

Poor pharmacokinetics have severely hindered the clinical use of anti-HIV peptides. Different small molecules, such as lipid, cholesterol, and small PEG, were designed to modify peptides to improve their pharmacokinetics. In this study, we incorporated large branched PEG to anti-HIV peptide and obtained a conjugate with extended half-life and improved in vivo efficacy. The strategy we developed in this study can also be applicable for the development of other peptide candidates.
KEYWORDS: HIV inhibitor discovery, peptide therapeutics chemical modification to extend half-life, anti-HIV fusion peptide, pharmacokinetics
ABSTRACT
Anti-human immunodeficiency virus type 1 (anti-HIV-1) fusion peptides have been studied for nearly 2 decades, but few candidates have found useful clinical applications. One factor underlying the failure of such agents to reach the clinic is their poor pharmacokinetic properties, and many efforts have been made to overcome this problem. In this study, we modified C34, a peptide inhibitor of HIV-1 fusion, at its conserved glycosylation site using polyethylene glycols (PEGs) of different molecular weights. PEG40-NC, a conjugate of C34 and branched PEG 40 kDa (PEG40), which has been previously shown to improve the pharmacokinetic profiles of proteins, showed a significantly extended half-life (t1/2; 10.39 h in rats), which compensated for decreased in vitro activity (50% effective concentration [EC50] of 18.51 nM). PEG40-NC also showed a mechanism of action similar to that of C34. PEG40-NC monotherapy in acutely simian-human immunodeficiency virus (SHIV)-infected rhesus monkeys significantly suppressed viral load compared with a control treatment. Efficacy was linked to the extended half-life and lymphatic exposure conferred by attached PEG40. These results highlight the potential of further clinical investigations of PEG40-NC in combination with antiretroviral therapy or other anti-HIV agents.
IMPORTANCE Poor pharmacokinetics have severely hindered the clinical use of anti-HIV peptides. Different small molecules, such as lipid, cholesterol, and small PEG, were designed to modify peptides to improve their pharmacokinetics. In this study, we incorporated large branched PEG to anti-HIV peptide and obtained a conjugate with extended half-life and improved in vivo efficacy. The strategy we developed in this study can also be applicable for the development of other peptide candidates.
INTRODUCTION
AIDS, caused by human immunodeficiency virus (HIV), remains a dangerous, progressive disease. About 37.9 million people globally were living with HIV in 2018, with ∼1.7 million new infections worldwide (1). Highly active antiretroviral therapy (HAART) is the standard of care for HIV infection. Three or more drugs targeting at least two enzymes are used in HAART regimens to suppress replication and reduce plasma viral loads below the limits of detection (<50 RNA copies/ml) of diagnostic tests (2–5). However, several issues limit the ability of HAART to eradicate HIV, including development of multidrug resistance (6), inability of drugs to reach latent HIV reservoirs (7, 8), and adverse effects. Thus, further efforts are required to discover and develop new anti-HIV drugs.
Binding and fusion between viral and host cell membranes mediated by the envelope glycoprotein (Env) (9) are essential for HIV-1 entry. Following Env binding to CD4, dissociation of gp120 from gp41 induces exposure of the N-terminal fusion peptide of gp41 and its insertion into the cell membrane. Then, three C-terminal heptad repeats fold in antiparallel fashion onto the trimeric NHR (N-terminal heptad repeats) coiled-coil to form a thermostable six-helix bundle (6-HB) (10–12), which brings together the viral and cellular membranes and lowers the free-energy barrier to fusion. HIV-1 fusion is an attractive target for drug discovery with several important advantages (13). However, only one HIV-1 fusion inhibitor, enfuvirtide (T20), has been approved, and its clinical use has been seriously limited by its short half-life (t1/2), which necessitates high-dose injections. Moreover, extended exposure can cause resistance (14, 15), with mutant virions emerging in about 1 month (16). Several new fusion inhibitors, such as T1249 (17), T1144 (16), sifuvirtide (18), SC22EK (15), HP23 (19), and T2635 (16), have been generated using the CHR (C-terminal heptad repeats) peptide C34 as a design template. However, rapid proteolysis and short half-lives are major limitations. This has prompted the development of half-life extension strategies to improve the pharmacokinetic profiles of anti-HIV-1 fusion peptides; these include polyethylene glycol (PEG)ylation (20–22), lipidation (23–25), glycosylation (26), modification with cholesterol (27), and addition of a maleimide group for covalent conjugation to human serum albumin (28–30). PEGylation is a common half-life extension strategy: more than 10 PEGylated therapeutics have entered the clinic since the early 1990s, and over 20 PEGylated drugs are undergoing clinical trials (31).
We previously developed a series of PEGylated T20 peptides with extended half-lives in vivo through site-specific attachment of small linear PEGs (2 and 5 kDa) (21). However, most PEGylated drugs use large branched PEGs (≥40 kDa) (31), as these show decreased immunogenicity and increased half-life compared with linear low-molecular-weight (MW) PEGs. Moreover, molecules of >16 to 20 kDa are thought to have increased lymphatic exposure and retention within lymph nodes (22, 32, 33), where HIV reservoirs are located (34). Based on our previous work with T20 (21, 26), in this study we attached large branched PEGs (40 kDa) to different C34 sites to obtain a more potent HIV-1 fusion inhibitor. C34 is a first-generation HIV-1 fusion inhibitor with more potent anti-HIV-1 activity than T20 (10). Furthermore, we have demonstrated C34 has a different resistance mechanism with T20 and retained high antiviral activity against T20-resistant strains (35). However, the low solubility of C34 undermines its potential use as a clinical drug (36).
RESULTS AND DISCUSSION
We constructed PEG-C34 conjugates using thiol-maleimide Michael reactions (Fig. 1) (21, 35). C34 is a 34-mer peptide derived from the gp41 CHR (residues 628 to 661) and includes Asn637, which forms a conserved glycosylation site in native gp41 (37). We hypothesized that a hydrophilic PEG chain attached at Asn637 could potentiate the antiviral activity of C34. A single Cys was introduced at the N or C terminus of the C34 sequence, or Asn637 was replaced with Cys. Linear methoxy-PEG-maleimides (mPEG-Mal) of 2 and 5 kDa were used to modify these three sites one at a time, and the optimal PEGylation site was identified using antiviral activity assays. Large branched mPEG-Mal (10, 20, and 40 kDa) was also used to investigate the impact of MW on antiviral activity. All reactions were completed in 10 min, with yields of >70%. Conjugates were purified using high-performance liquid chromatography (HPLC) with high purity (>95%), and matrix-assisted laser desorption/ionization (MALDI)–time of flight (TOF) mass spectra confirmed the attachment of a single PEG chain.
FIG 1.

Synthesis and characterization of PEG-C34 conjugates. (A) Reactions were conducted in phosphate buffer (pH 7.2) at room temperature. Reaction progress was monitored by analytic HPLC, and the products were purified by semipreparative HPLC. (B) The large branched mPEG-Mal structure. (C) HPLC chromatogram (reaction mixture). (D) MALDI-TOF mass spectrum of PEG40-NC.
The antiviral activities of peptides and conjugates against the laboratory-adapted HIV-1 strain NL4-3 were investigated. Anti-HIV-1 activity was measured using the Reed and Muench method (38) in TZM-b1 cells. All peptides and conjugates inhibited replication of HIV-1 NL4-3 at nanomolar concentrations (Fig. 2). C34NC, the internal Cys-conjugated C34, showed higher antiviral activity than the N- or C-terminally conjugated C34s and the parent C34 peptide, potentially because Cys and Asn are structurally similar residues. In contrast, terminal addition of Cys changes the length of the peptide and alters the structure of the terminal Trp and Leu residues. Consistently, the 50% effective concentrations (EC50s) for small linear PEGylated peptides (PEG2- and PEG5-modified C34) showed the same trends as Cys-incorporated C34s. Terminal PEG2 or PEG5 modification decreased antiviral activity significantly, but PEGylation with small linear PEGs at the internal Cys did not affect activity. Thus, the attached PEG chain at the native glycosylation site of C34 likely mimicked the native glycan as previously reported (39).
FIG 2.
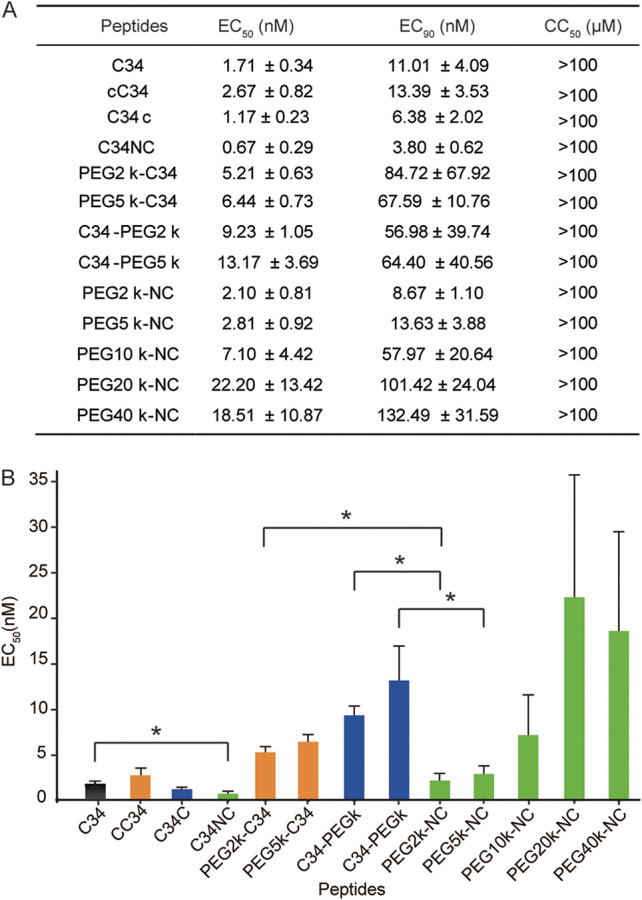
Antiviral activities of PEG-C34 conjugates against HIV-1 NL4-3. EC50 and EC90 values were determined from three independent experiments and reported as the means ± standard deviations. (A) EC50, EC90, and CC50 (50% cytotoxic concentration) values of all peptides. (B) EC50 values of all peptides. *, P < 0.05 (Student’s t test).
Asn637 was chosen as the modification site for large branched PEGs. C34NC was PEGylated with branched PEGs (10, 20, and 40 kDa) to assess the impact of PEG MW on antiviral activity. When PEG MW was increased from 10 to 20 kDa, the activity of PEGylated C34 decreased. However, a slight increase in inhibitory activity was observed when PEG MW was increased from 20 to 40 kDa. Because high-MW PEGs are optimal for peptides and protein drugs, we hypothesized that losses of in vitro activity with increasing PEG size would be compensated by prolonged residence in blood and higher lymphatic exposure in vivo.
We next used surface plasmon resonance (SPR) to assess binding of the internally PEGylated C34 conjugates. N36, a 36-mer peptide from the NHR of gp41, is the target of C34 (40). SPR showed that all conjugates bound N36 with affinities (Kd [dissociation constant]) of 124 to 424 nM, similar to that of C34 (Kd = 185 nM) (Fig. 3). PEG2-NC and PEG5-NC showed slightly stronger binding to N36 than C34, contrary to our experiences using PEGylated T20 (21). This result strongly suggested that Asn637 was an optimal site for C34 modification. Binding affinity decreased with increasing PEG MW (PEG10-NC, PEG20-NC, and PEG40-NC), consistent with the antiviral activity of these molecules. Although the MW of PEG40-NC was almost twice that of PEG20-NC, the binding affinities of these conjugates were similar. These results suggested that attachment of PEGs, including PEG40, at C34 Asn637 did not significantly affect the intrinsic affinity of C34 for gp41 NHR.
FIG 3.

Interaction of C34 and internally PEGylated C34 with N36 by SPR. The N36 peptide was immobilized on the CM5 sensor chip surface. Solutions containing different concentrations of C34 or conjugates were flowed over the chip surface. Concentrations of 1 μM are shown.
C34 has an almost featureless structure and can form a stable α-helical structure in complex with N36 in aqueous media (10, 40). We further evaluated the effect of the internally conjugated PEG chain on the conformation of C34 and its complex with N36 using circular-dichroism (CD) spectroscopy. Like C34, PEG-NC did not show minimum α-helical conformation at 222 nm (Fig. 4A), suggesting that the PEG chain did not alter the secondary structure of C34; all peptides displayed unordered coil structures in phosphate-buffered saline (PBS) with α-helicities ranging from 14% to 29%. In contrast, the C34/N36 complex formed a typical α-helix with an α-helical content of 91% (Fig. 4B). All PEG-NCs showed CD spectra similar to that of C34 in the presence of N36, except for PEG10-NC, which showed moderately increased helical content compared with C34-N36. These results suggested that even large internal PEG chains had little impact on the secondary conformation of the C34-N36 complex, which is essential for antiviral activity.
FIG 4.

CD spectra of C peptides and their complexes with N36. C peptides (40 μM) were incubated at 37°C for 30 min in PBS (5 mM, pH 7.2) in the absence (A) and presence (B) of equimolar amounts of N36, and then the CD spectra were recorded at 20°C.
The CD results were seemingly inconsistent with the antiviral activity and SPR analyses, in which the higher-MW PEGs had a negative impact on inhibitory activity and binding affinity for N36. We speculate that this discrepancy is largely due to different modes of action. In the CD experiment, PEG-NC bound to free N36 in aqueous solution, and the large internal PEG chain probably faced outside the 6-HB and did not affect the C34-N36 interaction. However, in the antiviral activity analyses, the N-terminal fusion peptide inserted into cell membrane. In this case, N36, which was included in the NHR and closed to the N-terminal fusion peptide, was fixed in high density on the surface of TZM-b1 cells, making binding more sensitive to steric hindrance. Similarly, the high density of N36 immobilized on the sensor chip in SPR experiments may have resulted in the weaker affinities observed for large PEGylated C34 than for PEG2-NC and PEG5-NC.
Although PEG40-NC, the largest PEG-modified C34 peptide in this study, showed a slightly decreased affinity for N36 and moderately weakened inhibition of HIV-1 NL4-3 in vitro, we hypothesized that the attached PEG40 would extend plasma circulation time, compensating for the loss of inhibitory activity. We next evaluated the in vivo stability of PEG40-NC using a simple, rapid, and sensitive system developed by Li et al. (41). PEG40-NC (1.7 μmol/kg of body weight) or C34 was subcutaneously injected into each rat, and sera were harvested at different times. The serum dilutions required for 50% inhibition of infection were calculated according to ex vivo antiviral activity analyses. As shown in Fig. 5A, serum activity in C34-treated rats peaked in 1 h, with a serum dilution that inhibited 50% of virus infection of 1:2,275, and then declined quickly. No inhibitory serum activity was detected after 4 h. These results showed that C34 was rapidly adsorbed and cleared from circulation. In contrast, PEG40-NC peaked in serum at 23 h postinjection, with a serum dilution that inhibited 50% of virus infection of 1:53.42. At this time, the plasma concentration of PEG40-NC, calculated based on the EC50, was about 7.50-fold above the EC90 determined for PEG40-NC (132.49 nM [Fig. 2A]).This plasma concentration is expected to be effective in vivo. The anti-HIV activity of sera dropped slowly; even after 72 h, PEG40-NC still exhibited potent inhibitory activity, with a serum dilution that inhibited 50% of virus infection of 1:4.29. Thus, the large branched PEG40-modified C34 showed a slower but much more sustained release profile from the injection site, which was attributable to the long-lasting effect of PEG40. The plasma concentrations in Fig. 5B were calculated from serum dilutions and the corresponding EC50 values (Fig. 2A). Significant differences were also observed between C34 and PEG40-NC. The half-life of PEG40-NC was about 10.39 h, significantly longer than that of C34 (t1/2 = 0.60 h). This improvement in the pharmacokinetic profile following attachment was in line with previous results (42–45).
FIG 5.

Pharmacokinetic analyses of C34 and PEG40-NC in rats. (A) The serum dilutions required for 50% inhibition of virus infection. (B) Plasma concentrations of peptides were calculated based on the serum dilutions and the corresponding EC50 values. Half-lives were estimated using the equation t1/2 = 0.693/k, where k is the rate constant for concentration decay. Four rats (two males and two females) were used for each peptide. Error bars represent standard deviations (SDs) from four independent experiments (four rats for each group).
These results encouraged us to further explore the therapeutic efficacy of PEG40-NC in vivo. Eight Chinese rhesus monkeys were infected with a strain of simian-human immunodeficiency virus, SHIVSF162P3, and randomly assigned to two groups (groups A and B, four monkeys each, subcutaneously treated with saline or PEG40-NC once daily from the 7th day to the 35th day postinfection). Blood was collected before infection and at different times postinfection, and the plasma SHIV RNA viral load was determined. As shown in Fig. 6A, the plasma viral loads of all monkeys in the control group reached peaks ranging from 7.8 to 8.2 log10 RNA copies/ml on day 11 postinfection, demonstrating the establishment of acute infection. Viral loads declined naturally thereafter but remained above 6 log10 RNA copies/ml between days 21 and 32, except for monkey A2, with a viral load of 5.55 log10 RNA copies/ml on day 28. However, viral loads in the PEG40-NC-treated group peaked lower than in group A, with SHIV RNA viral loads ranging from 6.23 to 7.12 log10 RNA copies/ml on day 11 (Fig. 6B).
FIG 6.
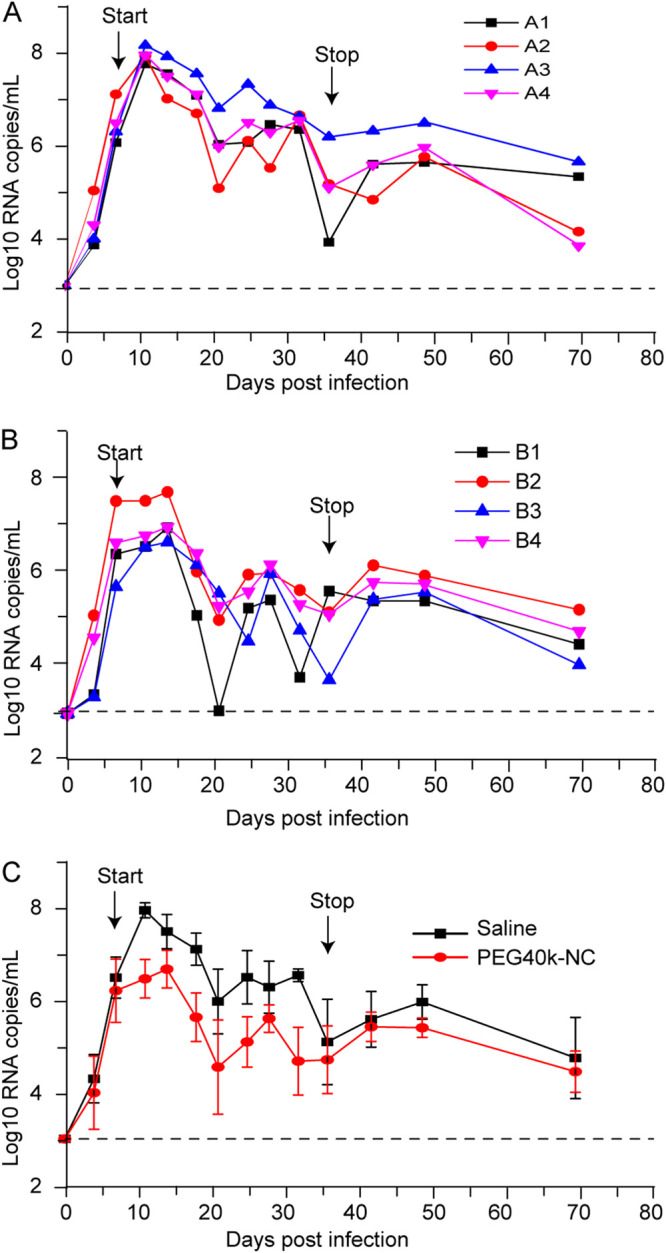
Treatment of SHIVSF162P3-infected rhesus monkeys with PEG40-NC. Four monkeys were subcutaneously treated with saline (A) or 1.38 μmol/kg of PEG40-NC (B) on the 7th day postinfection. Plasma viral loads in SHIV-infected monkeys were measured using a quantitative real-time reverse transcription-PCR (qRT-PCR) assay. The average viral loads of PEG40-NC-treated monkeys were compared with those of saline-treated monkeys (C). Error bars in panel C represent SDs from four independent experiments (four monkeys for each group).The horizontal dashed line at 3 log10 RNA copies/ml indicates the limit of detection of the assay. P = 0.0126 for the therapy period.
Thus, viral replication was suppressed by treatment with PEG40-NC. Thereafter, viral loads in the PEG40-NC-treated group decreased more dramatically than in group A. The plasma viral loads of monkey B1 declined to nearly undetectable levels (1,000 copies/ml) by 2 weeks following initial treatment, with a value of 3.02 log10 RNA copies/ml. Viral RNA levels in all treated monkeys were reduced to below 6 log10 RNA copies/ml between days 21 and 32 and then rebounded slightly after therapy was stopped. Thereafter, viral loads began to decline slowly from day 6 after dosage withdrawal until to the end of the observation period (Fig. 6C). The difference in viral loads for control group A and the PEG40-NC-treated group B was statistically significant (P = 0.0126) for the treatment period (days 7 to 36 postinfection). Subcutaneous administration of PEG40-NC was well tolerated at the dosage used, and the animals exhibited no clinical signs of toxicity.
In a previous study (24), T20 administered once daily showed no protective effect in SHIV-infected monkeys because of its short half-life. Viral kinetics in T20-treated monkeys were similar to those in control animals. We speculated that C34 would not show a therapeutic effect in SHIV-infected monkeys using the same protocol because C34 has a pharmacokinetic profile similar to that of T20. However, PEG40-NC exhibited clear therapeutic efficacy despite steric hindrance problems impeding binding to the NHR. Therapeutic efficacy mainly results from the extended half-life and extended lymphatic exposure. These results highlight the potential for clinical development of PEG40-NC.
Conclusion.
We developed a highly potent therapeutic against HIV with long-lasting efficacy in vivo by PEGylating C34 with a 40-kDa branched PEG. Covalent attachment of PEG40 to proteins prolongs their half-lives by preventing kidney filtration and proteolysis (31). Small peptides are often inactivated by modification with high-MW PEGs owing to impaired binding to their receptors. To date, no peptides PEGylated with PEG40 have been approved for clinical use. We have demonstrated that modification of C34 with PEG40 at a suitable site significantly improved its pharmacokinetic profile while retaining binding to its target protein. Monotherapy with this conjugate in SHIV-infected rhesus monkeys showed a significant protective effect. This conjugate may have clinical utility in combination with antiretroviral therapy regimens. Furthermore, PEGylation with large branched PEGs may also aid the clinical development of other peptides.
MATERIALS AND METHODS
Materials and instrumentation.
Peptides were synthesized using a standard solid-phase 9-fluorenylmethoxy carbonyl (Fmoc) protocol at Beijing Scilight Biotechnology LLC (Beijing, China) and purified by high-performance liquid chromatography (HPLC) (purity > 95%). Peptides were protected by C-terminal amidation and N-terminal acetylation and characterized by MALDI-TOF mass spectrometry. The laboratory-adapted strain NL4-3 used in the infectivity assay was isolated and propagated in the European Research Infrastructures for Poverty Related Diseases project. TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program (USA) and propagated in Dulbecco’s modified Eagle medium (DMEM) containing fetal calf serum (10%, vol/vol), penicillin (100 U/ml), and streptomycin (100 μg/ml). SHIVSF162P3 was obtained through the NIH AIDS Research and Reference Reagent Program. Sprague-Dawley rats (male, 7 weeks old, 150 to 180 g) were purchased from Beijing HFK Bioscience (Beijing, China) and fed for 2 days before pharmacokinetic analyses. mPEG-MAL (2, 5, 10, 20, and 40 kDa) was purchased from Beijing Kaizheng Biotech Development (Beijing, China). All other chemicals were purchased from various commercial sources and were analytical grade. HPLC analyses were performed on an Agilent 1200 system (Agilent, USA) equipped with a UV detector. MALDI-TOF mass spectra were recorded on a Bruker BIFLEX III spectrometer (Bruker Daltonics, Germany). All experiments with viruses were performed in an approved biosafety level 3 (BSL3) containment laboratory.
Synthesis of PEGylated C34.
The mono-Cys-incorporated C34 (cC34, C34c, or C34NC; 10 mg) and mPEG-MAL (1.50 equivalent) were dissolved in sodium phosphate buffer (10 ml, 5 mM; pH 7.5) at room temperature. Reactions were monitored by analytical HPLC on an Agilent C18 column (4.6 by 250 mm) at 40°C with a linear gradient (10% to 90% MeCN containing 0.1% trifluoroacetic acid [TFA] over 25 min, flow rate of 1 ml/min, for mPEG2KDMAL, mPEG5KDMAL, and mPEG10KDMAL modifications). The reaction was monitored by analytical HPLC on a Komasil column (4.6 × 250 mm) at 40°C with a linear gradient (35% to 45% MeCN containing 0.1% TFA over 25 min, flow rate of 1 ml/min, for mPEG40KDMAl modification). When the peptides disappeared, the products were purified by HPLC on the corresponding semipreparative column to yield PEG2-C34 (12 mg, 82%), C34-PEG2 (13 mg, 85%), PEG5-C34 (15 mg, 70%), C34-PEG5 (16 mg, 75%), PEG2-NC (13 mg, 88%), PEG5-NC (19 mg, 89%), PEG10-NC (31 mg, 87%), PEG20-NC (49 mg, 90%) or PEG40-NC (96 mg, 92%) as a white powder. The products were confirmed by MALDI-TOF mass spectrometry; the average molecular weights of C34-PEG2, C34-PEG5, PEG2-NC, PEG5-NC, PEG10-NC, PEG20-NC, and PEG40-NC were 6,645, 9,305, 6,487, 9,147, 15,001, 23,413, and 44,647, respectively.
HIV inhibition assay.
The antiviral activities of PEG-NC conjugates were measured as previously described (21, 35). Briefly, TZM-b1 cells were incubated on a tissue culture plate (96 well, 1 × 104 cells/well) at 37°C overnight, then titrated PEG-NC conjugates were added (100 μl in DMEM), followed by 200 50% tissue culture infective doses (TCID50) of HIV-1NL4-3. Cells (100 μl) were harvested after incubation at 37°C for 48 h and lysed using a lysis reagent (Promega; 100 μl). Luciferase activity was then measured, and the EC50 and EC90 values were calculated using the Reed and Muench method (38) based on the percent inhibition of luciferase activity.
CD analysis.
The secondary structures of peptides and peptide-N36 complexes were measured using a Chirascan spectropolarimeter (Applied Photophysics Ltd., UK) as described previously (21, 26). Briefly, each peptide or complex was dissolved in PBS (5 mM, pH 7.2) at a concentration of 40 μM for both peptides and N36. The solution was incubated at 37°C for 30 min, and then the CD spectra were recorded at 20°C with a bandwidth of 1.0 nm and a scanning speed of 5 nm/min. The spectra were corrected by blank subtraction of the signals from solvent. The α-helicities of the compounds were determined according to the software CDNN (calculating the ratio of protein secondary structure).
SPR analysis.
The binding affinities of C34 and PEG-NC conjugates for N36 peptide were analyzed using a Biacore biosensor system (Biacore 3000; Biacore Co., Ltd.) according to a previously described procedure (21). Briefly, N36 was immobilized on a carboxymethyl dextran-coated sensor chip (CM-5, research grade) using standard primary amine coupling. C34 or PEG-NC conjugates in PBS containing Tween 20 (0.05%, vol/vol) at graded concentrations (0.125 to 4 μM) were flowed over the sensor chip surface at a rate of 20 μl/min. C34 was used as a positive control. Bovine serum albumin was used as a negative control instead of peptide N36. Kinetic parameters were calculated using Biacore evaluation software (version 4.1).
Pharmacokinetic analysis.
The pharmacokinetic profile of PEG40-NC was studied in 7-week-old Sprague-Dawley rats (n = 4; 180 to 200 g) as described previously (21, 24, 35). A single subcutaneous injection of PEG40-NC or C34 (1.7 μmol/kg in physiological saline) was given to each rat (35). Blood samples (300 μl) were collected from the tail vein before administration and at different intervals after injection (3, 6, 9, 20, 23, 29, 48, 72, and 96 h). All blood samples were collected into microcentrifuge tubes containing heparin (5 μl, 5,000 IU/ml) and aprotinin (6 μl, 50 trypsin inhibitor units [TIU]/ml). Plasma samples were obtained by centrifugation of a whole-blood sample for ex vivo anti-HIV-1NL4-3 activity evaluation as described previously (24, 35). The highest dilution of plasma samples that inhibited 50% of virus infection was identified based on the single-cycle assay. The plasma concentrations of C34 and PEG40-NC were calculated according to the dilutions and EC50 values. Half-lives were estimated using the equation t1/2 = 0.693/k, where k is the rate constant for concentration decay.
Evaluation of therapeutic efficacy.
The in vivo therapeutic efficacy of PEG40-NC was evaluated in acutely SHIVSF162P3-infected Chinese rhesus macaques as reported previously (24). Briefly, eight adult Chinese rhesus macaques (3 to 4 kg) were screened and inoculated intravenously with SHIVSF162P3 (100 TCID50). The TCID50 was determined by infection of macaque peripheral blood mononuclear cells. Macaques were divided randomly into the acute infection treatment group (n = 4) and the control group (n = 4). Macaques in the former and latter groups received PEG40-NC (1.38 μmol/kg in saline) and 0.9% saline solution (1 ml), respectively, by subcutaneous injection once daily for 28 days beginning on day 7 post-virus inoculation. Blood samples were collected on days 1, 4, 7, 11, 14, 18, 21, 25, 28, 32, 36, 42, 49, and 70 postinoculation. Viral RNA was extracted from cell-free plasma using the QIAmp viral RNA minikit (Qiagen, Valencia, CA). Plasma viral loads were measured using a One Step PrimeScript reverse transcription-PCR (RT-PCR) kit (TaKaRa) and a PerkinElmer ABI 7500 instrument. RNA levels were determined using SHIV-gag primers and probes following primary infection. Primers and probes for real-time PCR were as previously described (24, 46). RNA standards (103 to 1013 copies/ml) were prepared in our laboratory. Reactions were performed in duplicate for each sample.
Ethics statement.
The protocol (no. SQIMCAS20) for the pharmacokinetic study in rats was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Microbiology, Chinese Academy of Sciences. The protocol (no. XJ19001) for the in vivo efficacy study in rhesus monkeys was approved by the IACUC of the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences, where the study were carried out. All experiments involving SHIV-infected animals were performed in an animal biosafety level 3 (ABSL-3) laboratory. All monkeys were anesthetized with ketamine hydrochloride prior to procedures (10 mg/kg).
ACKNOWLEDGMENTS
This work was financially supported by the National Key R&D Program of China (grants 2016YFE0205800 and 2017YFD0500201) and the National Natural Science Foundation of China (grants 21778074, 81871694, and 31770862).
We declare no competing financial interests.
REFERENCES
- 1.UNAIDS. 2018. Global HIV statistics for 2018. https://www.unaids.org/en/resources/fact-sheet.
- 2.CDC. 2019. HIV treatment. https://www.cdc.gov/hiv/basics/livingwithhiv/treatment.html.
- 3.Broder S. 2010. The development of anti- retroviral therapy and its impact on the HIV-1/AIDS pandemic. Antiviral Res 85:1–18. doi: 10.1016/j.antiviral.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMahon JH, Elliott JH, Bertagnolio S, Ku Biak R, Jordan MR. 2013. Viral suppression after 12 months of antiretroviral therapy in low- and middle-income countries: a systematic review. Bull World Health Organ 91:377–385E. doi: 10.2471/BLT.12.112946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson MA, Aberg JA, Cahn P, Montaner JSG, Rizzardini G, Telenti A, Gatell JM, Günthard HF, Hammer SM, Hirsch MS, Jacobsen DM, Reiss P, Richman DD, Volberding PA, Yeni P, Schooley RT. 2010. Antiretroviral treatment of adult HIV infection: 2010 recommendations of the International AIDS Society–USA panel. JAMA 304:321–333. doi: 10.1001/jama.2010.1004. [DOI] [PubMed] [Google Scholar]
- 6.Hogg RS, Bangsberg DR, Lima VD, Alexander C, Bonner S, Yip B, Wood E, Dong WW, Montaner JS, Harrigan PR. 2006. Emergence of drug resistance is associated with an increased risk of death among patients first starting HAART. PLoS Med 3:e356. doi: 10.1371/journal.pmed.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 8.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Lexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 9.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. 1996. HIV-1 entry into CD4 cells is mediated by the chemokine receptor CC-CK R-5. Nature 381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 10.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 11.Tan K, Liu J, Wang J, Shen S, Lu M. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A 94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 13.Eggink D, Bontjer I, de Taeye SW, Langedijk JPM, Berkhout B, Sanders RW. 2019. HIV-1 anchor inhibitors and membrane fusion inhibitors target distinct but overlapping steps in virus entry. J Biol Chem 294:5736–5746. doi: 10.1074/jbc.RA119.007360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan JJ, Ma XT, Liu C, Zhang XY, Wang CX. 2013. The current status and challenges in the development of fusion inhibitors as therapeutics for HIV-1 infection. Curr Pharm Des 19:1810–1817. doi: 10.2174/1381612811319100005. [DOI] [PubMed] [Google Scholar]
- 15.Chong H, Yao X, Qiu Z, Sun J, Zhang M, Waltersperger S, Wang M, Liu SL, Cui S, He Y. 2013. Short-peptide fusion inhibitors with high potency against wild-type and enfuvirtide-resistant HIV-1. FASEB J 27:1203–1213. doi: 10.1096/fj.12-222547. [DOI] [PubMed] [Google Scholar]
- 16.Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. 2007. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc Natl Acad Sci U S A 104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eron JJ, Gulick RM, Bartlett JA, Merigan T, Arduino R, Kilby JM, Yangco B, Diers A, Drobnes C, Demasi R, Greenberg M, Melby T, Raskino C, Rusnak P, Zhang Y, Spence R, Miralles GD. 2004. Short-term safety and antiretroviral activity of T-1249, a second-generation fusion inhibitor of HIV. J Infect Dis 189:1075–1083. doi: 10.1086/381707. [DOI] [PubMed] [Google Scholar]
- 18.Wang RR, Yang LM, Wang YH, Pang W, Tam SC, Tien P, Zheng YT. 2009. Sifuvirtide, a potent HIV fusion inhibitor peptide. Biochem Biophys Res Commun 382:540–544. doi: 10.1016/j.bbrc.2009.03.057. [DOI] [PubMed] [Google Scholar]
- 19.Chong H, Qiu Z, Su Y, Yang L, He Y. 2015. Design of a highly potent HIV-1 fusion inhibitor targeting the gp41 pocket. AIDS 29:13–21. doi: 10.1097/QAD.0000000000000498. [DOI] [PubMed] [Google Scholar]
- 20.Danial M, van Dulmen TH, Aleksandrowicz J, Potgens AJ, Klok HA. 2012. Site-specific PEGylation of HR2 peptides: effects of PEG conjugation position and chain length on HIV-1 membrane fusion inhibition and proteolytic degradation. Bioconjug Chem 23:1648–1660. doi: 10.1021/bc3002248. [DOI] [PubMed] [Google Scholar]
- 21.Cheng S, Wang Y, Zhang Z, Lv X, Gao GF, Shao Y, Ma L, Li X. 2016. Enfuvirtide-PEG conjugate: a potent HIV fusion inhibitor with improved pharmacokinetic properties. Eur J Med Chem 121:232–237. doi: 10.1016/j.ejmech.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaminskas LM, Williams CC, Leong NJ, Chan LJ, Butcher NJ, Feeney OM, Porter CJH, Tyssen D, Tachedjian G, Ascher DB. 2019. A 30 kDa polyethylene glycol-enfuvirtide complex enhances the exposure of enfuvirtide in lymphatic viral reservoirs in rats. Eur J Pharm Biopharm 137:218–226. doi: 10.1016/j.ejpb.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Chong H, Wu X, Su Y, He Y. 2016. Development of potent and long-acting HIV-1 fusion inhibitors. AIDS 30:1187–1196. doi: 10.1097/QAD.0000000000001073. [DOI] [PubMed] [Google Scholar]
- 24.Chong H, Xue J, Xiong S, Cong Z, Ding X, Zhu Y, Liu Z, Chen T, Feng Y, He L, Guo Y, Wei Q, Zhou Y, Qin C, He Y. 2017. A lipopeptide HIV-1/2 fusion inhibitor with highly potent in vitro, ex vivo, and in vivo antiviral activity. J Virol 91:e00288-17. doi: 10.1128/JVI.00288-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chong H, Xue J, Zhu Y, Cong Z, Chen T, Guo Y, Wei Q, Zhou Y, Qin C, He Y. 2018. Design of novel HIV-1/2 fusion inhibitors with high therapeutic efficacy in rhesus monkey models. J Virol 92:e00775-18. doi: 10.1128/JVI.00775-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng S, Chang X, Wang Y, Gao GF, Shao Y, Ma L, Li X. 2015. Glycosylated enfuvirtide: a long-lasting glycopeptide with potent anti-HIV activity. J Med Chem 58:1372–1379. doi: 10.1021/jm5016582. [DOI] [PubMed] [Google Scholar]
- 27.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. 2009. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A 106:5801–5806. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H, Jin R, Yao C, Zhang T, Wang M, Xia W, Peng H, Wang X, Lu R, Wang C, Xie D, Wu H. 2016. Combination of long-acting HIV fusion inhibitor albuvirtide and LPV/r showed potent efficacy in HIV-1 patients. AIDS Res Ther 13:8. doi: 10.1186/s12981-016-0091-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoddart CA, Nault G, Galkina SA, Thibaudeau K, Bakis P, Bousquet-Gagnon N, Robitaille M, Bellomo M, Paradis V, Liscourt P, Lobach A, Rivard ME, Ptak RG, Mankowski MK, Bridon D, Quraishi O. 2008. Albumin-conjugated C34 peptide HIV-1 fusion inhibitor: equipotent to C34 and T-20 in vitro with sustained activity in SCID-hu Thy/Liv mice. J Biol Chem 283:34045–34052. doi: 10.1074/jbc.M805536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie D, Yao C, Wang L, Min W, Xu J, Xiao J, Huang M, Chen B, Liu B, Li X, Jiang H. 2010. An albumin-conjugated peptide exhibits potent anti-HIV activity and long in vivo half-life. Antimicrob Agents Chemother 54:191–196. doi: 10.1128/AAC.00976-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swierczewska M, Lee KC, Lee S. 2015. What is the future of PEGylated therapies? Expert Opin Emerg Drugs 20:531–536. doi: 10.1517/14728214.2015.1113254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trevaskis NL, Kaminskas LM, Porter CJ. 2015. From sewer to saviour—targeting the lymphatic system to promote drug exposure and activity. Nat Rev Drug Discov 14:781–803. doi: 10.1038/nrd4608. [DOI] [PubMed] [Google Scholar]
- 33.Kaminskas LM, Kota J, McLeod VM, Kelly BD, Karellas P, Porter CJ. 2009. PEGylation of polylysine dendrimers improves absorption and lymphatic targeting following SC administration in rats. J Control Release 140:108–116. doi: 10.1016/j.jconrel.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 34.Kuo HH, Ahmad R, Lee GQ, Gao C, Chen HR, Ouyang Z, Szucs MJ, Kim D, Tsibris A, Chun TW, Battivelli E, Verdin E, Rosenberg ES, Carr SA, Yu XG, Lichterfeld M. 2018. Anti-apoptotic protein BIRC5 maintains survival of HIV-1-infected CD4+ T cells. Immunity 48:1183–1194. doi: 10.1016/j.immuni.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C, Cheng S, Zhang Y, Ding Y, Chong H, Xing H, Jiang S, Li X, Ma L. 2019. Long-acting HIV-1 fusion inhibitory peptides and their mechanisms of action. Viruses 11:e811. doi: 10.3390/v11090811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qi Z, Shi W, Xue N, Pan C, Jing W, Liu K, Jiang S. 2008. Rationally designed anti-HIV peptides containing multifunctional domains as molecule probes for studying the mechanisms of action of the first and second generation HIV fusion inhibitors. J Biol Chem 283:30376–30384. doi: 10.1074/jbc.M804672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perrin C, Fenouillet E, Jones IM. 1998. Role of gp41 glycosylation sites in the biological activity of human immunodeficiency virus type 1 envelope glycoprotein. Virology 242:338–345. doi: 10.1006/viro.1997.9016. [DOI] [PubMed] [Google Scholar]
- 38.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- 39.Araman C, Thompson RE, Wang S, Hackl S, Payne RJ, Becker CF. 2017. Semisynthetic prion protein (PrP) variants carrying glycan mimics at position 181 and 197 do not form fibrils. Chem Sci 8:6626–6632. doi: 10.1039/c7sc02719b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan DC, Chutkowski CT, Kim PS. 1998. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A 95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, Qian H, Miyamoto F, Naito T, Kawaji K, Kajiwara K, Hattori T, Matsuoka M, Watanabe K, Oishi S, Fujii N, Kodama EN. 2012. A simple, rapid, and sensitive system for the evaluation of anti-viral drugs in rats. Biochem Biophy Res Commun 424:257–261. doi: 10.1016/j.bbrc.2012.06.097. [DOI] [PubMed] [Google Scholar]
- 42.Fishburn CS. 2008. The pharmacology of PEGylation: balancing PD with PK togenerate novel therapeutics. J Pharm Sci 97:4167–4183. doi: 10.1002/jps.21278. [DOI] [PubMed] [Google Scholar]
- 43.Rajender Reddy K, Modi MW, Pedder S. 2002. Use of peginterferon alfa-2a (40 KD) (Pegasys) for the treatment of hepatitis C. Adv Drug Deliv Rev 54:571–586. doi: 10.1016/S0169-409X(02)00028-5. [DOI] [PubMed] [Google Scholar]
- 44.Algranati NE, Sy S, Modi M. 1999. A branched methoxy 40 kDa polyethylene glycol (PEG) moiety optimises the pharmacokinetics (PK) of peginterferon α-2a (PEG-IFN) and may explain its enhanced efficacy in chronic hepatitis C (CHC). Hepatology 40:190A. [Google Scholar]
- 45.Blick SK, Curran MP. 2007. Certolizumab pegol: in Crohn’s disease. BioDrugs 21:195–201. doi: 10.2165/00063030-200721030-00006. [DOI] [PubMed] [Google Scholar]
- 46.Xue J, Cong Z, Xiong J, Wang W, Jiang H, Chen T, Wu F, Liu K, Su A, Ju B, Chen Z, Couto MA, Wei Q, Qin C. 2013. Repressive effect of primary virus replication on superinfection correlated with gut-derived central memory CD4+ T cells in SHIV-infected Chinese rhesus macaques. PLoS One 8:e72295. doi: 10.1371/journal.pone.0072295. [DOI] [PMC free article] [PubMed] [Google Scholar]