

Abstract
Glycosaminoglycans (GAGs) are important biological molecules that are highly anionic and occur in nature as complex mixtures. A platform that combines capillary zone electrophoresis (CZE) separations with mass spectrometry (MS) and gas-phase sequencing by using negative electron transfer dissociation (NETD) is shown to be efficacious for the structural analysis of GAG mixtures. CZE is a separation method well suited to the highly negatively charged nature of GAGs. NETD is an electron-based ion activation method that enables the generation of informative fragments with retention of the labile sulfate half-ester modification that determine specific GAG function. Here we combine for the first time NETD and CZE for assigning the structures of GAG oligomers present in mixtures. The speed of ion activation by NETD is found to couple well with the narrow peaks resulting from CZE migration. The platform was optimized with mixtures of GAG tetrasaccharide standards. The potential of the platform is demonstrated by the analysis of enoxaparin, a complex mixture of low molecular weight heparins, which was separated by CZE within 30 minutes and characterized by NETD MS/MS in one online experiment. 37 unique molecular compositions have been identified in enoxaparin using CZE-MS and 9 structures have been assigned with CZE-NETD-MS/MS.
Graphical Abstract:
INTRODUCTION
Sulfated glycosaminoglycan (GAG) carbohydrates serve a wide array of biological functions that are modulated by their structural modifications [1–5]. For example, heparin’s pharmaceutical efficacy as an anti-blood clotting agent is tied to a well-defined pattern of sulfation in a pentasaccharide sequence, and binds the protein antithrombin III (ATIII) with high specificity [6, 7]. Determining GAG-protein binding interactions that rely on specific GAG sequence motifs is nontrivial and currently requires considerable effort and sample. There is considerable interest in developing sensitive and rapid analytical methods to enable the identification of biologically active GAG sequences. Sulfated GAG carbohydrates are challenging targets for analysis due to variability in chain length as well as degree and placement of modifications. These structural variations are a result of their biosynthesis through a series of enzymatic reactions that do not go to completion and lead to highly heterogeneous carbohydrate chains of high molecular weight [8, 9]. The most complex and highly modified GAG is heparin/heparan sulfate (Hep/HS), which consists of a repeating disaccharide unit of a glucosamine, which can be modified with up to three sulfo-modifications, and a uronic acid, which can host one sulfo-modification. Additional complexity results from the epimerization of the carboxyl group on C5 of the uronic acid [10, 11]. The occurrence of carboxyl groups and the sulfo-modifications on GAGs also makes them highly anionic.
The structural characterization of GAG oligomers by mass spectrometry has shown great promise in recent years. Tandem mass spectrometry techniques such as sodium-hydrogen exchange for threshold activation and electron based activation techniques like electron detachment dissociation (EDD) have made structural assignment of the labile sulfo-half-ester modification possible [12, 13]. Native GAG polysaccharides are too long and complex for typical top down analysis and significant effort is required to characterize even the shortest and least complex intact GAG chains [14, 15]. Bottom up approaches where polysaccharides are digested to disaccharides simplify characterization of base constituents but any knowledge of extended sequence is lost [16–19]. Typically, a small segment of 4–12 monosaccharides in a chain is required for protein binding, and a quasi-bottom up approach where polysaccharides are partially digested is highly amenable to mass spectrometry-based analysis.
Partial digestion of GAG polysaccharides leads to complex mixtures of isomers and variable chain lengths that overlap in mass-to-charge that are difficult, if not impossible, to differentiate by MS alone. Liquid chromatography mass spectrometry (LC-MS) is not well suited to the separation of GAGs [20–22]. Reverse phase ion-pairing (RPIP) LC-MS has been utilized to investigate disaccharide mixtures and low molecular weight heparin (LMWH) mixtures, like enoxaparin, but requires specific ion pairing reagents that add spectral complexity and suppress signal [19, 23–25]. Hydrophilic interaction chromatography (HILIC) LC-MS can be applied to the separation of polar molecules such as GAGs and is well suited for the separation of complex N-glycans by size and degree of modification, but less useful for separating isomers [17, 26, 27].
Capillary zone electrophoresis (CZE) is an ideal technique for the separation of GAG polysaccharide mixtures because the separation mechanism acts upon the charge and shape of a molecule. Not only can CZE resolve structures that differ in mass, such as those that differ in amino modification, but CZE has been utilized to resolve positional isomers as well as enantiomers with baseline resolution [28]. CZE has been under investigation for some time because of its powerful and robust mechanism that separates molecules based on their size, shape, and charge [18, 29–34]. A new commercial interface has made sheathed CZE paired with MS (CZE-MS) more sensitive by lowering the dilution factor associated with these interfaces, providing the capability for nanospray [35]. This feature of the interface is especially useful because it allows for injection and detection of nanoliter sample volumes, making it compatible with the limited amount of a GAG sample when it is isolated from a natural source. CZE-MS has proven very effective at separating purified HS disaccharides, purified HS tetrasaccharides, and low molecular weight heparin pharmaceuticals [28, 36].
CZE-MS analysis has recently been used to assign the degree of polymerization, sulfo-modification, and to show the presence of isomers [28]. In order to determine specific sites of modification and assign structural motifs for GAG oligomers tandem MS is necessary. Threshold activation methods such as collision induced dissociation (CID/HCD) are well matched with the CZE peak widths (tens of seconds) but are sub-optimal due to their propensity to decompose labile sulfo-modifications [16]. These can be stabilized by forming sodium adducts, or direct deprotonation of the sulfate [12, 13]. During CZE-MS, NaOH can be added to the sheath liquid of the interface to exchange ionizable protons with sodium cations but this results in added experimental and spectral complexity, and reduced sensitivity. The direct deprotonation of all sulfo-groups to generate higher charge state precursors for ion activation would be ideal, but it is difficult to achieve, particularly when employing CZE for online separations.
Electron activation techniques, specifically electron detachment dissociation (EDD), have previously been shown to provide highly informative fragmentation of GAGs [13, 37–39]. However, the duty cycle for this approach is too slow for the narrow (~30 s) peaks generated by CZE separations as several seconds are required for external accumulation and electron irradiation due to poor conversion efficiencies. Fewer than 10 EDD spectra can be generated and averaged together under these constraints. Alternatively, negative electron transfer dissociation (NETD), the negative ion compliment to electron transfer dissociation (ETD), can provide electron activation through comparatively fast ion-ion interactions [40–42] on the order of ~50–100 ms. NETD has previously been implemented to characterize highly purified and synthetic GAG samples from dp4 to dp10 and up to 8 sulfo-modifications [43–46].
Although CE-MS has been previously shown to effectively separate GAG oligomers and provide compositional data, this approach has not been combined with tandem mass spectrometry for structure assignment. The current work provides the first data for CE-MS/MS of GAGs. We show NETD to be a compatible activation technique for sequencing GAGs that have been separated by CZE-MS [28]. The rationale for this combination is that NETD is much more rapid than EDD, while yielding many of the same products, and so is well suited for GAG mixture analysis. The CZE-MS/MS NETD technique was optimized with mixtures of naturally occurring and synthetic GAG tetrasaccharides, and then applied to enoxaparin, a complex low molecular weight heparin mixture.
MATERIALS & METHODS
Materials.
Bare fused silica (BFS) capillary for CZE (360 μm o.d. × 50 μm i.d.) was purchased from PolyMicro Technologies (Phoenix, AZ), borosilicate glass capillaries (1.0 mm OD × 0.75 mm i.d.) and pulled coated electrospray emitters (1.0 mm OD × 0.75 mm i.d.) were obtained from CMP scientific (Brooklyn, NY). Coating reagent N-(6-aminohexyl) aminomethyltriethoxysilane (AHS, Gelest, Morrisville, PA) was prepared in toluene. Ammonium acetate, water, and methanol were of HPLC grade (Fisher Scientific, Hampton, NH). Sodium hydroxide, acetone, toluene, and fluoranthene were purchased from Sigma-Aldrich (St. Louis, MO).
GAG Samples.
GAG tetrasaccharide samples were prepared by enzymatic depolymerization of heparan sulfate and purified using strong anion exchange high-pressure liquid chromatography (SAX-HPLC), using methods described previously [8, 36]. Epimer pair heparan sulfate tetrasaccharides were chemically synthesized and purified as described in the literature [47]. Low molecular weight heparin, enoxaparin, was purchased from USP (Rockville, MD). All samples were desalted with 3 kDa Amicon Ultra centrifugal filter (Millipore, Temecula, CA) prior to CZE separation and mass spectrometry analysis. Filters were conditioned with water, and the sample washed with two filter volumes of HPLC grade water (14,000 × g for 25 min each). Before analysis, GAG samples were diluted to 50 μg/mL in water, and enoxaparin was diluted to 0.1 mg/mL in water.
Instrumentation
CZE separations were conducted with an Agilent HP 3D capillary electrophoresis instrument (Wilmington, DE). A bare fused silica capillary was used for CZE of GAG analyte, and its inner surface was modified with AHS to reduce observed analyte migration times. The total length of the capillary ranged from 58–60 cm, and its inner diameter was 50 μm with an internal volume of approximately 1 μL. Ammonium acetate (25 mM) was used as both the background electrolyte (BGE) and the sheath liquid (SL) for reverse polarity experiments. Aqueous GAG samples were injected for 3 s at 950 mbar followed by a background electrolyte (BGE) injection for 10 s at 10 mbar. The injected volume was 0.1 nL. The ionic strength of the injected sample plug is 2–3 orders of magnitude less than that of the background electrolyte, and sample stacking is expected under these conditions which provides a sharp sample front [29]. The inlet of the capillary was then placed into a vial of BGE prior to separation. A voltage of −30 kV was applied to the capillary to drive the separation.
An EMASS-II (CMP Scientific, Brooklyn, NY) CZE-MS interface was employed to couple the CZE with a Thermo Fisher Scientific Orbitrap Elite mass spectrometer (Bremen, Germany) [35, 36, 48]. A separation capillary with a beveled outlet was nested inside a glass emitter sheath with a 0.75 mm inner diameter and a 30 μm tip orifice (CMP Scientific, Brooklyn, NY). The etched end of the capillary was positioned 0.3–0.5 mm from the tip of the sheath emitter orifice to create a mixing volume of ca. 1–5 nL and the emitter tip was filled with sheath liquid (SL) consisting of 25 mM ammonium acetate in 70 % methanol. An external power supply provided a voltage of −1.9 kV to the sheath liquid reservoir through a platinum wire, which produces electrospray at the emitter tip. MS detection was performed in negative ion mode. Prior to CZE-MS experiments, a semi-automatic optimization of source parameters was performed using sucrose octasulfate to improve sensitivity of sulfated GAGs and reduce sulfate decomposition during ion transfer prior to MS analysis. The Orbitrap Elite was scanned from m/z 150–2000 in negative ion mode for GAG oligosaccharides with a specified resolution of 120,000 for full MS and 60,000 for tandem MS experiments.
Fluoranthene radical cation produced by electron ionization was used as the reagent ion for NETD experiments. 110 ms activation times were used for doubly charged tetrasaccharides and triply charged hexasaccharides. 50 ms activation times were used for triply charged tetrasaccharides. Emission current was set to 75 μA and electron energy set to −70 V. Experiments were manually optimized. Preliminary CZE-MS was acquired to identify potential precursor ions, followed by individual NETD tandem experiments for each precursor, and then methods that split spectra acquisition evenly between MS1 and NETD MS2 of a single selected precursor. A data dependent method was designed to apply NETD fragmentation to the two most intense m/z in the MS1 throughout the electropherogram, excluding the BGE peaks.
RESULTS & DISCUSSION
With CZE, application of a strong electric field (ca. 30 kV) causes ions to migrate through an electrolyte solution. Small differences in mobility based on charge and molecular shape result in separation. This method is ideal for separating GAGs, which have a repeating disaccharide backbone structure with a high degree of charge arising from ionizable sulfo-modifications and carboxyl groups. CZE separations are rapid, with typical migration times less than 20–30 minutes (column lengths of 55 cm, and applied potential of 30 kV). Migration times vary too much to use this parameter to assign structures due to capillary heating effects, changes in the degree of modification of the interior wall of the separation capillary, and differences in capillary lengths between runs. Peaks in the electropherogram representing unique oligosaccharide structures are approximately 30 s wide. NETD activation is accomplished within 120 ms for low charge state precursors and 60 ms for higher charge state precursors and allows spectral averaging for a selected precursor ion or even for multiple co-migrating ions of different mass-to-charge. NETD operates near the speed of CID/HCD but with the informative ion activation of EDD. For an NETD experiment at a mass resolving power of 60,000 and a 110 ms activation time, 10–20 spectra can be recorded and averaged within a 30 s window corresponding to the peak width in CZE. NETD can also effectively fragment low charge state precursors without prior modification and produce fragmentation that provides sufficient information to assign sites of sulfo-modification to specific residues.
CZE has sufficient resolution to separate the enantiomeric iduronic acid (IdoA) and glucuronic acid (GlcA) residues, which differ only in the stereochemistry of C5. Figure 1A shows CZE separation of the synthetic tetrasaccharides GlcA-GlcNAc6S-IdoA-GlcNAc6S (GI) and GlcA-GlcNAc6S-GlcA-GlcNAc6S (GG), which both have two sulfo-modifications. A simple binary mixture of known tetrasaccharide epimers is a good test of CZE separation efficiency and NETD sequencing. Figure 1B shows the NETD fragmentation of the [M-2H]2- precursor of GI, a tetrasaccharide with two sulfo-modifications on each of the 6-O positions of the glucosamine residues. Glycosidic bond fragment coverage is nearly complete across the GI ion with only B1 and C1 missing. The two sulfo-modifications can be confidently assigned to the glucosamine residues and cross-ring cleavages allow the assignment of the sulfo-modification on the reducing end at the 6-O position. The second sulfo-modification, assigned to the inner hexosamine residue cannot be differentiated between the 6-O- and 3-O- position; however, sulfo-modification of the 3-O position is rare [49]. Figure 1C is the NETD fragmentation of GG. The fragmentation patterns for both GG and GI are almost identical except for a 0,2A3 peak present in GG and not in GI. This diagnostic ion has been shown previously to indicate the presence of glucuronic acid [50]. With this method it is possible to distinguish enantiomers as well as anomers. The latter often appear as a small peak of similar migration time to the dominant anomeric conformation, leading to the peak splitting observed in Figure 1A. Previous work using differential ion mobility gave similar results for resolving anomers [51]. The MS/MS spectra for the two structures can be differentiated by inspection in this case, though multivariate statistical approaches like principal component analysis are required for more highly sulfated HS tetramers [52, 53].
Figure 1.

A) Capillary zone electrophoresis mass spectrometry (CZE-MS) separation and detection of a binary mixture of synthetically produced heparan sulfate (HS) tetrasaccharide epimers with two sulfo-modifications, with GlcA-GlcNAc6S-IdoA-GlcNAc6S (GI) on the left, and GlcA-GlcNAc6S-GlcA-GlcNAc6S (GG) on the right. B) The negative electron transfer dissociation (NETD) spectrum for the peak at 20 min in the CZE-MS electropherogram corresponding to GI, with structural assignments. C) The NETD spectrum for the peak at 21 min in the CZE-MS electropherogram corresponding to GG, with structural assignments.
The degree of sulfo-modification has an observable effect on the charge state distribution that impacts precursor selection. The difference in the CZE-MS spectra is demonstrated for a tetrasaccharide with two sulfo-modifications (Figure 2A top) compared to a tetrasaccharide with six sulfo-modifications (Figure 2A bottom). The tetrasaccharide with two sulfo-modifications generated only a 2- charge state peak, while the tetrasaccharide with six sulfo-modifications generated both 2- and a 3- charge state peak for precursor selection. By direct infusion, one would expect to see a larger difference in the charge state distributions for these two HS tetrasaccharides, but we find that the degree of ionization is lower with our CZE interface.
Figure 2.

A) Comparison of peak intensity measured for the 2- charge state and 3- charge state between a HS tetrasaccharide with two sulfo-modifications (top) and a tetrasaccharide with six sulfo-modifications (bottom). B) The NETD spectrum for the 2- charge state precursor of the six sulfo-modified HS tetrasaccharide, with structural assignments. C) The NETD spectrum for the 3- charge state precursor for the six-sulfo modified HS tetrasaccharide, with structural assignments.
The higher charge state enables more complete sequence information by NETD. Figure 2B shows the NETD fragmentation of the [M-2H]2- precursor for the tetrasaccharide (HS6) with six sulfo-modifications. The quality and coverage of NETD fragmentation has decreased compared to GI, due to the high degree of sulfo-modification on the low charge state precursor, but there is enough information to assign sulfo-modifications to each residue. The molecular weight of the molecule indicates that the amine group of both glucosamine residues are sulfated. The glycosidic cleavages also indicate that each uronic acid residue contains a sulfo-modification, which should be in the 2-O position, based on the known details of the biosynthesis of HS/Hp. This leaves a sulfate group on either the 6-O or 3-O position of each glucosamine residue, with the former occurring far more commonly than the latter.
In contrast, the NETD mass spectrum of the [M-3H]3- precursor for the same tetrasaccharide (HS6) is far more informative, as seen in Figure 2C. Glycosidic fragmentation coverage is nearly complete, with only the B1 fragment missing, and all sulfo-modifications can be confidently assigned to specific residues. Cross-ring cleavage is sufficient to assign sites of modification, and the 3,5A peak present on both glucosamine residues can be utilized to differentiate between 6-O and 3-O sulfo-modifications.
Unlike threshold activation techniques, such as CID or HCD, NETD does not require sodium replacement of protons to stabilize the labile sulfo-modifications. However, it has been shown that a certain degree of Na+/H+ exchange can improve electron based fragmentation [12]. Figure 3 shows annotated structures for a 2- precursor series of a HS tetrasaccharide with four sulfo-modifications and A) [M-2H]2-, B) [M-4H+2Na]2-, and C) [M-6H+4Na]2-. Overall, fragmentation for this tetrasaccharide is informative, with the production of large set of glycosidic and cross-ring fragments. Annotated spectra for the three tetrasaccharides are provided in the supplemental information (Figures S1–S3). The addition of two sodium adducts to the precursor has a minor effect; two glycosidic fragments are lost, C1 and Y2, while two cross-ring fragments are gained overall, though the observed cross-ring fragments are not entirely comparable. Fully ionizing the molecule by replacing all ionizable protons with sodium ions reduces overall fragmentation; few glycosidic bond fragments remain in comparison, and cross-ring fragmentation is shunted towards the outer residues with only two of the nine cross-ring fragments located on an inner residue. Adducting sodium to low charge state precursors presented by CZE-MS changes NETD fragmentation pathways and rearrangement options, and in some cases reduces the number of product ions once the molecule is fully ionized. In general, sodium adduction does not benefit NETD activation of heparin/HS after CZE separation.
Figure 3.

Comparison of NETD fragmentation patterns for different degrees of Na+ stabilization of a four sulfo-modified HS tetrasaccharide. A) Two of six sites are ionized with no Na+ adduction. B) Four of six sites are ionized with two Na+ adductions. C) All six sites are ionized with 4 Na+ adductions.
A more stringent test of the CZE-NETD MS/MS platform is the analysis of complex mixtures of GAGs. Enoxaparin, a low molecular weight heparin (LMWH) pharmaceutical, is a complex mixture derived from heparin polysaccharides that have been chemically digested to oligomers with up to 30 saccharide rings reported by others [54–56], and 3–12 saccharide rings observed with CZE-MS. We have examined this mixture by CZE-MS previously, and found that the components are detected within a 6 min wide window and present a series of well-defined peaks [28]. We have also identified compositions in enoxaparin with an extra water loss that is consistent with chemical digestion pathways that lead to a 1,6-anhydro bicyclic reducing end glucosamine residue [57, 58]. Here, we have integrated NETD with CZE-MS for the first time to assign structures to the components of enoxaparin. Figure 4 shows the assignment of the top five most intense peaks found in the CZE separation of enoxaparin and characterized using NETD fragmentation. Mass lists of assigned peaks for all structures found in enoxaparin are in supplemental tables 1–7 (ST1–7). The Orbitrap Elite was set to acquire data dependent MS/MS, selecting the two most intense peaks from the MS1 as a precursor to isolate and activate with NETD. The NETD spectra of the five most prominent masses were manually averaged and utilized to assign structure and sulfo-modification sites. The five prominent structures in enoxaparin are found to be highly sulfated, as one would expect for these heparin-derived fragments. Two of these five components, m/z 526.98 (2-) and 543.31 (3-), are found to have a molecular weight that indicates the presence of a 1,6-anhydro bicyclic ring on the reducing end residue, which is an expected byproduct caused by the base-catalyzed digestion process used in the production of enoxaparin.
Figure 4.

An electropherogram of the low molecular weight heparin pharmaceutical enoxaparin with structure assignments for the 5 most intense peaks determined by means of NETD activation.
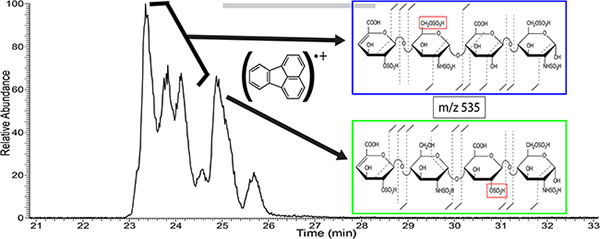
Figure 5 shows extracted ion electropherograms for the five most prominent mass-to-charge values found in the CE separation of enoxaparin. Multiple peaks in the extracted ion electropherograms indicate that isomeric species exist for many of the prominent mass-to-charge values. These isomers can include GlcA/IdoA diastereomers as well as different combinations of sulfo-modification. NETD fragmentation was used to examine structural differences for these isomeric species. As an example, in Figure 6A more than five peaks were observed for m/z 535.982- throughout the electropherogram. These five peaks were selected for NETD analysis. Figure 6B shows the NETD spectra for the first peak in the grouping of three peaks from 23–24 min, which are partially resolved. Annotated NETD spectra and structural assignments for the three peaks in the grouping are shown for comparison in supplemental figure S4, and can be seen to be nearly identical. High intensity peaks in the spectra that are unassigned are uninformative neutral losses and are detailed in the supplemental tables. The NETD spectra suggest a common pattern of sulfo-modification that differs from the peak that migrates at 25 min. The molecular structures in this grouping must differ by uronic acid stereochemistry (for the uronic acid residue closest to the reducing end) or by anomer state of the reducing end. NETD of the fourth peak at 25 min, Figure 6C, reveals a different pattern of sulfo-modification; on the 2-O position of the inner uronic acid residue instead of the 6-O position of the glucosamine observed for the previous three peaks, indicated by the red box in Figure 6. A peak list for the spectra in 6B and 6C, along with the assigned structures, is provided in supplementary table ST5. The fifth peak, 25.7 min, is too low in intensity to generate informative fragmentation with NETD. Multiple peaks indicating isomeric structures appear for many compositions found in Enoxaparin. Figure 7A shows that m/z 526 appears as only two main peaks in the electropherogram. Characterization of these two peaks with NETD reveals that they differ in sulfo-modification pattern. The annotated spectrum for the first peak at 24.53 min, Figure 7B, shows that the sulfo-modification is located on the reducing end glucosamine residue, and the 1,6-anhydro bicyclic ring structure of the reducing end glucosamine residue indicates that the sulfo-modification is most likely located on the rare 3-O position. The annotated spectrum for the second peak at 25.08 min, Figure 7C, shows that the sulfo-modification is located on the inner uronic acid residue, most likely at the 2-O position. NETD characterization of the two peaks for m/z 543, supplemental Figure S5, shows that the hexasaccharide has two different sites of sulfo-modification as well, with the late migrating peak most likely having a sulfo-modification in the rare 3-O position of the non-reducing end glucosamine due to the 1,6-anhydro bicyclic ring structure. Annotated spectra for the last remaining prominent mass-to-charge value m/z 415, a trisaccharide, as well as a separate low intensity tetrassacharide, m/z 549, are provided in the supplemental information (Figures S6 and S7).
Figure 5.

Extracted ion electropherograms with the mass-to-charge of the five most intense components found in enoxaparin, showing the presence of isomers at each of these m/z values. Composition assignments are represented as [dHexA, HexA, GlcN, NAc, S, water loss].
Figure 6.

A) Electropherogram for NETD activation of m/z 535.98 (2-) within enoxaparin, with structural assignment for the four most intense peaks. B) NETD spectrum for the first of the three peaks (23–24 min) with annotations and structural assignment. C) NETD spectrum for the fourth peak (25 min) with annotation and structural assignments. Annotated spectra for the second and third peak can be found in the supplemental information.
Figure 7.

A) Selected ion electropherogram of m/z 526.98 in enoxaparin. B) Annotated spectrum and structure for m/z 526 left peak (24.53 min). C) Annotated spectrum and structure for m/z 526.98 right peak (25.08 min).
CONCLUSIONS
CZE-NETD MS/MS provides a promising platform for analyzing complex GAG mixtures, to derive both chemical composition and structural information. GAG mixtures can be effectively separated by CZE. The speed of NETD activation (50–100 ms) is a good match for the CZE peak width (30 s), allowing improved signal-to-noise by signal averaging, and generating informative fragmentation spectra. In general, lower charge states are produced in the online CZE-MS experiment compared to direct infusion of GAGs. For this reason, CID is not a good match for CZE-MS sequencing of GAGs, as these lower charge states readily undergo sulfate decomposition. In contrast to CID, NETD of these lower charge state precursors produced by CZE separation generates fragmentation detail that is sufficient to assign sulfo-modification to specific GAG residues. Higher charge state precursors become more intense as the degree of sulfo-modifications increases and provides even more detailed fragmentation for the assignment of sulfo-modification to specific sites within GAG residues. With enough sulfo-modifications the higher charge state peak is sufficiently intense for NETD activation, and this provides near total coverage of the molecule. CZE-MS with NETD activation of complex mixtures like enoxaparin heparin results in structural assignment of multiple compositions. Isomeric compositions within enoxaparin are resolved by CZE and unique structures are assigned when there are differences in sulfo-modification. CZE is found to resolve diastereomers resulting from differences in uronic acid stereochemistry, but their assignment by tandem mass spectrometry (IdoA versus GlcA) remains a challenge in most cases.
Supplementary Material
Table 1.
The structure of four HS tetrasaccharide standards used for experiments are shown using symbol notation as defined in the table.
| Sample 1 | |
| Sample 2 | |
| Sample 3 | |
| Sample 4 |
 Hexuronic Acid
Hexuronic Acid
 Iduronic Acid
Iduronic Acid
 Glucoronic Acid
Glucoronic Acid
 Glucosamine
Glucosamine
 N-acetyl Glucosamine
N-acetyl Glucosamine
Mixtures of glycosaminoglycans can be resolved and structurally characterized by combining capillary zone electrophoresis (CZE) and ion-ion chemistry for tandem mass spectrometry analysis
Glycosaminoglycans exist as negative ions in solution, and require reverse polarity for CZE analysis, negative ionization conditions, and negative electron transfer dissociation (NETD) for tandem mass spectrometry analysis.
The successful combination of CZE and NETD is demonstrated for the analysis of a complex mixture, enoxaparin, a pharmaceutical compound produced by chemical depolymerization of heparin.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge generous financial support from the National Institutes of Health, grants R21HL136271, U01CA231074, and P41GM103390. The authors are grateful for the GAG tetrasaccharide standards that were provided by Prof. Geert-Jan Boons.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Linhardt RJ, Toida T, Role of Glycosaminoglycans in Cellular Communication, Accounts of Chemical Research, 37 (2004) 431–438. [DOI] [PubMed] [Google Scholar]
- [2].Aquino RS, Park PW, Glycosaminoglycans and infection, Frontiers in bioscience (Landmark edition), 21 (2016) 1260–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Iozzo RV, Zoeller JJ, Nyström A, Basement membrane proteoglycans: Modulators Par Excellence of cancer growth and angiogenesis, Molecules and Cells, 27 (2009) 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Linhardt RJ, 2003 Claude S Hudson Award Address in Carbohydrate Chemistry. Heparin: Structure and Activity, Journal of Medicinal Chemistry, 46 (2003) 2551–2564. [DOI] [PubMed] [Google Scholar]
- [5].Xu D, Esko JD, Demystifying Heparan Sulfate–Protein Interactions, Annual Review of Biochemistry, 83 (2014) 129–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Choay J, Petitou M, Lormeau JC, Sinaÿ P, Casu B, Gatti G, Structure-activity relationship in heparin: A synthetic pentasaccharide with high affinity for antithrombin III and eliciting high anti-factor Xa activity, Biochemical and Biophysical Research Communications, 116 (1983) 492–499. [DOI] [PubMed] [Google Scholar]
- [7].Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW, The anticoagulant activation of antithrombin by heparin, Proceedings of the National Academy of Sciences, 94 (1997) 14683–14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Singh A, Kett WC, Severin IC, Agyekum I, Duan J, Amster IJ, Proudfoot AEI, Coombe DR, Woods RJ, The Interaction of Heparin Tetrasaccharides with Chemokine CCL5 Is Modulated by Sulfation Pattern and pH, The Journal of Biological Chemistry, 290 (2015) 15421–15436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen Y, Lin L, Agyekum I, Zhang X, St Ange K, Yu Y, Zhang F, Liu J, Amster IJ, Linhardt RJ, Structural Analysis of Heparin-Derived 3-O-Sulfated Tetrasaccharides: Antithrombin Binding Site Variants, Journal of pharmaceutical sciences, 106 (2017) 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Casu B, Naggi A, Torri G, Re-visiting the structure of heparin, Carbohydrate Research, 403 (2015) 60–68. [DOI] [PubMed] [Google Scholar]
- [11].Laremore TN, Ly M, Solakyildirim K, Zagorevski DV, Linhardt RJ, High-resolution preparative separation of glycosaminoglycan oligosaccharides by polyacrylamide gel electrophoresis, Analytical Biochemistry, 401 (2010) 236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kailemia MJ, Li L, Ly M, Linhardt RJ, Amster IJ, Complete Mass Spectral Characterization of a Synthetic Ultralow-Molecular-Weight Heparin Using Collision-Induced Dissociation, Analytical Chemistry, 84 (2012) 5475–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wolff JJ, Chi L, Linhardt RJ, Amster IJ, Electron Detachment Dissociation of Gycosaminoglycan Tetrasaccharides, Journal of the American Society for Mass Spectrometry, 18 (2007) 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ly M, Leach Iii FE, Laremore TN, Toida T, Amster IJ, Linhardt RJ, The proteoglycan bikunin has a defined sequence, Nature Chemical Biology, 7 (2011) 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yu Y, Duan J, Leach FE, Toida T, Higashi K, Zhang H, Zhang F, Amster IJ, Linhardt RJ, Sequencing the Dermatan Sulfate Chain of Decorin, Journal of the American Chemical Society, 139 (2017) 16986–16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zaia J, Glycosaminoglycan Glycomics Using Mass Spectrometry, Molecular & Cellular Proteomics: MCP, 12 (2013) 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gill VL, Aich U, Rao S, Pohl C, Zaia J, Disaccharide Analysis of Glycosaminoglycans Using Hydrophilic Interaction Chromatography and Mass Spectrometry, Analytical Chemistry, 85 (2013) 1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hitchcock AM, Bowman MJ, Staples GO, Zaia J, Improved Workup for Glycosaminoglycan Disaccharide Analysis using Capillary Electrophoresis with Laser-Induced Fluorescence Detection, Electrophoresis, 29 (2008) 4538–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yang B, Weyers A, Baik JY, Sterner E, Sharfstein S, Mousa SA, Zhang F, Dordick JS, Linhardt RJ, Ultra-performance ion-pairing liquid chromatography with on-line electrospray ion trap mass spectrometry for heparin disaccharide analysis, Analytical Biochemistry, 415 (2011) 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zaia J, On-Line Separations Combined with MS for Analysis of Glycosaminoglycans, Mass spectrometry reviews, 28 (2009) 254–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Staples GO, Zaia J, Analysis of Glycosaminoglycans Using Mass Spectrometry, Current proteomics, 8 (2011) 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang Z, Li D, Sun X, Bai X, Jin L, Chi L, Liquid chromatography–diode array detection–mass spectrometry for compositional analysis of low molecular weight heparins, Analytical Biochemistry, 451 (2014) 35–41. [DOI] [PubMed] [Google Scholar]
- [23].Yang B, Chang Y, Weyers AM, Sterner E, Linhardt RJ, Disaccharide analysis of glycosaminoglycan mixtures by ultra-high-performance liquid chromatography-mass spectrometry, J Chromatogr A, 1225 (2012) 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jones CJ, Beni S, Larive CK, Understanding the Effect of the Counterion on the Reverse-Phase Ion-Pair High-Performance Liquid Chromatography (RPIP-HPLC) Resolution of Heparin-Related Saccharide Anomers, Analytical Chemistry, 83 (2011) 6762–6769. [DOI] [PubMed] [Google Scholar]
- [25].Fasciano JM, Danielson ND, Ion chromatography for the separation of heparin and structurally related glycoaminoglycans: A review, Journal of Separation Science, 39 (2016) 1118–1129. [DOI] [PubMed] [Google Scholar]
- [26].Ruhaak LR, Zauner G, Huhn C, Bruggink C, Deelder AM, Wuhrer M, Glycan labeling strategies and their use in identification and quantification, Analytical and Bioanalytical Chemistry, 397 (2010) 3457–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wuhrer M, de Boer AR, Deelder AM, Structural glycomics using hydrophilic interaction chromatography (HILIC) with mass spectrometry, Mass Spectrometry Reviews, 28 (2009) 192–206. [DOI] [PubMed] [Google Scholar]
- [28].Sanderson P, Stickney M, Leach FE, Xia Q, Yu Y, Zhang F, Linhardt RJ, Amster IJ, Heparin/heparan sulfate analysis by covalently modified reverse polarity capillary zone electrophoresis-mass spectrometry, Journal of Chromatography A, 1545 (2018) 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Whatley H, Basic Principles and Modes of Capillary Electrophoresis, in: Petersen JR, Mohammad AA (Eds.) Clinical and Forensic Applications of Capillary Electrophoresis, Humana Press, Totowa, NJ, 2001, pp. 21–58. [Google Scholar]
- [30].Albin M, Grossman PD, Moring SE, Sensitivity enhancement for capillary electrophoresis, Analytical Chemistry, 65 (1993) 489A–497A. [Google Scholar]
- [31].Smith RD, Olivares JA, Nguyen NT, Udseth HR, Capillary zone electrophoresis-mass spectrometry using an electrospray ionization interface, Analytical Chemistry, 60 (1988) 436–441. [Google Scholar]
- [32].Ruiz-Calero V, Moyano E, Puignou L, Galceran MT, Pressure-assisted capillary electrophoresis— electrospray ion trap mass spectrometry for the analysis of heparin depolymerised disaccharides, Journal of Chromatography A, 914 (2001) 277–291. [DOI] [PubMed] [Google Scholar]
- [33].Zaia J, Capillary electrophoresis-mass spectrometry of carbohydrates, Methods in molecular biology (Clifton, N.J.), 984 (2013) 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ucakturk E, Cai C, Li L, Li G, Zhang F, Linhardt RJ, Capillary electrophoresis for total glycosaminoglycan analysis, Analytical and Bioanalytical Chemistry, 406 (2014) 4617–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sun L, Zhu G, Zhang Z, Mou S, Dovichi NJ, A third-generation electro-kinetically pumped sheath flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests, Journal of proteome research, 14 (2015) 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lin L, Liu X, Zhang F, Chi L, Amster IJ, Leach FE, Xia Q, Linhardt RJ, Analysis of heparin oligosaccharides by capillary electrophoresis-negative-ion electrospray ionization mass spectrometry, Analytical and bioanalytical chemistry, 409 (2017) 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wolff JJ, Laremore TN, Aslam H, Linhardt RJ, Amster IJ, Electron-Induced Dissociation of Glycosaminoglycan Tetrasaccharides, Journal of the American Society for Mass Spectrometry, 19 (2008) 1449–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Oh HB, Leach FE, Arungundram S, Al-Mafraji K, Venot A, Boons G-J, Amster IJ, Multivariate Analysis of Electron Detachment Dissociation and Infrared Multiphoton Dissociation Mass Spectra of Heparan Sulfate Tetrasaccharides Differing Only in Hexuronic acid Stereochemistry, Journal of The American Society for Mass Spectrometry, 22 (2011) 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Leach FE, Ly M, Laremore TN, Wolff JJ, Perlow J, Linhardt RJ, Amster IJ, Hexuronic Acid Stereochemistry Determination in Chondroitin Sulfate Glycosaminoglycan Oligosaccharides by Electron Detachment Dissociation, Journal of The American Society for Mass Spectrometry, 23 (2012) 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].McLuckey SA, Stephenson JL, O’Hair RAJ, Decompositions of odd- and even-electron anions derived from deoxy-polyadenylates, Journal of the American Society for Mass Spectrometry, 8 (1997) 148–154. [Google Scholar]
- [41].Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF, Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry, Proceedings of the National Academy of Sciences of the United States of America, 101 (2004) 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Coon JJ, Shabanowitz J, Hunt DF, Syka JEP, Electron transfer dissociation of peptide anions, Journal of the American Society for Mass Spectrometry, 16 (2005) 880–882. [DOI] [PubMed] [Google Scholar]
- [43].Wolff JJ, Leach FE, Laremore TN, Kaplan DA, Easterling ML, Linhardt RJ, Amster IJ, Negative Electron Transfer Dissociation of Glycosaminoglycans, Analytical Chemistry, 82 (2010) 3460–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Leach FE, Wolff JJ, Xiao Z, Ly M, Laremore TN, Arungundram S, Al-Mafraji K, Venot A, Boons G-J, Linhardt RJ, Amster IJ, Negative electron transfer dissociation Fourier transform mass spectrometry of glycosaminoglycan carbohydrates, European Journal of Mass Spectrometry (Chichester, England), 17 (2011) 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Leach FE, Riley NM, Westphall MS, Coon JJ, Amster IJ, Negative Electron Transfer Dissociation Sequencing of Increasingly Sulfated Glycosaminoglycan Oligosaccharides on an Orbitrap Mass Spectrometer, Journal of The American Society for Mass Spectrometry, 28 (2017) 1844–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wu J, Wei J, Hogan JD, Chopra P, Joshi A, Lu W, Klein J, Boons G-J, Lin C, Zaia J, Negative Electron Transfer Dissociation Sequencing of 3-O-Sulfation-Containing Heparan Sulfate Oligosaccharides, Journal of The American Society for Mass Spectrometry, 29 (2018) 1262–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Arungundram S, Al-Mafraji K, Asong J, Leach FE, Amster IJ, Venot A, Turnbull JE, Boons G-J, Modular Synthesis of Heparan Sulfate Oligosaccharides for Structure−Activity Relationship Studies, Journal of the American Chemical Society, 131 (2009) 17394–17405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sun L, Zhu G, Zhao Y, Yan X, Mou S, Dovichi NJ, Ultrasensitive and Fast Bottom-up Analysis of Femtogram Amounts of Complex Proteome Digests, Angewandte Chemie International Edition, 52 (2013) 13661–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Thacker BE, Xu D, Lawrence R, Esko JD, Heparan sulfate 3-O-sulfation: a rare modification in search of a function, Matrix biology: journal of the International Society for Matrix Biology, 35 (2014) 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wolff JJ, Chi L, Linhardt RJ, Amster IJ, Distinguishing Glucuronic from Iduronic Acid in Glycosaminoglycan Tetrasaccharides by Using Electron Detachment Dissociation, Analytical Chemistry, 79 (2007) 2015–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kailemia MJ, Park M, Kaplan DA, Venot A, Boons G-J, Li L, Linhardt RJ, Amster IJ, Highfield asymmetric-waveform ion mobility spectrometry and electron detachment dissociation of isobaric mixtures of glycosaminoglycans, Journal of The American Society for Mass Spectrometry, 25 (2014) 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Agyekum I, Patel AB, Zong C, Boons G-J, Amster IJ, Assignment of hexuronic acid stereochemistry in synthetic heparan sulfate tetrasaccharides with 2-O-sulfo uronic acids using electron detachment dissociation, International Journal of Mass Spectrometry, 390 (2015) 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Agyekum I, Zong C, Boons G-J, Amster IJ, Single Stage Tandem Mass Spectrometry Assignment of the C-5 Uronic Acid Stereochemistry in Heparan Sulfate Tetrasaccharides using Electron Detachment Dissociation, Journal of The American Society for Mass Spectrometry, 28 (2017) 1741–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mourier PAJ, Agut C, Souaifi-Amara H, Herman F, Viskov C, Analytical and statistical comparability of generic enoxaparins from the US market with the originator product, Journal of Pharmaceutical and Biomedical Analysis, 115 (2015) 431–442. [DOI] [PubMed] [Google Scholar]
- [55].Guerrini M, Rudd TR, Mauri L, Macchi E, Fareed J, Yates EA, Naggi A, Torri G, Differentiation of Generic Enoxaparins Marketed in the United States by Employing NMR and Multivariate Analysis, Analytical Chemistry, 87 (2015) 8275–8283. [DOI] [PubMed] [Google Scholar]
- [56].Zaia J, Khatri K, Klein J, Shao C, Sheng Y, Viner R, Complete Molecular Weight Profiling of Low-Molecular Weight Heparins Using Size Exclusion Chromatography-Ion Suppressor-High-Resolution Mass Spectrometry, Analytical Chemistry, 88 (2016) 10654–10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mascellani G, Guerrini M, Torri G, Liverani L, Spelta F, Bianchini P, Characterization of di- and monosulfated, unsaturated heparin disaccharides with terminal N-sulfated 1,6-anhydro-beta-D-glucosaminSe or N-sulfated 1,6-anhydro-beta-D-mannosamine residues, 2007. [DOI] [PubMed]
- [58].Li D, Chi L, Jin L, Xu X, Du X, Ji S, Chi L, Mapping of low molecular weight heparins using reversed phase ion pair liquid chromatography–mass spectrometry, Carbohydrate Polymers, 99 (2014) 339–344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.