

Abstract

Selective direct ruthenium-catalyzed semihydrogenation of diaryl alkynes to the corresponding E-alkenes has been achieved using alcohols as the hydrogen source. The method employs a simple ruthenium catalyst, does not require external ligands, and affords the desired products in > 99% NMR yield in most cases (up to 93% isolated yield). Best results were obtained using benzyl alcohol as the hydrogen donor, although biorenewable alcohols such as furfuryl alcohol could also be applied. In addition, tandem semihydrogenation–alkylation reactions were demonstrated, with potential applications in the synthesis of resveratrol derivatives.
Introduction
The alkene motif is present in a variety of important molecules, including natural products, pharmaceuticals, and fragrances (Figure 1).1 Stereoselective installation of this functionality therefore remains central to organic synthesis. Semihydrogenation of alkynes is a natural synthetic transformation to obtain alkenes. However, E-selective alkyne semihydrogenations have historically been more difficult to achieve than Z-selective. The former transformation has typically been limited to alkynes bearing alcohols, amines, or ketones in the propargylic position, generally requiring stoichiometric reagents2 or proceeding via two-step methods such as trans-hydrosilylation followed by protodesilylation.3
Figure 1.

Selected E-alkenes.
Lately, hydrogenation based on homogeneous transition-metal catalysis has begun to offer a remedy to these limitations.2 For example, iron,4 cobalt,5 nickel,6 palladium,7 manganese,8 and iridium9 have been used to obtain alkenes from alkynes with E-selectivity. In particular, an iridium-catalyzed method for alkyne semihydrogenation recently reported by Yang et al. deserves highlighting9a as it allows the selective formation of either the E- or the Z-alkene isomer simply by adding a bulkier ligand (COD) to the reaction system in the latter case. In addition, an inexpensive and sustainable alcohol (ethanol) is used as the hydrogen source. A few accounts of direct ruthenium-based E-selective semihydrogenations of alkynes have also been published in the past decade (Scheme 1).10 Several of the reported ruthenium systems require elevated temperatures (145–180 °C)10c,10h or stoichiometric or excess amounts of organic acids (1–50 equiv).10c,10k Despite displaying good substrate scope in the presence of other reductive-sensitive functional groups, these harsh reaction conditions could limit the utility of these methods. Milder methods for semihydrogenations based on ruthenium have recently been described.10d,10j Fürstner and co-workers used high pressures of dihydrogen (10 bar) with silver triflate as an additive at ambient temperatures10d or propargylic alcohols as substrates at lower pressures (1 bar),10l while Lindhardt recently published a method proceeding at 45 °C with dihydrogen generated in situ in a closed two-chamber system.10j Djukic et al. have shown that μ-chlorido and μ-hydroxobridged ruthenacycles can affect the hydrogenation of triple bonds using isopropanol as the hydrogen donor at 90 °C,10b while Gelman has reported an elegant semihydrogenation of alkynes involving ligand–metal cooperation as the mode of action, using a ruthenium catalyst and a mixture of formic acid and sodium formate as the hydrogen source.10i By adding D2O, this procedure could also be applied toward deuterium labeling.
Scheme 1. Ruthenium-Catalyzed Methods for Alkyne Semihydrogenation to E-Alkenes.

While conducting a ruthenium-catalyzed “borrowing hydrogen” reaction involving alcohols and amines in the presence of an alkyne functionality,11 we noticed that small amounts of the corresponding alkene were formed. We envisioned that a transfer hydrogenation between the alcohol and the alkyne competed with the borrowing hydrogen reaction to a minor extent. Indeed, in 1981, Shvo and co-workers presented a ruthenium-catalyzed oxidative ester formation from alcohols using diphenylacetylene as a hydrogen acceptor.10a Despite recent reports on alkyne semihydrogenations, the scope of ruthenium-catalyzed transfer hydrogenation between alcohols and alkynes has, to the best of our knowledge, not been investigated in detail.10b We herein present a relatively mild semihydrogenation of alkynes, which can be performed without the necessity of external ligands or stoichiometric amounts of organic/inorganic acids or bases. The procedure performs well with diaryl acetylenes and is experimentally facile, using only commercially available reagents and without the need for any special equipment.
Results and Discussion
For the initial investigation of the transfer hydrogenation between alcohols and alkynes, diphenylacetylene (1a) was used as a model substrate (Scheme 2). A selection of different alcohols were screened for efficiency, E/Z selectivity, and their ability to avoid over-reduction. The reaction was performed in the presence of a simple ruthenium catalyst, Ru3(CO)12, and initially with stoichiometric amounts of tBuOK as a base, using toluene as the solvent and heating the reactions in a heating block. Experiments were analyzed by 1H NMR using 2,5-dimethylfuran as an internal standard.12 Of the screened hydrogen donors, benzylic alcohols (benzyl and furfuryl alcohol, 1-phenylethanol) stood out both in terms of selectivity and efficiency. In particular, benzyl alcohol produced E-stilbene ((E)-2a) with 100% selectivity over Z-stilbene ((Z)-2a) while only generating ∼2% bibenzyl (3) via over-reduction. A number of other alcohols also displayed good compatibility with the reaction. Cyclopentanol generated the semihydrogenation product in good yields, with only minor over-reduction, while longer noncyclic secondary aliphatic alcohols (2-butanol, 3-pentanol) reacted sluggishly. Isopropyl alcohol and ethanol both showed a good conversion to alkene, while the more hindered neopentyl alcohol and glycerol reacted slowly. Interestingly, the reactivity of isopropyl alcohol could be greatly enhanced by introducing a methoxy group in the 1-position, generating a substantial amount of bibenzyl. Control experiments were also performed. Excluding the base from the reaction significantly lowered the efficiency, affording 13% of (Z)-2a and no other products. The alcohol and catalyst, as expected, proved to be essential to the reaction, with no products formed in their absence.
Scheme 2. Screening of Alcohols as Hydrogen Donors for Ru-Catalyzed Alkyne Hydrogenation.

NMR yield (2,5-dimethylfuran as an internal standard). Reactions were heated in a heating block.
While benzyl alcohol outperformed the other hydrogen donors, the generation of reactive benzaldehyde in situ could under some circumstances be problematic due to its potential reactivity with nucleophiles. Isopropyl alcohol, on the other hand, forms acetone, which is less prone to adduct formation with nucleophiles. Additionally, compared to benzyl alcohol and benzaldehyde, both isopropyl alcohol and acetone can be easily removed through evaporation, thus expediting the purification of the product. Further optimization was thus performed using isopropyl alcohol as the hydrogen donor, aiming to improving the E/Z selectivity and yield.
In addition to Ru3(CO)12, nine other commercially available ruthenium catalysts were screened using isopropyl alcohol as the hydrogen donor (Table 1). The reactivity of the catalysts varied from very low when using RuCl3 (entry 2), Cp*RuCl(COD) (entry 5), or the Shvo catalyst (entry 9 and Figure 2) to being higher but unselective for the semihydrogenation product when RuCl2(PPh3)3 was employed (entry 7). The Grubbs first-generation catalyst (Figure 2) gave the fully reduced bibenzyl product 3 with nearly complete selectivity in a good yield (entry 4). However, our interest lays in the selective semihydrogenation to form the (E)-2a. In this context, catalyst RuCl2(DMSO)4 displayed good properties, with a combined yield of 91% and 6:1 in terms of the E/Z selectivity (entry 3). RuCl(CO)H(PPh3)3 also performed well, affording only (E)-2a in a good yield, albeit with some concomitant over-reduction to 3 (entry 6). Viable catalysts for the E-selective semihydrogenation of diphenylacetylene, using isopropyl alcohol as the hydrogen donor, were thus found to be Ru3(CO)12, RuCl2(DMSO)4, and RuCl(CO)H(PPh3)3. RuCl2(DMSO)4 was selected for further studies when using isopropyl alcohol as the hydrogen donor.
Table 1. Catalyst Screening in the Ru-Catalyzed Reduction of Phenylacetylene (1a)a.
Figure 2.

Structures of the Grubbs first-generation and Shvo catalysts.
With two catalyst systems in hand, that is, Ru3(CO)12/benzyl alcohol and RuCl2(DMSO)4/iPrOH, further studies concerning the loading of catalyst, base, and hydrogen donor were performed (Table 2). For Ru3(CO)12/benzyl alcohol, using a catalyst amount corresponding to 2 mol % Ru and reducing the amount of base to 0.2 equiv did not affect the yield (entries 1 and 4), while lowering the amount of alcohol (entry 5) or temperature (entry 6) had a negative effect on the yield and E/Z selectivity. Interestingly, reducing the amount of catalyst while maintaining the base at 1 equiv decreased the yield of the alkene (entry 2). Hence, the activity of the catalyst is related to the relative amount of base. The same behavior was observed when using a 5 mol % catalyst (entry 3). RuCl2(DMSO)4/iPrOH was also evaluated but displayed a much slower reaction rate. Reducing the amount of catalyst, base, and alcohol dramatically reduced the yield within the investigated time frame of 24 h (entries 7–10).
Table 2. Optimization of Reaction Conditions Using Phenylacetylene (1a)a.
| entry | catalyst (mol % Ru) | tBuOK (equiv) | yieldb2 (%) | E/Z2 |
|---|---|---|---|---|
| 1c,d | Ru3(CO)12 (10) | 1 | >99 | 1:0 |
| 2c,d | Ru3(CO)12 (2) | 1 | 93 | 1:0c |
| 3d | Ru3(CO)12 (5) | 1 | 89 | 1:0 |
| 4d | Ru3(CO)12 (2) | 0.2 | >99 | 1:0 |
| 5d,e | Ru3(CO)12 (2) | 0.2 | 17 | 1:2.4 |
| 6d,f | Ru3(CO)12 (2) | 0.2 | 79 | 2.3:1 |
| 7g | RuCl2(DMSO)4 (10) | 1 | 91 | 6:1 |
| 8g | RuCl2(DMSO)4 (2) | 1 | 19 | 1:1 |
| 9g | RuCl2(DMSO)4 (2) | 0.2 | 19 | 1:1 |
| 10e,g | RuCl2(DMSO)4 (2) | 0.2 | 5 | 4:1 |
Reactions performed at 100 °C (heating block) with 10 equiv hydrogen donor for 24 h unless otherwise indicated. Only trace bibenzyl (3) formed unless otherwise indicated.
NMR yield (2,5-dimethylfuran as an internal standard).
4% bibenzyl (3) formed.
Benzyl alcohol as a hydrogen donor.
2 equiv hydrogen donor.
Reaction performed at 80 °C.
Isopropyl alcohol as a hydrogen donor.
The optimized conditions for Ru3(CO)12/benzyl alcohol (Method A) were then applied to a series of alkynes (1a–n, Scheme 3) to investigate the scope. Diaryl acetylenes with varying electronic properties were well tolerated and formed their corresponding hydrogenated E-isomers selectively, with close to quantitative conversion (as determined by 1H NMR) and high isolated yields (compounds 2a–f). Electron-rich compounds such as 1c reacted slightly slower, and longer reaction times were needed to achieve full conversion. Primary amines and pyridines (1g–i) proved to be more challenging substrates. The hydrogenation of the p-amino derivative proceeded sluggishly under the standard conditions. Increasing the catalytic loading 4-fold gave a satisfactory hydrogenation yield, accompanied, however, by the formation of substantial amounts of another compound (Scheme 4). Interestingly, further analysis showed that this compound resulted from a hydrogen borrowing process13 between benzaldehyde, formed in situ from the benzyl alcohol hydrogen donor and the primary amine, to form an intermediate imine that could be reduced to the corresponding amine 4 (Scheme 4) using a second equivalent of hydrogen. The fact that a concomitant semihydrogenation–amine alkylation process is feasible is not surprising as Ru3(CO)12 has been employed for the direct amination of alcohols via hydrogen borrowing under similar conditions.14 This tandem process could potentially be applied toward the synthesis of resveratrol derivatives such as 5, reported as a promising lead compound for the treatment of Alzheimer’s disease.15 Switching to different reaction conditions, utilizing iPrOH as the hydrogen donor with RuCl2(DMSO)4 as the catalyst (Method B), suppressed the competing hydrogen borrowing reaction, allowing isolation of alkene 2g in a moderate yield. The more sterically challenging ortho-amine could be reduced using Method A but required a higher catalytic loading to proceed (compound 2h). In this case, the hydrogen borrowing product was not observed, most likely owing to the more hindered position of the amino group in the substrate. Similar to the other nitrogen-containing compounds, 3-(phenylethynyl)pyridine also required a higher catalytic loading and also a longer reaction time but afforded 2i in a high NMR yield. The lack of reactivity is most likely due to deactivation of the catalyst through coordination by nitrogen. This could also explain the lack of reports on the ruthenium-catalyzed semihydrogenation of aniline-containing compounds. The protons ortho to the nitrogen displayed broad signals in 1H NMR after completion of the reaction, indicating coordination. The stability of this interaction was further validated as it was maintained even after column chromatography on silica. The ruthenium could be removed by chromatography on amine-functionalized silica, supplying the pure semihydrogenation product with some loss in yield due to the more elaborate purification required. Other heterocyclic alkyne substrates were more successful, with indole- and thiophene-derivatives 2j and 2k formed in 56 and 76% yields, respectively. A ferrocenyl-substituted E-alkene (2l) could be obtained in a moderate yield, while appending an ester substituent to diphenyl acetylene was unproblematic (2m), although transesterification occurs if the corresponding methyl ester is used as the precursor instead. Exchanging the ester for a ketone gave interesting results. Method A afforded the benzylated ketone 6 (Figure 3), instead of the expected semihydrogenation product. This product is most likely also the result of a hydrogen borrowing-type mechanism (as for 4) but in this case involving carbon–carbon bond formation instead of amine alkylation. Method B instead affected concomitant alkyne semihydrogenation and transfer hydrogenation of the ketone, producing alcohol 2n in a moderate yield. In terms of limitations of the reaction, alkyl/aryl substitution of alkynes and dialkylacetylenes was unsuccessful, showing both low reactivity and formation of byproducts. Analysis of the crude products by 1H NMR showed that while some alkene was formed in the reaction, double bond isomerization had also occurred, resulting in a mixture of products. In addition, while a p-CF3 substituent on diphenylacetylene was well tolerated (2d), the corresponding p-NO2 compound afforded a complex mixture, where some concomitant reduction of the nitro group had taken place. Terminal alkynes such as 1-ethynyl-4-methoxybenzene afforded a complex mixture, with only trace amounts of products.
Scheme 3. Scope of the Semihydrogenation Reaction–

Prepared using Method A unless otherwise stated; reactions were heated in a heating block. See the Experimental Section for deviations in terms of reaction time. Yields in parentheses refer to NMR yields of E-alkene (for 2g–i and 2k a mixture of E- and Z-alkenes). Isolated yields refer to E-alkene only.
Prepared using Method B. See the Supporting Information for deviations in terms of reaction time. Product 2n is a result of semihydrogenation with concomitant reduction of the carbonyl group.
3.33 mol % Ru3(CO)12 used.
Product contains 3% of the (Z)-isomer.
20 mol % RuCl2(DMSO)4 used.
Scheme 4. Tandem Alkyne Semihydrogenation and Direct Amine Alkylation.

Figure 3.

Product of tandem alkyne semihydrogenation and ketone alkylation (Method A).
The reaction of diphenylacetylene with benzyl alcohol, using Ru3(CO)12 as the catalyst, could be monitored over time using 1H NMR, which revealed an initial hydrogenation to form the Z-isomer that underwent an isomerization process to the E-isomer (Figure 4). This observation is in line with previous reports.10c,10k,16
Figure 4.

Compound distribution over time.
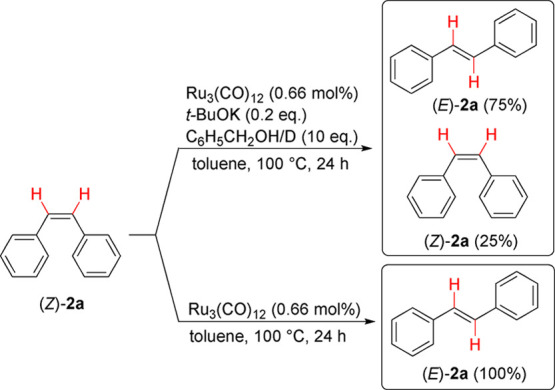
The isomerization was further investigated by subjecting cis-stilbene to the standard reaction conditions in the presence of deuterated benzyl alcohol (Bn-OD). Z-Stilbene ((Z)-2a) was isomerized into E-stilbene ((E)-2a) under these conditions but without incorporation of deuterium (Scheme 5). This observation differs from the recent study by Lindhardt and co-workers10j in which they found that isomerization of (Z)-2a in the presence of a ruthenium catalyst and D2 results in incorporation of deuterium at the alkenylic positions. We further found that the isomerization to (E)-2a occurred in the presence of the catalyst alone. These results indicate that the isomerization process does not proceed via a hydrogenation/rotation/β-hydride elimination route. No isomerization was observed when omitting the catalyst while including the other reactants. Both benzaldehyde and benzyl benzoate were observed as side products after the transfer–hydrogenation reaction. Benzyl benzoate is likely formed via a second reaction between benzaldehyde and benzyl alcohol with subsequent oxidation, as previously reported by Shvo.10a
Scheme 5. Investigation of the Isomerization Process.
Conclusions
In conclusion, a methodology for the selective semihydrogenation of diaryl alkynes to E-alkenes was developed, involving the use of a simple Ru catalyst, a low catalyst loading, ligand-free conditions, and alcohols as the source of hydrogen. While benzyl alcohol gave the most favorable E-selectivity and conversion, renewable alcohols such as furfuryl alcohol could also be applied as hydrogen donors with good results. A tandem semihydrogenation–amine alkylation reaction, the latter via hydrogen borrowing, was also demonstrated, using 4-(phenylethynyl)aniline (1g) as the substrate. Reaction monitoring indicates that the high E-selectivity in the semihydrogenation is due to isomerization of initially formed Z-alkene by the catalyst, rather than a result of the semihydrogenation process itself.
Experimental Section
General Remarks
All reactions were carried out under an argon atmosphere with dry solvents in oven-dried glassware, unless otherwise noted. Toluene, triethylamine (Et3N), ethanol (EtOH), ethyl acetate (EtOAc), and petroleum ether were bought from commercial vendors. Toluene was purchased in anhydrous form and used without further purification. Et3N was dried over molecular sieves (3 Å). EtOH, EtOAc, and petroleum ether were used as received. Reagents as well as alkynes 1a and 1c were purchased from commercial vendors and used as received, unless otherwise stated. For the Sonogashira reaction, oxygen-free Et3N was obtained by bubbling argon through the solvent for 15 min. Reactions were monitored by thin-layer chromatography carried out on 0.25 mm E. Merck silica gel plates (60F-254) using UV light as the visualizing agent. Flash chromatography was performed on a Biotage Isolera One using Biotage KP-Sil columns (packed with 50 μm irregular silica) using 254 nm and 280 nm UV light for monitoring. NMR spectra were recorded on samples in deuterated chloroform (CDCl3) or DMSO (DMSO-d6) on an Agilent 400 MHz (101 MHz for 13C) instrument. Residual undeuterated chloroform (1H: δ = 7.26 ppm, 13C: δ = 77.2 ppm) or DMSO (1H: δ = 2.50 ppm, 13C: δ = 39.5 ppm) were used as the internal reference. The following abbreviations, or a combination thereof, were used to characterize the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad. Melting points (mp) were recorded on a Mettler FP 90/82 melting point apparatus and were uncorrected. IR spectra were recorded with a PerkinElmer Spectrum ONE FT-IR spectrometer using KBr pellet sample preparation. High-resolution mass determinations were obtained with an Agilent QTOF 6520 with Infinity UHPLC and electrospray ionization.
General Procedure for the Preparation of Internal Alkynes 1b and 1d–n via Sonogashira Reaction
Arylhalide, bis(triphenylphosphine)palladium(II) dichloride (Pd(PPh3)2Cl2), and copper(I) iodide (CuI) (see each compound for amounts) were transferred to a dry 20 mL Biotage microwave reaction vial equipped with a cross-shaped magnetic stirring bar. The vial was sealed using a cap with septum, evacuated of air, and refilled with argon (three cycles). The alkyne and dry deoxygenated Et3N were thereafter transferred to the vial. The obtained mixture was further deoxygenated by bubbling argon through for 5 min while stirring. The argon inlet was removed and the reaction was heated in a Radleys Heat-On block to 80 °C for an indicated amount of time. The reaction was cooled to room temperature and concentrated under reduced pressure. The crude product was taken up in approximately 5 mL CH2Cl2 and the slurry was transferred to a 3 g Biotage KP-Sil samplet. After allowing the samplet to dry, it was transferred to a 25 g column and purified by flash chromatography.
1-Methoxy-4-(phenylethynyl)benzene (1b)17
The reaction was performed according to the general procedure using 4-iodoanisole (1.17 g, 5.0 mmol), Pd(PPh3)2Cl2 (105 mg, 0.15 mmol), CuI (28 mg, 0.15 mmol), phenylacetylene (0.81 mL, 7.4 mmol), and Et3N (13 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 95:5 (10 column volumes) to 95:5 (10 column volumes). Product 1b was obtained as a light orange crystalline solid (991 mg, 95%): 1H NMR (400 MHz, CDCl3): δ 7.54–7.49 (m, 2H), 7.47 (XX′ signal of AA′XX′ spin system, 2H), 7.37–7.30 (m, 3H), 6.88 (AA′ signal of AA′XX′ spin system, 2H), 3.83 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 159.7, 133.2, 131.6, 128.4, 128.1, 123.7, 115.5, 114.1, 89.5, 88.2, 55.5.
1-(Phenylethynyl)-4-(trifluoromethyl)benzene (1d)18
The reaction was performed according to the general procedure using 1-iodo-4-(trifluoromethyl)benzene (554 mg, 2.0 mmol), Pd(PPh3)2Cl2 (28 mg, 0.04 mmol), CuI (7.6 mg, 0.04 mmol), phenylacetylene (0.26 mL, 2.4 mmol), and Et3N (6 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (5 column volumes) to 85:15 (15 column volumes). Product 1d was obtained as a white crystalline solid (512 mg, >99%): 1H NMR (400 MHz, CDCl3): δ 7.68–7.59 (m, 4H), 7.58–7.52 (m, 2H), 7.41–7.35 (m, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 132.0, 131.9, 130.0 (q, J = 32.7 Hz), 129.0, 128.6, 127.3 (q, J = 1.5 Hz), 125.4 (q, J = 3.8 Hz), 124.1 (q, J = 272.2 Hz), 122.7, 91.9, 88.1.
1-((4-Chlorophenyl)ethynyl)-3-methoxybenzene (1e)19
The reaction was performed according to the general procedure using 4-bromochlorobenzene (957 mg, 5 mmol), Pd(PPh3)2Cl2 (105 mg, 0.15 mmol), CuI (28 mg, 0.15 mmol), 3-ethynylanisole (0.94 mL, 7.4 mmol), and Et3N (16 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (15 column volumes). Product 1e was obtained as a white crystalline solid (573 mg, 47%): 1H NMR (400 MHz, CDCl3): δ 7.47 (XX′ signal of AA′XX′ spin system, 2H), 7.33 (AA′ signal of AA′XX′ spin system, 2H), 7.29–7.24 (m, 1H), 7.12 (ddd, J = 7.6, 1.5, 1.0 Hz, 1H), 7.05 (ddd, J = 2.7, 1.4, 0.4 Hz, 1H), 6.91 (ddd, J = 8.3, 2.6, 1.0 Hz, 1H), 3.83 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 159.5, 134.5, 133.0, 129.6, 128.8, 124.3, 124.0, 121.8, 116.5, 115.3, 90.4, 88.2, 55.5.
1-Methoxy-4-((4-(trifluoromethyl)phenyl)ethynyl)-benzene (1f)20
The reaction was performed according to the general procedure using 1-iodo-4-(trifluoromethyl)benzene (272 mg, 1 mmol), Pd(PPh3)2Cl2 (21 mg, 0.03 mmol), CuI (5.7 mg, 0.03 mmol), 4-ethynylanisole (0.13 mL, 1.02 mmol), and Et3N (3 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 95:5 (20 column volumes) to 95:5 (10 column volumes). Product 1f was obtained as a white crystalline solid (264 mg, 96%): 1H NMR (400 MHz, CDCl3): δ 7.63–7.57 (m, 4H), 7.49 (XX′ signal of AA′XX′ spin system, 2H), 6.90 (AA′ signal of AA′XX′ spin system, 2H), 3.84 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 160.2, 133.4, 131.7, 129.7 (q, J = 32.6 Hz), 127.6 (q, J = 1.5 Hz), 125.4 (q, J = 3.8 Hz), 124.2 (q, J = 272.1 Hz), 114.8, 114.2, 92.1, 87.0, 55.4.
4-(Phenylethynyl)aniline (1g)21
The reaction was performed according to the general procedure using 4-iodoaniline (1.1 g, 5 mmol), Pd(PPh3)2Cl2 (70 mg, 0.1 mmol), CuI (19 mg, 0.1 mmol), phenylacetylene (0.66 mL, 6 mmol), and Et3N (15 mL). Flash chromatography gradient: petroleum ether/EtOAc, 98:2 to 93:7 (10 column volumes) to 93:7 (10 column volumes) to 4:1 (10 column volumes). Product 1g was obtained as an orange crystalline solid (822 mg, 85%): 1H NMR (400 MHz, CDCl3): δ 7.52–7.47 (m, 2H), 7.39–7.28 (m, 5H), 6.65 (AA′ signal of AA′XX′ spin system, 2H), 3.82 (br s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 146.8, 133.1, 131.5, 128.4, 127.8, 124.0, 114.9, 112.8, 90.2, 87.5.
2-(Phenylethynyl)aniline (1h)22
The reaction was performed according to the general procedure using 2-iodoaniline (1.1 g, 5 mmol), Pd(PPh3)2Cl2 (70 mg, 0.1 mmol), CuI (19 mg, 0.1 mmol), phenylacetylene (0.66 mL, 6 mmol), and Et3N (15 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (5 column volumes) to 85:15 (15 column volumes). Product 1h was obtained as a yellow crystalline solid (769 mg, 80%): 1H NMR (400 MHz, CDCl3): δ 7.56–7.50 (m, 2H), 7.40–7.32 (m, 4H), 7.14 (ddd, J = 8.1, 7.4, 1.6 Hz, 1H), 6.75–6.70 (m, 2H), 4.28 (br s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 147.9, 132.2, 131.6, 129.8, 128.5, 128.3, 123.4, 118.1, 114.4, 108.0, 94.8, 86.0.
3-(Phenylethynyl)pyridine (1i)23
The reaction was performed according to the general procedure using 3-bromopyridine (0.48 mL, 5 mmol), Pd(PPh3)2Cl2 (105 mg, 0.15 mmol), CuI (28 mg, 0.15 mmol), phenylacetylene (0.81 mL, 7.4 mmol), and Et3N (16 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 9:1 (10 column volumes) to 9:1 (15 column volumes). Product 1i was obtained as a light brown crystalline solid (546 mg, 61%): 1H NMR (400 MHz, CDCl3): δ 8.77 (dd, J = 2.2, 0.9 Hz, 1H), 8.55 (dd, J = 4.9, 1.7 Hz, 1H), 7.81 (ddd, J = 7.9, 2.2, 1.7 Hz, 1H), 7.59–7.52 (m, 2H), 7.40–7.35 (m, 3H), 7.29 (ddd, J = 7.9, 4.9, 0.9 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 152.4, 148.7, 138.6, 131.8, 129.0, 128.6, 123.2, 122.7, 120.6, 92.8, 86.1.
5-(Phenylethynyl)-1H-indole (1j)24
The reaction was performed according to the general procedure using 5-iodoindole (1.22 g, 5 mmol), Pd(PPh3)2Cl2 (70 mg, 0.1 mmol), CuI (19 mg, 0.1 mmol), phenylacetylene (0.6 g, 6 mmol), and Et3N (15 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (10 column volumes) to 9:1 (20 column volumes) to 9:1 (20 column volumes). Product 1j was obtained as a light yellow crystalline solid (882 mg, 81%): 1H NMR (400 MHz, DMSO-d6): δ 11.35 (br s, 1H), 7.80 (dt, J = 1.5, 0.7 Hz, 1H), 7.58–7.51 (m, 2H), 7.47–7.35 (m, 5H), 7.27 (dd, J = 8.4, 1.6 Hz, 1H), 6.48 (ddd, J = 2.9, 1.9, 0.9 Hz, 1H); 13C{1H} NMR (101 MHz, DMSO-d6): δ 135.7, 131.1, 128.7, 128.1, 127.6, 126.7, 124.3, 123.9, 123.2, 112.3, 111.9, 101.4, 91.6, 86.6.
3-(Phenylethynyl)thiophene (1k)25
The reaction was performed according to the general procedure using 3-bromothiophene (815 mg, 5 mmol), Pd(PPh3)2Cl2 (70 mg, 0.1 mmol), CuI (19 mg, 0.1 mmol), phenylacetylene (0.6 g, 6 mmol), and Et3N (15 mL). Flash chromatography: petroleum ether (10 column volumes). Product 1k was obtained as a clear oil that crystallized in matter of days (788 mg, 86%). The compound turns orange upon air exposure: 1H NMR (400 MHz, CDCl3): δ 7.57–7.49 (m, 3H), 7.39–7.32 (m, 3H), 7.31 (dd, J = 5.0, 3.0 Hz, 1H), 7.21 (dd, J = 5.0, 1.2 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 131.7, 130.0, 128.7, 128.5, 128.4, 125.5, 123.3, 122.4, 89.0, 84.6.
(4-Chlorophenylethynyl)ferrocene (1l)26
The reaction was performed according to the general procedure using 4-bromochlorobenzene (0.618 mL, 3.2 mmol), Pd(PPh3)2Cl2 (105 mg, 0.15 mmol), CuI (28 mg, 0.15 mmol), ethynylferrocene (1.0 g, 4.8 mmol), and Et3N (16 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 98:2 (10 column volumes) to 98:2 (10 column volumes). Product 1l was obtained as a red crystalline solid (634 mg, 61%): 1H NMR (400 MHz, CDCl3): δ 7.40 (XX′ signal of AA′XX′ spin system, 2H), 7.30 (AA′ signal of AA′XX′ spin system, 2H), 4.51–4.49 (m, 2H), 4.26–4.24(m, 7H); 13C{1H} NMR (101 MHz, CDCl3): δ 133.7, 132.7, 128.7, 122.6, 89.6, 84.8, 71.6, 70.2, 69.2, 65.1.
Benzyl 4-(phenylethynyl)benzoate (1m)
This compound was prepared in two steps via the corresponding methyl ester. Step 1, Sonogashira reaction: methyl 4-(phenylethynyl)benzoate27 was first prepared according to the general procedure using methyl 4-iodoacetophenone (1.23 g, 5 mmol), Pd(PPh3)2Cl2 (70 mg, 0.1 mmol), CuI (19 mg, 0.1 mmol), phenylacetylene (0.61 g, 6 mmol), and Et3N (15 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (10 column volumes) to 95:5 (10 column volumes) to 9:1 (5 column volumes) to 9:1 (15 column volumes). Product was obtained as a light yellow crystalline solid (689 mg, 58%): 1H NMR (400 MHz, CDCl3): δ 8.03 (XX′ signal of AA′XX′ spin system, 2H), 7.59 (AA′ signal of AA′XX′ spin system, 2H), 7.57–7.52 (m, 2H), 7.40–7.34 (m, 3H), 3.93 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 166.7, 131.9, 131.6, 129.64, 129.59, 128.9, 128.6, 128.1, 122.8, 92.5, 88.8, 52.4. Step 2, transesterification: a dry 5 mL reaction vial containing methyl 4-(phenylethynyl)benzoate (71 mg, 0.3 mmol), tBuOK (17 mg, 0.15 mmol), benzyl alcohol (0.31 mL, 3 mmol), and toluene (0.7 mL) was heated in a Radleys Heat-On block to 100 °C for 24 h under an atmosphere of argon. The reaction was cooled to room temperature and the toluene was evaporated under a stream of N2. The resulting mixture was taken up in ∼0.5 mL DCM and transferred to a 1 g Biotage KP-Sil samplet. After allowing the samplet to dry, it was transferred to a 10 g column and purified through flash chromatography. Gradient: petroleum ether/EtOAc, 1:0 to 1:0 (5 column volumes) to 9:1 (25 column volumes). Product 1m was obtained as a white crystalline solid: mp = 103–105 °C; νmax/cm–1 3031 (C–H), 2958 (C–H), 2213 (C≡C), 1709 (C=O), 1604 (C=C); 1H NMR (400 MHz, CDCl3): δ 8.07 (XX′ signal of AA′XX′ spin system, 2H), 7.59 (AA′ signal of AA′XX′ spin system, 2H), 7.57–7.53 (m, 2H), 7.49–7.45 (m, 2H), 7.44–7.34 (m, 6H), 5.38 (s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 166.0, 136.0, 131.9, 131.6, 129.8, 129.6, 128.9, 128.8, 128.6, 128.5, 128.4, 128.3, 122.8, 92.6, 88.8, 67.0; HRMS (ESI + QTOF): [M + H]+ calcd for C22H17O2, 313.1223; found, 313.1221.
1-(4-(Phenylethynyl)phenyl)ethan-1-one (1n)28
The reaction was performed according to the general procedure using methyl 4-iodobenzoate (1.3 g, 5 mmol), Pd(PPh3)2Cl2 (105 mg, 0.15 mmol), CuI (28 mg, 0.15 mmol), phenylacetylene (0.81 g, 7.4 mmol), and Et3N (16 mL). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (5 column volumes) to 99:1 (3 column volumes) to 9:1 (11 column volumes). Product 1n was obtained as an off-white crystalline solid (337 mg, 31%): 1H NMR (400 MHz, CDCl3): δ 7.93 (XX′ signal of AA′XX′ spin system, 2H), 7.61 (AA′ signal of AA′XX′ spin system, 2H), 7.58–7.52 (m, 2H), 7.41–7.34 (m, 3H), 2.60 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 197.4, 136.3, 131.8, 131.8, 128.9, 128.6, 128.4, 128.3, 122.7, 92.8, 88.7, 26.7.
Semihydrogenation of Internal Alkynes, Method A
To an oven-dried 5 mL Biotage microwave reaction vial equipped with a magnetic stirring bar was transferred alkyne, Ru3(CO)12, and tBuOK (see each compound for amounts). The vial was sealed with a Biotage cap and connected to a Schlenk line. The atmosphere was evacuated and the vial was refilled with argon (three cycles). Dry toluene and benzyl alcohol were subsequently transferred (no special precautions were taken to exclude air from these components). The Schlenk connection was removed and the sealed system was heated in a Radleys Heat-On block to 100 °C. After being stirred at that temperature for an indicated period of time, the reaction was allowed to cool to room temperature and toluene was removed under a stream of N2. The reported NMR yields were obtained using 2,5-dimethylfuran as the internal standard.29 Everything was taken up in ∼2 mL of CDCl3 and a 1H-NMR was recorded (no. of transients: 2, relaxation delay: 60 s). The spectrum was phase-corrected and baseline-corrected before being integrated. The amount of product was calculated as previously described using the 2,5-dimethylfuran HAr peak at δ 5.87 ppm and appropriate product peaks. As an example, the hydrogenation of phenylacetylene with cyclopentanol as the hydrogen donor can be found in the Supporting Information (Figure S1). The CDCl3 was after analysis evaporated and the crude mixture was taken up in ∼1–3 mL of DCM and transferred to either a 1 g or 3 g Biotage KP-Sil samplet. After drying the samplet, it was transferred to a 10 g or a 25 g column and purified through flash chromatography.
Method B
Method B was the same as Method A but used RuCl2(DMSO)4 (10 mol %) as the catalyst, iPrOH (10 equiv) as the hydrogen donor, and 50 mol % tBuOK as the base instead.
(E)-Stilbene ((E)-2a)30
The reaction was performed according to Method A using diphenylacetylene (107 mg, 0.60 mmol), Ru3(CO)12 (2.6 mg, 0.004 mmol), tBuOK (14 mg, 0.12 mmol), benzyl alcohol (0.62 mL, 6.0 mmol), and toluene (1.4 mL) with a reaction time of 24 h. NMR-yield (E/Z %): 100/0. Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (10 column volumes). Product (E)-2a was obtained as a white crystalline solid (95 mg, 88%): 1H NMR (400 MHz, CDCl3): δ 7.57–7.51 (m, 4H), 7.42–7.35 (m, 6H), 7.31–7.25 (m, 1H), 7.12 (s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 137.5, 128.8 (two signals overlap), 127.8, 126.6.
(E)-1-Methoxy-4-styrylbenzene (2b)6
The reaction was performed according to Method A using 1-methoxy-4-(phenylethynyl)benzene (187 mg, 0.90 mmol), Ru3(CO)12 (3.8 mg, 0.006 mmol), tBuOK (20 mg, 0.18 mmol), benzyl alcohol (0.93 mL, 9.0 mmol), and toluene (2.1 mL) with a reaction time of 44 h. NMR yield (E/Z %): 100/0. Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 95:5 (20 column volumes) to 95:5 (10 column volumes). Product 2b was obtained as an off-white crystalline solid, which was contaminated with 11% benzyl benzoate (198 mg total, 177 mg only considering product, 93%). An analytically pure sample could be obtained through recrystallization from EtOH: 1H NMR (400 MHz, CDCl3): δ 7.54–7.49 (m, 2H), 7.47 (XX′ signal of AA′XX′ spin system, 2H), 7.36 (dd, J = 8.4, 6.9 Hz, 2H), 7.28–7.23 (m, 1H), 7.09 (d, J = 16.3 Hz, 1H), 6.99 (d, J = 16.3 Hz, 1H), 6.92 (AA′ signal of AA′XX′ spin system, 2H), 3.84 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 159.4, 137.8, 130.3, 128.8, 128.3, 127.9, 127.3, 126.7, 126.4, 114.3, 55.5.
(E)-1-Chloro-4-styrylbenzene (2c)10j
The reaction was performed according to Method A using 1-chloro-4-(phenylethynyl)benzene (128 mg, 0.60 mmol), Ru3(CO)12 (2.6 mg, 0.004 mmol), tBuOK (14 mg, 0.12 mmol), benzyl alcohol (0.62 mL, 6.0 mmol), and toluene (1.4 mL) with a reaction time of 42 h. NMR yield (E/Z %): 100/0. Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 95:5 (10 column volumes) to 95:5 (10 column volumes). Product 2c was obtained as a white crystalline solid (109 mg, 85%): 1H NMR (400 MHz, CDCl3): δ 7.55–7.49 (m, 2H), 7.45 (XX′ signal of AA′XX′ spin system, 2H), 7.42–7.36 (m, 2H), 7.34 (AA′ signal of AA′XX′ spin system, 2H), 7.32–7.27 (m, 1H), 7.10 (d, J = 16.4, 1H), 7.05 (d, J = 16.4, 1H); 13C NMR (101 MHz, CDCl3): δ 137.1, 136.0, 133.3, 129.4, 129.0, 128.9, 128.0, 127.8, 127.5, 126.7.
(E)-1-Styryl-4-(trifluoromethyl)benzene (2d)31
The reaction was performed according to Method A using 1-(2-phenylethynyl)-4-(trifluoromethyl)benzene (195 mg, 0.79 mmol), Ru3(CO)12 (3.4 mg, 0.005 mmol), tBuOK (18 mg, 0.16 mmol), benzyl alcohol (0.82 mL, 7.9 mmol), and toluene (1.8 mL) with a reaction time of 24 h. NMR yield (E/Z %): ∼100/0 (product peaks overlap with benzyl alcohol, rendering exact measurements difficult). Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (5 column volumes) to 99:1 (5 column volumes). Product 2d was obtained as a white crystalline solid (175 mg, 89%): 1H NMR (400 MHz, CDCl3): δ 7.66–7.59 (m, 4H), 7.58–7.54 (m, 2H), 7.44–7.39 (m, 2H), 7.37–7.31 (m, 1H), 7.22 (d, J = 16.3 Hz, 1H), 7.13 (d, J = 16.3 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 140.8 (q, J = 1.5 Hz), 136.6, 131.2, 129.2 (q, J = 32.4 Hz), 128.8, 128.3, 127.1 (q, J = 0.8 Hz), 126.8, 126.6, 125.6 (q, J = 3.8 Hz), 124.3 (q, J = 272 Hz).
(E)-1-(4-Chlorostyryl)-3-methoxybenzene (2e)32
The reaction was performed according to Method A using 1-chloro-4-[2-(3-methoxyphenyl)ethynyl]benzene (218 mg, 0.9 mmol), Ru3(CO)12 (3.8 mg, 0.006 mmol), tBuOK (20 mg, 0.18 mmol), benzyl alcohol (0.93 mL, 9.0 mmol), and toluene (2.1 mL) with a reaction time of 24 h. NMR yield (E/Z %): 100/0. Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 97:3 (20 column volumes) to 97:3 (10 column volumes). Product 2e was obtained as a white crystalline solid (191 mg, 87%): mp = 71 °C; νmax/cm–1 3006 (C–H), 2835 (C–H), 1605 (C=C); 1H NMR (400 MHz, CDCl3): δ 7.48–7.42 (XX′ signal of AA′XX′ spin system, 2H), 7.34–7.31 (AA′ signal of AA′XX′ spin system, 2H), 7.31–7.26 (m, 1H), 7.10 (ddd, J = 7.7, 1.6, 0.9 Hz, 1H), 7.05 (s, 2H), 7.04 (dd, J = 2.5, 1.5 Hz, 1H), 6.84 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 3.85 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 160.0, 138.6, 135.9, 133.4, 129.8, 129.3, 129.0, 127.8, 127.8, 119.4, 113.6, 111.9, 55.4; HRMS (ESI + QTOF): [M + H]+ calcd for C15H14ClO, 245.0728; found, 245.0730.
(E)-1-Methoxy-4-(4-(trifluoromethyl)styryl)benzene (2f)10j
The reaction was performed according to Method A using 1-methoxy-4-((4-(trifluoromethyl)phenyl)ethynyl)-benzene (83 mg, 0.3 mmol), Ru3(CO)12 (1.3 mg, 0.002 mmol), tBuOK (7 mg, 0.06 mmol), benzyl alcohol (0.31 mL, 3.0 mmol), and toluene (0.69 mL) with a reaction time of 42 h. NMR yield (E/Z %): 100/0. Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (10 column volumes) to 95:5 (10 column volumes). Product 2f was obtained as a white crystalline solid contaminated with a small amount of benzyl benzoate (69 mg, 83%): 1H NMR (400 MHz, CDCl3): δ 7.63–7.55 (m, 4H), 7.48 (XX′ signal of AA′XX′ spin system, 2H), 7.15 (d, J = 16.3 Hz, 1H), 6.98 (d, J = 16.3 Hz, 1H), 6.93 (AA′ signal of AA′XX′ spin system, 2H), 3.84 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 159.9, 141.1, 130.7, 129.4, 128.8 (q, J = 32.4 Hz), 128.1, 126.3, 125.6 (q, J = 3.9 Hz), 124.9, 124.3 (q, J = 271 Hz), 114.2, 55.3.
(E)-4-Styrylaniline (2g)33
The reaction was performed according to Method B using 4-(phenylethynyl)aniline (58 mg, 0.3 mmol), RuCl2(DMSO)4 (14.5 mg, 0.03 mmol), tBuOK (17 mg, 0.15 mmol), 2-propanol (0.23 mL, 3.0 mmol), and toluene (0.77 mL) with a reaction time of 24 h. NMR yield (E/Z %): 65/14. Flash chromatography gradient: petroleum ether/EtOAc, 95:5 to 85:15 (30 column volumes). Product 2g was obtained as a light yellow crystalline solid (30 mg, 51%): 1H NMR (400 MHz, CDCl3): δ 7.52–7.45 (m, 2H), 7.39–7.29 (m, 4H), 7.25–7.18 (m, 1H), 7.03 (d, J = 16.3 Hz, 1H), 6.93 (d, J = 16.3 Hz, 1H), 6.67 (AA′ signal of AA′XX′ spin system, 2H), 3.75 (br s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 146.3, 138.1, 128.8, 128.7, 128.2, 127.9, 127.0, 126.2, 125.2, 115.3.
(E)-2-Styrylaniline (2h)34
The reaction was performed according to Method A using 2-(2-phenylethynyl)aniline (174 mg, 0.90 mmol), Ru3(CO)12 (19 mg, 0.03 mmol), tBuOK (20 mg, 0.18 mmol), benzyl alcohol (0.93 mL, 9.0 mmol), and toluene (2.1 mL) with a reaction time of 28 h. NMR yield (E/Z %): 45/14. Flash chromatography gradient: petroleum ether/EtOAc, 98:2 to 9:1 (30 column volumes). Product 2h was obtained as a white crystalline solid that rapidly turned brown upon air exposure (60 mg, 34%). A sample was obtained for analytical purposes through recrystallization from EtOH: 1H NMR (400 MHz, CDCl3): δ 7.57–7.52 (m, 2H), 7.44 (dd, J = 7.7, 1.5 Hz, 1H), 7.43–7.37 (m, 2H), 7.33–7.27 (m, 1H), 7.20 (d, J = 16.1 Hz, 1H), 7.17–7.12 (m, 1H), 7.02 (d, J = 16.1 Hz, 1H), 6.89–6.82 (m, 1H), 6.74 (dd, J = 8.0, 1.2 Hz, 1H), 3.82 (br s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 144.1, 137.7, 130.4, 128.80, 128.78, 127.7, 127.4, 126.5, 124.4, 123.9, 119.7, 116.4.
(E)-3-Styrylpyridine (2i)35
The reaction was performed according to Method A using 3-(2-phenylethynyl)pyridine (54 mg, 0.30 mmol), Ru3(CO)12 (6.3 mg, 0.01 mmol), tBuOK (6.7 mg, 0.06 mmol), benzyl alcohol (0.31 mL, 3.0 mmol), and toluene (0.69 mL) with a reaction time of 70 h. NMR yield (E/Z %): 70/30. The crude product was transferred to an amino-functionalized 1 g samplet instead to the unfunctionalized samplet described in Method A. Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 85:15 (15 column volumes) to 85:15 (10 column volumes). Product 2i was obtained as a light yellow crystalline solid (26 mg, 48%): 1H NMR (400 MHz, CDCl3): δ 8.72 (d, J = 2.3 Hz, 1H), 8.49 (dd, J = 4.8, 1.6 Hz, 1H), 7.83 (dddd, J = 8.0, 2.2, 1.6, 0.6 Hz, 1H), 7.56–7.50 (m, 2H), 7.41–7.35 (m, 2H), 7.33–7.26 (m, 2H), 7.17 (d, J = 16.4 Hz, 1H), 7.07 (d, J = 16.4 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 148.6, 136.7, 133.1, 132.8, 130.9, 128.9, 128.3, 126.8, 125.0, 123.7.
(E)-5-Styryl-1H-indole (2j)36
The reaction was performed according to Method B using 5-(phenylethynyl)-1H-indole (131 mg, 0.6 mmol), RuCl2(DMSO)4 (29 mg, 0.06 mmol), tBuOK (34 mg, 0.3 mmol), 2-propanol (0.46 mL, 6.0 mmol), and toluene (1.54 mL) with a reaction time of 72 h. NMR yield: 75%. Flash chromatography gradient: petroleum ether/EtOAc, 96:4 to 92:8 (10 column volumes) to 92:8 (8 column volumes). Product 2j was obtained as an off-white crystalline solid (74 mg, 56%): 1H NMR (400 MHz, CDCl3): δ 8.15 (br s, 1H), 7.77 (dt, J = 1.6, 0.8 Hz, 1H), 7.56–7.52 (m, 2H), 7.46 (dd, J = 8.5, 1.7 Hz, 1H), 7.41–7.34 (m, 3H), 7.25 (d, J = 16.3 Hz, 1H), 7.26–7.23 (m, 1H), 7.21 (dd, J = 3.3, 2.3 Hz, 1H), 7.09 (d, J = 16.3 Hz, 1H), 6.57 (ddd, J = 3.1, 2.0, 1.0 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 138.1, 135.7, 130.2, 129.6, 128.8, 128.3, 127.1, 126.3, 126.2, 124.9, 120.8, 119.6, 111.4, 103.1.
(E)-3-Styrylthiophene (2k)37
The reaction was performed according to Method A using 3-(phenylethynyl)thiophene (166 mg, 0.90 mmol), Ru3(CO)12 (3.8 mg, 0.006 mmol), tBuOK (20 mg, 0.18 mmol), benzyl alcohol (0.93 mL, 9.0 mmol), and toluene (2.1 mL) with a reaction time of 48 h. NMR yield (E/Z %): 89/10. Flash chromatography: petroleum ether (10 column volumes). Product 2k was obtained as a white crystalline solid, which was contaminated with 3% (Z)-3-styrylthiophene (127 mg, 76%): 1H NMR (400 MHz, CDCl3): δ 7.53–7.48 (m, 2H), 7.41–7.31 (m, 4H), 7.30–7.25 (m, 2H), 7.15 (d, J = 16.3 Hz, 1H), 6.98 (d, J = 16.3 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 140.2, 137.5, 128.79, 128.78, 127.6, 126.4, 126.3, 125.0, 123.0, 122.5.
(E)-(4-Chlorophenyl)ferrocenylethene (2l)
The reaction was performed according to Method A using 1-chloro-4-(ferroceneethynyl)benzene (192 mg, 0.60 mmol), Ru3(CO)12 (2.6 mg, 0.004 mmol), tBuOK (14 mg, 0.12 mmol), benzyl alcohol (0.62 mL, 6.0 mmol), and toluene (1.4 mL) with a reaction time of 24 h. NMR yield (E/Z %): 60/n.d. Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (10 column volumes) to 97:3 (10 column volumes). Product 2l was obtained as a red crystalline solid (95 mg, 49%): mp = 154–158 °C; νmax/cm–1 3083 (C–H), 2956 (C–H), 2924 (C–H), 2854 (C–H), 1632 (C=C); 1H NMR (400 MHz, CDCl3): δ 7.34 (XX′ signal of AA′XX′ spin system, 2H), 7.29 (AA′ signal of AA′XX′ spin system, 2H), 6.85 (d, J = 16.1 Hz, 1H), 6.64 (d, J = 16.1 Hz, 1H), 4.47–4.45 (t, J = 1.9 Hz, 2H), 4.29 (t, J = 1.8 Hz, 2H), 4.14 (s, 5H); 13C{1H} NMR (101 MHz, CDCl3): δ 136.5, 132.3, 128.9, 127.9, 127.0, 124.8, 83.0, 69.7, 69.3, 67.1; HRMS (ESI + QTOF): [M + H]+ calcd for C18H16ClFe, 323.0284; found, 323.0268.
Benzyl (E)-4-styrylbenzoate (2m)
The reaction was performed according to Method A using benzyl 4-(phenylethynyl)benzoate (84 mg, 0.27 mmol), Ru3(CO)12 (1.2 mg, 0.0018 mmol), tBuOK (6.1 mg, 0.054 mmol), benzyl alcohol (0.28 mL, 0.27 mmol), and toluene (0.65 mL) with a reaction time of 24 h. NMR yield (E/Z %): 100/0. Flash chromatography gradient: petroleum ether/EtOAc, 1:0 to 1:0 (5 column volumes) to 9:1 (20 column volumes). Product 2m was obtained as a white crystalline solid (60 mg, 71%): mp = 123–129 °C; νmax/cm–1 3027 (C–H), 2952 (C–H), 2892 (C–H), 1708 (C=O), 1603 (C=C); 1H NMR (400 MHz, CDCl3): δ 8.10–8.05 (m, 2H), 7.60–7.52 (m, 4H), 7.50–7.45 (m, 2H), 7.44–7.34 (m, 5H), 7.33–7.28 (m, 1H), 7.22 (d, J = 16.3 Hz, 1H), 7.13 (d, J = 16.4 Hz, 1H), 5.38 (s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 166.3, 142.1, 136.8, 136.2, 131.4, 130.3, 129.0, 128.9, 128.7, 128.4, 128.4, 128.3, 127.7, 126.9, 126.5, 66.8; HRMS (ESI + QTOF): [M + H]+ calcd for C22H19O2, 315.1380; found, 315.1378.
(E)-1-(4-Styrylphenyl)ethan-1-ol (2n)
The reaction was performed according to Method B using 1-(4-(phenylethynyl)phenyl)ethan-1-one (66 mg, 0.3 mmol), RuCl2(DMSO)4 (29 mg, 0.006 mmol), tBuOK (17 mg, 0.15 mmol), 2-propanol (0.23 mL, 3.0 mmol), and toluene (0.77 mL) with a reaction time of 70 h. NMR yield (E/Z %): 52/0. Flash chromatography gradient: petroleum ether/EtOAc, 100:0 to 85:15 (20 column volumes) to 85:15 (10 column volumes). Product 2n was obtained as a white crystalline solid (29 mg, 44%): mp = 118–120 °C; νmax/cm–1 3307 (O–H), 3024 (C–H), 2973 (C–H); 1H NMR (400 MHz, CDCl3): δ 7.56–7.48 (m, 4H), 7.41–7.33 (m, 4H), 7.29–7.24 (m, 1H), 7.11 (s, 2H), 4.92 (qd, J = 6.4, 2.9 Hz, 1H), 1.81 (d, J = 2.5 Hz, 1H), 1.52 (d, J = 6.4 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 145.3, 137.4, 136.8, 128.8, 128.8, 128.4, 127.8, 126.8, 126.6, 125.9, 70.4, 25.3; HRMS (ESI + QTOF): [M + H–[H2O]]+ calcd for C16H15, 207.1168; found, 207.1178.
Tandem Semihydrogenation/Hydrogen Borrowing
(E)-N-Benzyl-4-styrylaniline (4)38
The reaction was performed according to Method A using 4-(phenylethynyl)aniline (39 mg, 0.2 mmol), Ru3(CO)12 (4.2 mg, 0.007 mmol), tBuOK (4.5 mg, 0.04 mmol), benzyl alcohol (0.21 mL, 2.0 mmol), and toluene (0.46 mL) with a reaction time of 24 h. NMR yield (4-[(E)-styryl]aniline/N-benzyl-4-[(E)-styryl]aniline %): 23/51. Flash chromatography gradient: petroleum ether/EtOAc, 98:2 to 9:1 (30 column volumes). Product 4 was obtained as an off-white crystalline solid (17 mg, 30%): 1H NMR (400 MHz, CDCl3): δ 7.51–7.46 (m, 2H), 7.41–7.28 (m, 9H), 7.24–7.19 (tt, J = 1.3, 7.3 Hz, 1H), 7.04 (d, J = 16.3 Hz, 1H), 6.92 (d, J = 16.3 Hz, 1H), 6.65 (AA′ signal of AA′XX′ spin system, 2H), 4.38 (s, 2H), 4.20 (br s, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 147.9, 139.3, 138.2, 128.9, 128.8, 128.7, 127.9, 127.6, 127.5, 127.2, 126.9, 126.2, 124.7, 113.1, 48.3.
(E)-3-Phenyl-1-(4-styrylphenyl)propan-1-one (6)
The reaction was performed according to Method A using 1-(4-(phenylethynyl)phenyl)ethan-1-one (66 mg, 0.3 mmol), Ru3(CO)12 (1.3 mg, 0.002 mmol), tBuOK (6.7 mg, 0.06 mmol), benzyl alcohol (0.31 mL, 3.0 mmol), and toluene (0.69 mL) with a reaction time of 24 h. NMR yield: 45%. Flash chromatography gradient: petroleum ether/EtOAc, 100:0 to 100:0 (5 column volumes) to 80:20 (25 column volumes). A mixture of product 6 and benzyl alcohol was obtained after chromatography. The mixture was recrystallized from boiling EtOAc, yielding pure 6 as white crystals. The mother liquid was recrystallized again yielding another batch of pure 6 (33 mg, 35%): mp = 150–155 °C; νmax/cm–1 3026 (C–H), 2927 (C–H), 1681 (C=O), 1602 (C=C); 1H NMR (400 MHz,) δ 7.96 (XX′ signal of AA′XX′ spin system, 2H), 7.58 (AA′ signal of AA′XX′ spin system, 2H), 7.56–7.52 (m, 2H), 7.42–7.36 (m, 2H), 7.35–7.27 (m, 4H), 7.26–7.20 (m, 1H), 7.23 (d, J = 16.4 Hz, 1H), 7.13 (d, J = 16.3 Hz, 1H), 3.35–3.28 (m, 2H), 3.09 (dd, J = 8.5, 6.9 Hz, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 198.7, 142.1, 141.5, 136.8, 135.8, 131.6, 128.9, 128.7, 128.7, 128.6, 128.4, 127.6, 126.9, 126.7, 126.3, 40.6, 30.4; HRMS (ESI + QTOF): [M + H]+ calcd for C23H21O, 313.1592; found, 313.1592.
Acknowledgments
The Swedish Research Council Formas (N.K., grant no. 2015-1106) and the Swedish Research Council (N.K., grant no. 2015-05360) are gratefully acknowledged for funding.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.9b02721.
1H and 13C{1H} NMR spectra for all compounds (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Kale A. P.; Kumar G. S.; Kapur M. Palladium-Catalyzed Synthesis of 2-Alkenyl-3-Arylindoles via a Dual α-Arylation Strategy: Formal Synthesis of the Antilipemic Drug Fluvastatin. Org. Biomol. Chem. 2015, 13, 10995–11002. 10.1039/C5OB01632K. [DOI] [PubMed] [Google Scholar]; b Sarkic A.; Stappen I. Essential Oils and Their Single Compounds in Cosmetics—a Critical Review. Cosmetics 2018, 5, 11. 10.3390/cosmetics5010011. [DOI] [Google Scholar]; c Satoh H.; Takeuchi K. Management of NSAID/Aspirin-Induced Small Intestinal Damage by GI-Sparing NSAIDs, Anti-Ulcer Drugs and Food Constituents. Curr. Med. Chem. 2012, 19, 82–89. 10.2174/092986712803413980. [DOI] [PubMed] [Google Scholar]; d Keylor M. H.; Matsuura B. S.; Stephenson C. R. J. Chemistry and Biology of Resveratrol-Derived Natural Products. Chem. Rev. 2015, 115, 8976–9027. 10.1021/cr500689b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frihed T. G.; Fürstner A. Progress in the trans-Reduction and trans-Hydrometalation of Internal Alkynes. Applications to Natural Product Synthesis. Bull. Chem. Soc. Jpn. 2016, 89, 135–160. 10.1246/bcsj.20150317. [DOI] [Google Scholar]
- Trost B. M.; Ball Z. T. Markovnikov Alkyne Hydrosilylation Catalyzed by Ruthenium Complexes. J. Am. Chem. Soc. 2001, 123, 12726–12727. 10.1021/ja0121033. [DOI] [PubMed] [Google Scholar]
- Srimani D.; Diskin-Posner Y.; Ben-David Y.; Milstein D. Iron Pincer Complex Catalyzed, Environmentally Benign, E-Selective Semi-Hydrogenation of Alkynes. Angew. Chem. Int. Ed. 2013, 52, 14131–14134. 10.1002/anie.201306629. [DOI] [PubMed] [Google Scholar]
- Li K.; Khan R.; Zhang X.; Gao Y.; Zhou Y.; Tan H.; Chen J.; Fan B. Cobalt Catalyzed Stereodivergent Semi-Hydrogenation of Alkynes Using H2O as the Hydrogen Source. Chem. Commun. 2019, 55, 5663–5666. 10.1039/C9CC01970G. [DOI] [PubMed] [Google Scholar]
- Richmond E.; Moran J. Ligand Control of E/Z Selectivity in Nickel-Catalyzed Transfer Hydrogenative Alkyne Semireduction. J. Org. Chem. 2015, 80, 6922–6929. 10.1021/acs.joc.5b01047. [DOI] [PubMed] [Google Scholar]
- Zhao C. Q.; Chen Y. G.; Qiu H.; Wei L.; Fang P.; Mei T. S. Water as a Hydrogenating Agent: Stereodivergent Pd-Catalyzed Semihydrogenation of Alkynes. Org. Lett. 2019, 21, 1412–1416. 10.1021/acs.orglett.9b00148. [DOI] [PubMed] [Google Scholar]
- Zhou Y.-P.; Mo Z.; Luecke M.-P.; Driess M. Stereoselective Transfer Semi-Hydrogenation of Alkynes to E-Olefins with N-Heterocyclic Silylene-Manganese Catalysts. Chem. – Eur. J. 2018, 24, 4780–4784. 10.1002/chem.201705745. [DOI] [PubMed] [Google Scholar]
- a Yang J.; Wang C.; Sun Y.; Man X.; Li J.; Sun F. Ligand-Controlled Iridium-Catalyzed Semihydrogenation of Alkynes with Ethanol: Highly Stereoselective Synthesis of E- and Z-Alkenes. Chem. Commun. 2019, 55, 1903–1906. 10.1039/C8CC09714C. [DOI] [PubMed] [Google Scholar]; b Wang Y.; Huang Z.; Huang Z. Catalyst as Colour Indicator for Endpoint Detection to Enable Selective Alkyne trans-Hydrogenation with Ethanol. Nat. Catal. 2019, 2, 529–536. 10.1038/s41929-019-0299-2. [DOI] [Google Scholar]; c Takemoto S.; Kitamura M.; Saruwatari S.; Isono A.; Takada Y.; Nishimori R.; Tsujiwaki M.; Sakaue N.; Matsuzaka H. Bis(Bipyridine) Ruthenium(II) Bis(Phosphido) Metalloligand: Synthesis of Heterometallic Complexes and Application to Catalytic (E)-Selective Alkyne Semi-Hydrogenation. Dalton Trans. 2019, 48, 1161–1165. 10.1039/C8DT04646H. [DOI] [PubMed] [Google Scholar]
- a Blum Y.; Reshef D.; Shvo Y. H-Transfer Catalysis with Ru3(CO)12. Tetrahedron Lett. 1981, 22, 1541–1544. 10.1016/S0040-4039(01)90372-7. [DOI] [Google Scholar]; b Djukic J. P.; Parkhomenko K.; Hijazi A.; Chemmi A.; Allouche L.; Brelot L.; Pfeffer M.; Ricard L.; Le Goff X. F. μ-Chlorido, μ-Hydroxo-Bridged Dicarbonyl Ruthenacycles: Synthesis, Structure and Catalytic Properties in Hydrogen Atom Transfer. Dalton Trans. 2009, 2695–2711. 10.1039/b820426h. [DOI] [PubMed] [Google Scholar]; c Li J.; Hua R. Stereodivergent Ruthenium-Catalyzed Transfer Semihydrogenation of Diaryl Alkynes. Chem. – Eur. J. 2011, 17, 8462–8465. 10.1002/chem.201003662. [DOI] [PubMed] [Google Scholar]; d Radkowski K.; Sundararaju B.; Fürstner A. A Functional-Group-Tolerant Catalytic trans Hydrogenation of Alkynes. Angew. Chem. Int. Ed. 2013, 52, 355–360. 10.1002/anie.201205946. [DOI] [PubMed] [Google Scholar]; e Schabel T.; Belger C.; Plietker B. A Mild Chemoselective Ru-Catalyzed Reduction of Alkynes, Ketones, and Nitro Compounds. Org. Lett. 2013, 15, 2858–2861. 10.1021/ol401185t. [DOI] [PubMed] [Google Scholar]; f Fuchs M.; Fürstner A. trans-Hydrogenation: Application to a Concise and Scalable Synthesis of Brefeldin A. Angew. Chem. Int. Ed. 2015, 54, 3978–3982. 10.1002/anie.201411618. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Leutzsch M.; Wolf L. M.; Gupta P.; Fuchs M.; Thiel W.; Farès C.; Fürstner A. Formation of Ruthenium Carbenes by gem-Hydrogen Transfer to Internal Alkynes: Implications for Alkyne trans-Hydrogenation. Angew. Chem. Int. Ed. 2015, 54, 12431–12436. 10.1002/anie.201506075. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Karunananda M. K.; Mankad N. P. E-Selective Semi-Hydrogenation of Alkynes by Heterobimetallic Catalysis. J. Am. Chem. Soc. 2015, 137, 14598–14601. 10.1021/jacs.5b10357. [DOI] [PubMed] [Google Scholar]; i Musa S.; Ghosh A.; Vaccaro L.; Ackermann L.; Gelman D. Efficient E-Selective Transfer Semihydrogenation of Alkynes by Means of Ligand-Metal Cooperating Ruthenium Catalyst. Adv. Synth. Catal. 2015, 357, 2351–2357. 10.1002/adsc.201500372. [DOI] [Google Scholar]; j Neumann K. T.; Klimczyk S.; Burhardt M. N.; Bang-Andersen B.; Skrydstrup T.; Lindhardt A. T. Direct trans-Selective Ruthenium-Catalyzed Reduction of Alkynes in Two-Chamber Reactors and Continuous Flow. ACS Catal. 2016, 6, 4710–4714. 10.1021/acscatal.6b01045. [DOI] [Google Scholar]; k Kusy R.; Grela K. E- and Z-Selective Transfer Semihydrogenation of Alkynes Catalyzed by Standard Ruthenium Olefin Metathesis Catalysts. Org. Lett. 2016, 18, 6196–6199. 10.1021/acs.orglett.6b03254. [DOI] [PubMed] [Google Scholar]; l Guthertz A.; Leutzsch M.; Wolf L. M.; Gupta P.; Rummelt S. M.; Goddard R.; Farès C.; Thiel W.; Fürstner A. Half-Sandwich Ruthenium Carbene Complexes Link trans-Hydrogenation and gem-Hydrogenation of Internal Alkynes. J. Am. Chem. Soc. 2018, 140, 3156–3169. 10.1021/jacs.8b00665. [DOI] [PubMed] [Google Scholar]; m Ji S.; Chen Y.; Zhao S.; Chen W.; Shi L.; Wang Y.; Dong J.; Li Z.; Li F.; Chen C.; Peng Q.; Li J.; Wang D.; Li Y. Atomically Dispersed Ruthenium Species inside Metal-Organic Frameworks: Combining the High Activity of Atomic Sites and the Molecular Sieving Effect of MOFs. Angew. Chem. Int. Ed. 2019, 58, 4271–4275. 10.1002/anie.201814182. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Wang Q.; Yu Z. Substitution of Alcohols by N-Nucleophiles via Transition Metal-Catalyzed Dehydrogenation. Chem. Soc. Rev. 2015, 44, 2305–2329. 10.1039/C4CS00496E. [DOI] [PubMed] [Google Scholar]
- Gerritz S. W.; Sefler A. M. 2,5-Dimethylfuran (DMFu): An Internal Standard for the “Traceless” Quantitation of Unknown Samples via 1H NMR. J. Comb. Chem. 2000, 2, 39–41. 10.1021/cc990041v. [DOI] [PubMed] [Google Scholar]
- a Irrgang T.; Kempe R. 3d-Metal Catalyzed N- and C-Alkylation Reactions via Borrowing Hydrogen or Hydrogen Autotransfer. Chem. Rev 2019, 119, 2524–2549. 10.1021/acs.chemrev.8b00306. [DOI] [PubMed] [Google Scholar]; b Nixon T. D.; Whittlesey M. K.; Williams J. M. J. Transition Metal Catalysed Reactions of Alcohols Using Borrowing Hydrogen Methodology. Dalton Trans. 2009, 753–762. 10.1039/B813383B. [DOI] [PubMed] [Google Scholar]
- Pingen D.; Müller C.; Vogt D. Direct Amination of Secondary Alcohols Using Ammonia. Angew. Chem. Int. Ed. 2010, 49, 8130–8133. 10.1002/anie.201002583. [DOI] [PubMed] [Google Scholar]
- Lu C.; Guo Y.; Yan J.; Luo Z.; Luo H.-B.; Yan M.; Huang L.; Li X. Design, Synthesis, and Evaluation of Multitarget-Directed Resveratrol Derivatives for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2013, 56, 5843–5859. 10.1021/jm400567s. [DOI] [PubMed] [Google Scholar]
- Cho C. S.; Kim D. T.; Shim S. C. Ruthenium-Catalyzed Transfer Hydrogenation of Alkynes by Tributylamine. Bull. Korean Chem. Soc. 2009, 30, 1931–1932. 10.5012/bkcs.2009.30.9.1931. [DOI] [Google Scholar]
- Park S. B.; Alper H. Recyclable Sonogashira Coupling Reactions in an Ionic Liquid, Effected in the Absence of Both a Copper Salt and a Phosphine. Chem. Commun. 2004, 1306–1307. 10.1039/b402477j. [DOI] [PubMed] [Google Scholar]
- Najman R.; Cho J. K.; Coffey A. F.; Davies J. W.; Bradley M. Entangled Palladium Nanoparticles in Resin Plugs. Chem. Commun. 2007, 5031–5033. 10.1039/b711978j. [DOI] [PubMed] [Google Scholar]
- Wang S.; Min Y.; Zhang X.; Xi C. External Oxidant-Free Cross-Coupling of Arylcopper and Alkynylcopper Reagents Leading to Arylalkyne. RSC Adv. 2017, 7, 28308–28312. 10.1039/C7RA03348F. [DOI] [Google Scholar]
- Mouriès V.; Waschbüsch R.; Carran J.; Savignac P. A Facile and High Yielding Synthesis of Symmetrical and Unsymmetrical Diarylalkynes Using Diethyl Dichloromethylphosphonate as Precursor. Synthesis-Stuttgart 1998, 271–274. 10.1055/s-1998-2037. [DOI] [Google Scholar]
- Adjabeng G.; Brenstrum T.; Frampton C. S.; Robertson A. J.; Hillhouse J.; McNulty J.; Capretta A. Palladium Complexes of 1,3,5,7-Tetramethyl-2,4,8-Trioxa-6-Phenyl-6-Phosphaadamantane: Synthesis, Crystal Structure and Use in the Suzuki and Sonogashira Reactions and the Alpha-Arylation of Ketones. J. Org. Chem. 2004, 69, 5082–5086. 10.1021/jo049474x. [DOI] [PubMed] [Google Scholar]
- Bernini R.; Cacchi S.; Fabrizi G.; Forte G.; Petrucci F.; Prastaro A.; Niembro S.; Shafir A.; Vallribera A. Perfluoro-Tagged, Phosphine-Free Palladium Nanoparticles Supported on Silica Gel: Application to Alkynylation of Aryl Halides, Suzuki-Miyaura Cross-Coupling, and Heck Reactions under Aerobic Conditions. Green Chem. 2010, 12, 150–158. 10.1039/B915465E. [DOI] [Google Scholar]
- Chen H. J.; Lin Z. Y.; Li M. Y.; Lian R. J.; Xue Q. W.; Chung J. L.; Chen S. C.; Chen Y. J. A New, Efficient, and Inexpensive Copper(II)/Salicylic Acid Complex Catalyzed Sonogashira-Type Cross-Coupling of Haloarenes and Iodoheteroarenes with Terminal Alkynes. Tetrahedron 2010, 66, 7755–7761. 10.1016/j.tet.2010.07.072. [DOI] [Google Scholar]
- Hénon H.; Anizon F.; Golsteyn R. M.; Léonce S.; Hofmann R.; Pfeiffer B.; Prudhomme M. Synthesis and Biological Evaluation of New Dipyrrolo [3,4-a:3,4-c] Carbazole-1,3,4,6-Tetraones, Substituted with Various Saturated and Unsaturated Side Chains via Palladium Catalyzed Cross-Coupling Reactions. Bioorg. Med. Chem. 2006, 14, 3825–3834. 10.1016/j.bmc.2006.01.030. [DOI] [PubMed] [Google Scholar]
- Mochida S.; Hirano K.; Satoh T.; Miura M. Rhodium-Catalyzed Regioselective Olefination Directed by a Carboxylic Group. J. Org. Chem. 2011, 76, 3024–3033. 10.1021/jo200509m. [DOI] [PubMed] [Google Scholar]
- Carollo L.; Floris B. Metallation of Alkynes Part 10. Acetoxymercuration of Arylferrocenylethynes. J. Organomet. Chem. 1999, 583, 80–85. 10.1016/S0022-328X(99)00110-2. [DOI] [Google Scholar]
- Zhao H.; Wang Y.; Sha J.; Sheng S.; Cai M. MCM-41-Supported Bidentate Phosphine Palladium(0) Complex as an Efficient Catalyst for the Heterogeneous Stille Reaction. Tetrahedron 2008, 64, 7517–7523. 10.1016/j.tet.2008.05.120. [DOI] [Google Scholar]
- Kakusawa N.; Yamaguchi K.; Kurita J. Palladium-Catalyzed Cross-Coupling Reaction of Ethynylstibanes with Organic Halides. J. Organomet. Chem. 2005, 690, 2956–2966. 10.1016/j.jorganchem.2005.03.021. [DOI] [Google Scholar]
- Ekebergh A.; Lingblom C.; Sandin P.; Wennerås C.; Mårtensson J. Exploring a Cascade Heck-Suzuki Reaction Based Route to Kinase Inhibitors Using Design of Experiments. Org. Biomol. Chem. 2015, 13, 3382–3392. 10.1039/C4OB02694B. [DOI] [PubMed] [Google Scholar]
- Meić Z.; Vikić-topić D.; Güsten H. Unusual Deuterium-Isotope Effects in 13C NMR Spectra of Trans-Stilbene. Org. Magn. Reson. 1984, 22, 237–244. 10.1002/mrc.1270220409. [DOI] [Google Scholar]
- Selvakumar K.; Zapf A.; Beller M. New Palladium Carbene Catalysts for the Heck Reaction of Aryl Chlorides in Ionic Liquids. Org. Lett. 2002, 4, 3031–3033. 10.1021/ol020103h. [DOI] [PubMed] [Google Scholar]
- Schmidt B.; Elizarov N.; Berger R.; Hölter F. Scope and Limitations of the Heck-Matsuda-Coupling of Phenol Diazonium Salts and Styrenes: A Protecting-Group Economic Synthesis of Phenolic Stilbenes. Org. Biomol. Chem. 2013, 11, 3674–3691. 10.1039/c3ob40420j. [DOI] [PubMed] [Google Scholar]
- Barder T. E.; Walker S. D.; Martinelli J. R.; Buchwald S. L. Catalysts for Suzuki-Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure. J. Am. Chem. Soc. 2005, 127, 4685–4696. 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- Shen M.; Leslie B. E.; Driver T. G. Dirhodium(II)-Catalyzed Intramolecular C-H Amination of Aryl Azides. Angew. Chem. Int. Ed. 2008, 47, 5056–5059. 10.1002/anie.200800689. [DOI] [PubMed] [Google Scholar]
- Alacid E.; Nájera C. Aqueous Sodium Hydroxide Promoted Cross-Coupling Reactions of Alkenyltrialkoxysilanes under Ligand-Free Conditions. J. Org. Chem. 2008, 73, 2315–2322. 10.1021/jo702570q. [DOI] [PubMed] [Google Scholar]
- Ferla S.; Gomaa M. S.; Brancale A.; Zhu J.; Ochalek J. T.; DeLuca H. F.; Simons C. Novel Styryl-Indoles as Small Molecule Inhibitors of 25-Hydroxyvitamin D-24-Hydroxylase (CYP24A1): Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2014, 87, 39–51. 10.1016/j.ejmech.2014.09.035. [DOI] [PubMed] [Google Scholar]
- Truong T.; Daugulis O. Transition-Metal-Free Alkynylation of Aryl Chlorides. Org. Lett. 2011, 13, 4172–4175. 10.1021/ol2014736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elangovan S.; Neumann J.; Sortais J. B.; Junge K.; Darcel C.; Beller M. Efficient and Selective N-Alkylation of Amines with Alcohols Catalysed by Manganese Pincer Complexes. Nat. Commun. 2016, 7, 12641. 10.1038/ncomms12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.