doi: 10.1016/j.molcel.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghossoub R, Lembo F, Rubio A, Gaillard CB, Bouchet J, Vitale N, Slavík J, Machala M, Zimmermann P. 2014. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat Commun 5:445–456. doi: 10.1038/ncomms4477. [DOI] [PubMed] [Google Scholar]
- 44.Lacalle RA, De Karam JC, Martínez-Muñoz L, Artetxe I, Peregil RM, Sot J, Rojas AM, Goñi FM, Mellado M, Mañes S. 2015. Type I phosphatidylinositol 4-phosphate 5-kinase homo- and heterodimerization determines its membrane localization and activity. FASEB J 29:2371–2385. doi: 10.1096/fj.14-264606. [DOI] [PubMed] [Google Scholar]
- 45.Doughman RL, Firestone AJ, Wojtasiak ML, Bunce MW, Anderson RA. 2003. Membrane ruffling requires coordination between type I alpha phosphatidylinositol phosphate kinase and Rac signaling. J Biol Chem 278:23036–23045. doi: 10.1074/jbc.M211397200. [DOI] [PubMed] [Google Scholar]
- 46.Booth AM, Fang Y, Fallon JK, Yang J-M, Hildreth JEK, Gould SJ. 2006. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol 172:923–935. doi: 10.1083/jcb.200508014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kemler I, Meehan A, Poeschla EM. 2010. Live-cell coimaging of the genomic RNAs and Gag proteins of two lentiviruses. J Virol 84:6352–6366. doi: 10.1128/JVI.00363-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ling K, Schill NJ, Wagoner MP, Sun Y, Anderson RA. 2006. Movin’ on up: the role of PtdIns(4,5)P(2) in cell migration. Trends Cell Biol 16:276–284. doi: 10.1016/j.tcb.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Pelchen-Matthews A, Giese S, Mlčochová P, Turner J, Marsh M. 2012. β2 integrin adhesion complexes maintain the integrity of HIV-1 assembly compartments in primary macrophages. Traffic 13:273–291. doi: 10.1111/j.1600-0854.2011.01306.x. [DOI] [PubMed] [Google Scholar]
- 50.Wu Z, Li X, Sunkara M, Spearman H, Morris AJ, Huang C. 2011. PIPKIγ regulates focal adhesion dynamics and colon cancer cell invasion. PLoS One 6:e24775. doi: 10.1371/journal.pone.0024775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inlora J, Chukkapalli V, Bedi S, Ono A. 2016. Molecular determinants directing HIV-1 Gag assembly to virus-containing compartments in primary macrophages. J Virol 90:8509–8519. doi: 10.1128/JVI.01004-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joshi A, Ablan SD, Soheilian F, Nagashima K, Freed EO. 2009. Evidence that productive human immunodeficiency virus type 1 assembly can occur in an intracellular compartment. J Virol 83:5375–5387. doi: 10.1128/JVI.00109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kisseleva M, Feng Y, Ward M, Song C, Anderson RA, Longmore GD. 2005. The LIM protein Ajuba regulates phosphatidylinositol 4,5-bisphosphate levels in migrating cells through an interaction with and activation of PIPKI alpha. Mol Cell Biol 25:3956–3966. doi: 10.1128/MCB.25.10.3956-3966.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P. 2002. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1γ by the FERM domain of talin. Nature 420:85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- 55.Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, Voeltz GK. 2010. ER sliding dynamics and ER–mitochondrial contacts occur on acetylated microtubules. J Cell Biol 190:363–375. doi: 10.1083/jcb.200911024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK. 2014. ER contact sites define the position and timing of endosome fission. Cell 159:1027–1041. doi: 10.1016/j.cell.2014.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.El Meshri SE, Dujardin D, Godet J, Richert L, Boudier C, Darlix JL, Didier P, Mély Y, De Rocquigny H. 2015. Role of the nucleocapsid domain in HIV-1 gag oligomerization and trafficking to the plasma membrane: a fluorescence lifetime imaging microscopy investigation. J Mol Biol 427:1480–1494. doi: 10.1016/j.jmb.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 58.MüLler B, Daecke J, Fackler OT, Dittmar MT, Zentgraf H, Kräusslich H-G, 2004. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. JVI 78:10803–10813. doi: 10.1128/JVI.78.19.10803-10813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Micucci F, Capuano C, Marchetti E, Piccoli M, Frati L, Santoni A, Galandrini R. 2008. PI5KI-dependent signals are critical regulators of the cytolytic secretory pathway. Blood 111:4165–4172. doi: 10.1182/blood-2007-08-108886. [DOI] [PubMed] [Google Scholar]
- 60.Xu Q, Zhang Y, Xiong X, Huang Y, Salisbury JL, Hu J, Ling K. 2014. PIPKI gamma targets to the centrosome and restrains centriole duplication. J Cell Sci 127:1293–1305. doi: 10.1242/jcs.141465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Padrón D, Wang YJ, Yamamoto M, Yin H, Roth MG. 2003. Phosphatidylinositol phosphate 5-kinase Iβ recruits AP-2 to the plasma membrane and regulates rates of constitutive endocytosis. J Cell Biol 162:693–701. doi: 10.1083/jcb.200302051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hübner W, Chen P, Del Portillo A, Liu Y, Gordon RE, Chen BK. 2007. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J Virol 81:12596–12607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schill NJ, Anderson RA. 2009. Two novel phosphatidylinositol-4-phosphate 5-kinase type Igamma splice variants expressed in human cells display distinctive cellular targeting. Biochem J 422:473–482. doi: 10.1042/BJ20090638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van den Bout I, Divecha N. 2009. PIP5K-driven PtdIns(4,5)P2 synthesis: regulation and cellular functions. J Cell Sci 122:3837–3850. doi: 10.1242/jcs.056127. [DOI] [PubMed] [Google Scholar]