

Abstract
With approval rates <5% and the probability of success in oncology clinical trials of 3.4%, more physiologically relevant in vitro three-dimensional models are being deployed during lead generation to select better drug candidates for solid tumors. Multicellular tumor spheroids (MCTSs) resemble avascular tumor nodules, micrometastases, or the intervascular regions of large solid tumors with respect to morphology, cell–cell and cell–extracellular matrix contacts, and volume growth kinetics. MCTSs develop gradients of nutrient and oxygen concentration resulting in diverse microenvironments with differential proliferation and drug distribution zones. We produced head and neck squamous cell carcinoma (HNSCC) MCTSs in 384-well U-bottom ultra-low-attachment microtiter plates and used metabolic viability and imaging methods to measure morphologies, growth phenotypes and the effects of 19 anticancer drugs. We showed that cell viability measurements underestimated the impact of drug exposure in HNSCC MCTS cultures, but that incorporating morphology and dead-cell staining analyses increased the number of drugs judged to have substantially impacted MCTS cultures. A cumulative multiparameter drug impact score enabled us to stratify MCTS drug responses into high-, intermediate-, and low-impact tiers, and maximized the value of these more physiologically relevant tumor cultures. It is conceivable that the viable cells present in MCTS cultures after drug exposure arise from drug-resistant populations that could represent a source of drug failure and recurrence. Long-term monitoring of treated MCTS cultures could provide a strategy to determine whether these drug-resistant populations represent circumstances where tumor growth is delayed and may ultimately give rise to regrowth.
Keywords: cancer and cancer drugs, high-content screening, image analysis, multicellular tumor spheroids, head and neck squamous cell carcinoma
Introduction
Historically, new cancer drug leads are identified in high-throughput screening (HTS) growth inhibition (GI) assays conducted in panels of tumor cell lines maintained and assayed in two-dimensional (2D) cultures.1–3 Cytotoxic compounds progress to antitumor efficacy studies in mice, and mechanism of action studies are instigated for compounds that demonstrate in vivo efficacy.2,4,5 For molecular agents targeting specific oncogenic alterations in tumor cells, biochemical or cell-based screens usually precede these steps.1–3 However, <5% of new small-molecule cancer drugs that enter phase I clinical trials gain Food and Drug Administration (FDA) approval, and between 2000 and 2015, the overall probability for success in oncology clinical trials was a dismal 3.4%.2,4–8 To improve clinical development success rates for solid tumors, more physiologically relevant in vitro three-dimensional (3D) models are being deployed at the lead generation stage to select better cancer drug candidates.9–24 Multicellular tumor spheroids (MCTSs) resemble avascular tumor nodules, micrometastases, or the intervascular regions of large solid tumors with respect to morphology, cell–cell and cell–extracellular matrix (ECM) contacts, and volume growth kinetics that develop gradients of nutrient distribution and oxygen concentration that give rise to diverse microenvironments with differential zones of proliferation and drug distribution.9,10,16,20,22,23,25
Head and neck cancers (HNCs) are the eighth leading cause of cancer worldwide, with an estimated 600,000 new cases and 300,000 deaths per annum.26–28 In 2019 it is projected that 53,000 people in the United States will develop oral cavity or pharynx cancer and 10,860 will die of these cancers. Seven drugs are approved for HNC therapy: methotrexate, 5-fluorouracil (5-FU), bleomycin, cisplatin, docetaxel, cetuximab, and pembrolizumab. However, HNC cure rates have hovered at around 50% for >30 years, and patients with advanced, recurrent, or metastatic HNC have median survival rates of 6–12 months.26–31 Only 10%–25% of HNC patients respond to monotherapy with the approved chemotherapeutics or cetuximab, an epidermal growth factor receptor (EGFR) blocking antibody, and they have not improved 5-year survival or cure rates.26,27 Although pembrolizumab (Keytruda) was well tolerated in patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC) and produced clinically relevant antitumor activity, only 16% of HNSCC patients responded to Keytruda treatment.32–34 The low response rates and limited efficacy of existing drugs for HNSCC underscores the need to discover new and effective HNC therapies.
We have described the production of HNSCC MCTSs in 384-well U-bottom ultra-low-attachment microtiter plates (ULA-plates) at a scale compatible with HTS cancer drug discovery.9,20,22,25 We developed cell viability and imaging methods to characterize HNSCC MCTS morphologies, viability, and growth phenotypes, and to analyze the effects of anticancer drug exposure.9,20,22,25 The present paper describes the implementation of five HNSCC MCTS cultures with different morphologies and growth phenotypes to screen a set of 19 cancer drugs (Suppl. Table S1). The set includes the five chemotherapy drugs that are approved for HNC: methotrexate, 5-FU, bleomycin, cisplatin, and docetaxel. The EGFR tyrosine kinase inhibitors gefitinib and erlotinib were used as surrogates for cetuximab. The set contains three other tyrosine kinase inhibitors: sunitinib, a multitargeted receptor tyrosine kinase inhibitor; dasatinib, an inhibitor of SRC family protein tyrosine kinases; and ruxolitinib, a Janus kinase inhibitor. Two phosphatidylinositol 3-kinase (PI3K) inhibitors, buparlisib (a pan-PI3K inhibitor) and dactolisib (a dual PI3K and mTOR inhibitor), were selected because genes in the PI3K signaling pathway were among the most commonly mutated in HNC.28 The set contains three topoisomerase inhibitor chemotherapeutics, etoposide, topotecan, and doxorubicin, and four molecularly targeted anticancer agents: the molecular chaperone heat shock protein 90 (Hsp90) ATPase inhibitor ganetespib (STA-9090), the 26S proteasome inhibitor bortezomib, the histone deacetylase (HDAC) inhibitor romidepsin, and the mTOR inhibitor everolimus. Drugs not approved for HNC either are approved for other cancer types or are in clinical evaluation.
To provide a benchmark for comparison, we also screened the 19 drugs in established 2D monolayer GI assays using the same HNSCC cell lines.35,36 In 95 possible pairwise cancer drug × HNSCC cell line experiments performed using the CellTiter-Blue (CTB) metabolic activity viability reagent, 97.9% of the 2D monolayer cultures exhibited concentration-dependent GI responses compared with only 35.8% of the matching MCTS cultures. Only 24.4% of the pairwise drug × HNSCC cell line combinations reduced MCTS calcein AM (CAM) live-cell staining by ≥50%, whereas 67.8% increased the ethidium homodimer (EHD) dead-cell staining by ≥50%. When MCTS morphology parameters including size, shape, the type of perimeter, and the density or compactness of spheroids were considered, 89.5% of the drug × HNSCC cell line conditions altered more than one morphology attribute. Our results demonstrate that the application of multiple analysis methods is required to accurately determine the impact of cancer drugs on HNSCC MCTS cultures and to maximize the value of these more physiologically relevant tumor cultures.
Materials and Methods
Reagents
Thirty-seven percent formaldehyde was purchased from Sigma-Aldrich (St. Louis, MO). Hoechst 33342 was purchased from Life Technologies (Thermo Fisher Scientific, Waltham, MA). DMSO 99.9% high-performance liquid chromatography grade was obtained from Alfa Aesar (Ward Hill, MA). Dulbecco’s Mg2+- and Ca2+-free phosphate-buffered saline (PBS) was purchased from Gibco (Grand Island, NY). Dulbecco’s modified Eagle’s medium (DMEM) and Dulbecco’s modified Eagle’s medium/Ham’s F12 50/50 (DMEM/F12) was purchased from Corning (Manassas, VA). Fetal bovine serum (FBS), l-glutamine, and penicillin/streptomycin (P/S) were purchased from Thermo Fisher Scientific. CTB was purchased from Promega Corporation (Madison, WI). CAM and EHD were purchased from Life Technologies (Thermo Fisher Scientific).
Anticancer Drug Test Set and Mechanisms of Action
Nineteen FDA-approved cancer drugs were obtained from commercial sources and shipped to the University of Pittsburgh by the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP).37,38 Supplemental Table S1 lists the 19 drugs, their mechanisms of action, and the cancers for they have been either approved or under evaluation in clinical trials. Five of the drugs are approved for HNC among other cancers. The test set includes two EGFR tyrosine kinase inhibitors as surrogates for cetuximab, three additional tyrosine kinase inhibitors, two PI3K family inhibitors, three topoisomerase inhibitors, and four molecularly targeted anticancer agents.
Cells and Tissue Culture
Five human HNSCC cell lines were provided by Dr. Jennifer Grandis of the Head and Neck Cancer Spore at the University of Pittsburgh Medical Center Hillman Cancer Center and were maintained in a humidified incubator at 37 °C, 5% CO2, and 95% humidity: Cal33, FaDu, UM-22B, BICR56, and OSC-19. All cell lines were cultured in DMEM supplemented with 10% FBS, 1% l-glutamine, and 1% P/S. The culture medium for the FaDu and OSC-19 cell lines was also supplemented with 1% nonessential amino acids, and the medium for the BICR56 cell line was supplemented with 0.4 μg/mL hydrocortisone. HNSCC cell lines were passaged or used to generate MCTSs after isolated cell suspensions were prepared from tissue culture flasks by dissociating cells with trypsin and centrifugation at 270g for 5 min at room temperature, and resuspension in growth media. The number of viable trypan blue-excluding cells in the cell suspension was counted using a hemocytometer.
Generation of HNSCC MCTSs in ULA-Plates
We have previously described the production of MCTSs by seeding HNSCC cell lines into 384-well ULA-plates (cat. 4516; Corning, Tewksbury, MA).9,20,22,25 Briefly, 384-well ULA-plates were rehydrated by the addition of 50 μL of serum-free culture medium to each well and incubated in a humidified incubator for 15 min. Media was removed from the wells of the ULA-plates and 45 μL of a single-cell suspension of the HNSCC cell lines at a seeding density of 2500 cells/well in the appropriate growth medium was transferred into each well using a Matrix automated multi-channel pipette (Thermo Fisher Scientific). ULA-plates were centrifuged at 17g for 1 min and then placed in an incubator at 37 °C, 5% CO2, and 95% humidity for the indicated time periods. If HNSCC MCTS cultures were maintained in the ULA-plates beyond 3 days, spent media was exchanged for fresh medium every 3 days using a Janus MDT Mini (PerkinElmer, Waltham, MA) automated liquid handler platform equipped with a 384-well transfer head. Each medium exchange cycle consisted of 2 × 20 μL aspiration and discard steps, followed by 2 × 20 μL fresh media dispense steps. Three media exchange cycles were performed to achieve ~85% exchange of fresh medium for spent medium and a uniform volume of 45 μL/well.
Investigation of HNSCC MCTS Morphology, Viability, and Growth in ULA-Plates by High-Content Imaging
We used the ImageXpress Micro (IXM) automated wide-field high-content imaging platform integrated with MetaXpress Imaging and Analysis software (Molecular Devices LLC, Sunnyvale, CA) to acquire and analyze images of HNSCC MCTSs. The IXM optical drive uses a 300 W Xenon lamp broad-spectrum white light source and a 1.4-megapixel 2/3-inch-chip Cooled CCD Camera and optical train for standard fluorescence imaging and transmitted light (TL) phase-contrast imaging. The IXM is equipped with Zero Pixel Shift (ZPS) filter sets: DAPI, FITC/ALEXA 488, CY3/TRITC, CY5, and Texas Red. Single images of HNSCC MCTSs were sequentially acquired using a 4× Plan Apo 0.20 NA objective in both the TL and fluorescent image acquisition modes; DAPI, FITC, and Texas Red.9,20,22,25,39 To acquire the best focus images of MCTSs, we used the IXM automated image-based focus algorithm to obtain a coarse-focus (large-micrometer steps) set of images of Hoechst stained objects in the DAPI channel for the first MCTS to be imaged, followed by a fine-focus (small-micrometer steps) set of images. In all subsequent wells and channels only a fine-focus set of images were acquired to select the best focus Z plane.9,20,22,25,39 MCTS morphology and growth were assessed daily by the acquisition of 4× TL images on the IXM, and we used the line-scan tool of the MetaXpress image analysis software to measure HNSCC MCTS diameters.25,39
To label viable and/or dead cells within HNSCC 2D monolayer and MCTS cultures, we used the CAM live-cell and EHD dead-cell reagents. The nonfluorescent cell-permeant CAM dye is converted to green fluorescent calcein after acetoxymethyl ester hydrolysis by intracellular esterase enzymes in live cells.9,20,25 The cell-impermeant EHD-1 dye is excluded from live cells but stains the DNA of dead cells.9,20,25 HNSCC 2D monolayer and MCTS cultures were incubated with a cocktail of the Hoechst (8 μg/mL) DNA stain, the CAM (2.5 μM) live reagent, and the EHD (5 μM) dead reagent for 1 h, and single images of HNSCC MCTSs were sequentially acquired on the IXM using a 4× objective in both the TL and fluorescent image acquisition modes: DAPI, FITC, and Texas Red channels. We used the multiwavelength cell scoring (MWCS) image analysis module to analyze HNSCC MCTS fluorescent images as described previously.9,20,22 To create a whole MCTS mask, we set the approximate minimum width of the Hoechst stained nuclei of the MCTS to be 150 μm, with an approximate maximum width of 550 μm, and applied a threshold intensity above the local background of 70. The total MCTS mask from the Hoechst channel was used to count the number of MCTSs per image, typically one. After applying user-defined background average intensity thresholds, typically 50–70 in both the FITC and Texas Red channels, the MWCS module image segmentation then created total MCTS masks in all three fluorescent channels. The derived HNSCC total MCTS masks were used to quantify the mean integrated fluorescence intensity (MIFI) of the CAM live-cell signal in the FITC channel and the EHD dead-cell signal in the Texas Red channel. MIFI values represent the total pixel fluorescent intensities in channel 1, 2, or 3 within MCTS masks of positively stained MCTSs above preset background thresholds.
Analysis of HNSCC 2D Monolayer and MCTS Culture Viability and GI Using the CTB Reagent
The homogeneous CTB cell viability reagent provides a fluorescent method for monitoring cell viability and/or GI that is based on the ability of living cells to convert the redox dye resazurin into a fluorescent end-product resorufin. 2D monolayer HNSCC GI assays were performed as described previously, save that CTB was used instead of CellTiter-Glo (CTG) as the detection reagent.35,36 Each of the five HNSCC cell lines was harvested, counted, and seeded into 384-well assay plates at 500 cells/well in 45 μL of tissue culture media and cultured overnight at 37 °C, 5% CO2, and 95% humidity. On the next day, 5 μL of test compounds at the indicated concentrations or plate controls diluted in SFM was transferred to assay plates on the Janus MDT Mini liquid handler equipped with a 384-well transfer head, and assay plates were then cultured for an additional 72 h in an incubator at 37 °C, 5% CO2, and 95% humidity. Maximum control wells (max controls, n = 32) received 5 μL of DMSO (0.25% DMSO final) and minimum control wells (min controls, n = 32) received 200 μM doxorubicin plus 0.25% DMSO. After the 72 h compound exposure period, 10 μL of the CTB cell viability detection reagent was dispensed into the wells of HNSCC assay plates and incubated for 4 h at 37 °C, 5% CO2, and 95% humidity before capturing the relative fluorescent unit (RFU) signals (excitation 560 nm/emission 590 nm) on a SpectraMax M5e (Molecular Devices, LLC, Sunnyvale, CA) microiter plate reader platform.
For MCTS GI assays HNSCC cell lines were seeded at 2500 cells/well into 384-well ULA-plates in 45 μL of growth medium incubated at 37 °C, 5% CO2, and 95% humidity for 3 days.25 After 3 days, spent media was exchanged for fresh media as described above and test compounds at the indicted concentrations were transferred to the wells using a Janus MDT Mini platform equipped with a 384-well transfer head, and the plates were returned to the incubator for an additional 72 h. Maximum and minimum control wells received 0.25% DMSO and 200 μM doxorubicin plus 0.25% DMSO, respectively. After the 72 h compound exposure period, 10 μL of the CTB cell viability detection reagent was dispensed into the wells of HNSCC MCTS assay plates and incubated for 4 h at 37 °C, 5% CO2, and 95% humidity before capturing RFU signals (excitation 560 nm/emission 590 nm) on a SpectraMax M5e microtiter plate reader platform.
Data Processing, Analysis, and Curve Fitting
For HNSCC 2D monolayer and MCTS GI (GI50) assays, the mean maximum DMSO control wells (max controls, n = 32) and 200 μM doxorubicin mean minimum plate control wells (min controls, n = 32) were used to normalize the data from compound-treated wells and to represent uninhibited growth and 100% cytotoxicity, respectively. The GI50 data were fit to a nonlinear sigmoidal log (inhibitor) versus normalized response variable slope model using the following equation: Y = 100/(1 + 10^((LogIC50 – X)*Hillslope)), where Y is the percent GI and X is the corresponding log10 of the compound concentration. GI50 is the concentration of compound that gives a 50% response, halfway between 0% and 100%. Hillslope describes the steepness of the curve. All curve fitting, linear regression analysis, and graphs were created using the GraphPad Prism 6 software.
Drug Impact Scoring
To facilitate the analysis of the effects of cancer drugs on HNSCC cultures, we used a drug impact score to summarize our observations for each of the readouts that we acquired: HNSCC GI, MCTS live/dead staining, and MCTS morphology. If a drug produced a calculable GI50 in CTB GI assays, it scored 1; if it failed to produce a calculable GI50 but exhibited a concentration-dependent GI response and achieved ≥25% GI, it scored 0.5; and it received a score of 0 if it achieved neither of these attributes. In MCTS HNSCC cultures stained with the CAM live reagent, if the maximum concentration of the drug reduced the CAM MIFI by ≥50% relative to DMSO controls, it scored 1; if it reduced the CAM MIFI by ≥20% but <50%, it scored 0.5; and if it reduced the CAM MIFI by <20%, it scored 0. In MCTS HNSCC cultures stained with the EHD dead reagent, if the maximum concentration of the drug increased the EHD MIFI by ≥50% relative to DMSO controls, it scored 1; if it increased the EHD MIFI by ≥20% but <50%, it scored 0.5; and if it increased the EHD MIFI by <20%, it scored 0. We analyzed the effects of drugs on three MCTS morphology readouts: MCTS perimeters and shape, MCTS density or compactness, and MCTS size indicated by diameter measurements. If a drug altered ≥2 of the 3 morphology readouts, it scored 1; if it only altered 1 readout, it scored 0.5; and if it did not change any of the 3 readouts, it scored 0. The maximum cumulative MCTS drug impact score for an individual HNSCC cell line was 4, and 20 across all five HNSCC cell lines.
Results
Cancer Drug Induced GI in 2D Monolayer and MCTS HNSCC Cultures
Supplemental Figure S1 shows representative TL and color composite images of CAM (FITC channel) live-cell and EHD (Texas Red channel) dead-cell stained MCTSs from DMSO control wells captured on days 3 and 6 of a typical drug screening experiment. Consistent with our previous studies, after 3 days in culture FaDu and Cal33 cell lines formed condensed MCTSs with a smooth and even periphery, OSC-19 and BICR56 cell lines produced rounded MCTSs with uneven perimeters, and the UM-22B cell line formed cell aggregates with irregular outer margins (Suppl. Fig. S1).9,20,22,25 The BICR56 cell line formed smaller MCTSs ~200–250 μm in diameter, while the other cell lines produced larger MCTSs ~350–400 μm in diameter. FaDu, UM-22B, and Cal33 MCTSs were larger on day 6 than on day 3, while the sizes of BIRC56 and OSC-19 MCTSs were unchanged. On day 3, MCTSs and aggregates exhibited strong CAM staining with little or no EHD staining, indicating that nearly all cells were viable. By day 6, UM-22B cell aggregates coalesced into more condensed MCTSs with irregular outer margins. MCTSs also exhibited strong CAM staining on day 6, indicating that most cells were viable, although FaDu and OSC-19 MCTSs exhibited EHD staining in their interior regions, suggestive of the emergence of necrotic cores.25
We used the CTB metabolic activity reagent to monitor MCTS cell viability, growth, and drug-induced GI.25 In 2D HNSCC monolayer GI assays run for comparison, cell lines were seeded at 500 cells/well in 384-well flat-bottom tissue culture-treated plates and cultured overnight before 72 h compound exposure.35,36 HNSCC cell lines cultured in 2D monolayers exhibited exponential growth rates (r2 > 0.93), and their doubling times provided the following rank order of growth: Cal33 (33.6 h) = UM-22B (35.5 h) = FaDu (37.4 h) < BIRC56 (39.5 h) << OSC-19 (61.8 h) (Suppl. Fig. S2). We have shown that growth rates for HNSCC cell lines in MCTS cultures are linear with a rank order of FaDu < UM-22B << Cal33, with BIRC56 remaining static but viable, and OSC-19 slowly declining over time in culture.25 To define assay signal windows and normalize the data for GI50 determinations, we used 0.2% DMSO control wells to represent uninhibited growth (0% GI, max controls, n = 32), and 200 μM doxorubicin + 0.2% DMSO control wells to represent fully inhibited growth (100% GI, min controls, n = 32), respectively, and to calculate signal-to-background (S/B) ratios and Z′-factor coefficient assay performance statistics (Suppl. Table S2). S/B ratios for 2D monolayer cultures were ~2-fold higher than those for MCTS cultures of the same cell lines, with OSC-19 having the closest S/B ratios for 2D and 3D cultures. S/B ratios ranged between 12- and 18-fold in 2D monolayer cultures, and between 5- and 10-fold in MCTS cultures. 2D and MCTS CTB GI assays produced Z′-factor coefficients ≥0.5, indicating that they were robust and reproducible enough for HTS.
Figure 1 shows representative GI50 curves produced in 2D monolayer and MCTS cultures exposed to the approved HNC drugs: 5-FU, methotrexate, bleomycin, docetaxel, and cisplatin. In 2D monolayer cultures, cisplatin, docetaxel, and 5-FU exhibited complete concentration–response curves (two asymptotes) with good-quality curve fits (r2 > 0.95), efficacies >80%, and calculable GI50 values (Fig. 1, Table 1). However, bleomycin and methotrexate exhibited incomplete curves (only one asymptote) in 2D monolayer cultures with lesser-quality curve fits (r2 = 0.6–0.9), efficacies in the 50%–80% range, and calculable GI50 values (Fig. 1, Table 1). Only cisplatin and docetaxel produced complete concentration–response curves with good-quality curve fits and GI50 values in most MCTS cultures (Fig. 1). Docetaxel did not, however, exhibit concentration-dependent GI in Cal33 MCTSs. 5-FU and bleomycin produced incomplete curves with lesser-quality curve fits in some MCTS cultures and efficacies in the 25%–80% range, but only produced GI50 values in UM-22B MCTSs (Fig. 1, Table 1). In some MCTS cultures, however, 5-FU and bleomycin failed to achieve >25% efficacy or exhibit concentration-dependent GI. Methotrexate did not achieve >25% efficacy or show evidence of concentration-dependent GI in any MCTS cultures (Fig. 1).
Figure 1.

2D monolayer and MCTS CTB GI curves for FDA-approved HNSCC drugs. For 2D monolayer cultures, the five HNSCC cell lines were seeded into 384-well assay plates and cultured for 24 h before they were exposed to the indicated concentrations of cisplatin, docetaxel, 5-FU, bleomycin, or methotrexate for 72 h prior to the addition of the CTB and measurement of the RFU signals. For MCTS cultures, the five HNSCC cell lines were seeded in 384-well ULA-plates, and after 3 days in culture, the MCTSs were exposed to the indicated concentrations of the drugs for 72 h prior to the addition of CTB and measurement of the RFU signals. The mean maximum (0.5% DMSO) and minimum (200 μM doxorubicin + 0.5% DMSO) plate control CTB RFUs were used to normalize the RFU data from the compound treated wells as percent inhibition of growth and the GI50 data were fitted to a nonlinear sigmoidal log inhibitor concentration versus the normalized response variable slope model using the GraphPad Prism 6 software. The normalized mean ± SD (n = 3) GI data from triplicate wells for each compound concentration are presented. The data and curve fits for 2D monolayer (●) and MCTS (●) cultures are indicated in blue and red, respectively. Representative experimental data from one of three or four independent experiments are shown.
Table 1.
HNSCC 2D Monolayer and MCTS Culture GI50 Determinations.
| Compound | 2D Monolayer HNSCC Culture GI50 Values (μM) |
MCTS HNSCC Culture GI50 Values (μM) |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FaDu |
UM-22B |
Cal33 |
BIRC56 |
OSC-19 |
FaDu |
UM-22B |
Cal33 |
BIRC56 |
OSC-19 |
|||||||||||
| M | SD | M | SD | M | SD | M | SD | M | SD | M | SD | M | SD | M | SD | M | SD | M | SD | |
| 5-FU* | 24.6 | 7.03 | 10 | 3.9 | 2.8 | 1.07 | 2.37 | 1.07 | 14.8 | 4.0 | >500 | 135.4 | 54.1 | >500 | >500 | 0.0 | >500 | |||
| Methotrexate* | 10.6 | 6.3 | 6.2 | 4.4 | 4.7 | 1.0 | 3.3 | 2.1 | >20 | >20 | >20 | >20 | >20 | >20 | ||||||
| Bleomycin* | 16.5 | 2.3 | 5.2 | 1.0 | 12.4 | 1.5 | 6.6 | 1.5 | 18.3 | 1.9 | >20 | 18.1 | 1.2 | >20 | >20 | >20 | ||||
| Docetaxel* | 1.2 | NA | 1.4 | NA | 0.7 | NA | 0.8 | NA | 2.3 | NA | 20.6 | 6.1 | >100 | 3.73 | 17.7 | |||||
| Cisplatin* | 11.1 | 3.9 | 8.7 | 2.8 | 6.0 | 0.6 | 6.7 | 1.1 | 17.0 | 3.7 | 99.8 | 40.3 | 52.2 | 29.1 | 88.4 | 8.6 | 80.5 | 20.9 | 114 | 32.2 |
| Gefitinib | 6.9 | 1.6 | 55.6 | 31.5 | 8.5 | 2.4 | 7.3 | 3.8 | 67.5 | 24.8 | >100 | >100 | >100 | >100 | >100 | |||||
| Erlotinib | >500 | 433 | 21.6 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | |||||||||
| Dasatinib | 13.1 | 3.1 | 17.0 | 2.8 | 4.5 | 1.0 | 4.1 | 2.5 | >20 | >20 | >20 | >20 | >20 | >20 | ||||||
| Sunitinib | 5.4 | 1.7 | 5.2 | 1.0 | 11.5 | 2.4 | 5.9 | 2.3 | 8.6 | 2.0 | 21.7 | 3.4 | 14.2 | 0.8 | 12.5 | 2.4 | 15.3 | 8.9 | 19.8 | 5.5 |
| Ruxolitinib | 30.9 | 2.1 | 38.1 | 7.9 | 35.5 | 4.4 | 43.5 | 7.2 | 75.5 | 6.4 | >200 | >200 | >200 | 111 | 15.1 | >200 | ||||
| Doxorubicin | 0.017 | 0.004 | 0.013 | 0.01 | 0.025 | 0.007 | 0.03 | 0.01 | 0.01 | 0.00 | 3.45 | 1.17 | 0.89 | 0.63 | 1.13 | 0.65 | 5.40 | 2.97 | 5.15 | 2.98 |
| Etoposide | 32.6 | 6.0 | 36.2 | 9.1 | 50.2 | 10.7 | 31.5 | 11.6 | >100 | >100 | >100 | >100 | >100 | >100 | ||||||
| Topotecan | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | ||||||||||
| Dactolisib | 7.6 | 4.1 | >50 | 1.4 | 0.4 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | ||||||||
| Buparlisib | >20 | >20 | 13.3 | 2.4 | 16.7 | 6.4 | >20 | >20 | >20 | >20 | >20 | >20 | ||||||||
| Romidepsin | >0.5 | >0.5 | >0.5 | >0.5 | >0.5 | >0.5 | >0.5 | >0.5 | >0.5 | >0.5 | ||||||||||
| Bortezomib | 0.3 | 0.0 | 0.1 | 0.0 | 0.1 | 0.02 | 0.2 | 0.1 | 0.3 | 0.14 | >0.5 | >0.5 | >0.5 | >0.5 | >0.5 | |||||
| Everolimus | 29.8 | 6.2 | 28.8 | 4.7 | 34.0 | 6.2 | 28.5 | 8.7 | 40.1 | 3.1 | 88.7 | 6.8 | 32.5 | 9.1 | 48.0 | 8.4 | 66.9 | 19.4 | 90.2 | 5.19 |
| Ganetespib | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | >1 | ||||||||||
M = mean GI50 from three or four independent experiments; SD = standard deviation of the mean GI50; > = greater than the maximum concentration tested.
Drugs approved by the FDA for HNSCC therapy.
Table 1 summarizes the CTB assay GI50 values for the 19 test drugs in 2D monolayer and MCTS HNSCC cultures assayed at the top starting concentrations listed in Supplemental Table S1. Four drugs effectively inhibited the growth of both culture formats, nine were selective for 2D monolayer cultures, and three were ineffective in both culture conditions (Table 1). Of the 19 drugs tested, 14 produced GI50 values in 2D cultures of the FaDu and Cal33 cell lines, UM-22B and BIRC56 were sensitive to 13 of the agents, and OSC-19 only responded to 10 drugs (Table 1). In MCTS cultures of the same cell lines, 7 produced GI50 values in UM-22B MCTSs, 6 in BIR56 MCTSs, 5 in FaDu and OSC-19 MCTSs, and only 4 in Cal33 MCTSs (Table 1). The four compounds that inhibited the growth of all five HNSCC cell lines in both culture formats were cisplatin, sunitinib, doxorubicin, and everolimus. Docetaxel inhibited the growth of all HNSCC cultures except for Cal33 MCTSs (Table 1). On average, the GI50 values for doxorubicin, cisplatin, and docetaxel were substantially (≥8.6-fold) lower in 2D monolayers than in MCTS cultures, while sunitinib and everolimus were only 2.5- and 2-fold lower, respectively. Overall, the CTB GI data indicate that HNSCC MCTS cultures were substantially more resistant than 2D monolayer cultures to GI induced by the 19 cancer drugs (Fig. 1, Table 1).
Cancer Drug Effects on HNSCC MCTS Cell Viability and/or Death
Supplemental Table S4A summarizes the CAM live- and EHD dead-cell MIFI values, normalized and expressed as percent of DMSO controls, in MCTS cultures exposed to the top concentrations of the 19 drugs listed in Supplemental Table S1. Figure 2A shows color composite fluorescent images of CAM and EHD stained MCTSs exposed to DMSO, 5-FU, methotrexate, bleomycin, docetaxel, or cisplatin for 72 h. Figure 2B shows the MIFI values of the CAM and EHD staining in 6-day HNSCC MCTS DMSO controls, and Figure 2C–G shows the normalized CAM and EHD MIFI values for MCTS exposed to the five HNC drugs. Exposure to 100 μM docetaxel or 500 μM cisplatin dramatically increased the EHD staining apparent in the CAM/EHD composite images of HNSCC MCTSs (Fig. 2A) and their EHD MIFI values (Fig. 2F,G). Although exposure to docetaxel and cisplatin increased EHD staining by three- to sixfold over DMSO controls, the corresponding reductions in CAM MIFI values were not as substantial (Fig. 2F,G, Suppl. Table S4A). For example, docetaxel increased EHD staining by three-fold in FaDu MCTSs but only reduced CAM staining by twofold. Exposure to 500 μM 5-FU increased EHD staining in UM-22B, Cal33, and OSC-19 MCTSs between 1.5- and 4-fold, but only reduced CAM staining by >20% in FaDu MCTSs (Fig. 3A,C, Suppl. Table S4A). Although neither methotrexate nor bleomycin exposure substantially (>50%) increased EHD staining in any MCTSs at 20 μM, they both decreased CAM staining by >50% in FaDu and UM-22B MCTSs (Fig. 3A,D,E, Suppl. Table S4A). Although 68.7% of the 95 drug–HNSCC MCTS pairings substantially increased MCTS EHD dead staining by ≥50%, only 24.4% reduced CAM live staining by ≥50% (Suppl. Table S4A).
Figure 2.

Live/dead staining of HNSCC MCTS cultures exposed to DMSO or HNSCC approved drugs. (A) Color composite fluorescent images of live-cell CAM (green) and dead-cell EHD (red) stained HNSCC MCTSs exposed to DMSO or the top concentrations of HNSCC approved drugs for 72 h. Representative images from multiple independent experiments are presented. All scale bars represent 300 μm. (B) The MIFI values of the CAM (■) and EHD (■) staining in HNSCC MCTS DMSO controls. The MIFI CAM (■) and EHD (■) signals of HNSCC MCTSs exposed to the top concentration of the HNSCC approved drugs were normalized and expressed as percent of DMSO controls for (C) 5-FU, (D) methotrexate, (E) bleomycin, (F) docetaxel, and (G) cisplatin. The MIFI or normalized MIFI data for each compound are presented as mean ± SD (n = 3) from triplicate wells. Representative data from one of three independent experiments are shown.
Figure 3.

Morphologies of HNSCC MCTS cultures exposed to DMSO or HNSCC approved drugs. (A) TL images of HNSCC MCTS cultures exposed to DMSO or the top concentrations of the HNSCC approved drugs for 72 h. Representative images from multiple independent experiments are presented. All scale bars represent 300 μm. (B) The line-scan tool of the MetaXpress image analysis software was used to measure the diameters (μm) of the five HNSCC MCTSs in the TL images of day 3 (□) and day 6 (■) DMSO controls. MCTS diameters (μm) extracted from the TL images of HNSCC MCTS cultures exposed to DMSO (□) or the top concentrations (■) of (C) 5-FU, (D) methotrexate, (E) bleomycin, (F) docetaxel, and (G) cisplatin for 72 h. The mean ± SD (n = 3) diameter data from triplicate wells for DMSO controls and each compound are presented. Representative data from one of three independent experiments are shown.
Cancer Drug Effects on HNSCC MCTS Morphologies
To evaluate the effects of drug exposure on MCTS morphology, we acquired TL images and qualitatively compared the perimeters, shapes, and compactness of DMSO controls to treated MCTSs, and measured spheroid or aggregate diameters using the line-scan tool of the MetaXpress image analysis software. Supplemental Table S5A summarizes the changes in MCTS morphology parameters relative to DMSO controls in MCTS cultures exposed to the top concentrations of the 19 drugs. Figure 3A shows the TL images of MCTSs exposed to DMSO, 5-FU, methotrexate, bleomycin, docetaxel, or cisplatin. The changes in MCTS architecture and morphology caused by drug exposure varied with both the specific agent and the HNSCC cell line (Fig. 3, Suppl. Table S5A). For example, although exposure to docetaxel produced diffuse irregular cell aggregates in all five MCTSs, four of five exhibited larger diameters than control MCTSs, with Cal33 MCTSs being smaller (Fig. 3A,F, Suppl. Table S5A). In contrast, cisplatin exposure produced diffuse irregular cell aggregates in only UM-22B and OSC-19 MCTSs, distorted the shape and increased the size of Cal33 MCTSs, produced uniformly dense/darker FaDu MCTSs that lacked the lighter outer regions of control MCTSs, and decreased the diameters of BIRC56 MCTSs (Fig. 3A,G, Suppl. Table S5A). Exposure of MCTS to 5-FU, methotrexate, or bleomycin reduced MCTS diameters relative to DMSO controls and induced a variety of changes in their perimeters, shapes, and compactness (Fig. 3A,C–E, Suppl. Table S5A). Across 25 possible pairings of HNSCC MCTSs with FDA-approved HNC drugs, 7 (28%) reduced, 5 (20%) increased, and 13 (52%) did not alter MCTS or aggregate diameters relative to DMSO controls (Fig. 3A,C–E, Suppl. Table S5A). Exposure to the 19 drugs altered at least one morphological feature in 89.5% of the 95 drug–HNSCC MCTS pairings (Suppl. Table S5A).
Effects of Exposure to Sunitinib and Everolimus on HNSCC MCTS Cultures
Figure 4A and B shows the TL, Hoechst, CAM, and EHD images of MCTSs exposed to 100 μM sunitinib or everolimus, respectively, for 72 h, alongside DMSO controls. Representative 2D monolayer and MCTS CTB GI50 curves produced by sunitinib (Fig. 4C–G) or everolimus (Fig. 4H–L) are presented together with their corresponding normalized CAM and EHD MIFI values (Fig. 4M,N), and MCTS diameter measurements (Fig. 4O,P). MCTSs exposed to sunitinib displayed altered morphologies, reduced Hoechst and CAM staining, and increased EHD staining (Fig. 4A). In 2D monolayer cultures, sunitinib produced complete CTB concentration–response curves with good-quality curve fits and GI50 values in the 5–11 μM range (Fig. 4C–G, Table 1). Sunitinib also produced complete CTB concentration–response curves in MCTS cultures with GI50 values in the 12.5–22 μM range, except for OSC-19, with lesser-quality curve fits. Sunitinib exposure reduced CAM staining and increased EDH staining by ≥50% in four of five HNSCC MCTS cultures (Fig. 4M, Suppl. Table S4A) and altered ≥1 morphology parameter in all MCTSs (Fig. 4O, Suppl. Table S5A). MCTSs exposed to everolimus also displayed changed morphologies, reduced Hoechst and CAM staining, and increased EHD staining (Fig. 4B). HNSCC cultures exposed to everolimus produced incomplete CTB curves with lesser-quality curve fits that produced GI50 values in the 28–40 μM range for 2D monolayers and in the 32–90 μM range for MCTSs (Fig. 4H–L, Table 1). Everolimus increased EDH staining ≥50% in all five MCTS cultures, but reduced CAM staining by ≥50% in only two of five (Fig. 4N, Suppl. Table S4A). Everolimus changed ≥1 morphology parameter in all MCTS cultures (Fig. 4P, Suppl. Table S5A). There was reasonably good consensus among the different analysis methods that both sunitinib and everolimus effectively inhibited HNSCC MCTS growth in a concentration-dependent manner (Fig. 4, Table 1, Suppl. Tables S4A and S5A).
Figure 4.
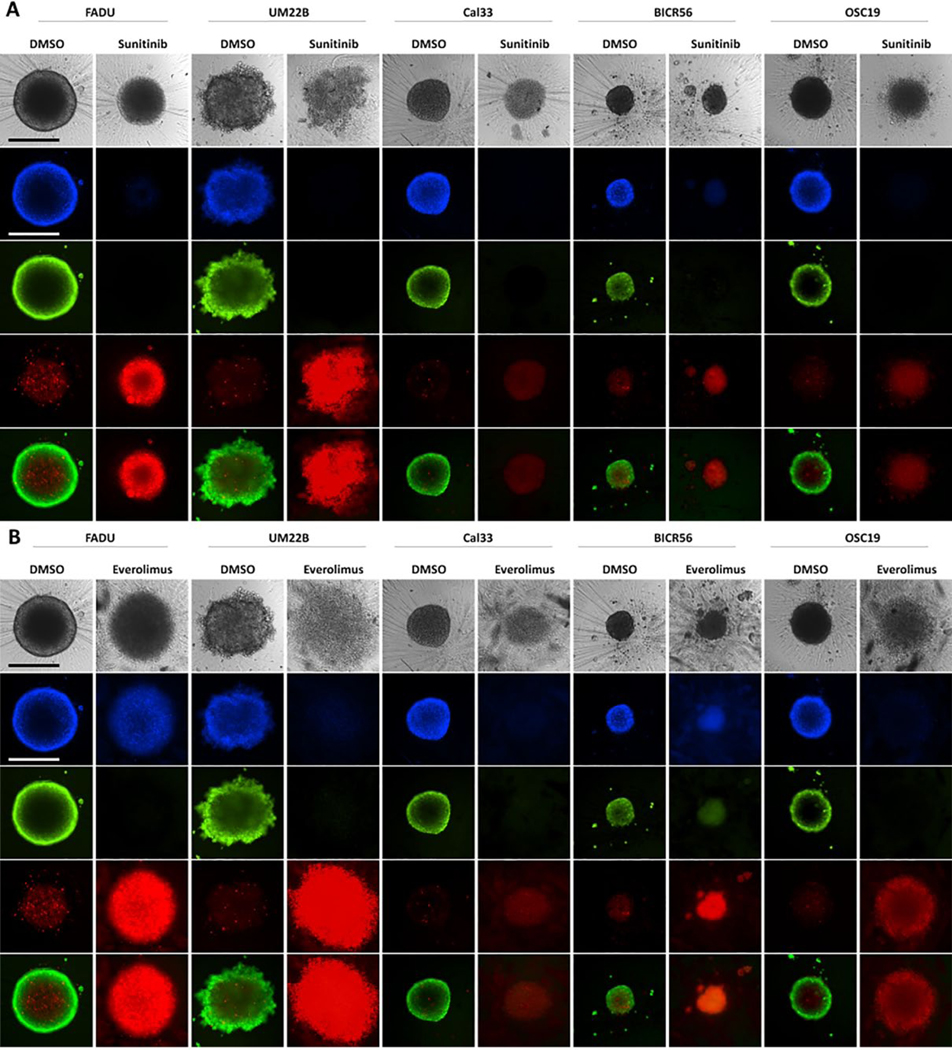

Impact of sunitinib and everolimus exposure on HNSCC MCTS cultures: TL (grayscale), Hoechst (blue), CAM (green), EHD (red), and live/dead composite images of HNSCC MCTS cultures exposed to DMSO or the top concentrations of (A) sunitinib or (B) everolimus for 72 h. All scale bars represent 300 μm. The CTB GI data and curve fits for 2D monolayer (●) and MCTS (●) HNSCC cultures exposed to the indicated concentrations of (C–G) sunitinib or (H–L) everolimus are indicated in blue and red, respectively. The normalized mean ± SD (n = 3) GI data from triplicate wells for each compound concentration are presented. The MIFI CAM (■) and EHD (■) signals of HNSCC MCTSs exposed to the top concentration of (M) sunitinib or (N) everolimus were normalized and expressed as percent of DMSO controls. The normalized MIFI data for compounds are presented as mean ± SD (n = 3) from triplicate wells. MCTS diameters (μm) extracted from the TL images of HNSCC MCTS cultures exposed to DMSO (□) or the top concentration (■) of (O) sunitinib or (P) everolimus for 72 h are presented. The mean ± SD (n = 3) diameter data from triplicate wells for DMSO controls and compounds are presented. Representative data from one of three or four independent experiments for each of the analyses are shown.
Effects of Exposure to 5-FU and Etoposide on Cal33 HNSCC Cultures
Figure 5 summarizes the effects of 72 h exposure to 500 μM 5-FU or 100 μM etoposide on Cal33 2D monolayer and MCTS cultures. Figure 5A shows TL, Hoechst, CAM, and EHD images of Cal33 monolayers and MCTS cultures exposed to DMSO or to 18.5 or 500 μM 5-FU. Cal33 monolayers exposed to 18.5 μM 5-FU contained much fewer cells, Hoechst stained nuclei, and CAM-positive cells than DMSO controls. Even with fewer cells, the number of EDH-positive cells in Cal33 monolayers treated with 18.5 μM 5-FU increased. In contrast, images of Cal33 MCTSs treated with 18.5 μM 5-FU were not noticeably different from DMSO controls. At 500 μM 5-FU, however, Cal33 MCTS morphology was changed and EHD staining was substantially higher. In 2D Cal33 monolayers, 5-FU produced complete CTB concentration–response curves with good-quality curve fits and a mean GI50 of 2.8 ± 1.07 μM (Fig. 5C, Table 1). In contrast, 5-FU produced no CTB GI response in Cal33 MCTSs. Although exposure to 500 μM did not significantly reduce CAM staining in Cal33 MCTSs, it increased EHD staining by 300% over DMSO controls (Fig. 5D, Suppl. Table S4A). Exposure of Cal33 MCTSs to 500 μM 5-FU altered the MCTS perimeter from smooth to irregular and changed the apparent density or compactness (Fig. 5A, Suppl. Table S5A). Neither the CTB nor the CAM viability measurements correlated with the changes in morphology and increased EHD staining apparent in Cal33 MCTSs exposed to 500 μM 5-FU (Fig. 5A,C,D, Table 1, Suppl. Tables S4A and S5A). Figure 5B shows TL, Hoechst, CAM, and EHD images of Cal33 monolayers and MCTS cultures exposed to DMSO or to 3.7 or 100 μM etoposide. Cal33 monolayers exposed to 3.7 μM etoposide contained much fewer cells, Hoechst stained nuclei, and CAM-positive cells than DMSO controls. The number of EDH-positive cells in Cal33 monolayers treated with 3.7 μM etoposide increased. In contrast, images of Cal33 MCTSs treated with 3.7 μM etoposide were not noticeably different from DMSO controls. At 100 μM etoposide, however, Cal33 MCTSs were smaller and EHD staining was substantially higher. Cal33 monolayers exposed to etoposide exhibited incomplete CTB GI curves with lesser-quality curve fits that produced a mean GI50 of 50.2 ± 10.7 μM (Fig. 5F, Table 1). In contrast, etoposide produced no CTB GI response in Cal33 MCTSs. Exposure to 100 μM etoposide did not reduce CAM staining in Cal33 MCTSs, but it increased EHD staining by 200% over DMSO controls and reduced MCTS diameters (Fig. 5B,G,H, Suppl. Table S4A,B). 5-FU and etoposide are examples of drugs whose effects on CTB and CAM viability measurements did not correlate with their impact on MCTS morphology or EHD staining.
Figure 5.
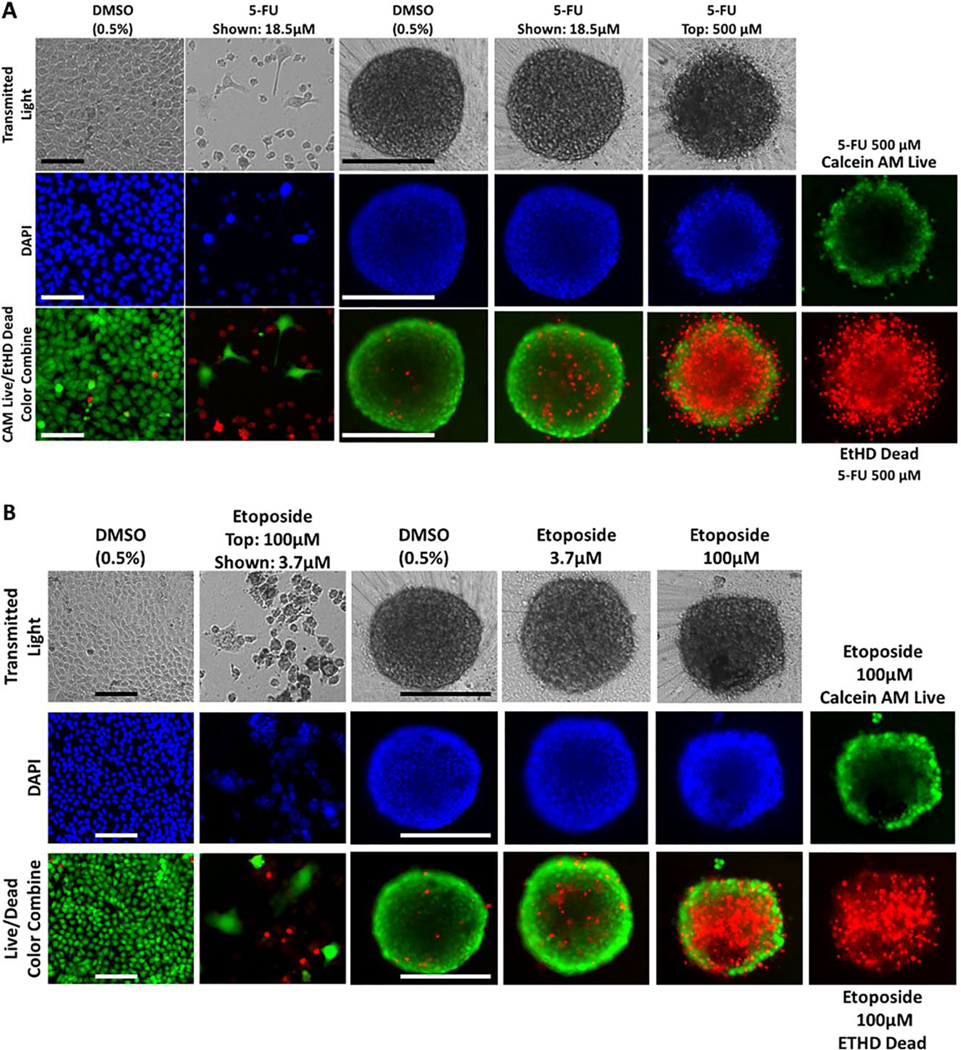

Impact of 5-FU and etoposide exposure on Cal33 HNSCC 2D monolayer and MCTS cultures: TL (grayscale), Hoechst (blue), CAM (green), EHD (red), and live/dead composite images of 2D monolayer and MCTS HNSCC cultures exposed to DMSO or the top concentrations of (A) 5-FU or (B) etoposide for 72 h. Scale bars for 2D monolayer and MCTS cultures represent 100 and 300 μm, respectively. The CTB GI data and curve fits for 2D monolayer (●) and MCTS (●) HNSCC cultures exposed to the indicated concentrations of (C) 5-FU or (F) etoposide are indicated in blue and red, respectively. The normalized mean ± SD (n = 3) GI data from triplicate wells for each compound concentration are presented. The MIFI CAM (■) and EHD (■) signals of HNSCC MCTSs exposed to the top concentration of (D) 5-FU or (G) etoposide were normalized and expressed as percent of DMSO controls. The normalized MIFI data are presented as mean ± SD (n = 3) from triplicate wells. MCTS diameters (μm) extracted from the TL images of HNSCC MCTS cultures exposed to DMSO (□) or the top concentration (■) of (E) 5-FU or (H) etoposide for 72 h are presented. The mean ± SD (n = 3) diameter data from triplicate wells for DMSO controls and compounds are presented. Representative data from one of three or four independent experiments for each of the analyses are shown.
HNSCC MCTS Drug Impact Scores
We wanted to explore whether a consolidated drug impact score that incorporated all these parameters might be a better indicator of cancer drug responses in HNSCC MCTS cultures. CTB GI drug impact scores for the 19 test drugs in both culture formats are presented in Supplemental Table S3. If a drug produced a calculable GI50 in CTB GI assays, it scored 1; if it did not produce a calculable GI50 but exhibited a concentration-dependent GI response and achieved ≥25% GI, it scored 0.5; and if it achieved neither of these attributes, it received a score of 0. In 95 possible pairwise cancer drug × HNSCC cell line GI experiments performed with CTB, 71.6% of the 2D monolayer cultures produced calculable GI50 values compared with only 27.4% of MCTS cultures. If we include concentration-dependent GI activities ≥25%, 97.9% of the 2D monolayer cultures responded compared with only 35.8% of MCTS cultures. The much lower CTB GI drug impact scores of MCTSs suggest that they were substantially more resistant than their corresponding 2D monolayers to cancer drug-induced GI (Suppl. Table S3). Supplemental Table S4B lists the CAM and EHD drug impact scores for the 95 drug HNSCC MCTS pairs at the maximum drug concentrations tested. In MCTS cultures stained with the CAM reagent, if the drug reduced the CAM MIFI by ≥50% relative to DMSO controls, it scored 1; if it reduced the CAM MIFI by ≥20% but <50%, it scored 0.5; and if it reduced the CAM MIFI by <20%, it scored 0. In MCTS cultures stained with EHD reagent, if the drug increased the EHD MIFI by ≥50% relative to DMSO controls, it scored 1; if it increased the EHD MIFI by ≥20% but <50%, it scored 0.5; and if it increased the EHD MIFI by <20%, it scored 0. Only 24.4% of the 95 drug–MCTS pairs reduced CAM MIFI values by ≥50%, and only 58.9% reduced staining intensity by ≥20% (Suppl. Table S4B). In contrast, 67.8% of the 95 drug–MCTS pairs increased EHD MIFI by ≥50%, and 76.7% increased staining intensity by ≥20%. The increased sensitivity of MCTS EDH dead-cell staining over CAM live-cell staining to drug exposure resulted in higher drug impact scores for the EHD analysis. Supplemental Table S5B lists the morphology drug impact scores for the 95 drug–MCTS pairs. If a drug altered ≥2 of the 3 MCTS morphology parameters, it scored 1; if it only altered 1 of 3, it scored 0.5; and if it did not change any of the parameters, it scored 0. Overall, 63.2% of the 95 drug–MCTS pairs exhibited alterations in ≥2 of the morphology parameters, and 89.5% displayed changes in ≥1 morphology parameter. Substantially more test compounds altered HNSCC MCTS morphology and EHD dead staining than were detected by either the CTB or CAM viability reagents (Suppl. Tables S3–S5).
The cumulative HNSCC MCTS drug impact scores for the 19 drugs are presented in Table 2. Doxorubicin attained a perfect cumulative drug impact score of 20, consistent with its ability to disrupt and effectively kill all five HNSCC MCTS cultures.22,25 None of the drugs achieved an MCTS drug impact score of 0, indicating that they all produced measurable responses in at least some of the cultures. Even the 9 (47.4%) cancer drugs that failed to display evidence of concentration-dependent CTB GI against any of the 5 HNSCC MCTS cultures (Table 1, Suppl. Table S3) produced scorable responses in the other analyses (Suppl. Tables S4A,B and S5A,B). The test drugs segregated into three tiers of cumulative HNSCC MCTS drug impact scores: the scores for the high-impact drugs doxorubicin, sunitinib, everolimus, cisplatin, and docetaxel ranged from 20 to 16 (100% to 80%); the scores for the intermediate-impact drugs gefitinib, ruxolitinib, dasatinib, buparlisib, romidepsin, ganetespib, 5-FU, etoposide, and bortezomib ranged from 13.5 to 8.5 (67.5% to 42.5%); and the scores for the low-impact drugs bleomycin, topotecan, dactolisib, erlotinib, and methotrexate ranged from 7.5 to 6 (37.5% to 30%) (Table 2). With respect to the HNSCC cell lines, UM-22B and FaDu MCTSs were the most responsive to drug exposure with the highest cumulative drug impact scores (67.8% and 66.4%), OSC-19 MCTSs were intermediate (54.6%), and BIRC56 and Cal33 MCTSs were the least responsive (48% and 47.4%). For the five drugs approved for HNC, cisplatin and docetaxel were in the high-impact tier, 5-FU was in the intermediate-impact tier, and bleomycin and methotrexate were in the low-impact tier. The cumulative drug impact scores indicate that studies that rely solely on metabolic viability endpoints to measure the effects of drugs in MCTS cultures may seriously underestimate responses to those agents.
Table 2.
Cumulative HNSCC MCTS Drug Impact Scores.
| Compound | FaDu | UM-22B | Cal33 | BIRC56 | OSC-19 | Total | Rank |
|---|---|---|---|---|---|---|---|
| 5-FU* | 2.5 | 3.0 | 1.0 | 1.0 | 2.0 | 9.5 | 10 |
| Methotrexate* | 2.0 | 2.0 | 0.5 | 1.0 | 0.5 | 6.0 | 16 |
| Bleomycin* | 1.0 | 3.5 | 1.0 | 0.5 | 1.5 | 7.5 | 13 |
| Docetaxel* | 4.0 | 3.5 | 2.0 | 3.5 | 3.5 | 16.5 | 4 |
| Cisplatin* | 3.5 | 3.5 | 3.0 | 3.0 | 3.0 | 16.0 | 5 |
| Gefitinib | 3.0 | 3.0 | 2.5 | 2.0 | 3.0 | 13.5 | 6 |
| Erlotinib | 1.5 | 2.5 | 1.5 | 0.0 | 1.0 | 6.5 | 15 |
| Dasatinib | 2.5 | 2.5 | 1.5 | 2.0 | 2.0 | 10.5 | 8 |
| Sunitinib | 4.0 | 4.0 | 3.0 | 3.5 | 3.5 | 18.0 | 2 |
| Ruxolitinib | 3.0 | 2.0 | 1.5 | 3.5 | 2.5 | 12.5 | 7 |
| Doxorubicin | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 | 20.0 | 1 |
| Etoposide | 2.5 | 2.5 | 1.5 | 0.5 | 2.0 | 9.0 | 11 |
| Topotecan | 2.5 | 2.5 | 1.5 | 0.0 | 0.5 | 7.0 | 14 |
| Dactolisib | 1.0 | 2.0 | 1.5 | 2.0 | 0.5 | 7.0 | 14 |
| Buparlisib | 2.5 | 2.5 | 1.5 | 2.0 | 2.0 | 10.5 | 8 |
| Romidepsin | 2.5 | 1.5 | 2.0 | 2.5 | 2.0 | 10.5 | 8 |
| Bortezomib | 2.0 | 2.0 | 1.5 | 1.0 | 2.0 | 8.5 | 12 |
| Everolimus | 4.0 | 3.0 | 3.0 | 3.0 | 4.0 | 17.0 | 3 |
| Ganetespib | 2.5 | 2.0 | 2.0 | 1.5 | 2.0 | 10.0 | 9 |
Cumulative HNSCC MCTS drug impact scores are the sum of GTB GI, live/dead staining, and morphology drug impact scores in Supplemental Tables S3–S5. Maximum drug impact score per HNSCC MCTS culture = 4, and across all cultures = 20. Minimum drug impact score per HNSCC MCTS culture = 0, and across all cultures = 0.
Approved for HNSCC therapy.
Discussion
We investigated the use of HNSCC MCTS cultures to screen for cancer drug leads that could be developed into candidates with a better probability of success in in vivo animal models and patients. The five HNSCC cell lines selected form xenografts in immunodeficient mice, have genetic profiles similar to those of patient tumors, and have been used for HNSCC drug discovery.30,35,36 These HNSCC cell lines produce MCTS cultures with distinct morphologies, sizes, and growth phenotypes (Fig. 3A, Suppl. Fig. S1, Suppl. Table S5A) that develop microenvironments and exhibit drug penetration and distribution gradients that coincide with enhanced resistance to several anticancer agents.9,22,25 We present here the results of our analysis of the impact of 19 cancer agents of different drug classes on multiple HNSCC MCTS viability and morphology parameters.
Cellular metabolic pathways convert components of viability reagents into forms that produce easily detectable increases in absorbance or fluorescence, or they generate ATP that promotes the luminescence signal of added luciferase. Larger signals correlate with increased cell numbers that are used as surrogate measurements of proliferation- and/or drug-induced GI in 2D and 3D cultures. We used CTB for these studies because the RFUs produced by the same number of cells in 2D and 3D HNSCC cultures were comparable, whereas the relative light units (RLUs) produced by the CTG ATP detection reagent were ~10-fold lower in 3D than in 2D cultures.25 In 2D monolayer HNSCC cultures, 71.6% of the 95 drug–tumor cell line pairwise combinations produced GI50 values and 97.9% exhibited concentration-dependent GI (Figs. 1, 4, and 5, Table 1, Suppl. Table S3). In contrast, only 27.4% of the 95 drug–cell line pairs produced GI50 values in the corresponding MCTS cultures, and only 35.8% exhibited concentration-dependent GI. Only cisplatin, sunitinib, doxorubicin, and everolimus inhibited the growth of all five HNSCC cell lines in both culture formats, and docetaxel produced GI50 values in all cultures except for Cal33 MCTSs. The GI50 values for sunitinib in 2D monolayer (5–11 μM range) and MCTS (12.5–22 μM range) HNSCC cultures were well beyond the clinical achievable Cmax of 0.181 μM, suggesting that it is unlikely to have therapeutic application for HNC. Similarly, the GI50 values for everolimus in 2D monolayer (28–40 μM range) and MCTS (32–90 μM range) HNSCC cultures were well beyond the clinically achievable Cmax of 0.064 μM. 2D HNSCC monolayers were between 2- and 366-fold more sensitive than MCTSs to these five drugs (Table 1). Eleven of the 19 drugs produced maximum CTB impact scores of 5, and 8 scored ≥2.5 in 2D monolayer cultures (Suppl. Table S3). In MCTS cultures, however, only four drugs produced CTB impact scores of 5 and only two scored ≥2.5. Nine of the drugs had CTB impact scores of zero and four scored ≤2 in MCTS cultures. 3D tumor cultures have variously demonstrated greater resistance, enhanced sensitivity, or equipotency for cancer drugs when compared with matching 2D cultures.10,12,13,40–46 3D spheroids formed by U87 glioblastoma, PC3 prostate, T47D breast, and HCT116 colon cancer cell lines were more sensitive to GI by the PI3K inhibitor PX-866 than their matching 2D cultures.47 PI3K and AKT pathway inhibitors were selectively cytotoxic toward 3D prostate cancer cell line cultures compared with the corresponding 2D monolayers.12 Although EGFR and cMET inhibitors were more potent in 3D lung cancer spheroids than 2D monolayers, other drugs were equipotent in 2D and 3D.10 In pilot screens of the FDA and NCI drug collections in HT-29 colon cancer 2D monolayer and 3D spheroid cultures, between 66% and 76% of the drugs were selectively cytotoxic toward 2D cultures, with fewer drugs showing selectivity for 3D cultures or equipotency in both 2D and 3D.17 3D spheroid cultures of 11 epithelial ovarian cancer cell lines were overwhelmingly more resistant than 2D monolayer cultures to cisplatin and paclitaxel in resazurin formation assays.44 In 2D and 3D culture models of breast cancer, three cell lines that formed dense MCTSs showed more resistance to paclitaxel and doxorubicin than their matching 2D cultures in CTG ATP detection assays.43 However, paclitaxel and doxorubicin were equipotent with 2D cultures in three cell lines that formed looser MCTSs.43 IC50 values for doxorubicin, carboplatin, and 5-FU were significantly higher in 3D than 2D cultures of MCF-7 breast cancer cells.46 2D monolayer cultures of cells isolated from a triple-negative breast cancer patient-derived xenograft (PDX) displayed sensitivity to several drug classes, whereas matching 3D spheroid cultures were resistant.45 Although diminished drug responses were observed in many NCI-60 cell lines grown as 3D spheroids versus 2D monolayers, some drugs were equally cytotoxic in 2D and 3D cultures.21 The CTB viability data presented here support a conclusion that HNSCC MCTSs cultures are substantially more resistant than their corresponding 2D monolayer cultures to 16 of the 19 test drugs.
Tumor cell lines adapted to long-term 2D culture conditions proliferate faster than cells isolated from primary tumors, tend to underrepresent self- and population-renewing tumor stem cells that contribute to recurrence and metastasis, and display enhanced sensitivity to antiproliferative agents.10,12,13,22,40,41 In regions of MCTSs with reduced nutrient and/or oxygen concentrations, dormant or quiescent tumor cells that have either stopped replicating or are replicating slowly display resistance to molecules targeting cell proliferation mechanisms.48–50 Only the cells in the outer layers of FaDu and UM-22B MCTSs contained actively proliferating cells detectable by 5-ethynyl-2′-deoxyuridine (EdU) incorporation.25 Based on Ki-67 staining, the proliferation rates of eight of nine HNSCC MCTS cultures formed in 96-well ULA-plates declined substantially compared with their matching 2D cultures.51 The 2D monolayer HNSCC cultures used in these studies were proliferating exponentially throughout the 96 h culture period and underwent >2 but <3 doublings (Suppl. Fig. S1).35,36 In contrast, FaDu, UM-22B, and Cal33 MCTS cultures had linear growth rates, BIRC56 MCTSs were dormant, and OSC-19 had a slow progressive death phenotype.25 Faster cell proliferation and growth rates may be contributing to the apparent increased drug sensitivity of 2D monolayers versus MCTS HNSCC cultures (Table 1, Suppl. Table S3). However, the reduced proliferation and growth rates of HNSCC MCTS cultures may be better aligned to solid tumor growth in vivo, which may be critical for finding leads and candidates with potential for improved efficacy in in vivo animal models and in patients.
Cells in solid tumors and MCTSs experience adhesive, topographical, and mechanical cues that alter their underlying biology and responses to stimuli.10–14,19–26 Tumor cell packing densities and adhesion junctions between adjacent cells and with the ECM constitute permeability barriers that can restrict cancer drug penetration, distribution, and efficacy.48–50,52–54 Drug physiochemical properties also affect tissue distribution, and cells that are distal to blood vessels in solid tumors experience lower concentrations due to limited drug penetration.50,52,53 Reduced drug penetration in solid tumors contributes to the resistance toward several chemo-therapeutic agents, including doxorubicin, epirubicin, daunorubicin, mitoxantrone, methotrexate, 5-FU, vinblastine, paclitaxel, and docetaxel.27–29,44–54 Three breast cancer cell lines that formed dense MCTSs showed greater resistance to paclitaxel and doxorubicin than cell lines that formed loose MCTSs or their matching 2D cultures.43 The UM-22B cell line produced the least densely packed HNSCC MCTS cultures and was the most sensitive to drug-induced GI, with 7 of the 19 drugs producing GI50 values, while only 4 drugs produced GI50 values in the more densely packed Cal33 MCTSs (Table 1). Ellipticine, idarubicin, daunorubicin, and doxorubicin accumulation in 2D monolayer and MCTS HNSCC cultures increases linearly with respect to drug concentration.22 2D monolayers accumulate uniform drug levels independent of cell number and/or cell seeding density.22 In MCTS cultures, however, drug accumulation increases as cell numbers and MCTS sizes become larger.22 Initially, drugs accumulate preferentially in cells in the outer layers of HNSCC MCTSs compared with those in the inner cores.22,25 Doxorubicin and other anthracycline drugs may become sequestered in the peripheral layers of MCTSs and solid tumors because they are trapped in acidic compartments such as endosomes or lysosomes, or because they bind so avidly to DNA.49,50,55,56 Similarly, paclitaxel binding to tubulin may lead to its sequestration in the peripheral layers of MCTSs and human solid tumor histocultures.57 After 24 h of drug exposure, however, drugs penetrated throughout HNSCC MCTSs that were smaller than DMSO controls and displayed other changes in morphology; outer cell layers were either missing, smaller, or disintegrating, and inner core regions were darker.22,25 Cal33 MCTSs were 6-, 20-, 10-, and 16-fold more resistant than 2D monolayers to GI by ellipticine, idarubicin, daunorubicin, and doxorubicin, respectively.22 Drug-induced cell death of peripheral cell layers is a major determinant of solid tumor drug penetration.22,25,49,52,53,57–59 In our studies, FaDU, UM-22B, Cal33, BIRC56, and OSC-19 MCTS cultures were 199-, 67-, 44-, 213-, and 366-fold more resistant than 2D monolayers to GI by doxorubicin. MCTS cultures were 9-, 6-, 15-, 12-, and 7-fold more resistant than 2D monolayers to cisplatin. FaDU, UM-22B, BIRC56, and OSC-19 MCTS cultures were 18-, 4-, 5-, and 7.5-fold more resistant than 2D monolayers to docetaxel. On average, MCTS HNSCC cultures were 2.5- and 2-fold more resistant than 2D monolayers to sunitinib and everolimus, respectively. Reduced drug penetration and distribution in HNSCC MCTS cultures may also be contributing to the increased resistance of some drugs versus 2D monolayer cultures (Table 1, Suppl. Table S3).
CAM live- and EHD dead-cell staining in conjunction with fluorescence imaging and image analysis have been used to evaluate and characterize MCTS cell viability, growth, and the consequences of cancer drug exposure.9,20,21,25,45,60 In HNSCC MCTS cultures exposed to the maximum concentrations of the test drugs, only 24.4% of the 95 drug–cell line pairwise combinations reduced CAM MIFI values by ≥50% (Suppl. Table S4A). The CAM drug impact scores show that the HNSCC cell lines with the fastest MCTS growth rates, FaDu and UM-22B, were the most sensitive to drug exposure (Suppl. Table S4B), while Cal33 MCTSs were the least affected. Across the MCTS cultures, none of the drugs produced a maximum CAM drug impact score of 5, and only six scored ≥2.5. Bortezimib produced a minimum CAM drug impact score of 0, and 10 drugs scored ≤2. The CAM data were consistent with the CTB data in that both metabolic viability measurements indicated that HNSCC MCTS cultures were resistant to most of the 19 drugs (Figs. 1 and 2, Table 1, Suppl. Tables S3 and S4A,B). However, 67.8% of the 95 drug–cell line pairwise combinations increased EHD MIFI values by ≥50% (Suppl. Table S4A,B). Across MCTS cultures, 7 of the 19 drugs produced maximum EDH drug impact scores of 5, 7 scored ≥2.5, and only 4 scored ≤2. The robust drug-induced increases in HNSCC MCTS EHD staining of many drugs deviated from their relatively minor effects on CAM and CTB viability measurements (Figs. 1 and 2, Table 1, Suppl. Tables S3 and S4A,B). Although topotecan exposure induced a concentration- and time-dependent increase in EHD staining of HT29 colon carcinoma spheroids, CAM staining was not always reduced to the same extent in these cultures.21 Also consistent with our observations in HNSCC MCTSs, drug-induced increases in EHD staining were not always matched by similar decreases in CAM staining in 3D spheroid cultures of cells from a triple-negative PDX model.45 The average fluorescent intensity ratio of CAM to propidium iodide was used to rank the cytotoxic effects of 14 anticancer agents in U87MG glioblastoma MCTS cultures, and several of the compounds that induced substantial increases in EHD staining retained considerable levels of CAM staining.60 Thirty-seven (39%) of the 95 drug–MCTS pairwise combinations failed to reduce CAM staining by ≥20%, while only 21 (22%) failed to increase EHD staining by ≥20% (Fig. 2, Suppl. Table S4A,B).
Spheroid integrity and volume growth kinetics can be rapidly and easily visualized by noninvasive phase-contrast and/or bright-field imaging, and both have been used to monitor the effects of cancer drug exposure.9,10,12,20–23,25,41,43,44,51,60 We used TL images to characterize the distinct morphologies and growth of the five HNSCC MCTS cultures.9,22,25 HNSCC develops in the paranasal sinuses, nasal cavity, oral cavity, pharynx, larynx, salivary gland, and thyroid, and histology, molecular characteristics, and clinical outcomes vary widely across different anatomical sites.28,61–63 Cal33 and BICR56 HNSCC cell lines were established from oral cavity tumors, the OSC-19 cell line was established from a tumor in the tongue that had metastasized to a cervical lymph node, and the FaDu and UM-22B cell lines were established from hypopharynx tumors.30 The different HNSCC MCTS morphologies displayed here (Fig. 3, Suppl. Fig. S1, Suppl. Table S5A) were consistent with our previous observations9,20,22,25 and those of HNSCC MCTSs produced in 96-well ULA-plates.23,51 We examined the effects of cancer drug exposure on the perimeters, shapes, density or compactness, and diameters of treated MCTS versus DMSO controls (Fig. 3, Suppl. Table S5A). In 95 pairwise cancer drug–MCTS experiments, 63.2% displayed changes in ≥2 of 3 morphology parameters, and ≥1 morphology parameter was altered in 89.5% of the tests. 3D spheroid cultures of 11 epithelial ovarian cancer cell lines were subcategorized based on histological differentiation features and gross morphology into large dense aggregates, large loose aggregates, or small aggregates.44 NCI-60 tumor cell lines cultured in 96-well ULA-plates produced four spheroid categories: condensed spheroids with a smooth and even periphery, less condensed spheroids with a rougher-appearing perimeter, clumped cell aggregates with highly irregular outer perimeters, and loose cell aggregates with highly irregular shape.21 Three breast cancer cell lines that formed dense MCTSs exhibited greater resistance to paclitaxel and doxorubicin than cell lines that produced loose MCTSs or their matching 2D cultures.43 Time-course bright-field images and diameter measurements were used to monitor the growth of U87MG glioblastoma MCTS cultures and to determine the growth inhibitory effects of 14 anticancer agents.60 The GI effects of carfilzomib in U87MG MCTSs were correlated with changes in morphology from a tight dark spheroid to a loose cell aggregate.60 Control U87MG MCTSs had a layer of lighter-intensity cells surrounding a darker MCTS core, and some concentrations of carfilzomib induced shedding of the outer layer, while others induced the formation of a halo of loose cells surrounding a small spheroid.60 Consistent with the EHD dead-cell staining results, more test compounds altered HNSCC MCTS morphology endpoints than were detected by either the CTB or CAM viability reagents (Figs. 3–5, Tables 1 and 2, Suppl. Tables S3–S5).
Our studies demonstrate that CTB and CAM viability parameters were the least sensitive indicators of MCTS drug responses and that relying solely on cell viability measurements can seriously underestimate the impact of cancer drug exposure on HNSCC MCTS cultures. Incorporating changes in MCTS morphology and increased EHD dead-cell staining into the analysis more than doubled the number of drugs that were considered to have caused substantial changes in MCTS cultures. The cumulative drug impact score allowed us to stratify MCTS test drug responses into high-, intermediate-, and low-impact tiers (Table 4). A similar multiparametric drug scoring strategy was employed to categorize and classify the responses of 14 anticancer agents in U87MG glioblastoma MCTS cultures grown in 384-well ULA-plates.60 Changes in U87MG MCTS diameters over time in culture were used as an indicator of growth and compound-mediated cytotoxicity. Changes in the area of U87MG MCTS cultures embedded in Matrigel were used to measure invasion and drug effects. U87MG MCTS cultures were also stained with Hoechst, CAM, propidium iodide, and caspase 3/7 reagents.60 Compounds were ranked into high, medium, low, or no effect groups based on the sum of scores assigned for their relative effects on growth, invasion, viability, and apoptosis.60 Although CTB and CAM viability readouts indicated that Cal33 MCTS cultures were resistant to 5-FU and etoposide, these parameters did not correlate with the changes in morphology and robust increases in MCTS EHD dead-cell staining produced by the top concentrations of these drugs (Fig. 5). Other examples where viability measurements did not correlate with changes in morphology and dead-cell staining included docetaxel in Cal33 MCTSs and 5-FU in OSC-19 MCTSs (Figs. 1–3, Tables 1 and 2, Suppl. Tables S3–S5). It is possible that the viable cells in MCTSs exposed to these drugs emanate from intrinsically drug-resistant cell populations present in MCTS cultures that represent the source of drug failure and recurrence. Tumor intrinsic drug resistance factors include coexistent genetic alterations to drug targets, mutations in other signaling pathway genes, and/or inactivation of pro-apoptotic pathways.64,65 Drug resistance in MCTS cultures might also be due to their slower growth rates and/or the presence of subpopulations of dormant or quiescent tumor cells that either have stopped or are replicating slowly. Some drugs may be less effective in hypoxic or acidic microenvironments within MCTSs, or adhesion junctions between adjacent cells, and with the ECM may restrict drug penetration and distribution such that drugs do not reach therapeutic concentrations in all cells. Long-term monitoring of treated MCTS cultures, with or without drug washout, would be one way to determine whether the remaining viable cells in drug-treated MCTS cultures represent circumstances where tumor growth is delayed and might ultimately give rise to regrowth.
Supplementary Material
Acknowledgments
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The studies were supported in part by a Development Research Project award (Johnston, PI) from the Head and Neck Cancer Spore P50 (Ferris and Grandis, CA097190) of the University of Pittsburgh Medical Center Hillman Cancer Center.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material is available online with this article.
References
- 1.Al-Lazikani B; Banerji U; Workman P Combinatorial Drug Therapy for Cancer in the Post-Genomic Era. Nat. Biotechnol 2012, 30, 679–692. [DOI] [PubMed] [Google Scholar]
- 2.Ocana A; Pandiella A; Siu LL; et al. Preclinical Development of Molecular-Targeted Agents for Cancer. Nat. Rev. Clin. Oncol 2011, 8, 200–209. [DOI] [PubMed] [Google Scholar]
- 3.Ocaña A; Pandiella A Personalized Therapies in the Cancer “Omics” Era. Mol. Cancer 2010, 9, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hait W Anticancer Drug Development: The Grand Challenges. Nat. Rev. Drug Discov 2010, 9, 253–254. [DOI] [PubMed] [Google Scholar]
- 5.Hutchinson L; Kirk R High Drug Attrition Rates—Where Are We Going Wrong? Nat. Rev. Clin. Oncol 2011, 8, 189–190. [DOI] [PubMed] [Google Scholar]
- 6.Toniatti C;Jones P;Graham H;et al. OncologyDrugDiscovery: Planning a Turnaround. Cancer Discov. 2014, 4, 397–404. [DOI] [PubMed] [Google Scholar]
- 7.Kamb A; Wee S; Lengauer C Why Is Cancer Drug Discovery So Difficult? Nat. Rev. Drug Discov 2007, 6, 115–120. [DOI] [PubMed] [Google Scholar]
- 8.Wong C; Siah KW; Lo AW Estimation of Clinical Trial Success Rates and Related Parameters. Biostatistics 2019, 20, 273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Close DA; Camarco DP; Shan F; et al. The Generation of Three-Dimensional Head and Neck Cancer Models for Drug Discovery in 384-Well Ultra-Low Attachment Microplates. Methods Mol. Biol 2018, 1683, 355–369. [DOI] [PubMed] [Google Scholar]
- 10.Ekert J; Johnson K; Strake B; et al. Three-Dimensional Lung Tumor Microenvironment Modulates Therapeutic Compound Responsiveness In Vitro—Implication for Drug Development. PLoS One 2014, 9, e92248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang Y; Eglen RM Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Härmä V; Virtanen J; Mäkelä R; et al. A Comprehensive Panel of Three-Dimensional Models for Studies of Prostate Cancer Growth, Invasion and Drug Responses. PLoS One 2010, 5, e10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hongisto V; Jernström S; Fey V; et al. High-Throughput 3D Screening Reveals Differences in Drug Sensitivities between Culture Models of JIMT1 Breast Cancer Cells. PLoS One 2013, 8, e77232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howes A; Richardson RD; Finlay D; et al. 3-Dimensional Culture Systems for Anti-Cancer Compound Profiling and High-Throughput Screening Reveal Increases in EGFR Inhibitor-Mediated Cytotoxicity Compared to Monolayer Culture Systems. PLoS One 2014, 9, e108283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lovitt C; Shelper TB; Avery VM Miniaturized Three-Dimensional Cancer Model for Drug Evaluation. Assay Drug Dev. Technol 2013, 11, 435–448. [DOI] [PubMed] [Google Scholar]
- 16.Lovitt C; Shelper TB; Avery VM Advanced Cell Culture Techniques for Cancer Drug Discovery. Biology 2014, 3, 345–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madoux F; Tanner A; Vessels M; et al. A 1536-Well 3D Viability Assay to Assess the Cytotoxic Effect of Drugs on Spheroids. SLAS Discov. 2017, 22, 516–524. [DOI] [PubMed] [Google Scholar]
- 18.Onozato Y; Kaida A; Harada H; et al. Radiosensitivity of Quiescent and Proliferating Cells Grown as Multicellular Tumor Spheroids. Cancer Sci. 2017, 108, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryan S; Baird AM; Vaz G; et al. Drug Discovery Approaches Utilizing Three-Dimensional Cell Culture. Assay Drug Dev. Technol 2016, 14, 19–28. [DOI] [PubMed] [Google Scholar]
- 20.Sant S; Johnston PA The Production of 3D Tumor Spheroids for Cancer Drug Discovery. Drug Discov. Today Technol 2017, 23, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selby M; Delosh R; Laudeman J; et al. 3D Models of the NCI60 Cell Lines for Screening Oncology Compounds. SLAS Discov. 2017, 22, 473–483. [DOI] [PubMed] [Google Scholar]
- 22.Shan F; Close DA; Camarco DP; et al. High-Content Screening Comparison of Cancer Drug Accumulation and Distribution in Two-Dimensional and Three-Dimensional Culture Models of Head and Neck Cancer. Assay Drug Dev. Technol 2018, 16, 27–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vinci M; Gowan S; Boxall F; et al. Advances in Establishment and Analysis of Three-Dimensional Tumor Spheroid-Based Functional Assays for Target Validation and Drug Evaluation. BMC Biol. 2012, 10, 29–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenzel C; Riefke B; Gründemann S; et al. 3D High-Content Screening for the Identification of Compounds That Target Cells in Dormant Tumor Spheroid Regions. Exp. Cell Res 2014, 323, 131–143. [DOI] [PubMed] [Google Scholar]
- 25.Kochanek S; Close DA; Johnston PA High Content Screening Characterization of Head and Neck Squamous Cell Carcinoma Multicellular Tumor Spheroid Cultures Generated in 384-Well Ultra-Low Attachment Plates to Screen for Better Cancer Drug Leads. Assay Drug Dev. Technol 2019, 17, 17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brockstein B Management of Recurrent Head and Neck Cancer: Recent Progress and Future Directions. Drugs 2011, 71, 1551–1559. [DOI] [PubMed] [Google Scholar]
- 27.Goerner M; Seiwert TY; Sudhoff H Molecular Targeted Therapies in Head and Neck Cancer—An Update of Recent Developments. Head Neck Oncol. 2010, 2, 8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stransky N; Egloff AM; Tward AD; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denaro N; Russi EG; Adamo V; et al. Postoperative Therapy in Head and Neck Cancer: State of the Art, Risk Subset, Prognosis and Unsolved Questions. Oncology 2011, 81, 21–29. [DOI] [PubMed] [Google Scholar]
- 30.Li H; Wawrose JS; Gooding WE; et al. Genomic Analysis of Head and Neck Squamous Cell Carcinoma Cell Lines and Human Tumors: A Rational Approach to Preclinical Model Selection. Mol. Cancer Res 2014, 12, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez-Ordonez B; Beauchemin M; Jordan RCK Molecular Biology of Squamous Cell Carcinoma of the Head and Neck. J. Clin. Pathol 2006, 59, 445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Addeo R; Caraglia M; Iuliano G Pembrolizumab: The Value of PDL1 Biomarker in Head and Neck Cancer. Expert Opin. Biol. Ther 2016, 16, 1075–1078. [DOI] [PubMed] [Google Scholar]
- 33.Chow L; Haddad R; Gupta S; et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients with Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results from the Phase Ib KEYNOTE-012 Expansion Cohort. J. Clin. Oncol 2016, 34, 3838–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seiwert T; Burtness B; Mehra R; et al. Safety and Clinical Activity of Pembrolizumab for Treatment of Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-012): An Open-Label, Multicentre, Phase 1b Trial. Lancet Oncol. 2016, 17, 956–965. [DOI] [PubMed] [Google Scholar]
- 35.Johnston P; Sen M; Hua Y; et al. High-Content pSTAT3/1 Imaging Assays to Screen for Selective Inhibitors of STAT3 Pathway Activation in Head and Neck Cancer Cell Lines. Assay Drug Dev. Technol 2014, 12, 55–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnston P; Sen M; Hua Y; et al. HCS Campaign to Identify Selective Inhibitors of IL-6-Induced STAT3 Pathway Activation in Head and Neck Cancer Cell Lines. Assay Drug Dev. Technol 2015, 13, 356–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Close D; Wang AX; Kochanek SJ; et al. Implementation of the NCI-60 Human Tumor Cell Line Panel to Screen 2260 Cancer Drug Combinations to Generate >3 Million Data Points Used to Populate a Large Matrix of Anti-Neoplastic Agent Combinations (ALMANAC) Database. SLAS Discov. 2019, 24, 242–263 [DOI] [PubMed] [Google Scholar]
- 38.Kochanek S; Close DA; Wang AX; et al. Confirmation of Selected Synergistic Cancer Drug Combinations Identified in an HTS Campaign and Exploration of Drug Efflux Transporter Contributions to the Mode of Synergy. SLAS Discov. 2019, 24, 653–668. [DOI] [PubMed] [Google Scholar]
- 39.Singh M; Close DA; Mukundan S; et al. Production of Uniform 3D Microtumors in Hydrogel Microwell Arrays for Measurement of Viability, Morphology, and Signaling Pathway Activation. Assay Drug Dev. Technol 2015, 13, 570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker B; Chen CS Deconstructing the Third Dimension: How 3D Culture Microenvironments Alter Cellular Cues. J. Cell Sci 2012, 125, 3015–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedrich J; Seidel C; Ebner R; et al. Spheroid-Based Drug Screen: Considerations and Practical Approach. Nat. Protoc 2009, 4, 309–324. [DOI] [PubMed] [Google Scholar]
- 42.Wang C; Tang Z; Zhao Y; et al. Three-Dimensional In Vitro Cancer Models: A Short Review. Biofabrication 2014, 6, 022001. [DOI] [PubMed] [Google Scholar]
- 43.Imamura Y; Mukohara T; Shimono Y; et al. Comparison of 2D- and 3D-Culture Models as Drug-Testing Platforms in Breast Cancer. Oncol. Rep 2015, 33, 1837–1843. [DOI] [PubMed] [Google Scholar]
- 44.Lee J; Mhawech-Fauceglia P; Lee N; et al. A Three-Dimensional Microenvironment Alters Protein Expression and Chemosensitivity of Epithelial Ovarian Cancer Cells In Vitro. Lab. Invest 2013, 93, 528–542. [DOI] [PubMed] [Google Scholar]
- 45.Matossian M; Burks HE; Elliott S; et al. Drug Resistance Profiling of a New Triple Negative Breast Cancer Patient-Derived Xenograft Model. BMC Cancer 2019, 19, 205–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu H; Liu W; Zhang XZ; et al. Development of Three-Dimensional Breast Cancer Cell Culture Drug Resistance Model. Sheng Li Xue Bao 2016, 68, 179–184. [PubMed] [Google Scholar]
- 47.Howes A; Chiang GG; Lang ES; et al. The Phosphatidylinositol 3-Kinase Inhibitor, PX-866, Is a Potent Inhibitor of Cancer Cell Motility and Growth in Three-Dimensional Cultures. Mol. Cancer Ther 2007, 6, 2505–2514. [DOI] [PubMed] [Google Scholar]
- 48.Kerr D; Kaye SB Aspects of Cytotoxic Drug Penetration, with Particular Reference to Anthracyclines. Cancer Chemother. Pharmacol 1987, 19, 1–5. [DOI] [PubMed] [Google Scholar]
- 49.Minchinton A; Tannock IF Drug Penetration in Solid Tumours. Nat. Rev. Cancer 2006, 6, 583–592. [DOI] [PubMed] [Google Scholar]
- 50.Tannock I; Lee CM; Tunggal JK; et al. Limited Penetration of Anticancer Drugs through Tumor Tissue: A Potential Cause of Resistance of Solid Tumors to Chemotherapy. Clin. Cancer Res 2002, 8, 878–884. [PubMed] [Google Scholar]
- 51.Schmidt M; Scholz CJ; Polednik C; et al. Spheroid-Based 3-Dimensional Culture Models: Gene Expression and Functionality in Head and Neck Cancer. Oncol. Rep 2016, 35, 2431–2440. [DOI] [PubMed] [Google Scholar]
- 52.Grantab R; Sivananthan S; Tannock IF The Penetration of Anticancer Drugs through Tumor Tissue as a Function of Cellular Adhesion and Packing Density of Tumor Cells. Cancer Res. 2006, 66, 1033–1039. [DOI] [PubMed] [Google Scholar]
- 53.Grantab R; Tannock IF Penetration of Anticancer Drugs through Tumour Tissue as a Function of Cellular Packing Density and Interstitial Fluid Pressure and Its Modification by Bortezomib. BMC Cancer 2012, 12, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tredan O; Galmarini CM; Patel K; et al. Drug Resistance and the Solid Tumor Microenvironment. J. Natl. Cancer Inst 2007, 99, 1441–1454. [DOI] [PubMed] [Google Scholar]
- 55.Kyle A; Huxham LA; Chiam AS; et al. Direct Assessment of Drug Penetration into Tissue Using a Novel Application of Three-Dimensional Cell Culture. Cancer Res. 2004, 64, 6304–6309. [DOI] [PubMed] [Google Scholar]
- 56.Kyle A; Huxham LA; Yeoman DM; et al. Limited Tissue Penetration of Taxanes: A Mechanism for Resistance in Solid Tumors. Clin. Cancer Res 2007, 13, 2804–2810. [DOI] [PubMed] [Google Scholar]
- 57.Kuh H; Jang SH; Wientjes MG; et al. Determinants of Paclitaxel Penetration and Accumulation in Human Solid Tumor. J. Pharmacol. Exp. Ther 1999, 290, 871–880. [PubMed] [Google Scholar]
- 58.Au J; Jang SH; Zheng J; et al. Determinants of Drug Delivery and Transport to Solid Tumors. J. Control. Release 2001, 74, 31–46. [DOI] [PubMed] [Google Scholar]
- 59.Zheng J; Chen CT; Au JL; et al. Time- and Concentration-Dependent Penetration of Doxorubicin in Prostate Tumors. AAPS PharmSci. 2001, 3, E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cribbes S; Kessel S; McMenemy S; et al. A Novel Multiparametric Drug-Scoring Method for High-Throughput Screening of 3D Multicellular Tumor Spheroids Using the Celigo Image Cytometer. SLAS Discov. 2017, 22, 547–557. [DOI] [PubMed] [Google Scholar]
- 61.Kang H; Kiess A; Chung CH Emerging Biomarkers in Head and Neck Cancer in the Era of Genomics. Nat. Rev. Clin. Oncol 2015, 12, 11–26. [DOI] [PubMed] [Google Scholar]
- 62.The Cancer Genome Atlas Network. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun W; Califano JA Sequencing the Head and Neck Cancer Genome: Implications for Therapy. Ann. N.Y. Acad. Sci 2014, 1333, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holohan C; Van Schaeybroeck S; Longley DB; et al. Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [DOI] [PubMed] [Google Scholar]
- 65.Lovly C; Shaw AT Molecular Pathways: Resistance to Kinase Inhibitors and Implications for Therapeutic Strategies. Clin. Cancer Res 2014, 20, 2249–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.