

Abstract
Axon guidance molecules and neuronal activity have been implicated in the establishment and refinement of neural circuits during development. It is unclear, however, whether these guidance molecule- and activity-dependent mechanisms interact with one another to shape neural circuit formation. The formation of corticospinal (CS) circuits, which are essential for voluntary movements, involves both guidance molecule- and activity-dependent components during development. We previously showed that semaphorin6D (Sema6D)-plexinA1 (PlexA1) signaling eliminates ipsilateral projections of CS neurons in the spinal cord, while other studies demonstrate that CS projections to the spinal cord are eliminated in an activity-dependent manner. Here we show that inhibition of cortical neurons during postnatal development causes defects in elimination of ipsilateral CS projections in mice. We further show that mice that lack the activity-dependent Bax/Bak pathway or caspase-9 similarly exhibit defects in elimination of ipsilateral CS projections, suggesting that the activity-dependent Bax/Bak-caspase-9 pathway is essential for the removal of ipsilateral CS projections. Interestingly, either inhibition of neuronal activity in the cortex or deletion of Bax/Bak in mice causes a reduction in PlexA1 protein expression in corticospinal neurons. Finally, intracortical microstimulation induces activation of only contralateral forelimb muscles in control mice, whereas it induces activation of both contralateral and ipsilateral muscles in mice with cortical inhibition, suggesting that the ipsilaterally projecting CS axons that have been maintained in mice with cortical inhibition form functional connections. Together, these results provide evidence of a potential link between the repellent signaling of Sema6D-PlexA1 and neuronal activity to regulate axon elimination.
SIGNIFICANCE STATEMENT Both axon guidance molecules and neuronal activity regulate axon elimination to refine neuronal circuits during development. However, the degree to which these mechanisms operate independently or cooperatively to guide network generation is unclear. Here, we show that neuronal activity-driven Bax/Bak-caspase signaling induces expression of the PlexA1 receptor for the repellent Sema6D molecule in corticospinal neurons (CSNs). This cascade eliminates ipsilateral projections of CSNs in the spinal cord during early postnatal development. The absence of PlexA1, neuronal activity, Bax and Bak, or caspase-9 leads to the maintenance of ipsilateral projections of CSNs, which can form functional connections with spinal neurons. Together, these studies reveal how the Sema6D-PlexA1 signaling and neuronal activity may play a cooperative role in refining CS axonal projections.
Keywords: axon elimination, corticospinal neuron, motor neuron, plexin, semaphorin, spinal cord
Introduction
Formation of neural circuits requires multiple processes during development (Sanes and Yamagata, 2009; Kolodkin and Tessier-Lavigne, 2011; Riccomagno and Kolodkin, 2015). First, neuronal specification is determined by transcription factors; then axons and dendrites are navigated by axon guidance molecules and their receptors to form neuronal connections. Subsequently, many of the exuberant and/or inappropriate connections are eliminated through an activity-dependent process, thereby allowing neurons to maintain only appropriate and necessary connections. In addition, previous studies have shown that not only neuronal activity but also axon guidance molecules regulate axon elimination during development (Riccomagno et al., 2012; Koropouli and Kolodkin 2014). One unresolved question is whether neuronal activity and guidance molecules interact to control axon elimination or regulate this critical process via independent pathways.
The corticospinal (CS) circuit is a suitable model system to study how axon guidance molecules and neuronal activity control network formation during development since previous studies revealed that guidance molecule- and activity-dependent mechanisms are both involved in the establishment of CS circuits (Gu et al., 2017a,b; Martin, 2005). CS neurons (CSNs) are located in layer V of the sensorimotor cortex and project axons to the spinal cord (Lemon, 2008). After CS axons reach appropriate segmental levels of the spinal cord, they send collaterals from their main axon bundles in the white matter to connect to spinal neurons. It has been shown that ligands or receptors, such as netrin-1, semaphorins, wnts, and ephrins, control axon guidance of CSNs in the brain and spinal cord (Canty and Murphy, 2008). For example, netrin-1 and semaphorin3A/3C control axon guidance of CSNs in the brain (Bagnard et al., 1998; Finger et al., 2002), and Class 6 semaphorins are involved in the midline crossing of CS axons in the brainstem (Faulkner et al., 2008; Runker et al., 2008).
Led by a variety of guidance cues, CS axons grow and branch exuberantly in the direction of the spinal cord gray matter. Much of the excessive axon collateral branching formed at this stage will later be pruned in a neuronal activity-dependent manner to achieve their mature termination patterns (Martin, 2005). Previous studies using cats have shown that inhibition of neuronal activity in the motor cortex by the GABAA agonist muscimol causes deficits in CS axon termination in the developing spinal cord (Li and Martin, 2001; Friel and Martin, 2005; Martin et al., 2007). We recently showed that the Bax/Bak pathway regulates the reorganization of CS axon terminals and circuit formation (Gu et al., 2017b). Other studies have also shown that the nonapoptotic Bax-caspase signaling pathway, triggered by neuronal activity, controls some late aspects of neural circuit formation and synaptic plasticity in different regions of the CNS (Williams et al., 2006; Li et al., 2010; Ohsawa et al., 2010; Jiao and Li, 2011; Miura, 2012; Simon et al., 2012). More recently, we showed that the repellant signaling by semaphorin6D (Sema6D) and its receptor, plexinA1 (PlexA1), eliminates ipsilateral projections of CSNs in the spinal cord (Gu et al., 2017a). This raises the question of whether and how neuronal activity-driven Bax/Bak-caspase and Sema6D-PlexA1 signaling can function together to regulate the refinement of CS axon terminals.
Materials and Methods
Mouse lines
The following mouse strains were used in this study: Emx1-Cre (Gorski et al., 2002), PlexA1fl/fl (Yoshida et al., 2006), Baxfl/fl Bak–/– (Takeuchi et al., 2005), caspase-3–/– (Kuida et al., 1996), caspase-6–/– (Uribe et al., 2012), caspase-7–/– (Lakhani et al., 2006), and caspase-9 fl/fl (Simon et al., 2012). Mice were maintained on a C57BL/6J background. All animals were treated according to institutional and National Institutes of Health guidelines, with the approval of the Institutional Animal Care and Use Committee at Cincinnati Children's Hospital Medical Center and Burke Neurologic Institute/Weill Cornell Medicine.
Immunohistochemistry and imaging
Perfusion fixation (ice-cold PBS followed by 4% PFA) was used to harvest brains and spinal cords. Upon dissection, the brain and spinal cord were fixed overnight at 4°C, cryoprotected by immersion in 30% sucrose/PBS for 48 h, and sectioned using a cryostat at 50 and 80 µm thickness, respectively. Free-floating immunohistochemistry was performed by incubating brain and spinal cord sections with primary antibodies overnight at room temperature and then with fluorophore-conjugated secondary antibodies for 4 h at room temperature. Sections were mounted with VECTASHIELD Mounting Media (Vector Labs) and coverslipped for imaging. Confocal images were taken with a Nikon A1R GaAsP. The following primary antibodies were used in this study: chicken anti-eGFP (1:2000, Abcam), goat anti-mCherry (1:1000, Biorbyt), rabbit anti-Cre (1:4000, Covance), rabbit anti-c-fos (1:500, Cell Signaling Technology), and rat anti-Ctip2 (1:1000, Abcam). The fluorophore-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories and Invitrogen. To quantify the ventral and lateral CST (vlCST) in each mouse, images were acquired from several spinal cords (4-6 sections per mouse) from cervical level 4-6. ImageJ was used to quantify the area of the ventral and lateral funiculi that was occupied by vlCST. Then, an average area of the ventral and lateral funiculi that was occupied by fluorescently labeled vlCST was calculated for each mouse.
Surgery and virus injection
Mice were anesthetized with isoflurane during surgery and injections as follows:
Unilateral expression of designer receptor exclusively activated by designer drugs (DREADD) in the motor cortex. A 300 nl adeno-associated virus (AAV) mixture, which included AAV1-hSyn-Cre (Penn Vector Core), AAV8-hSyn-DIO-DREADDs (activation via hM3Dq and inhibition via hM4Di, UNC Viral Core), or AAV1-CAG-Flex-eGFP (Penn Vector Core), was injected into the right hemisphere of P4 mice using the following coordinates: AP: 1 mm from the bregma; ML: 0.7 mm; and depth, 0.4 mm below the surface of the brain. This injection was used to manipulate the activity of CSNs with DREADDs and label their descending axons with GFP simultaneously. AAV solutions were diluted to a final titer of 1 × 1012 genome copies/ml (GCs/ml) for injections. Brains and spinal cords were collected at P21 (for Figs. 1, 2).
Bilateral expression of DREADD in the motor cortex. Four injections of 400 nl AAV8-hSyn-DREADDs (activation via hM3Dq and inhibition via hM4Di, UNC Viral Core, 4 × 1012 GCs/ml) were made into two regions of the motor cortex of P2 mice using the following coordinates: AP: 1 mm from the bregma; ML: 0.7 mm; and depth, 0.4 mm below the surface of the brain; and AP: 0 mm from the bregma; ML: 0.7 mm; and depth, 0.4 mm below the surface of the brain. Brains were collected at P14 (see Fig. 2) or P8 (see Fig. 4) for analysis. Motor cortices were dissected out at P8, and protein lysis was performed for Western blot analysis (see Fig. 4). Intracortical microstimulation (ICMS; see Fig. 5) was performed using 3-month-old mice.
Bilateral injection of AAV1-hSyn-Cre in the motor cortex. Four injections of 400 nl of AAV1-hSyn-Cre, Penn Vector Core, 1 × 1012 GCs/ml) were made to the motor cortex of P2 Baxfl/fl and Baxfl/fl Bak−/− mice using the following coordinates: AP: 1 mm from the bregma; ML: 0.7 mm; and depth, 0.4 mm below the surface of the brain; and AP: 0 mm from the bregma; ML: 0.7 mm; and depth, 0.4 mm below the surface of the brain. Motor cortices were dissected out at P8, and protein lysis was performed for Western blot analysis (related to Fig. 4).
Unilateral injection of AAV1-hSyn-Cre in the motor cortex. A 200 nl AAV viral mixture that included AAV1-hSyn-Cre (Penn Vector Core) and AAV1-CAG-Flex-eGFP (Penn Vector Core) was injected into the right hemisphere of P4 WT, Baxfl/fl, Bak–/–, Baxfl/fl; Bak–/–, caspase-3+/–, caspase-3–/–, caspase-6–/–, caspase-7–/–, caspase-9 fl/fl, PlexA1fl/+, and PlexA1fl/fl mice using the following coordinates: AP: 1 mm from the bregma; ML: 0.7 mm; and depth, 0.4 mm below the surface of the brain. AAV solutions were diluted to a final titer of 1 × 1012 GCs/ml for injections. Spinal cords were collected at P21 (related to Figs. 2, 3).
Figure 1.
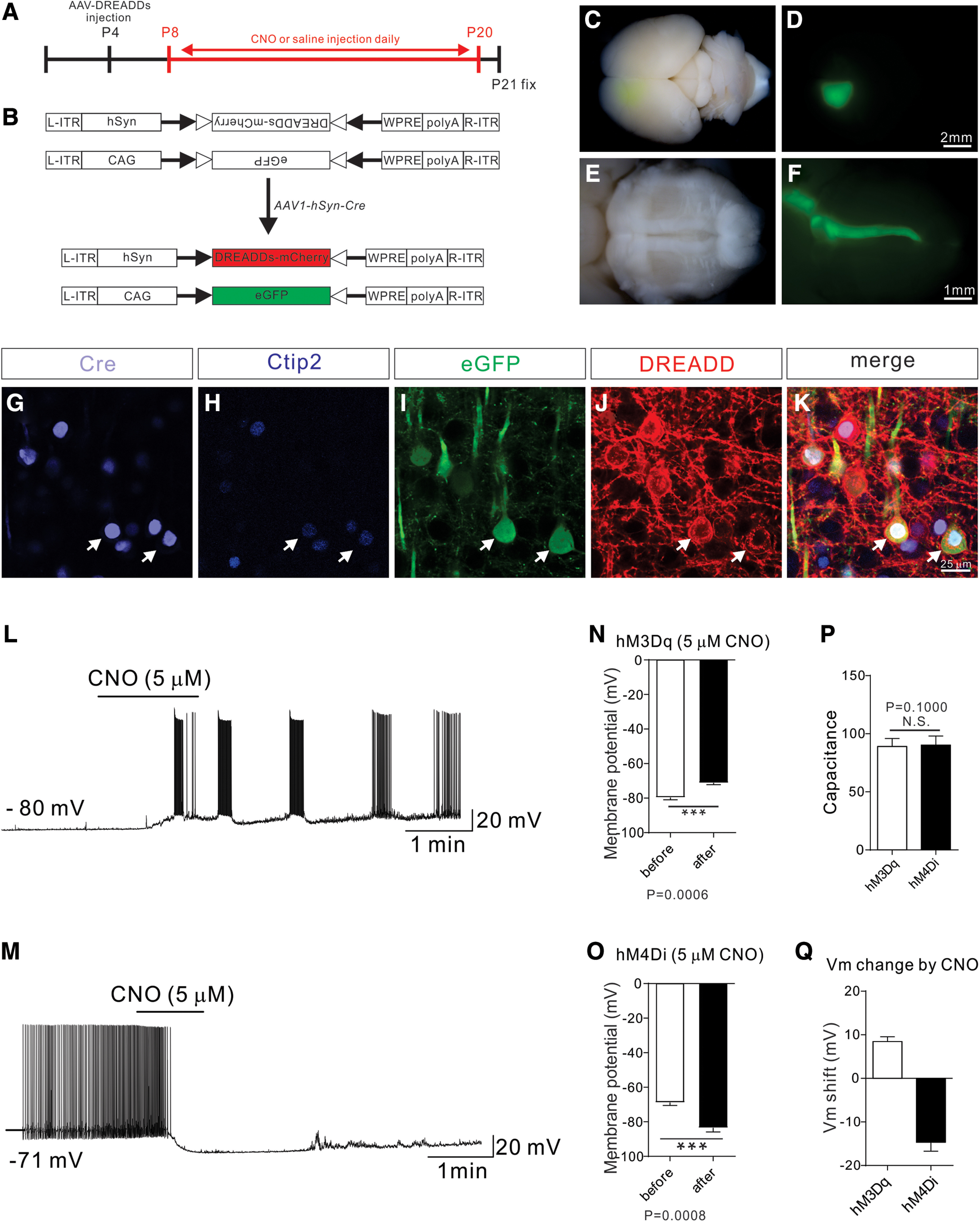
Establishment of a chemical genetic approach to manipulate neuronal activity in developing CS circuits. A, Experimental timeline: a DREADD-expressing AAV mixture was injected at P4, and CNO or saline (control) was injected daily from P8 to P20. Brains and spinal cords were collected at P21 for analysis. B, Experimental design: an AAV viral mixture consisting of AAV1-hSyn-Cre, AAV8-hSyn-DIO-DREADDs (activation via hM3Dq/inhibition via hM4Di), and AAV1-CAG-Flex-eGFP was injected to manipulate the activity of CSNs with DREADDs and simultaneously labels their descending axons with GFP. C-F, Representative images from injected mice showing eGFP labeling in the motor cortex (C,D) and descending CST fibers in the ventral brainstem (E,F). G-K, Representative images from the motor cortex showing immunostaining using antibodies targeting Cre (G; light blue), Ctip2 (H; blue), eGFP (I; green), or DREADD/mCherry (J; red) and a merged view (K). L, M, Whole-cell current-clamp recording from a CSN marked by GFP/mCherry fluorescence from mice injected with AAV8-DIO-hM3Dq-mCherry (L) and AAV8-DIO-hM4Di-mCherry (M). CNO (5 μm) elicited rapid depolarization of the membrane potential and greatly increased the firing rate of hM3Dq-expressing CSNs (n = 9). In contrast, CNO (5 μm) produced a robust inhibition in all sampled CSNs expressing hM4Di (n = 11). N, Bath application of CNO led to significant membrane depolarization (before: −79.91 ± 1.40 mV; after: −70.43 ± 1.05 mV; p = 0.0006, paired t test) in CSNs expressing hM3Dq (n = 9). O, In contrast, bath-applied CNO led to significant hyperpolarization of Vm (before: −68.14 ± 1.76 mV; after: −82.53 ± 2.44 mV; p = 0.0008, paired t test) in CSNs expressing hM4Di (n = 11). P, There was no significant difference in the membrane capacitance of the recorded neurons between the hM3Dq and hM4Di groups (p = 0.1000, Mann–Whitney test), suggesting that the two populations of cortical neurons sampled were comparable in size. Q, Quantification showing that the shift of membrane potential in the hM3Dq and hM4Di groups was 8.44 ± 1.12 mV and −14.66 ± 2.04 mV, respectively. Scale bars: D, 2 μm; F, 1 mm; K, 25 μm.
Figure 2.

Inhibition of CS neuronal activity during postnatal development leads to defects in elimination of vlCST. A, Schematic of a coronal section of the brain showing the injection of AAV8-DREADDs-mCherry into the motor cortex of P2 mice. B, Experimental design. AAV8-DREADDs-mCherry was injected into the motor cortex of P2 mice. Intraperitoneal injection of CNO or saline (as a control) was performed daily from P4 to P14. Brains were analyzed 10 h after intraperitoneal injection at P14. C-J, Representative images of coronal sections from the motor cortex showing immunostaining using antibodies targeting c-fos (green), mCherry (red for hM3Dq), and Ctip2 (blue) from saline- (C-F) or CNO-injected (O-J) hM3Dq-expressing mice at P14. K, Quantification showing the average number of c-fos+ neurons in CNO-injected, hM3Dq-expressing mice is significantly higher than in saline-injected mice (saline group, 44 ± 11.37; CNO group, 112 ± 3.58; p = 0.0013, unpaired t test, n = 4 mice in each group). L-S, Representative images of coronal sections from the motor cortex showing immunostaining using antibodies targeting c-fos (green), mCherry (red for hM4Di), and Ctip2 (blue) from saline- (L-O) or CNO-injected (P-S) hM4Di-expressing mice at P14. T, Quantification showing the average number of c-fos+ neurons in CNO-injected, hM4Di-expressing mice is significantly lower than in saline-injected mice (saline group, 42.75 ± 8.45; CNO group, 9 ± 4.97; p = 0.0013, unpaired t test, n = 4 mice in each group). U, V, Representative images of cervical spinal cords showing the absence of vlCST in the saline-injected (n = 7 mice) (U) group and the presence of vlCST (indicated by red arrows) in the CNO-injected (n = 9 mice) (V) group. W, Quantification of the average area occupied by vlCST in the ipsilateral ventral and lateral funiculi of CNO-injected hM4Di mice shows a significantly larger area than that in saline-injected hM4Di mice (saline group, 0.09999 ± 0.01633%; CNO group, 1.455 ± 0.1971%; p = 0.0003, unpaired t test). Scale bars: J, 50 μm; S, 50 μm; U, 200 μm.
Figure 4.
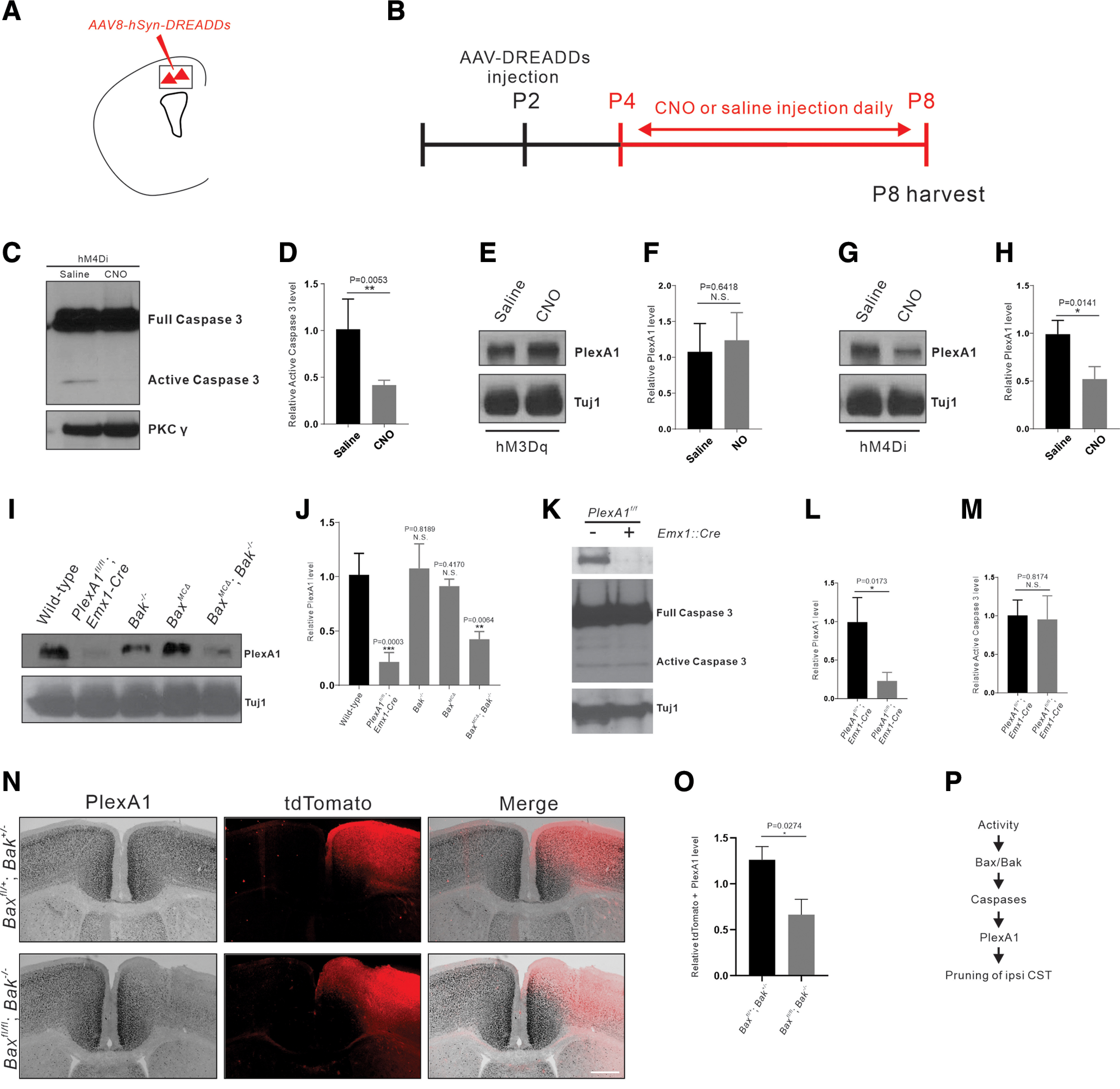
Neuronal inhibition or Bax/Bak loss of function decreases PlexA1 expression. A, Schematic diagram showing that DREADD-expressing AAVs (activation via hM3Dq/inhibition via hM4Di) were injected into the motor cortex to manipulate the activity of neurons. B, Experimental timeline: DREADD-expressing AAVs were injected at P2; CNO or saline (control) was injected daily from P4 to P8, and brains and spinal cords were collected at P8 for Western blot analysis. C, Western blot analyses of protein lysates from the motor cortex of P8 AAV8-hSyn-hM4Di-mCherry-injected mice using antibodies against full-length (inactive) caspase-3 and PKCγ. Full-length caspase-3 is present in both the saline and CNO groups, and the active form is absent in the CNO group. D, Quantification of Western blot showing that hM4Di-mediated inhibition resulted in significant reduction of the active (cleaved) form of caspase-3 (p = 0.0053, unpaired t test). E-H, Western blot analyses of protein lysates from the motor cortex of P8 AAV8-hSyn-hM3Dq-mCherry- (E) and AAV8-hSyn-hM4Di-mCherry-injected (G) mice using anti-PlexA1 and anti-Tuj antibodies. Quantification of Western blot showing that the expression of PlexA1 is comparable in the saline and CNO groups of AAV8-hSyn-hM3Dq-mCherry-injected mice (F; p = 0.6418, unpaired t test), whereas the expression of PlexA1 is markedly reduced in the CNO group of AAV8-hSyn-hM4Di-mCherry-injected mice (H; p = 0.0141, unpaired t test). I, Western blot analyses of protein lysates from the motor cortex of P8 WT, PlexA1fl/fl Emx1-Cre, Bak–/–, Baxfl/fl-AAV1-Cre, and Baxfl/fl Bak−/−-AAV1-Cre mice using anti-PlexA1 and anti-Tuj antibodies. J, Quantification of Western blot showing that the expression of PlexA1 is significantly decreased in PlexA1fl/fl Emx1-Cre mice (p = 0.0003, unpaired t test) and is markedly reduced in Baxfl/fl Bak−/−-AAV1-Cre mice (p = 0.0064, unpaired t test) but not in Bak (p = 0.8189, unpaired t test) or Bax (p = 0.4170, unpaired t test) single-mutant mice. K, Western blot analyses of protein lysates from the motor cortex of P8 PlexA1fl/fl and PlexA1fl/fl Emx1-Cre mice using anti-full-length caspase-3 and anti-Tuj antibodies. L, M, Quantification of Western blot showing that the expression of PlexA1 is significantly decreased in PlexA1fl/fl Emx1-Cre mice (p = 0.0173, unpaired t test), but the activation of caspase-3 is not significantly affected by the deletion of PlexA1 (p = 0.8174, unpaired t test). N, ISH of PlexA1 mRNA followed by immunostaining against tdTomato in P10 motor cortex of Baxfl/+ Bak+/− (n = 3) and Baxfl/fl Bak−/− (n = 3) mice injected with an AAV viral mixture (AAV1-hSyn-Cre and AAV1-CAG-Flex-tdTomato). O, Quantification of ISH signal in tdTomato-positive regions (p = 0.0274, unpaired t test). P, Proposed mechanism of PlexA1-mediated elimination of ipsilateral vlCST; neuronal activity induces the activation of the Bax/Bak-caspase pathway that affects the expression of PlexA1. Scale bar, 200 μm.
Figure 5.
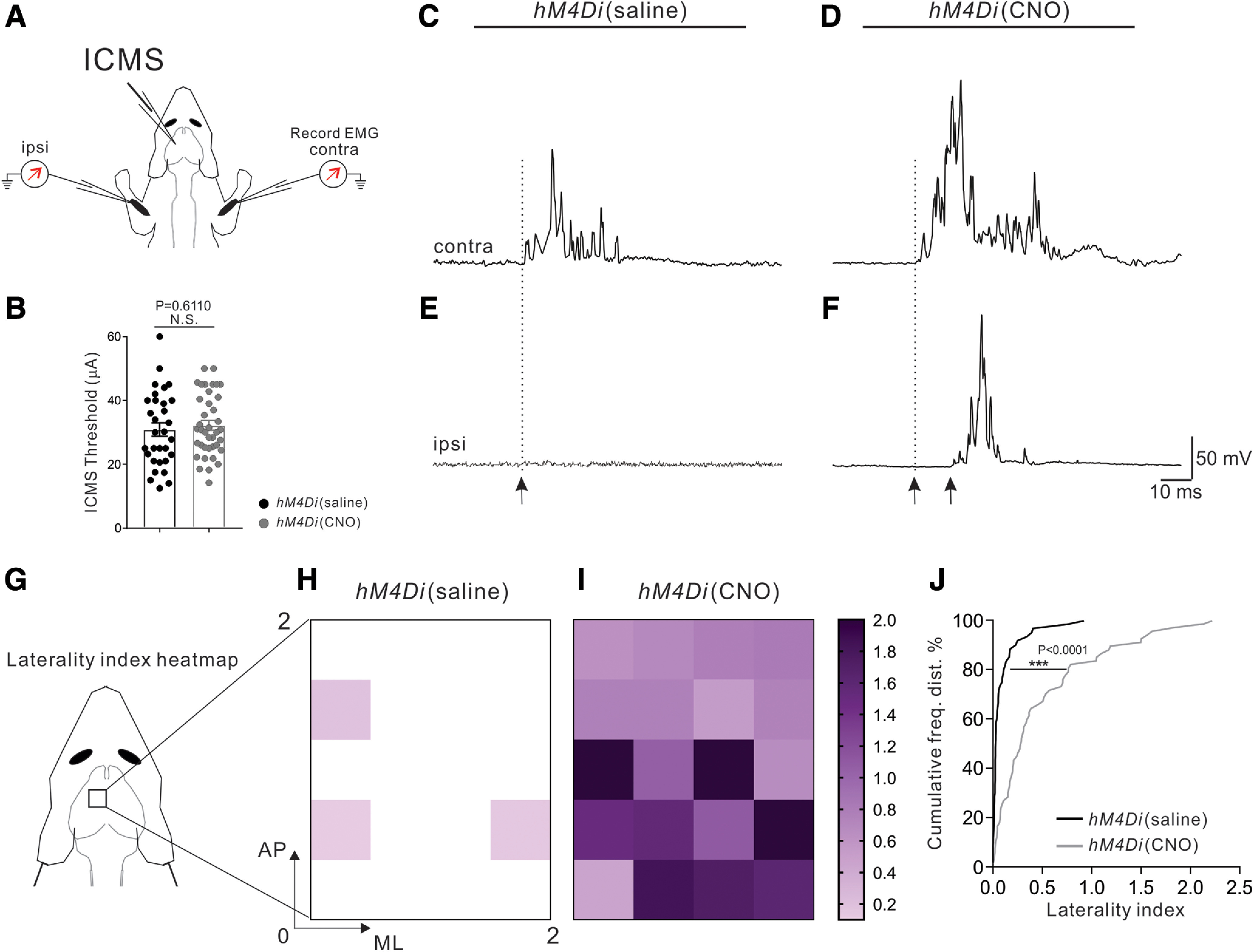
M1 stimulation-evoked bilateral forelimb response in CNO-injected hM4Di mice. A, Experimental strategy for recording ICMS-evoked muscle EMGs from Bi muscles bilaterally. B, Quantification showing that stimulation currents used for the 14-pulse ICMS from AAV8-hSyn-hM4Di-mCherry mice were similar in the CNO-injected group (39 sites from 3 mice) and saline-injected group (31 sites from 3 mice, p = 0.6110, unpaired t test). C-F, Representative examples of EMG responses demonstrate that threshold ICMS in the saline-injected group evoked strong contralateral (C) Bi responses but no activation on the ipsilateral side (E). In contrast, threshold ICMS in the CNO-injected group evoked strong contralateral (D) Bi responses and robust activation of ipsilateral Bi muscle (F). Black arrows indicate onset latencies. G-I, Heatmap plots of the laterality indices of CNO-injected mice (I; 39 sites from 3 mice) and saline-injected mice (H; 31 sites from 3 mice). J, Cumulative histograms of the laterality indices showing a shift from predominant contralateral activation in the saline-injected group (black) to robust coactivation of contralateral and ipsilateral Bi muscles in the CNO-injected mice (gray) (p < 0.0001, Kolmogorov-Smirnov test).
Figure 3.
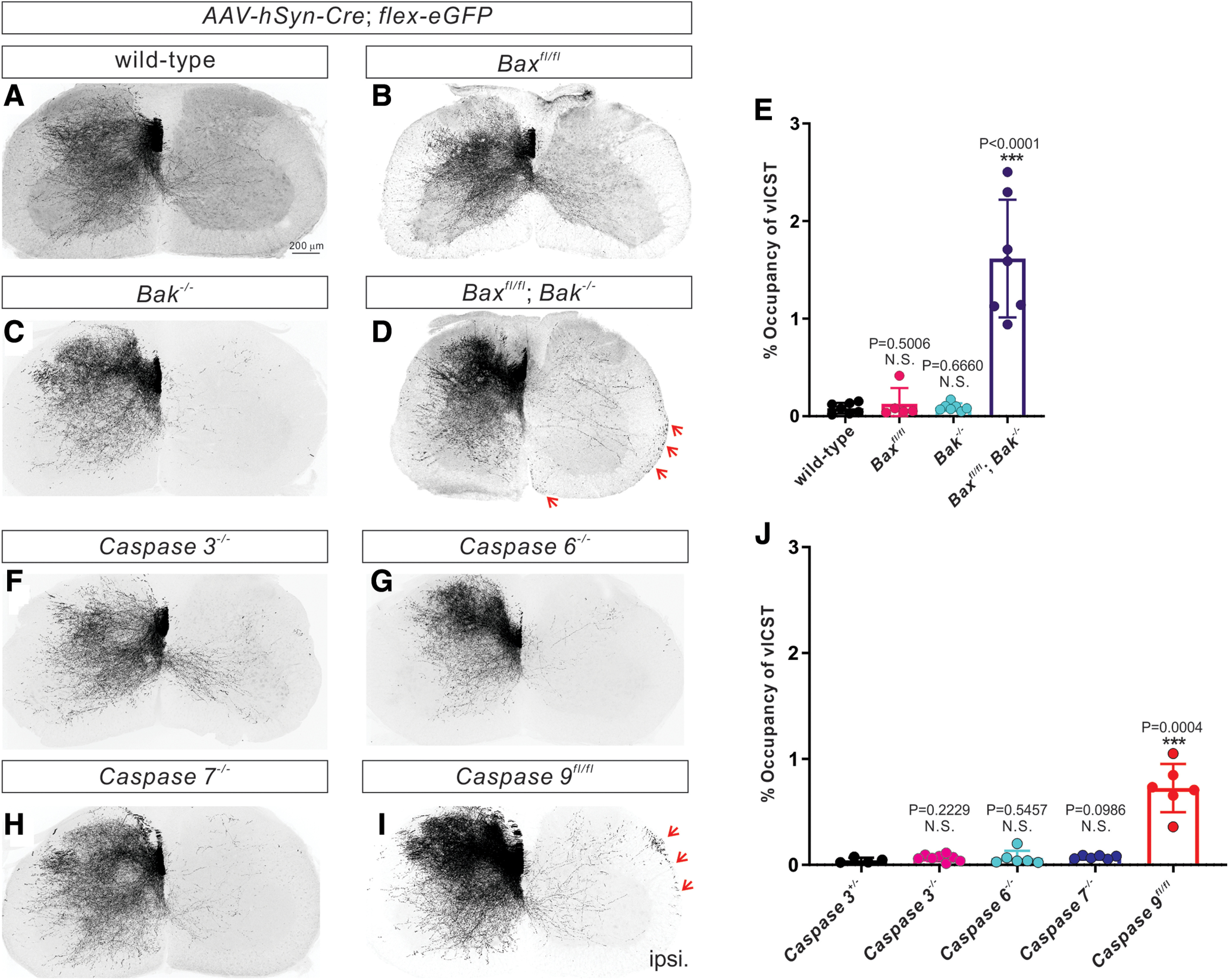
Bax/Bak and caspase-9 loss of function causes vlCST pruning defects. A-D, Representative images of cervical spinal cords showing the absence of vlCST in P21 WT (A, n = 7 mice), Baxfl/fl (B; n = 5 mice), and Bak–/– (C; n = 4 mice) mice and the presence of vlCST (indicated by red arrows) in Baxfl/fl Bak−/− mice (D; n = 9 mice). E, Quantification of the average area occupied by vlCST in the ipsilateral ventral and lateral funiculi, demonstrating that Baxfl/fl Bak−/− mice (1.615 ± 0.2278%; p < 0.0001, unpaired t test) but not Baxfl/fl (0.1271 ± 0.07238%, p = 0.5006, unpaired t test) or Bak–/– (0.09308 ± 0.01490%, p = 0.6660, unpaired t test) mice, shows a significantly greater presence of vlCST compared with control mice (0.08184 ± 0.02059%). F-I, Representative images of cervical spinal cords showing the absence of vlCST in P21 caspase-3–/– (F; n = 8 mice), caspase-6–/– (G; n = 6 mice), and caspase-7–/– (H; n = 6 mice) mice and the presence of vlCST (indicated by red arrows) in caspase-9fl/fl mice (I; n = 6 mice). J, Quantification showing that the average area occupied by vlCST in the ipsilateral ventral and lateral funiculi of caspase-9fl/fl mice (0.7251 ± 0.09310%, p = 0.0004, unpaired t test), but not caspase-3–/– (0.06655 ± 0.01128%, p = 0.2229, unpaired t test), caspase-6–/– (0.06549 ± 0.02768%, unpaired t test), or caspase-7–/– (0.06612 ± 0.006429%, unpaired t test) mice, is significantly higher than caspase-3+/– mice (0.04285 ± 0.01203%). Scale bar: A, 200 μm.
Clozapine N-oxide (CNO) injection
To activate hM3Dq and hM4Di in vivo, a 0.3 and 3 mg/kg CNO (Sigma Millipore, C0832-5MG) solution, respectively, was injected daily from P8 to P21 (see Figs. 1, 2, 5), P4 to P14 (see Fig. 2), or P4 to P8 (see Fig. 4). Saline injection was used as a control (related to Figs. 1, 2, 4, 5).
Quantification of c-fos immunostaining
Animals were killed 90 min after CNO injection. Animals were transcardially perfused, and brains were sectioned on a cryostat at 50 µm for c-fos immunostaining analysis. The forelimb motor cortex sections were imaged on a Nikon A1R GaAsP confocal microscope at 20× magnification for quantification. Cells with a visually identifiable nuclear c-fos label were considered c-fos+. Three brain sections were quantified for each mouse (related to Fig. 2).
Western blot analysis and quantification
Forelimb motor cortices were dissected 90 min after the CNO injection at P8. Tissues were homogenized and lysed in lysis buffer (50 mm Tris, pH 8.0, 1 mm MgCl2, 1% NP-40, 0.25% sodium deoxycholate, 150 mm NaCl, and protease inhibitors, Roche Diagnostics) at 4°C for 30 min. The lysates were centrifuged at 12,000 rpm at 4°C for 10 min, and then supernatants were collected. Western blot analyses were performed using standard methods with rabbit anti-full caspase-3 (1:1000, Cell Signaling Technology), rabbit anti-PKCγ (1:200, Santa Cruz Biotechnology), rabbit anti-PlexA1 (1:5000) (Yoshida et al., 2006), and mouse anti-Tuj1 (1:500, Covance) (related to Fig. 4). ImageJ was used to quantify Western blot.
ISH and quantification
ISH was performed as previously described by Gu et al. (2017a). Briefly, mice were perfused transcardially with 4% PFA in 0.1 M PB. Brains were dissected and postfixed in PFA overnight. The tissues were cryopreserved in 30% sucrose for 24-48 h and embedded in Tissue-Tek OCT compound (Sakura Finetek). Serial 50-μm-thick sections were made with a cryostat and mounted on SuperFrost Plus slides (Thermo Fisher Scientific).
Digoxigenin-labeled riboprobes were prepared by in vitro transcription from the cDNA fragments encoding Plexin A1. Signals were detected using alkaline phosphatase-conjugated anti-digoxigenin antibodies (Roche Diagnostics) with NBT/BCIP as substrates for the color reaction. Sections were further processed for immunohistochemistry with antibody to DsRed (1:1000 Clontech). Primer set was as follows: PlxnA1 forward: GCCAAGTACAAGGACGAGATTC; PlxnA1 reverse: GAAAACCACTAGCAGCTCTTGC. ImageJ was used to quantify sections.
In vitro brain slice preparations and patch-clamp recordings
At postnatal P21, DREADD-injected mice were deeply anesthetized with sodium pentobarbital (60 mg/kg) and perfused with ice-cold dissection solution (250 mm sucrose, 2.5 mm KCl, 25 mm NaHCO3, 1.0 mm NaH2PO4, 6 mm MgCl2, 0.5 mm CaCl2, and 25 mm glucose) continuously bubbled with 95% O2/5% CO2. Brains were quickly isolated, and transverse slices containing the motor cortex were sectioned (at 300 μm thickness) using a vibrating microtome (7000smz-2; Campden Instruments). Slices were incubated for 15-20 min in a recovery solution (92 mm NMDG, 2.5 mm KCl, 1.2 mm NaH2PO4, 30 mm NaHCO3, 20 mm HEPES, 25 mm glucose, 5 mm sodium ascorbate, 2 mm thiourea, 3 mm sodium pyruvate, 10 mm MgSO4, and 0.5 mm CaCl2) at room temperature and then allowed to recover further in an oxygenated aCSF solution (125 mm NaCl, 2.5 mm KCl, 25 mm NaHCO3, 1.0 mm NaH2PO4, 1.0 mm MgCl2, 2.0 mm CaCl2, and 25 mm glucose) for ≥1 h at room temperature. After recovery, brain slices were transferred to a submersion-type recording chamber (RC-22; Warner Instruments) and mounted onto the stage of an upright microscope (BX51WI, Olympus) equipped with optical filters to allow for the identification of eGFP-labeled neurons. Slices were then perfused at room temperature with oxygenated aCSF at a rate of 2-4 ml/min. Patch electrodes were constructed from thin-walled single-filamented borosilicate glass (1.5 mm outer diameter; World Precision Instruments) using a microelectrode puller (P-97; Sutter Instruments) and filled with an intracellular solution (130 mm K-gluconate, 10 mm KCl, 10 mm HEPES, 10 mm sodium phosphocreatine, 4 mm MgATP, and 0.3 mm Na2-GTP, pH 7.2, 295-300 mOsm). Pipette resistances ranged from 4 to 6 MΩ, and seal resistances were > 1 GΩ. Patch-clamp recordings were obtained from eGFP-labeled cortical neurons in layer V of the motor cortex (M1) using a MultiClamp 700B Amplifier (Molecular Devices). In the current-clamp mode, once a stable membrane potential was observed, the selective DREADD agonist CNO was bath-applied at 5 or 20 μm. To avoid the potential residual effects of CNO that may persist beyond the washout of the drug, only one cell was recorded from each slice. Membrane voltages were adjusted for liquid junction potentials (∼–14 mV) calculated using JPCalc software (P. Barry, University of New South Wales, Sydney, Australia; modified for Molecular Devices). Signals were filtered at 4-6 kHz through a –3 dB, four-pole low-pass Bessel filter and digitally sampled at 20 kHz using a commercially available data acquisition system (Digidata 1440A with pClamp 10.3 software; Molecular Devices) (related to Fig. 1).
ICMS and EMG recordings
Electrical responses were recorded from ipsilateral biceps brachii (Bi) muscles using percutaneous Ni-chrome wire electrodes (deinsulated 1 mm from the tip) in response to motor cortex threshold stimulation as previously reported (Gu et al., 2017a,b). EMG recording wires were inserted using a 27-gauge hypodermic needle guide. Using two wire electrodes within each muscle and a separate ground, we differentially recorded and verified the adequacy of the EMG recordings and muscle placements by noting increased EMG activity with passive joint rotation. EMG recordings were made with a differential AC amplifier with low- and high-pass filtration (model 1700; AM Systems). EMG signals were acquired using an analog-to-digital converter (CED) and processed using Signal software (version 6.05; CED). Averages of rectified EMGs were generated from individual sites from each animal. To quantify the results, we developed a laterality index that corresponded to the integrated EMG value of the ipsilateral Bi muscle divided by the integrated EMG value for the contralateral Bi muscle. Strongly contralateral responses have a coactivation value that is close to zero and bilateral coactivation responses that are close to 1 or larger. Laterality indices from all sites in each group were used to plot the cumulative frequency distribution (see Fig. 5J). To illustrate the laterality status of different cortical areas, a 2 mm × 2.5 mm portion of the motor cortex was divided equally into 20 squares. Individual sites were assigned to different squares based on their anterior to posterior and medial to lateral coordinates (relative to the bregma). An average laterality index was calculated from all sites in each square. Laterality index maps were generated from plots of the average laterality indices from all the squares in each map (see Fig. 5H,I).
Statistics
The results are expressed as the mean ± SEM. Cumulative frequency distributions of coactivation indices (see Fig. 5J) were analyzed by the Kolmogorov-Smirnov test. Student's t tests were used for other statistical analyses. Error bars indicate mean ± SEM.
Results
Establishment of a chemical genetic approach to manipulate neuronal activity in developing CS circuits
To probe the contribution of neuronal activity during the refinement of developing CS circuits, we used DREADDs to manipulate the neuronal activity of CSNs during early mouse postnatal (P) stages P8-P20 (Fig. 1A). We found that the expression of DREADD-mCherry was restricted to the soma and dendrites of CSNs (Fig. 1J,K) and was not detected in CS axons (data not shown). Therefore, we injected an AAV viral mixture consisting of AAV1-hSyn-Cre; AAV8-hSyn-DIO-DREADDs (hM3Dq for neuronal activation or hM4Di for neuronal inhibition) and AAV1-CAG-Flex-eGFP to manipulate the activity of CSNs with DREADD while simultaneously labeling their descending axons with GFP (Fig. 1A,B). Using this strategy, a small subset of CSNs and their projections to the spinal cord were labeled (Fig. 1D-G). We found that those CSNs were positive for Cre, Ctip2 (a layer V marker) (Arlotta et al., 2005; Chen et al., 2005), GFP, and DREADD (revealed by mCherry that was fused to DREADD) (Fig. 1G-K).
We then examined the effects of DREADD activation by the drug CNO on neuronal activity. For this, we isolated acute motor cortex slices from AAV-injected mice and performed whole-cell recordings from CSNs in the absence or presence of bath-applied CNO. Layer V cortical neurons were randomly selected within the motor cortex based on their expression of mCherry and GFP (Fig. 1I-K). We found that CNO induced significant membrane depolarization (8.44 ± 1.12 mV; p = 0.0006, paired t test; Fig. 1N,Q) and action potential discharge in all neurons (n = 9 from 2 mice, 6 cells at 5 μM CNO, 3 cells at 20 μM CNO) expressing hM3Dq (Fig. 1L). In contrast, the same doses of CNO inhibited the sampled neurons (n = 11 from 2 mice, 6 cells at 5 μM, 5 cells at 20 μM) expressing hM4Di (Fig. 1M), as evidenced by the significant hyperpolarization of Vm (−14.66 ± 2.04 mV; p = 0.0008, paired t test; Fig. 1O,Q). These experiments showed that the use of DREADDs is a robust approach to acutely and bidirectionally manipulate the activity of CSNs.
Inhibition of CS neuronal activity during postnatal development leads to defects in vlCST elimination
We next determined whether DREADD-expressing CSNs would respond to CNO in vivo by injecting AAVs that express DREADDs (AAV8-hSyn-hM3Dq-mCherry or AAV8-hSyn-hM4Di-mCherry) into the motor cortex of P2 mice (Fig. 2A). Daily injections of CNO or saline as a control were conducted from P4 to P14 (Fig. 2B), and the motor cortex was analyzed for c-fos expression at P14 (10 h after CNO injection at day P14). In hM3Dq-injected mice, we observed a marked increase (p = 0.0013, unpaired t test) in c-fos+ cortical neurons in the CNO-injected group (112 ± 3.58, n = 4 mice) compared with the saline group (44 ± 11.37, n = 4 mice) (Fig. 2C-K). In contrast, for the hM4Di group, we found a significant (p = 0.014, unpaired t test) decrease of c-fos+ cortical neurons in the CNO-injected group (9 ± 4.967, n = 4 mice) compared with the saline group (42.75 ± 8.499, n = 4 mice) (Fig. 2L-T). These findings suggest that CNO-induced activation of hM3Dq depolarizes and increases the firing of CSNs in vivo, whereas CNO-induced activation of hM4Di hyperpolarizes and decreases the firing of CSNs. Together, these data demonstrate that a chemical genetic approach can be used to manipulate the activity of a subset of CSNs during postnatal refinement of the CS system in vivo.
Next, we determined how inhibition of CSN activity affects CS projections in the spinal cord by expressing hM4Di in the motor cortex. CNO was injected daily from P8 to P20 to inhibit the activity of CSNs. Interestingly, we observed eGFP+ CST fibers in the ventral and lateral funiculi of the ipsilateral side of the cervical spinal cord (vlCST) in the CNO-injected group (n = 9) but not in the saline group (n = 7) (Fig. 2U,V). Quantitative analyses revealed that the unpruned vlCST occupied 1.455 ± 0.1971% of the ventral and lateral funiculi, which was significantly higher (p = 0.0003, unpaired t test) than that of controls (0.09999 ± 0.01633%) (Fig. 2W). Since we also recently showed that elimination of the vlCST is mediated by the Sema6D-PlexA1 pathway (Gu et al., 2017a), these data indicate that the postnatal elimination of vlCST is mediated by both CS neuronal activity and Sema6D-PlexA1 repellent signaling.
Role of the Bax/Bak-caspases pathway in elimination of the vlCST
Since our recent study showed that the activity-dependent, nonapoptotic Bax/Bak pathway is essential for the reorganization of CS circuits (Gu et al., 2017b), we decided to test whether this activity-dependent pathway is also involved in the elimination of the vlCST. To this end, we injected an AAV viral mixture (AAV1-hSyn-Cre and AAV1-CAG-Flex-eGFP) into the motor cortex of Baxfl/fl (n = 5) and Baxfl/fl; Bak−/− (n = 9) mice at P4 to delete floxed Bax alleles in CSNs and simultaneously label their descending axons with GFP. The viral injections were also performed in control (n = 7) and Bak−/− (n = 4) mice. This resulted in the labeling of a small subset of CSNs and their projections to the spinal cord. Spinal cords were collected at P21 to examine the vlCSTs. We found significantly (p < 0.0001, unpaired t test) more unpruned vlCSTs in Bax/Bak (1.615 ± 0.2278%) double-mutant mice compared with control mice (0.08184 ± 0.02059%) (Fig. 3A,D,E). In contrast, we did not find obvious defects in vlCST elimination in the Bax (0.1271 ± 0.07238%, p = 0.5006, unpaired t test) or Bak (0.09308 ± 0.01490%, p = 0.6660, unpaired t test) single-mutant mice (Fig. 3B,C,E). These data indicate that the Bax/Bak pathway mediates the elimination of the vlCST.
To examine whether caspases are also involved in the Bax/Bak-dependent elimination of the vlCST, we injected an AAV viral mixture (AAV1-hSyn-Cre and AAV1-CAG-Flex-eGFP) into the motor cortex of caspase-3+/− (n = 4), caspase-3−/− (n = 8), caspase-6−/− (n = 6), caspase-7−/− (n = 6), and caspase-9fl/fl (n = 6) mice at P4 to label a subset of CSNs with GFP or to delete floxed caspase-9 alleles in CSNs and label their descending CS axons with GFP simultaneously. Spinal cords were collected at P21 to assess whether the vlCST was properly eliminated or aberrantly maintained in any of the cohorts. We found defects in elimination of the vlCST only in caspase-9fl/fl; AAV1-Cre mice (0.7251 ± 0.09310%, p = 0.0004, unpaired t test) but not in caspase-3−/− (0.06655 ± 0.01128%, p = 0.2229, unpaired t test), caspase-6−/− (0.06549 ± 0.02768%, p = 0.5457, unpaired t test), or caspase-7−/− mice (0.06612 ± 0.006429%, p = 0.0986, unpaired t test) compared with controls (caspase-3+/−, 0.04285 ± 0.01203%) (Fig. 3F-J). Together, these data suggest that caspase-9 eliminates the vlCST during normal mouse development.
Inhibition of CSN activity or deletion of Bax/Bak decreases PlexA1 expression
Both the inhibition of CS neuronal activity and the absence of Sema6D-PlexA1 signaling caused defects in the elimination of the vlCST. This prompted us to examine the potential interactions between neuronal activity and the Sema6D-PlexA1 signaling pathway in this process.
We first examined the impact of neuronal inhibition on the expression of the activated caspase-3, which is an indicator of caspase-9 activation in sensory axons (Simon et al., 2012), and PlexA1 proteins. We injected AAVs that express DREADDs (AAV8-hSyn-hM3Dq-mCherry or AAV8-hSyn-hM4Di-mCherry) into the motor cortex of P2 mice (Fig. 4A). Daily injections of CNO or saline from P4 to P8 were performed as controls (Fig. 4B). Examination of protein lysates prepared from motor cortices revealed that hM4Di-mediated inhibition did not change the expression of the inactive (full-length) form of caspase-3 but led to a marked reduction of the active (cleaved) form of caspase-3 (p = 0.0053, unpaired t test), corroborating our recent study showing that nonapoptotic activation of the Bax/Bak-caspase pathway in CSNs is activity-dependent (Fig. 4C,D) (Gu et al., 2017b). We also observed that expression of the full-length and active forms of caspase-3 were unchanged in PlexA1 mutant mice (PlexA1fl/fl-Emx1-Cre; p = 0.8174, unpaired t test) (Fig. 4K-M). PlexA1 expression in the motor cortices of hM3Dq-expressing mice was similar (p = 0.6418, unpaired t test) between the saline-injected and CNO-injected groups (Fig. 4E,F). In contrast, in hM4Di-expressing mice, we found a marked decrease in PlexA1 expression in the CNO-injected group compared with the saline group (p = 0.0141, unpaired t test) (Fig. 4G,H). Using protein lysates prepared from the motor cortices of PlexA1 (PlexA1fl/fl; Emx1-Cre), Bak (Bak−/−), and Bax (Baxfl/fl-AAV1-Cre) single-mutant and Bax/Bak (Baxfl/fl Bak−/−-AAV1-Cre) double-mutant mice, we found significant reductions in PlexA1 expression in PlexA1 mutant mice (p = 0.0003, unpaired t test) and Bax/Bak double-mutant mice (p = 0.0064, unpaired t test) but not in Bak (p = 0.8189, unpaired t test) or Bax (p = 0.4170, unpaired t test) single-mutant mice (Fig. 4I,J). To determine whether PlexA1 mRNA expression is affected in Bax/Bak double-mutant mice, we injected an AAV viral mixture (AAV1-hSyn-Cre and AAV1-CAG-Flex-tdTomato) into the motor cortices of Baxfl/+; Bak+/− (n = 3) and Baxfl/fl; Bak−/− (n = 3) mice at P4, and analyzed PlexA1mRNA expression around the injection sites. We found a twofold reduction in PlexA1 mRNA levels specifically in regions marked by tdTomato expression (p = 0.0274, unpaired t test) (Fig. 4N,O). These findings strongly suggest that PlexA1 is a downstream effector of the Bax/Bak-signaling cascade (Fig. 4P).
M1 stimulation evoked a bilateral forelimb response in CNO-injected hM4Di mice
To determine whether the maintained ipsilateral CS projections in the CNO-injected hM4Di mice were functional, we performed ICMS in the motor cortex and recorded EMGs from the contralateral and Bi muscles in adult 3-month-old hM4Di mice that were injected with CNO or saline between P8 and P20 (Fig. 5A). We stimulated sites in the motor cortex (M1) that produced both forelimb flexion and contralateral Bi muscle contraction at threshold, then determined whether there was concurrent activation of the ipsilateral Bi muscle. We found no significant changes in the thresholds for ICMS (39 sites from three CNO-injected mice, 31 sites from three saline-injected mice, p = 0.6110, unpaired t test; Fig. 5B). Threshold ICMS in the saline-injected group evoked strong contralateral Bi responses but no or minimal activation from the ipsilateral side (Fig. 5C,E). In contrast, threshold ICMS in the CNO-injected group not only evoked a strong contralateral Bi response but also induced robust activation of the ipsilateral Bi muscle (Fig. 5D,F). To quantify these results, we developed a laterality index that corresponded to the integrated EMG values of the ipsilateral Bi muscle divided by the integrated EMG values for the contralateral Bi muscle (Fig. 5G-I). A topographic plot of the laterality index revealed no or only low activation of the ipsilateral Bi muscle throughout the forelimb motor cortex from the saline-injected group (Fig. 5H). However, the plot for the CNO-injected mice revealed widespread coactivation of contralateral and ipsilateral Bi muscle pairs in the forelimb motor cortex (Fig. 5I). Moreover, cumulative frequency distribution analyses resulted in distinct laterality index curves for the saline- and CNO-injected groups, showing a shift from no or little contralateral and ipsilateral coactivation in saline-injected mice to significantly strong coactivation in CNO-injected animals (p < 0.0001, Kolmogorov-Smirnov test) (Fig. 5J). These data revealed that unpruned ipsilateral CSTs in CNO-injected mice are functional.
Discussion
Axon guidance molecules and neuronal activity have been shown to be involved in axon elimination during development (Martin, 2005; Riccomagno and Kolodkin, 2015); however, it is unknown whether they are independent or interactive processes. In this study, we addressed whether the developmental elimination of ipsilateral axonal projections of CSNs, which is regulated by repellent Sema6D-PlexA1 signaling, is dependent on neuronal activity. We found that ipsilateral CS elimination is regulated by neuronal activity, Bax/Bak, and caspase-9. Interestingly, neuronal activity and Bax/Bak are required for full expression levels of PlexA1 proteins in CSNs. Our study provides two novel insights into our understanding of neural circuit formation. First, our results strongly suggest that ipsilateral CS axon elimination is regulated by both the neuronal activity-dependent Bax/Bak-caspase-9 pathway and the Sema6D-PlexinA1 signaling cascade. Intriguingly, we also found that neuronal activity and Bax/Bak are required for full expression levels of PlexA1 in CSNs, providing a potential link between these two pathways. The second novel insight concerns the novel mechanism underlying the Bax/Bak-caspase signaling-mediated axon elimination. Although the activity-dependent, nonapoptotic Bax/Bak-caspase pathway has been shown to control circuit maturation through localized caspase activity in areas such as axon terminals and dendritic spines, our data suggest that this pathway is activated within the axons, and then it sends the signal from the axons to the cell bodies to induce expression of PlexA1 in the cell bodies. This upregulation of PlexA1 mRNA levels by Bax/Bak-caspase may lead directly to the elimination of ipsilateral CS axons.
Activity-dependent Bax/Bak-caspase pathway in CS axon elimination
Activity-dependent Bax/Bak-caspase signaling has been shown to control the later steps of neural circuit formation through caspases in the CNS (Hollville and Deshmukh, 2018). Our genetic results indicate that only caspase-9, and not caspase-3, caspase-6, or caspase-7, mutant mice show defects in elimination of ipsilateral CS axon projections. This result suggests that caspase-9 is a downstream target of the neuronal activity-dependent Bax/Bak pathway in CSNs. However, this does not exclude possible roles for caspase-3, caspase-6, and caspase-7 in CS axon elimination since previous studies indicate that some caspases serve redundant functions (Zheng et al., 1999; Marsden et al., 2004; Lakhani et al., 2006; Kumar, 2007). Therefore, analysis of double- or triple-caspase mutant mice will be required to fully understand the roles of caspases in CSN axon elimination.
Here we show that neuronal activity in the sensorimotor cortex is required for full activation of caspase-3 (Fig. 4C,D), suggesting that neuronal activity in motor cortex CSNs is required for full caspase-3 activation. Since it is possible that neuronal activity in other cells aside from CSNs plays an important role in the activation of caspase-3, future experiments are needed to determine whether caspase-3 is indeed activated in CSNs in the motor cortex.
Although we were not able to examine the activation of caspase-9 in our system due to technical considerations, a previous study showed that caspase-9 mutant mice exhibited no caspase-3 activation in mouse sensory axons (Simon et al., 2012). Activation of caspase-3 may thus be an indicator of caspase-9 activation in CSNs. Further analysis of caspase-9 mutant mice will be required to determine the relationship between caspase-3 and caspase-9 in the developmental elimination of ipsilateral CSN axons.
The final remaining question is whether the elimination of ipsilateral CS projections mediated by the Bax/Bak-caspase and the Sema6D-PlexA1 signaling pathways is cell-autonomous. Since we found that PlexA1 mRNA expression is reduced only in Bax/Bak-deleted cortical neurons (Fig. 4N,O), it may indeed be a cell-autonomous process. Further detailed functional analyses will clarify this hypothesis.
Repellent semaphorin signaling and neuronal activity to eliminate CS axons
A large family of semaphorins and their receptors, neuropilins and plexins, have been shown to control axon elimination in the CNS (Riccomagno et al., 2012; Koropouli and Kolodkin, 2014; Riccomagno and Kolodkin, 2015). Our recent study further corroborates these data by showing that Sema6D-PlexA1 signaling eliminates ipsilateral projections of CSNs in the spinal cord (Gu et al., 2017a). However, it was unclear whether semaphorins control CS axon elimination independently of neuronal activity or whether there was some level of interaction between the two processes.
Our current study suggests that Sema6D-PlexA1 signaling and neuronal activity interactively control ipsilateral axon projections of CSNs, with Sema6D-PlexA1 signaling acting as a downstream signaling effector of the neuronal activity-dependent Bax/Bak pathway. Since we cannot exclude the possibility that Sema6D-PlexA1 and Bax/Bak-caspase signaling may also function via distinct, parallel pathways to eliminate ipsilateral CS projections, this possibility requires further investigation. Since PlexA1 expression has been shown to be regulated by neuronal activity in other regions of the CNS (Cheadle and Biederer, 2014), PlexA1 may have broad effects on neural circuit formation in the mammalian CNS.
Bilateral muscle activation as a consequence of defects in ipsilateral CS axon elimination
Our ICMS-EMG recording experiments previously showed that cortical stimulation induces activation of both flexor and extensor forelimb muscles in early postnatal mice, whereas it induces mainly flexor forelimb muscles in adult mice (Gu et al., 2017b). In contrast, cortical stimulation induces both flexor and extensor forelimb muscle activation in both early postnatal and adult Bax/Bak mutant mice (Gu et al., 2017b), as well as in hM4Di mice that were injected with CNO (Fig. 5). These data suggest that ipsilateral projections of CSNs in early postnatal mice form functional connections with spinal neurons and that those connections are eliminated during postnatal development. Only in adult Bax/Bak mutant mice or hM4Di mice with CNO injections were these connections maintained. The formation and elimination of functional connections between ipsilateral projections of CSNs and spinal neurons during development seem to be an energy-consuming process for structures that are ultimately unnecessary for the mouse. However, elimination of transient ipsilateral CS projections is reported in other mammalian species (Martin, 2005), suggesting that they may have some other functional benefit during development. Ipsilateral CS connections could, for example, serve as scaffolds in the formation of other neuronal connections in the spinal cord, or they may prevent aberrantly formed connections from establishing neural circuits.
In conclusion, we show that axon guidance molecules and the neuronal activity-dependent Bax/Bak-caspase pathway do, indeed, interact to control the elimination of ipsilateral axonal projections of CSNs. Based on our data, we propose a model in which neuronal activation of CSNs triggers the conversion of caspases to their active forms within CS axons. The activated caspases then cleave substrate proteins, which send signals back to the cell nucleus in the motor cortex to induce PlexA1 mRNA expression. The resulting increase in PlexA1 proteins then drives axon elimination in ipsilateral CSNs. Since it is possible that PlexA1 mRNA expression may be affected by elements other than neuronal activity or caspase activation, future studies are needed to explore these possibilities to create a fuller picture of this process. Such studies will also give insights into the degree to which dynamic, interactive pathways promote the refinement of neuronal circuits within the mammalian CNS.
Footnotes
Y.Y. was supported by National Institute of Neurological Disorders and Stroke Grants NS093002 and NS100772.
The authors declare no competing financial interests.
References
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD (2005) Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45:207–221. 10.1016/j.neuron.2004.12.036 [DOI] [PubMed] [Google Scholar]
- Bagnard D, Lohrum M, Uziel D, Puschel AW, Bolz J (1998) Semaphorins act as attractive and repulsive guidance signals during the development of cortical projections. Development 125:5043–5053. [DOI] [PubMed] [Google Scholar]
- Canty AJ, Murphy M (2008) Molecular mechanisms of axon guidance in the developing corticospinal tract. Prog Neurobiol 85:214–235. 10.1016/j.pneurobio.2008.02.001 [DOI] [PubMed] [Google Scholar]
- Cheadle L, Biederer T (2014) Activity-dependent regulation of dendritic complexity by semaphorin 3A through Farp1. J Neurosci 34:7999–8009. 10.1523/JNEUROSCI.3950-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JG, Rasin MR, Kwan KY, Sestan N (2005) Zfp312 is required for subcortical axonal projections and dendritic morphology of deep-layer pyramidal neurons of the cerebral cortex. Proc Natl Acad Sci USA 102:17792–17797. 10.1073/pnas.0509032102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner RL, Low LK, Liu XB, Coble J, Jones EG, Cheng HJ (2008) Dorsal turning of motor corticospinal axons at the pyramidal decussation requires plexin signaling. Neural Dev 3:21. 10.1186/1749-8104-3-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger JH, Bronson RT, Harris B, Johnson K, Przyborski SA, Ackerman SL (2002) The netrin 1 receptors Unc5h3 and Dcc are necessary at multiple choice points for the guidance of corticospinal tract axons. J Neurosci 22:10346–10356. 10.1523/JNEUROSCI.22-23-10346.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel KM, Martin JH (2005) Role of sensory-motor cortex activity in postnatal development of corticospinal axon terminals in the cat. J Comp Neurol 485:43–56. 10.1002/cne.20483 [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22:6309–6314. 10.1523/JNEUROSCI.22-15-06309.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Kalambogias J, Yoshioka S, Han W, Li Z, Kawasawa YI, Pochareddy S, Li Z, Liu F, Xu X, Wijeratne HR, Ueno M, Blatz E, Salomone J, Kumanogoh A, Rasin MR, Gebelein B, Weirauch MT, Sestan N, Martin JH, et al. (2017a) Control of species-dependent cortico-motoneuronal connections underlying manual dexterity. Science 357:400–404. 10.1126/science.aan3721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Serradj N, Ueno M, Liang M, Li J, Baccei ML, Martin JH, Yoshida Y (2017b) Skilled movements require non-apoptotic Bax/Bak pathway-mediated corticospinal circuit reorganization. Neuron 94:626–641.e624. 10.1016/j.neuron.2017.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollville E, Deshmukh M (2018) Physiological functions of non-apoptotic caspase activity in the nervous system. Semin Cell Dev Biol 82:127–136. 10.1016/j.semcdb.2017.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Li Z (2011) Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron 70:758–772. 10.1016/j.neuron.2011.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, Tessier-Lavigne M (2011) Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb Perspect Biol 3:a001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koropouli E, Kolodkin AL (2014) Semaphorins and the dynamic regulation of synapse assembly, refinement, and function. Curr Opin Neurobiol 27:1–7. 10.1016/j.conb.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA (1996) Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384:368–372. 10.1038/384368a0 [DOI] [PubMed] [Google Scholar]
- Kumar S. (2007) Caspase function in programmed cell death. Cell Death Differ 14:32–43. 10.1038/sj.cdd.4402060 [DOI] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA (2006) Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311:847–851. 10.1126/science.1115035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon RN. (2008) Descending pathways in motor control. Annu Rev Neurosci 31:195–218. 10.1146/annurev.neuro.31.060407.125547 [DOI] [PubMed] [Google Scholar]
- Li Q, Martin JH (2001) Postnatal development of corticospinal axon terminal morphology in the cat. J Comp Neurol 435:127–141. 10.1002/cne.1197 [DOI] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M (2010) Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141:859–871. 10.1016/j.cell.2010.03.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden VS, Ekert PG, Van Delft M, Vaux DL, Adams JM, Strasser A (2004) Bcl-2-regulated apoptosis and cytochrome c release can occur independently of both caspase-2 and caspase-9. J Cell Biol 165:775–780. 10.1083/jcb.200312030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JH. (2005) The corticospinal system: from development to motor control. Neuroscientist 11:161–173. 10.1177/1073858404270843 [DOI] [PubMed] [Google Scholar]
- Martin JH, Friel KM, Salimi I, Chakrabarty S (2007) Activity- and use-dependent plasticity of the developing corticospinal system. Neurosci Biobehav Rev 31:1125–1135. 10.1016/j.neubiorev.2007.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M. (2012) Apoptotic and nonapoptotic caspase functions in animal development. Cold Spring Harb Perspect Biol 4:a008664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa S, Hamada S, Kuida K, Yoshida H, Igaki T, Miura M (2010) Maturation of the olfactory sensory neurons by Apaf-1/caspase-9-mediated caspase activity. Proc Natl Acad Sci USA 107:13366–13371. 10.1073/pnas.0910488107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccomagno MM, Kolodkin AL (2015) Sculpting neural circuits by axon and dendrite pruning. Annu Rev Cell Dev Biol 31:779–805. 10.1146/annurev-cellbio-100913-013038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccomagno MM, Hurtado A, Wang H, Macopson JG, Griner EM, Betz A, Brose N, Kazanietz MG, Kolodkin AL (2012) The RacGAP beta2-Chimaerin selectively mediates axonal pruning in the hippocampus. Cell 149:1594–1606. 10.1016/j.cell.2012.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runker AE, Little GE, Suto F, Fujisawa H, Mitchell KJ (2008) Semaphorin-6A controls guidance of corticospinal tract axons at multiple choice points. Neural Dev 3:34. 10.1186/1749-8104-3-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Yamagata M (2009) Many paths to synaptic specificity. Annu Rev Cell Dev Biol 25:161–195. 10.1146/annurev.cellbio.24.110707.175402 [DOI] [PubMed] [Google Scholar]
- Simon DJ, Weimer RM, McLaughlin T, Kallop D, Stanger K, Yang J, O'Leary DD, Hannoush RN, Tessier-Lavigne M (2012) A caspase cascade regulating developmental axon degeneration. J Neurosci 32:17540–17553. 10.1523/JNEUROSCI.3012-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ (2005) Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci USA 102:11272–11277. 10.1073/pnas.0504783102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe V, Wong BK, Graham RK, Cusack CL, Skotte NH, Pouladi MA, Xie Y, Feinberg K, Ou Y, Ouyang Y, Deng Y, Franciosi S, Bissada N, Spreeuw A, Zhang W, Ehrnhoefer DE, Vaid K, Miller FD, Deshmukh M, Howland D, et al. (2012) Rescue from excitotoxicity and axonal degeneration accompanied by age-dependent behavioral and neuroanatomical alterations in caspase-6-deficient mice. Hum Mol Genet 21:1954–1967. 10.1093/hmg/dds005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW (2006) Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci 9:1234–1236. 10.1038/nn1774 [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Han B, Mendelsohn M, Jessell TM (2006) PlexinA1 signaling directs the segregation of proprioceptive sensory axons in the developing spinal cord. Neuron 52:775–788. 10.1016/j.neuron.2006.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng TS, Hunot S, Kuida K, Flavell RA (1999) Caspase knockouts: matters of life and death. Cell Death Differ 6:1043–1053. 10.1038/sj.cdd.4400593 [DOI] [PubMed] [Google Scholar]