

Abstract
Amyloid-β (Aβ) deposition occurs years before cognitive symptoms appear and is considered a cause of Alzheimer's disease (AD). The imbalance of Aβ production and clearance leads to Aβ accumulation and Aβ deposition. Increasing evidence indicates an important role of astrocytes, the most abundant cell type among glial cells in the brain, in Aβ clearance. We explored the role of low-density lipoprotein receptor-related protein 4 (LRP4), a member of the LDLR family, in AD pathology. We show that Lrp4 is specifically expressed in astrocytes and its levels in astrocytes were higher than those of Ldlr and Lrp1, both of which have been implicated in Aβ uptake. LRP4 was reduced in postmortem brain tissues of AD patients. Genetic deletion of the Lrp4 gene augmented Aβ plaques in 5xFAD male mice, an AD mouse model, and exacerbated the deficits in neurotransmission, synchrony between the hippocampus and PFC, and cognition. Mechanistically, LRP4 promotes Aβ uptake by astrocytes likely by interacting with ApoE. Together, our study demonstrates that astrocytic LRP4 plays an important role in Aβ pathology and cognitive function.
SIGNIFICANCE STATEMENT This study investigates how astrocytes, a type of non-nerve cells in the brain, may contribute to Alzheimer's disease (AD) development. We demonstrate that the low-density lipoprotein receptor-related protein 4 (LRP4) is reduced in the brain of AD patients. Mimicking the reduced levels in an AD mouse model exacerbates cognitive impairment and increases amyloid aggregates that are known to damage the brain. We show that LRP4 could promote the clearance of amyloid protein by astrocytes. Our results reveal a previously unappreciated role of LRP4 in AD development.
Keywords: Alzheimer's disease, amyloid-β, astrocyte, LRP4
Introduction
Alzheimer's disease (AD) is the most common form of dementia among the elderly and one of the leading contributors to global disease burden (Alzheimer's Association, 2019). Patients with AD suffer from a decline in episodic memory with an inability to recall the recent past and eventual loss of long-term memories, loss of cognitive function, and changes in personality (Albert et al., 2011). Pathologic hallmarks of AD include the accumulation of extracellular amyloid-β (Aβ) peptide into plaques and intracellular neurofibrillary tangles containing hyperphosphorylated tau, together with increased glial activation and neuronal loss (Hardy and Selkoe, 2002). Genetic risk factors of AD include APP, PSEN 1/2, as well as ApoE (Hardy and Selkoe, 2002). Recent studies have revealed how ApoE regulates Aβ metabolism (Yamazaki et al., 2019). In particular, it could enhance Aβ uptake and clearance by astrocytes (Kounnas et al., 1995; Beffert et al., 1999; Koistinaho et al., 2004; Jiang et al., 2008; Lee et al., 2012). In mice, ApoE4 impairs Aβ clearance without altering Aβ production (Castellano et al., 2011). On the other hand, Aβ uptake is more efficient in the presence of the ApoE3, compared with ApoE4 (Beffert et al., 1999). These observations suggest that impaired Aβ clearance may be a mechanism for ApoE4 in increasing the vulnerability to AD. Nevertheless, it remains unclear exactly how ApoE's function is regulated.
Increasing evidence indicates an important role of astrocytes, the most abundant cell type among glial cells in the brain, in Aβ clearance. First, in human AD brains, astrocytes contain Aβ-positive granules (Funato et al., 1998; Thal et al., 2000). In agreement, astrocytes are able to bind and uptake soluble and insoluble Aβ (Wisniewski and Wegiel, 1991; Funato et al., 1998; Wyss-Coray et al., 2003; Nielsen et al., 2009; Chung et al., 2013; Jones et al., 2013; R. X. Liu et al., 2016) and degrade them via the late endolysosomal pathway (Shaffer et al., 1995; Wyss-Coray et al., 2003; Nielsen et al., 2009; Basak et al., 2012). Second, astrocytes of AD model mice are less efficient in clearing extracellular Aβ (R. X. Liu et al., 2016), whereas enhancing the astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis (Xiao et al., 2014; Iram et al., 2016). Third, Aβ clearance is regulated by LDLR family members in particular LDLR and LRP1 (LDLR-related protein 1) (Lane-Donovan et al., 2014; Lane-Donovan and Herz, 2017). Considering the abundance of astrocytes in the brain and their modulatory functions on multiple brain cells, they may represent a major mechanism for Aβ clearance.
LRP4 is a core member of the LDLR family, which have been implicated in AD pathogenesis by modulating Aβ metabolism (Holtzman et al., 2012; Lane-Donovan and Herz, 2017). LRP4 serves as a receptor for agrin, a factor used by motor neurons, and plays a critical role in the formation and maintenance of the neuromuscular junction (N. Kim et al., 2008; B. Zhang et al., 2008; Wu et al., 2012; Zong et al., 2012; Barik et al., 2014). In the brain, LRP4 has been implicated in synaptic transmission, LTP, and cognitive function (Gomez et al., 2014; Sun et al., 2016) and adult hippocampal neurogenesis (H. Zhang et al., 2019). Recently, we found that Lrp4 is highly, perhaps specifically, expressed in astrocytes in the PFC and hippocampus, regions vulnerable in AD (Sun et al., 2016). Considering astrocytes' roles in Aβ metabolism, we explored the role of LRP4 in AD pathology. Intriguingly, the levels of Lrp4 in astrocytes were higher than those of Ldlr and Lrp1, but were reduced in postmortem brain tissues of AD patients. Remarkably, genetic deletion of the Lrp4 gene augmented Aβ plaques in 5xFAD mice, an AD mouse model and exacerbated the deficits in neurotransmission, synchrony between the hippocampus and PFC, and cognition. Further mechanistic studies suggest that LRP4 promotes astrocytic Aβ uptake by serving as a receptor for ApoE. These observations reveal a novel function of LRP4 and a potential pathologic mechanism in AD development.
Material and Methods
Animals
5xFAD transgenic mice (B6SJL-Tg (APPSwFlLon, PSEN1*M146L*L286V) 6799Vas/Mmja) were from The Jackson Laboratory (MMRRC stock #34 840-JAX) (Oakley et al., 2006), which express human APP and PSEN1 transgenes with five AD-linked mutations (the Swedish [K670N/M671L], Florida [I716V], and London [V717I] mutation in APP, and the M146L and L286V mutation in PSEN1) under the mouse Thy1 promoter. 5xFAD mice were maintained as hemizygotes on a C57BL/6 background. Lrp4f/f (Wu et al., 2012), GFAP-Cre (Zhuo et al., 2001), Ai9 (Madisen et al., 2010), GFAP-CreERT2 (Casper et al., 2007), Neurod6-Cre (Goebbels et al., 2006), Thy1-GFP (Feng et al., 2000), and Slc1a2-EGFP (Gong et al., 2003) were as described previously (Sun et al., 2016). Mice were group-housed no more than 5 per cage in a room with a 12 h light/dark cycle with ad libitum access to water and rodent chow diet (Diet 1/4 7097, Harlan Teklad). The Lrp4-CreERT2 mice were generated by Lin Gan (University of Rochester). Briefly, a targeting vector containing a 3′ FRT flanked neo cassette and P2A-GFP-CreERT2 cassette was inserted to exon 38. The construct was electroporated into C57BL/6J-derived embryonic stem cells. Correctly targeted embryonic stem cells were injected into blastocysts. The resulting chimeric animals were tested for germline transmission by crossing to C57BL/6J. Heterozygous animals were crossed subsequently to FLP recombinase expressing mice to remove the FRT site flanked PGK-neo cassette. Mice that no longer contained the FRT flanked PGK-neo cassette were then backcrossed to C57BL/6J to remove the FLP recombinase transgene. PCR genotyping with the following primers: 2AGCEF (5′- GGCAT CGTGC CCATC CTGAT CGAGC TGA -3'), and 2AGCER (5′- GCCAG CTGCA CGCTG CCATC CTCGA TGT -3') will reveal a fragment size 500 bp. Male mice were used. Some mice were injected as previously described (Sun et al., 2016; Appel et al., 2018). Tamoxifen (10 mg/ml, Sigma Millipore, catalog #T5648) was prepared in corn oil (Sigma Millipore, catalog #C8267) mixed with ethanol (9:1 ratio). Mice were injected with 100 mg/kg tamoxifen (i.p. every other day 3 times).
Experiments with animals were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University.
Antibodies
The information of primary antibodies used was as follows: rabbit anti-GFAP (Dako, catalog #Z0334, PRID:AB_10013382, 1:1000 for immunohistochemistry [IHC]), mouse anti-NeuN (Millipore, catalog #MAB377, PRID:AB_2341188, 1:200 for IHC), chicken anti-GFP (AVES, catalog #GFP-1020, PRID:AB_10000240,1:1000 for IHC), mouse anti-β-actin (Cell Signaling Technology, catalog #12262, PRID:AB_2566811, 1:5000 for Western blot), mouse anti-LRP4 (UC Davis/NIH NeuroMab Facility, catalog #75-221, PRID:AB_2139030, 1:1000 for Western blot), goat anti-Iba1 (Abcam, catalog #ab5076, RRID:AB_2224402, 1:250 for IHC), mouse anti-APP (6E10) (BioLegend, catalog #803001, PRID:AB_2564653, 1:1000 for Western blot), rabbit anti-APP (Sigma Millipore, catalog #A8717, PRID:AB_258409,1:1000 for Western blot), rabbit anti-aldolase C (Abcam, catalog #ab87122, PRID:AB_10673854, 1:200 for IHC), rabbit anti-S100 (Abcam, catalog #ab868, PRID:AB_306716, 1:200 for IHC), rabbit anti-Oligo2 (Novus, catalog #NBP128667, PRID:AB_1914109, 1:200 for IHC), and goat anti-ApoE (Menidian Life Science, catalog #K74180B, PRID:AB_150544,1:1000 for Western blot). The information of secondary antibodies used was as follows: AlexaFluor-488-donke anti-mouse- IgG (Jackson ImmunoResearch Laboratories, catalog #715-547-003, PRID:AB_2340851), AlexaFluor-594-donkey-anti-rabbit-IgG (Jackson ImmunoResearch Laboratories, catalog #711-586-152, RRID:AB_2340622), AlexaFluor-488-donkey-anti-rabbit-IgG (Jackson ImmunoResearch Laboratories, catalog #711-547-003, RRID:AB_2340620), AlexaFluor-488-donkey-anti-goat-IgG (Jackson ImmunoResearch Laboratories, catalog#705-547-003, RRID:AB_2340431), AlexaFluor-488-donkey-anti-chicken-IgG (Jackson ImmunoResearch Laboratories, catalog #703-546-155, RRID:AB_2340376), all diluted by 10% donkey serum in PBS 1:200 (Jackson ImmunoResearch Laboratories) for IHC; RDye680RD donkey anti-rabbit-IgG (LI-COR Biosciences, catalog #926-68073, RRID:AB_10954442) and IRDye 800CW donkey anti-mouse-IgG (LI-COR Biosciences, catalog #926-32 212, RRID:AB_621847) secondary antibodies, both 1:10,000 were diluted by 10% milk in TBST for Western blot.
Cell culture and protein purification
Mouse primary astrocytes were cultured from postnatal day 3 mouse pups as previously described with modification (Sun et al., 2016). In brief, cortices were dissected in ice-cold calcium- and magnesium-free Hanks Balanced Salt Solution (HBSS) to remove meninges. Brain tissues were washed with HBSS, digested in 0.25% trypsin-EDTA and 0.2 mg/ml DNase for 10 min at 37°C, and dissociated by trituration with fire-polished pipettes in HBSS containing 0.4 mg/ml DNase. The mixture was filtered through a 40 µm nylon mesh, pelleted at 900 × g for 5 min, and resuspended in a growth media containing DMEM, 10% FBS, 1× Glutamax, and 1× penicillin/streptomycin. Cells were then plated on a poly-L-lysine-coated T75 flask. The culture medium was changed after 8 h, and every 3 d following the initial change. Once the cells reached confluence, they were shaken at 220 rpm for 3 h, and the medium was aspirated to remove less adherent microglial cells. Astrocytes were washed with PBS, detached from the plate using 0.05% trypsin-EDTA, and seeded on 12-well plates or 10 cm plates.
HEK293T cells (ATCC, PRID:CVCL_0063) were cultured in DMEM supplemented with 10% FBS, 1× penicillin/streptomycin. They were transfected with Flag-tagged LRP4-ECD constructs with 0.1% polyethyleneimine (Polysciences, catalog #24765, molecular weight 40,000) as previously described (H. Zhang et al., 2019). To collect secreted ECD proteins, the culture medium was changed to serum-free for 36 h before collection. The medium was concentrated using Amicon centrifugal filter unit (Millipore, catalog #UFC803008), and Flag-tagged proteins were purified with Flag M2 beads (Sigma Millipore A2220), eluted with 100 mm glycine buffer, pH 3.8, and neutralized with Tris-HCl buffer.
Immortalized mouse astrocytes expressing human ApoE2, ApoE3, and ApoE4 were gifts from David Holtzman (Washington University) and cultured as previously described (Morikawa et al., 2005). The culture medium was conditioned with a serum-free medium for 36 h and concentrated 20 times with Amicon centrifugal filter unit (Millipore, catalog #UFC201024). The concentrated medium was run through a HiTrap heparin column on an AKTA Explorer A100 FPLC system (GE Healthcare). NaCl gradient from 0 to 1 M Tris buffer was used to elute ApoE. ApoE was concentrated and quantified by ELISA against a standard curve derived from serial dilutions of recombinant ApoE3.
qRT-PCR
Total RNA was purified from the hippocampus or cultured astrocytes with TRizol (Invitrogen, catalog #15596-026) according to the manufacturer's protocol. RNA (3 µg) was reverse-transcribed to cDNA with GoScript reverse transcription kit (Promega). qPCR was performed in the ABI Step-One Plus system (Applied Biosystems) using SYBR Green (QIAGEN). Relative expression was determined using the comparative Ct model (ΔΔCt) with glyceraldehyde 3-phosphate dehydrogenase (Gapdh) as a reference. Primers for individual genes were as follows: Vldlr (forward: 5′-ACGGC AGCGA TGAGG TCAAC TG-3′, reverse: 5′- CAGAG CCATC AACAC AGTCT CG-3′); Ldlr (forward: 5′-GAATC TACTG GTCCG ACCTG TC, reverse: 5′-CTGTC CAGTA GATGT TGCGG TG-3′); Lrp1 (forward: 5′-CGAGA GCCTT TGTGC TGGAT GA-3′, reverse: 5′-CGGAT GTCCT TCTCA ATGAG GG-3′); Lrp1b (forward: 5′-GACTA TGTCC TCACC TGTGA TGG-3′, reverse: 5′-ACAGC GACGA TTGTA GCACG GT-3′); Lrp2 (forward: 5′-CCAAT GGACT CACTC TGGAC CT-3′, reverse: 5′-GAATG GAAGG CAGTG CTGAT GAC-3′); Lrp4 (forward: 5′-GTGTG GCAGA ACCTT GACAG TC-3′, reverse: 5′-TACGG TCTGA GCCAT CCATT CC-3′); Lrp8 (forward: 5′-GTGGA AGTAG CCACC AATCG CA-3′, reverse: 5′-CTGCT CATCA ATGAG GACCA CC-3′); App (forward: 5′-TCCGT GTGAT CTACG AGCGC AT-3′, reverse: 5′-GCCAA GACAT CGTCG GAGTA GT-3′); APP (forward: 5′-CCTTC TCGTT CCTGA CAAGT GC-3′, reverse: 5′-GGCAG CAACA TGCCG TAGTC AT-3′); Bace1 (forward: 5′-TGCTG CCATC ACTGA ATCGG AC-3′, reverse: 5′-GGAAT GTGGG TCTGC TTCAC CA-3′); Bace2 (forward: 5′-GATTG GTGCG ACCGT GATGG AA-3′, reverse: 5′-GTTGC TGGCT ATGTC TTCCG TG-3′); Psen1 (forward: 5′-GAGAC TGGAA CACAA CCATA GCC-3′, reverse: 5′-AGAAC ACGAG CCCGA AGGTG AT-3′); Psen2 (forward: 5′-CTGGT GTTCA TCAAG TACCT GCC-3′, reverse: 5′-TTCTC TCCTG GGCAG TTTCC AC-3′); PSEN1 (forward: 5′-GCAGT ATCCT CGCTG GTGAA GA-3′, reverse: 5′-CAGGC TATGG TTGTG TTCCA GTC-3′); and Gapdh (forward: 5′-CATCA CTGCC ACCCA GAAGA CTG-3′, reverse: 5′-ATGCC AGTGA GCTTC CCGTT CAG-3′).
Tissue harvest, processing, staining, histology, and morphology analysis
Hippocampal, cortical, and entorhinal cortical samples from unaffected subjects (n = 9) and from subjects with AD (n = 9) were obtained from the Human Brain and Spinal Fluid Resource Center and the University of Miami Brain Endowment Bank. The diagnosis of AD was confirmed by pathologic and clinical criteria (Khachaturian, 1985) as well as Braak neuropathological staging of Alzheimer-related changes (Braak and Braak, 1991). The human brain samples were processed as previously described (Cohen et al., 2015). Human brain tissues were homogenized with Mini-bead beater 16 Cell Disrupter (Biospec) in 15% (w/v) TBS, pH 7.4, containing complete protease inhibitor cocktail (Roche Applied), 75 s three times. Samples were rehomogenized in 1% Sarkosyl (10% w/v) 75 s one time and cleaned by centrifugation at 500 × g for 5 min. Aliquots of the supernatant were stored at −80°C for future analysis. Protein concentration was measured by the BCA protein kit.
Mice were anesthetized and perfused transcardially with ice-cold PBS containing protease inhibitors. Brains were isolated and dissected into two hemibrains. Right hemibrains were dissected on ice to isolate cortex and hippocampus, which were snap-frozen in liquid nitrogen and stored at −80°C until use. Left hemibrains were drop-fixed in 4% PFA for 24 h and stored at 4°C in PBS containing 0.05% sodium azide. Tissues were sectioned (40 µm per section) using a vibratome (VT1000S, Leica Microsystems). Sections were sequentially collected and stored at −20°C in cryoprotectant solution (FD Section Storage Solution) before staining. Free-floating sections were washed three times in PBS, followed by 1 h blocking in PBS with 10% donkey serum. Sections were incubated in PBS with 10% donkey serum and primary antibodies for 48 h at 4°C, washed three times in PBS, and incubated in PBS with 10% donkey serum plus secondary antibodies for 2 h at room temperature.
Thioflavin S (Thio-S) staining for plaque and quantification were conducted as previously described (Wen et al., 2011; Youmans et al., 2012) by an investigator blind to the genotype beginning with the lateral-most section in the ROI, every sixth tissue section of six consecutive sections was used. Sections were washed in PBS (three times for 5 min each) and stained in 0.1% Thio-S (dissolved in 50% EtOH plus 50% PBS) for 10 min in the dark. Sections were destained in 80% EtOH (twice for 3 min each) and 95% EtOH (once for 3 min) in the dark, washed in water (three times, each for 3 min) in dark and mounted with antifade fluorescence mounting medium. Quantification of plaque deposition was performed as previously described (Youmans et al., 2012). Briefly, images were captured on a BZX slider scanner, exported with BZX viewer software, and converted to 8-bit grayscale using National Institutes of Health ImageJ software. Converted images were thresholded to highlight plaques and to diminish the background signal; each object identified after thresholding was examined individually to confirm it was indeed a plaque. Eight sections per mouse were quantified. The cortex and hippocampus in each image were outlined and analyzed with National Institutes of Health ImageJ software. Plaques >5.5 µm2 were quantified. Spine density was analyzed as previously described (H. Zhang et al., 2016).
Western and dot blot analysis
Brain tissues were homogenized in RIPA buffer containing the following (in mM): 50 Tris-HCl, pH 7.4, 150 NaCl, 2 EDTA, 1 PMSF, 50 sodium fluoride, 1 sodium vanadate, 1 DTT with 1% sodium deoxycholate, 1% SDS and 1× protease inhibitor cocktails as previously described (Hu et al., 2018). Homogenates were resolved by Nu-PAGE 4%-12% Bis-Tris gel (Invitrogen, catalog #NP0336BOX) and transferred to nitrocellulose membranes. Subsequently, blots were incubated in the TBS buffer containing 0.1% Tween-20 and 5% milk for 1 h at room temperature before the addition of primary antibody for incubation overnight at 4°C. After extensive wash, blots were incubated with secondary antibodies (LI-COR Biosciences) in the TBS buffer for 1 h at room temperature. Immunoreactive bands were imaged and quantified using the Odyssey Infrared Imaging System (LI-COR Biosciences, model 2800). Band density of interested proteins was normalized in relation to loading control.
Dot blot analysis was performed as previously described (Atagi et al., 2015). Proteins in PBS were spotted on a nitrocellulose membrane using a dot blot manifold apparatus (Bio-Rad). Membrane strips were blocked with 1% Block Ace (Bio-Rad) in PBS and incubated with the indicated proteins 12 h at 4°C. Bound proteins were detected with biotin-conjugated primary and IRDye-conjugated.
Brain homogenization
Brain tissues were homogenized as previously described with modification (Youmans et al., 2011). Briefly, frozen tissues (100 mg/1 ml ice-cold TBS/PI buffer) were homogenized with Dounce homogenizers until no visible pieces were seen. The mixture was centrifuged at 12,000 × g (4°C) for 30 min; TBS-insoluble fractions were added 5 M GDN/PI buffer at the same ratio and homogenized by sonication. The homogenates were rotated at room temperature for 6 h and subsequently centrifuged at 12,000 × g for 30 min. The supernatants were collected as the GDN-soluble fraction. All the procedures were performed on ice.
ELISA
Aβ1-40 and Aβ1-42 in TBS- and GDN-soluble brain homogenates were measured by the Aβ40 human ELISA kit (Invitrogen, catalog #KHB3481) and the Aβ42 Human ELISA kit (Invitrogen, catalog #KHB3441), respectively.
Behavioral analysis
The Morris water maze test was performed as previously described (H. Zhang et al., 2019). Briefly, the test was conducted in a circular water tank (with a diameter of 120 cm) filled with water (22°C) and in the presence of a constellation of spatial cues visible to the mice. Nontoxic white powder paints were added to the water to make the surface opaque to hide the escape platform. The maze was virtually divided into four quadrants, with one containing a hidden escape platform (diameter 10 cm) present 1 cm below the water surface. The training consisted of 5 consecutive days of testing with four trials per day; there was a recovery period of 20 min between the training trials. Mice were placed into the water with their nose pointing toward the wall in a random way to prevent strategy learning. Mice were given 60 s to search the platform, at which point the experimenter would guide the animal to the platform if necessary. Mice remained on the platform for 20 s and then were dried with a towel. For the probe test, 24 h after the last training period, the platform was removed and mice were given 60 s to swim; and the trajectory and amount of time spent in each quadrant were quantified using a video tracking system (Etho Vision, Noldus).
The Y maze test was performed as previously described (H. Zhang et al., 2016): mice were placed at one arm end of Y maze and allowed to freely explore the three arms for 8 min. During the multiple arm entries, mice showed a tendency to enter a less recently entered arm. The total number of arm entries and the number of triads were recorded to calculate the percentage of spontaneous alternation. The total distance is recorded to evaluate the locomotion. An entry count when all four limbs are within the arm, and the total entries >8 are included to calculate the spontaneous alternation.
Aβ42 uptake and binding assay
FAM-Aβ42 was purchased from AnaSpec, and the experiments were performed as previously described with modification (C. C. Liu et al., 2017). Briefly, primary astrocytes were incubated with FAM-Aβ42 (500 nm) at 37°C in DMEM with 10% FBS. For FACS analysis and Aβ42 internalization, astrocytes were dissociated from the plate, and washed and suspended in PBS containing 1.5% FBS, 1% sodium azide, and 1% PFA. Astrocytes were analyzed for fluorescence on a BD FACS caliber machine (BD Bioscience). For imaging analysis, astrocytes were washed with PBS three times, fixed with 4% PFA in room temperature for 10 min, and stained with the anti-GFAP antibody.
Oligomeric Aβ42 was prepared as previously described (Y. T. Lin et al., 2018). Briefly, Aβ42 (Bachem, catalog #H-1368) peptide was dissolved in 1% NH4OH at 1 mg/ml and sonicated. Lyophilized Aβ42 was further dissolved in water, filtered (0.22 µm), and incubated at 37°C for 24 h before use. Astrocytes derived from Lrp4f/f and GFAP-Lrp4f/f were seeded in 12-well plates for 4 d and incubated with oligomeric Aβ42 (200 ng/ml) for an additional 2 d. Oligomeric Aβ42 (200 ng/ml) was added to media only without astrocytes to measure the total levels of Aβ42. The measurement of Aβ42 in culture medium was performed after 2 d of incubation by human Aβ42 ELISA kit following the manufacturer's instructions. The relative remaining Aβ42 levels were normalized by the total protein level of cell lysate measured by the BCA protein assay kit.
Electrophysiological recording
The whole-cell patch-clamp recording was performed as previously described (Sun et al., 2016; Wang et al., 2018). Briefly, brain slices of 300 μm thickness were prepared with a vibratome (VT1200S, Leica Microsystems) in sectioning buffer of 4°C (in mM as follows: 120 choline-Cl, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 25 glucose). After incubation in oxygenated ACSF (in mM as follows: 124 NaCl, 2.5 KCl, 2.5 CaCl2, 2 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose) at 34°C for 30 min and recovery at room temperature for 1 h, slices were recorded under perfusion (2 ml/min, 32°C-34°C). The whole-cell recording was aided with infrared optics, using an upright microscope equipped with a 40× water-immersion lens (Axioskop 2 Plus, Carl Zeiss) and an infrared-sensitive CCD camera (C2400-75, Hamamatsu). For recording of mEPSCs, recording pipettes (input resistance: 3-5 MΩ) were filled with internal solution (in mM as follows: 125 K-gluconate, 5 KCl, 10 HEPES, 0.2 EGTA, 1 MgCl2, 4 Mg-ATP, 0.3 Na-GTP, 10 phosphocreatine, pH 7.25, 290-300 mm), while 1 μm TTX and 20 μm bicuculine methiodide were added to the perfusion buffer to block action potentials and GABAA receptor-mediated currents.
LTP was recorded as described previously (Chen et al., 2010; Sun et al., 2016). Briefly, the SC-CA1 pathway was stimulated with a concentric bipolar electrode, and the fEPSPs were recorded in CA1 neurons in current clamp with ACSF-filled glass pipettes (1-5 MΩ). Stimuli consisted of monophasic 100 μs pulses of constant currents (with intensity adjusted to produce ∼30% of maximal amplitudes) at a frequency of 0.033 Hz. The strength of synaptic transmission was determined by measuring the initial (10%-60% rising phase) slope of fEPSPs. LTP was induced by a train of 100 pulses at the same intensity in 1 s. The level of LTP was determined at an average of 50-60 min after tetanus stimulation.
In vivo recording as described previously (Tan et al., 2018). Mice were anesthetized with ketamine/xylazine (Sigma Millipore, 0.1 ml/10 × g body weight, i.p.), shaved on the skull, and positioned on a stereotaxic apparatus. After antiseptic treatment, the scalp was removed, and the exposed skull area was cleared using 1% H2O2. Craniotomy was performed unilaterally above the prelimbic PFC (1.8 mm anterior, 0.4 mm lateral, 1.4 mm ventral), and ventral hippocampus (vHPC) (3.1 mm posterior, 3.0 mm lateral, 3.9 mm ventral). Two sets of tetrodes were implanted in the PFC and vHPC, respectively. Tetrodes were made from 13-μm-diameter platinum (with 10% iridium) fine wire (California Fine Wire) and were attached to movable screw microdrives on a custom-made frame (L. Lin et al., 2006). Skull screws were placed over the cerebellum and olfactory bulb as ground and reference, respectively. After surgery, mice were allowed for recovery at least 1 week before recording. Local field potential (LFP) signals were recorded simultaneously from the vHPC and PFC at a sampling rate of 1 kHz. Movable microdrives were advanced at a rate of 30 μm per day to record a spectrum of neurons. LFP signals were amplified, bandpass filtered (0.5-1000 Hz), and digitized using the OmniPlex Neural Data Acquisition System (Plexon).
Data were imported into MATLAB (The MathWorks) for analysis using custom-written scripts. LFP recordings were processed in Neuroexplorer (Nex Technologies). Fourier transform of LFPs was performed, and data were then binned with a bin size as follows:
 |
and the number of bins equal to 2 times the number of frequency values, which was set to 1024 (Stujenske et al., 2014). The coherence for frequencies, f, was calculated for PFC and vHPC, x(t) and y(t), as follows:
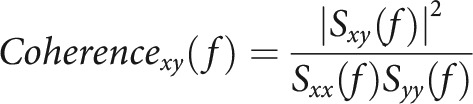 |
where is the cross-spectrum product of PFC and vHPC and and the auto-spectrum products from the two brain regions, respectively. The coherence was processed by a Gaussian filter with a bin size of 3.
Experimental design and statistical analysis
Male mice were used for all experiments, except either sex was used for culturing astrocytes. The sample sizes chosen are based on our previous studies (Wen et al., 2011; Sun et al., 2016; H. Zhang et al., 2019). Data are mean ± SEM. For two independent data comparisons, unpaired t test was used to determine statistical significance. For multiple comparisons, ANOVA was used. No statistical analysis was used to determine sample size prior. Statistical analyses were performed using Excel 2016 (Microsoft) or Prism 6.0 (GraphPad Software). The numbers of biological replicates are described in the figure legends.
Results
Reduced LRP4 protein in human AD brain tissues
Cumulative evidence indicates that LDLR family members can contribute to Aβ metabolism in astrocytes (Kanekiyo et al., 2013). To test the LDLR family members' expression in astrocytes, we performed qPCR assay. Interestingly, Lrp4 mRNA level related to Gapdh was the highest among LDLR family members, followed by Lrp2. The mRNA level of Lrp4 in astrocytes was 4- to 10-fold higher than that of Ldlr and Lrp1 (Fig. 1A), suggesting a potential role of LRP4 in astrocytes. Next, we analyzed LRP4 protein levels in the postmortem brains of AD patients. Remarkably, LRP4 levels were reduced in the hippocampus, cortex, and entorhinal cortex of AD patients, compared with unaffected controls (Fig. 1B,C). The ages of AD patients and control subjects were similar, suggesting the LRP4 reduction was not due to age difference (Fig. 1D). In addition, there was no difference in autolysis times of the two groups of samples (Fig. 1E). These results showed that LRP4 protein levels were reduced in the various regions of the brains from AD patients.
Figure 1.

Reduced LRP4 protein in the AD postmortem brains. A, qPCR of LDLR core members in primary cultured astrocytes. n = 4 for each group. B, Western analysis of brain tissues from unaffected Con and AD. Three independent replicated experiments were performed. Con, controls; AD, Alzheimer's disease; Hippo, Hippocampus; Ctx, cortex; EC, entorhinal cortex. C, Quantification of LRP4/β-actin ratio. n = 9 brain samples for AD and 9 samples for control. Hippo: t(16) = 5.446, p < 0.0001; Ctx: t(16) = 3.269, p = 0.0048; EC: t(16) = 4.974, p = 0.0001, unpaired t test. D, No difference in the ages of subjects between the two groups, n = 9 for each group, t(16) = 0.8205, p = 0.424, unpaired t test. E, No difference in autolysis time. n = 9 for each group, t(16) = 0.3709, p = 0.7156, unpaired t test. Data are mean ± SEM. **p < 0.01,. ***p < 0.001.
Exacerbated amyloid pathology in Lrp4 mutant 5xFAD mice
To examine whether LRP4 deficiency contributes to AD pathogenesis, we conditionally ablated the Lrp4 gene in 5xFAD mice, a well-characterized AD mouse model that recapitulated many AD-related phenotypes (Oakley et al., 2006). To confirm the deletion of Lrp4, we performed Western blot and found LRP4 was reduced in brain lysates of 5xFAD;GFAP-Lrp4f/f mice, compared with 5xFAD;Lrp4f/f mice (Fig. 2A,B). Next, we examined Aβ deposition by Thio-S staining, which was widely used to stain the amyloid plaques (Bussiere et al., 2004). No Aβ plaque was detected in mice at 1.5 months, in agreement with previous reports (Oakley et al., 2006). However, at 6 months, Aβ plaques were readily detected in 5xFAD;Lrp4f/f mice. Remarkably, Thio-S-positive area in 5xFAD;GFAP-Lrp4f/f mice was larger than that of 5xFAD;Lrp4f/f mice at 6 months in both cortex and hippocampus (Fig. 2C,D). In addition, soluble and insoluble of Aβ were assessed by ELISA following extraction of whole-brain tissues into TBS-soluble and insoluble (guanidine-HCl, GDN) fractions (Youmans et al., 2011). Consistent with increased Aβ deposition, insoluble Aβ1-40 and Aβ1-42 in guanidine fraction as well as the soluble fraction of Aβ1-42 were increased in 5xFAD;GFAP-Lrp4f/f mice (Fig. 2E,F). These results demonstrate increased Aβ deposition and Aβ levels in 5xFAD;GFAP-Lrp4f/f mice, suggesting that LRP4 deficiency exacerbates amyloid pathogenesis in 5xFAD mice.
Figure 2.
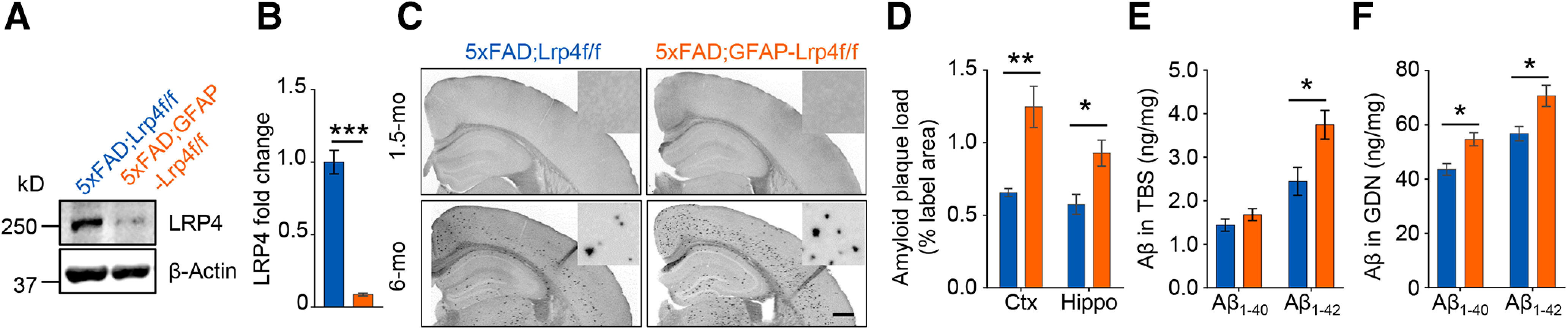
Exacerbated Aβ pathology in LRP4 deficiency 5xFAD mice. A, Reduced LRP4 protein in 5xFAD;GFAP-Lrp4f/f mice compared with 5xFAD;Lrp4f/f. B, Quantitative data in A. n = 3 for each group. t(4) = 11.2, p = 0.0004, unpaired t test. C, D, Increased Aβ deposition in 6-mo 5xFAD;GFAP-Lrp4f/f mice cortex and hippocampus, compared with 5xFAD;Lrp4f/f. Scale bar, 500 µm. Representative images (C) and quantification of area covered by Aβ plaque (D). n = 4 for each group. Ctx: t(6) = 4.061, p = 0.0066; Hippo: t(6) = 3.121, p = 0.0205, unpaired t test mo, month old. E, Increased TBS-soluble Aβ1-42 in 6-mo 5xFAD;GFAP-Lrp4f/f mice brain, compared with 5xFAD;Lrp4f/f. n = 4 for each group. Aβ1-40: t(6) = 1.231, p = 0.2642; Aβ1-42: t(6) = 2.822, p = 0.0303, unpaired t test. F, Increased GDN-soluble Aβ1-40 and Aβ1-42 levels in 6-mo 5xFAD;GFAP-Lrp4f/f mice brain, compared with 5xFAD;Lrp4f/f. n = 4 for each group. Aβ1-40: t(6) = 3.455, p = 0.0135; Aβ1-42: t(6) = 2.557, p = 0.0431, unpaired t test. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Worsened cognitive deficits in LRP4 deficiency 5xFAD mice
The clinical hallmark of AD is progressive memory loss (Selkoe and Hardy, 2016). To determine whether LRP4 deficiency alters cognitive deficits in 5xFAD mice, mice were subjected to the Y maze test. As reported previously (Oakley et al., 2006), spontaneous alternations in Y maze were reduced in 5xFAD mice, compared with Lrp4f/f mice (Fig. 3A,B). Intriguingly, these deficits were worsened by Lrp4 deletion (compare 5xFAD;GFAP-Lrp4f/f mice with 5xFAD;Lrp4f/f mice) (Fig. 3B). As control, Lrp4 mutation had little effect on the total distance or number of arm entries in Y maze, which were similar among the four genotypes (Fig. 3C,D). These results suggest that LRP4 deficiency precipitated the working memory deficits of 5xFAD mice. Next, we tested the spatial learning and memory using the Morris water maze. During the training phase, 5xFAD;Lrp4f/f and GFAP-Lrp4f/f mice had similar latency to locate the hidden platform, compared with the Lrp4f/f mice (Fig. 3E,F), in agreement with previous reports (Sun et al., 2016). However, 5xFAD;GFAP-Lrp4f/f mice were impaired in this task, exhibiting increased latency to locate the hidden platform (Fig. 3E,F). During the probe test, 5xFAD;GFAP-Lrp4f/f mice displayed reduced frequency crossing the hidden platform, spent less time in the target quadrant, compared with other groups (Fig. 3G,H). The velocity among the four was no different during the test (Fig. 3I). Thus, these data demonstrate that LRP4 deficiency exacerbates spatial learning and memory deficits in 5xFAD mice.
Figure 3.
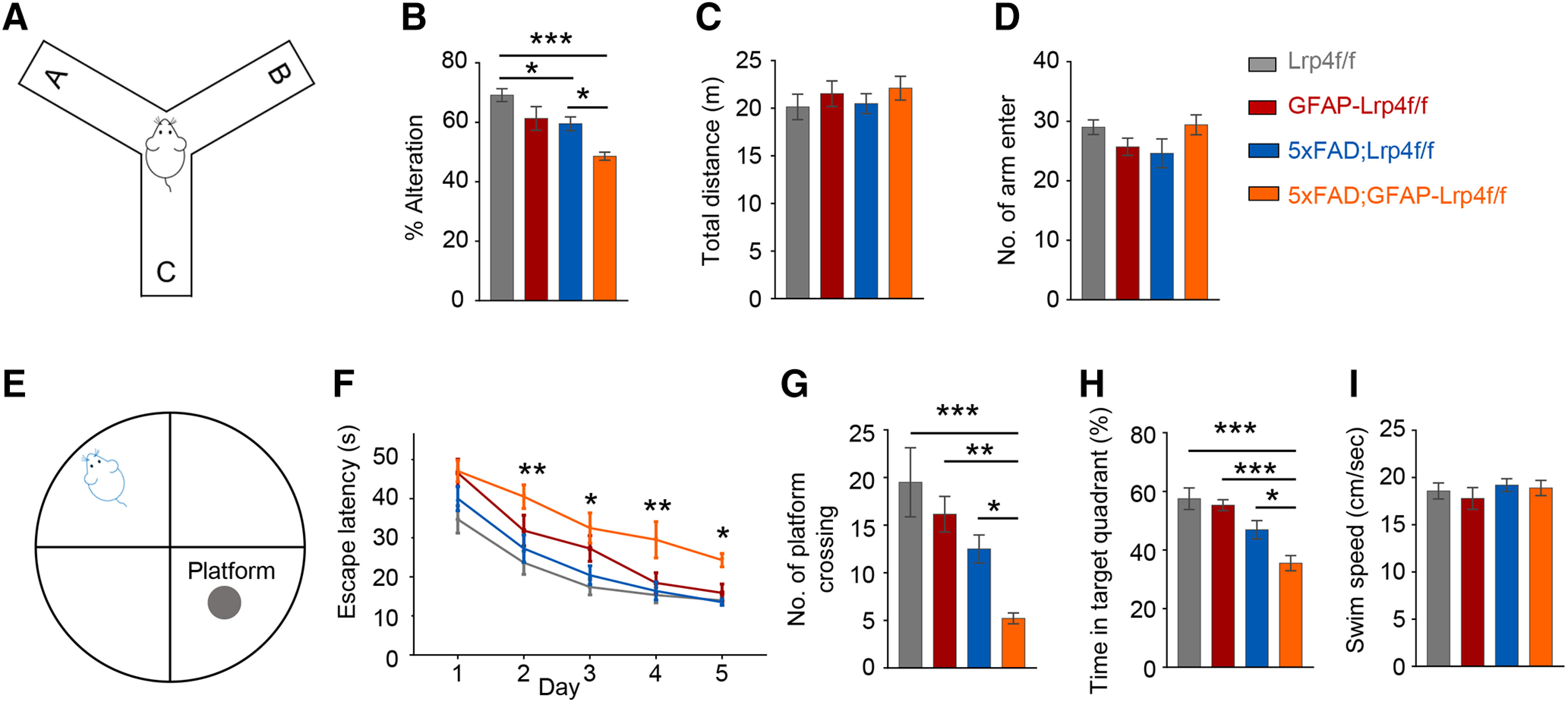
Precipitated cognitive deficits in Lrp4 mutant 5xFAD mice. A, Spatial working memory of Lrp4f/f (n = 13), GFAP-Lrp4f/f (n = 10), 5xFAD;Lrp4f/f (n = 10), and 5xFAD;GFAP-Lrp4f/f (n = 10) was assessed by spontaneous alternation in the Y maze. B, 5xFAD;GFAP-Lrp4f/f further reduced the spontaneous alternation compared with 5xFAD;Lrp4f/f at 6 months: F(3,39) = 11.21, p < 0.0001, ANOVA. C, The total distance explored in the Y maze was not different between the four groups: F(3,39) = 0.5198, p = 0.6711, ANOVA. D, The total number of arm entries was not different in the four groups: F(3,39) = 1.947, p = 0.1380, ANOVA. E, Spatial learning and memory of Lrp4f/f (n = 10), GFAP-Lrp4f/f (n = 13), 5xFAD;Lrp4f/f (n = 15), and 5xFAD;GFAP-Lrp4f/f (n = 12) was assessed in water maze. F-H, Six-mo 5xFAD;GFAP-Lrp4f/f exhibited increase the latency to the hidden platform (F), F(3,230) = 19.6, p < 0.0001, ANOVA; decrease the number of platform crossing (G), F(3,46) = 8.852, p< 0.0001, ANOVA; reduce the time in the target quadrant (H), F(3,46) = 11.35, p < 0.0001, ANOVA, compared with Lrp4f/f and 5xFAD;Lrp4f/f groups. I, Four groups of mice showed similar swim speed in the water maze: F(3,46) = 0.5068, p = 0.6795, ANOVA. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Exacerbated synaptic deficits in Lrp4 mutant AD mice
To determine whether LRP4 deficiency alters synaptic transmission, we measured mEPSCs of CA1 neurons in hippocampal slices. We found that mEPSC frequency, but not amplitude, was reduced in GFAP-Lrp4f/f slices, compared with littermate Lrp4f/f slices (Fig. 4A-C), in agreement with our previous report (Sun et al., 2016). mEPSC frequency and amplitude were reduced in 5xFAD;Lrp4f/f slices, compared with Lrp4f/f slices (Fig. 4A-C), which are in agreement with previous reports (Kimura and Ohno, 2009; Crouzin et al., 2013). Noticeably, LRP4 deficiency further reduced mEPSC frequency and amplitude, compared with 5xFAD;GFAP-Lrp4f/f slices and 5xFAD;Lrp4f/f slices (Fig. 4A-C). We further investigated the effect of LRP4 deficiency on LTP, a cellular model of learning and memory (Malenka and Bear, 2004). The slopes of fEPSPs in GFAP-Lrp4f/f and 5xFAD;Lrp4f/f slices were reduced compared with Lrp4f/f slices, suggesting impaired synaptic plasticity in CA1. The impairments were worsened in 5xFAD;GFAP-Lrp4f/f slices (Fig. 4D,E). The deficiency of LTP was further suppressed in 5xFAD;GFAP-Lrp4f/f slices, compared with 5xFAD;Lrp4f/f slices and Lrp4f/f slices (Fig. 4F). These results indicate that LRP4 deficiency exacerbates the synaptic plasticity deficits in 5xFAD mice.
Figure 4.
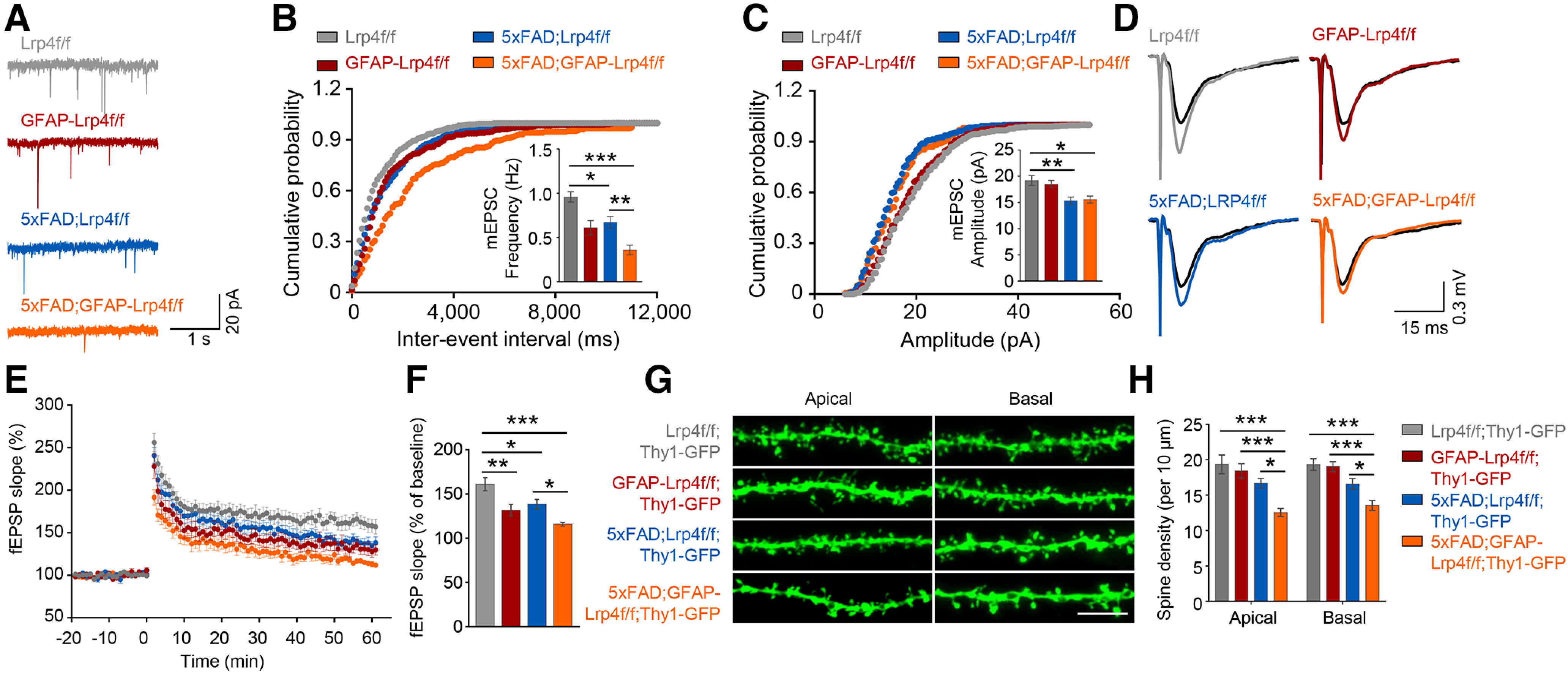
LRP4 deficiency further reduced mEPSC frequency, LTP, and spine density of 6-mo 5xFAD mice. A, Representative mEPSC traces. B, Cumulative distributions of mEPSC frequency, F(3,31) = 13.59, p < 0.0001, ANOVA. C, Cumulative distributions of mEPSC amplitude. Data are summarized in histograms. Data were from 3 mice of each genotype with 2 or 3 neurons per mouse, F(3,31) = 6.792, p = 0.0012, ANOVA. D, Representative fEPSP traces. E, The plot of fEPSP slope against time. F, Quantitative data in E. Shown were potentiation at 50-60 min after high-frequency stimulation. Data were from 4 mice of each genotype and >2 slices per mouse, F(3,29) = 10.41, p < 0.0001, ANOVA. G, Representative basal and apical dendritic spine. H, Quantitative data. The dendritic spine was collected from 4 mice of each genotype and >5 neurons per mouse: apical: F(3,78) = 11.78, p < 0.0001, ANOVA; basal: F(3,81) = 13.33, p < 0.0001, ANOVA. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
To examine whether LRP4 deficiency exacerbates synaptic loss in 5xFAD mice, we generated compound mice carrying Thy1-GFPm that enables labeling excitatory neurons by GFP (Feng et al., 2000). The spine density of both apical oblique and basal shaft dendrites of CA1 neurons were reduced in 5xFAD;Lrp4f/f mice; and this reduction was further decreased in 5xFAD;GFAP-Lrp4f/f hippocampus (Fig. 4G,H). Thus, these results demonstrate LRP4 deficiency exacerbates synapse loss in 5xFAD mice.
Further weakened hippocampal-PFC connectivity in 5xFAD mice by LRP4 deficiency
Hippocampus and PFC are vulnerable in AD and damages in these regions are thought to contribute to cognition decline (Sampath et al., 2017). First, we investigated whether the hippocampus-PFC connectivity was impaired in 5xFAD mice and whether the impairment could be exacerbated by LRP4 deficiency. To this end, we recorded LFPs in both the vHPC and PFC of free-moving mice by implanted tetrodes as described in our recent study (Tan et al., 2018). vHPC neurons are known to project to the PFC (Swanson, 1981; Jay et al., 1989); however, whether this projection is impaired in AD mouse models was unclear. Remarkably, we found that the coherence of vHPC and PFC LFPs at theta, beta, and gamma frequencies was reduced in 5xFAD mice, revealing a novel cellular mechanism of AD pathology (Fig. 5A). Importantly, the coherence at theta frequency was further reduced by LRP4 deficiency in 5xFAD mice (Fig. 5B,C). At delta frequency, although 5xFAD and LRP4 deficiency alone had no effect on the vHPC-PFC coherence, the dramatic reduction was observed in the 5xFAD;GFAP-Lrp4f/f mice. These results demonstrate that the hippocampal-PFC connectivity is compromised in 5xFAD mice, and such deficits are augmented by LRP4 deficiency (Fig. 5B,C).
Figure 5.
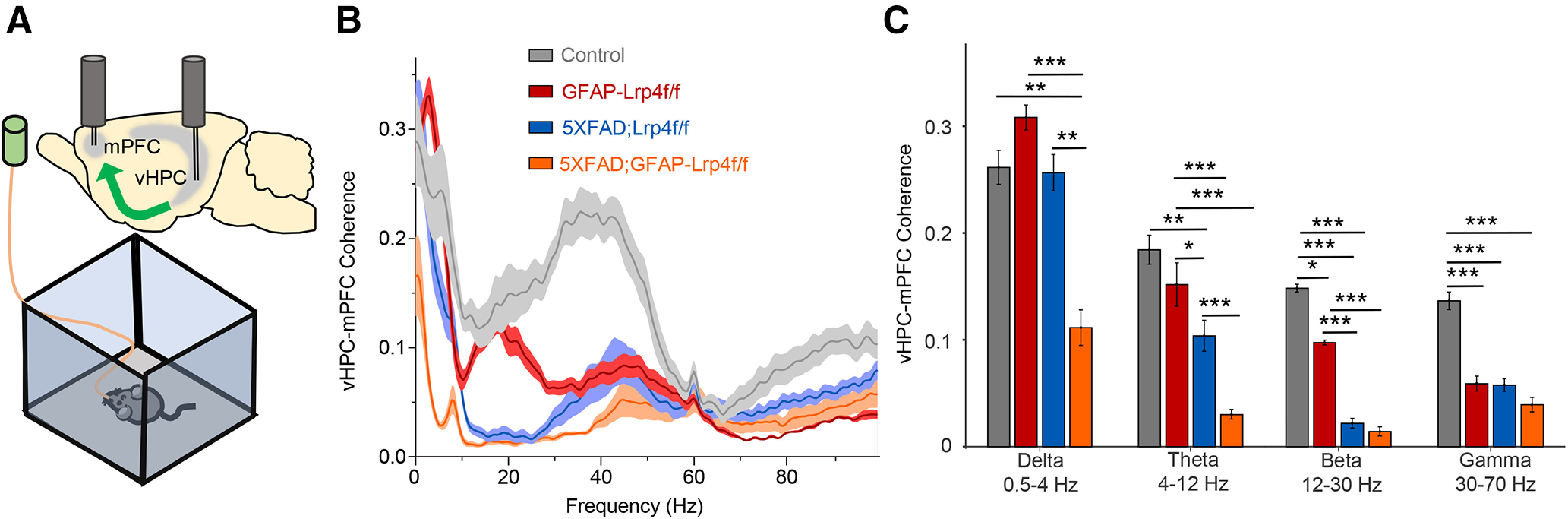
A further disconnect of the vHPC-PFC by Lrp4 mutation in 5xFAD mice. A, Schematic diagram of in vivo recording. Two tetrodes were implanted in vHPC and PFC, respectively. LFPs were recorded in free-moving 6-mo mice in an open field. B, Coherence between vHPC and PFC LFPs of Lrp4f/f (n = 10), GFAP-Lrp4f/f (n = 9), 5xFAD;Lrp4f/f (n = 14), and 5xFDA;GFAP-Lrp4f/f (n = 11) mice. C, vHPC-PFC synchrony at different frequencies: Delta, F(3,40) = 8.389, p = 0.0002, ANOVA; Theta, F(3,40) = 14.84, p < 0.0001, ANOVA; Beta, F(3,40) = 35.28, p < 0.0001, ANOVA; Gamma, F(3,40) = 15.76, p < 0.0001, ANOVA. Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Detection of the Lrp4 promoter activity in astrocytes, not pyramidal neurons, oligodendrocytes, or microglia
What are the mechanisms by which LRP4 regulates AD pathology in 5xFAD mice? To answer this question, it is necessary to know where Lrp4 is expressed. Recently, we demonstrated that Lrp4 is mainly expressed in astrocytes by Lrp4-LacZ reporter mice (Sun et al., 2016), although in vitro data suggest that it may be present in embryonic excitatory neurons (Handara et al., 2019). Because of the lack of reliable anti-LRP4 antibody for immunostaining analysis, we generated a knock-in mouse Lrp4-CreERT2, which expresses 2A-GFP-CreERT2 cassette under the promoter of the endogenous Lrp4 (Fig. 6A). Lrp4-CreERT2 mice were crossed with tdTomato reporter mice (Ai9), and then the compound mice were treated with tamoxifen (Tam) (100 mg/kg) at P42 every other day three times (Fig. 6B). As shown in Figure 6C-F, the tdTomato-labeled cells were distributed across the cortex and hippocampus and in areas where astrocytes are known to be enriched, such as stratum lacunosum-moleculare (Fig. 6F).
Figure 6.
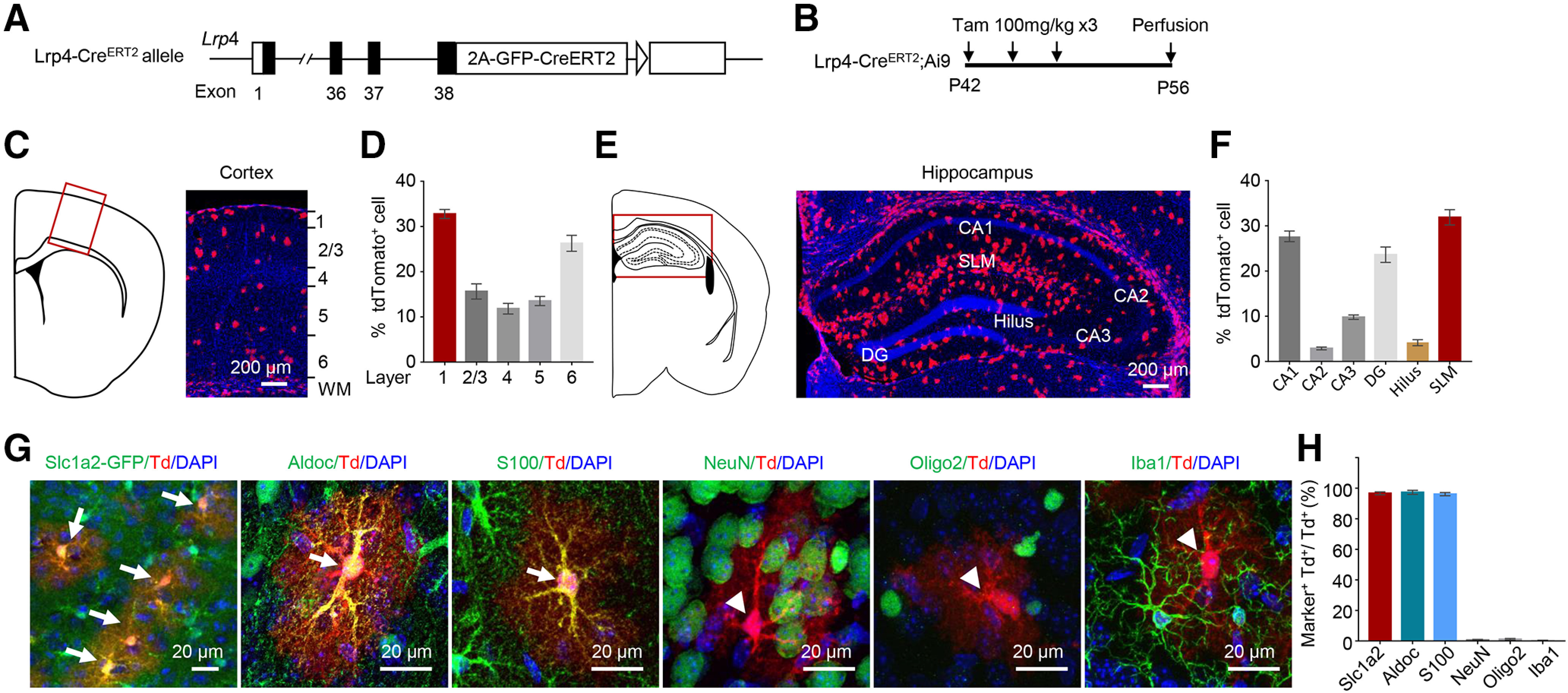
Lrp4 is mainly expressed in astrocytes, not neurons, oligodendrocytes, or microglia. A, Knock-in of the 2A-CreERT2 cassette in exon 38 of the Lrp4 allele. B, The protocol of Tam administration. Lrp4-CreERT2;Ai9 mice at P42 were treated with Tam (100 mg/kg, i.p., three times, once every other day) and killed at P56. Shown are brain sections of the cortex (C,D), hippocampus (E,F), and enlarged views of cortical cells costained with the different maker (G), except that the Slc1a2+ cells are labeled by Slc1a2-GFP (in Slc1a2-GFP; Lrp4-CreERT2;Ai9 mice). tdTomato+ cell costained with astrocytes marker Aldoc and S100β (arrow), but not the with NeuN, Oligo2, and Iba1 (arrowhead). Scale bar, 20 µm. H, Quantification of double-positive cells. n = 3 for each group. Td, tdTomato. Data are mean ± SEM.
To further study the identity of these tdTomato+ cells, we also cross the Lrp4-CreERT2;Ai9 mice with Tg(Slc1a2-EGFP) mice, which is labeling the astrocyte (Gong et al., 2003). We found that most of the tdTomato+ cells colabeled with the GFP-positive cell (Fig. 6G,H). The coimmunostaining study indicated that tdTomato+ cells could be labeled by astrocyte markers, including Aldoc and S100β, but not by neuronal marker NeuN, oligodendrocyte marker Oligo2, or microglia/macrophage marker Iba1 (Fig. 6G,H). Together, these results demonstrate that Lrp4 is mainly expressed in astrocytes, little in neurons, oligodendrocytes, or microglia.
Astrocyte-specific Lrp4 KO augmented Aβ load in 5xFAD mice
Knowing that Lrp4 is expressed in astrocytes, we next determined whether astrocytic LRP4 regulates Aβ deposition. Human GFAP-CreERT2 mice express CreERT2 by a 2 kb minimal promoter of the human GFAP gene (Casper et al., 2007). We and others have shown that CreERT2 is expressed in astrocytes on Tam induction (Casper et al., 2007; Sun et al., 2016). We generated GFAP-CreERT2;Lrp4f/f (referred to as iGFAP-Lrp4f/f) mice to specifically delete Lrp4 in astrocytes (Sun et al., 2016), which were crossed with 5xFAD to produce 5xFAD;iGFAP-Lrp4f/f compound mice. Tamoxifen was injected at 1.5 months (100 mg/kg, i.p., once every other day, 3 times) and mice were killed at 7 months. LRP4 protein levels were reduced to ∼20% in the whole-brain lysate of Tam-treated 5xFAD;iGFAP-Lrp4f/f mice, compared with Tam-treated 5xFAD;Lrp4f/f mice (Fig. 7A,B). Remarkably, Aβ deposition was higher in Tam-treated 5xFAD;iGFAP-Lrp4f/f mice, compared with Tam-treated 5xFAD;Lrp4f/f mice (Fig. 7C,D).
Figure 7.

Augmented Aβ deposition in astrocyte-specific Lrp4 KO 5xFAD mice. A, B, LRP4 protein was reduced in the brain of Tam-treated 5xFAD;iGFAP-Lrp4f/f mice, compared with Tam-treated 5xFAD;Lrp4f/f mice. Representative images (A) and quantification (B). n = 3 for each group, t(4) = 8.121, p = 0.0013, unpaired t test. C, D, Increase of Aβ deposition in Tam-treated 5xFAD;iGFAP-Lrp4f/f mice, compared with Tam-treated 5xFAD;Lrp4f/f mice at 7 months. Representative images (C) and quantification (D). Scale bar: C, 500 µm. n = 4 for each group: Ctx, t(6) = 3.312, p = 0.0162, unpaired t test; Hippo, t(6) = 3.433, p = 0.0139, unpaired t test. E, F, LRP4 protein level is similar between 5xFAD;Lrp4f/f and 5xFAD;Neurod6-Lrp4f/f brain. Representative images (E) and quantification (F). n = 3 for each group, t(4) = 0.5513, p = 0.6108, unpaired t test. G, H, Similar Aβ deposition in 5xFAD;Lrp4f/f and 5xFAD;Neurod6-Lrp4f/f mice at 6 months. Representative images (G) and quantification (H). Scale bar. G, 500 μm. n = 3 for each group: Ctx, t(4) = 0.5835, p = 0.5909, unpaired t test; Hippo, t(4) = 1.546, p = 0.1971, unpaired t test. Data are mean ± SEM. *p < 0.05, **p < 0.001.
In addition, to examine whether neuronal LRP4 has an effect on Aβ deposition, we generated neuron-specific Lrp4 KO mice by crossing 5xFAD;Lrp4f/f mice with Neurod6-Cre mice where Cre is expressed under the promoter of the gene of Neurod6 (Goebbels et al., 2006). Resulting Neurod6-Cre;Lrp4f/f;5xFAD (referred to as 5xFAD;Neurod6-Lrp4f/f) had similar LRP4 protein level in the whole-brain lysates, compared with 5xFAD;Lrp4f/f mice (Fig. 7E,F). These results agree with the notion that Lrp4 is mainly expressed in astrocytes, not neurons (Fig. 6) (Sun et al., 2016). Importantly, little changes in Aβ deposition was noticed in the cortex and hippocampus between 5xFAD;Lrp4f/f and 5xFAD;Neurod6-Lrp4f/f mice (Fig. 7G,H). Together, these results demonstrate that astrocytic LRP4 deficiency enhances Aβ deposition in 5xFAD mice.
Compromised Aβ42 uptake by Lrp4-deficient astrocytes
To investigate how Lrp4 mutation increases Aβ plaque formation, we first examined mRNA levels of App and various risk genes that may alter Aβ metabolism, including Bace1, Bace2, Psen1, and Psen2. We also assessed the impact of LRP4 deficiency on the expression of human APP and PSEN1 genes in 5xFAD mice. mRNA levels of mouse genes, as well as human genes, were similar between 5xFAD;GFAP-Lrp4f/f hippocampus and 5xFAD;Lrp4f/f hippocampus (Fig. 8A), excluding an effect of LRP4 deficiency on genes transcription. Next, we determined levels of APP and its degradation peptides CTFα and CTFβ. CTFα and CTFβ were detected by 8717, a polyclonal antibody that specifically recognizes the C-terminal of APP where CTFβ was detected by 6E10, a monoclonal antibody that specifically recognizes the N-terminal 1-16 amino acid residues of Aβ (Fig. 8B) (Wen et al., 2011). We found that APP (Fig. 8C,D), CTFβ (Fig. 8C,E), and CTFα (Fig. 8C,F) were not changed in the cortex and hippocampus between 5xFAD;GFAP-Lrp4f/f and 5xFAD;Lrp4f/f mice. Together, these results indicate that the deletion of Lrp4 does not change Aβ production in the cortex and hippocampus.
Figure 8.

No effect on Aβ production by Lrp4 mutation in 5xFAD mice. A, qPCR analysis of App and genes involved in its processing in 5xFAD;Lrp4f/f and 5xFAD;GFAP-Lrp4f/f mice at 1.5 months in the hippocampus. n = 3 for each group: App, t(4) = 0.3824, p = 0.7216; Bace1, t(4) = 0.3053, p = 0.7754; Bace2, t(4) = 1.109, p = 0.3296; Psen1, t(4) = 0.3639, p = 0.7343; Psen2, t(4) = 0.1852, p = 0.8621; APP, t(4) = 0.8909, p = 0.4233; PSEN1, t(4) = 0.3018, p = 0.7778, unpaired t test. B, Illustration of human APP structure and its cleavage sites. The indication of the epitopes for the antibodies 6E10 and 8717 and the cleavage fragments CTFβ and CTFα. C, Western blot analysis of APP-FL, βCTF, and αCTF levels using the indicated antibodies. Homogenates of cortex and hippocampus from 5xFAD;Lrp4f/f and 5xFAD;GFAP-Lrp4f/f mice at 1.5 months of age were subjected to Western blot. n = 3 for each group. Three independent replicated experiments were performed. D, Quantification of APP-FL: Ctx, t(4) = 0.0061, p = 0.9954; Hippo, t(4) = 0.9009, p = 0.4186, unpaired t test. E, Quantification of cleavage products βCTF: Ctx, t(4) = 0.2929, p = 0.7842; Hippo, t(4) = 0.8861, p = 0.4256, unpaired t test. F, Quantification of cleavage products αCTF: Ctx, t(4) = 0.8065, p = 0.4652; Hippo, t(4) = 0.9339, p = 0.4032, unpaired t test. Data are mean ± SEM. *p < 0.05.
Astrocytes can uptake oligomer and fibrillary Aβ42 in vitro and in vivo, which contributes to Aβ clearance and prevents the detrimental effect of Aβ accumulation in AD (Wyss-Coray et al., 2003; Koistinaho et al., 2004). Therefore, we next examined whether Lrp4 deletion in astrocytes affects cellular Aβ uptake. Primary astrocytes were incubated with 500 nm FAM-labeled Aβ42 for 2 h at 37°C and immunostained with an antibody against GFAP. As shown in Figure 9B, FAM-Aβ42 in GFAP-Lrp4f/f astrocytes was reduced, compared with Lrp4f/f astrocytes (Fig. 9A–C), suggesting that LRP4 deficiency may reduce Aβ uptake. To further test this hypothesis, we analyzed cell-associated FAM-Aβ42 by FACS. As shown in Figure 9D, FAM-Aβ42-associated cell numbers were reduced in Lrp4-deficient astrocytes, compared with controls (Fig. 9D), suggesting deficient Aβ uptake in Lrp4 mutant astrocytes. Finally, we measured oligomeric Aβ42 in media of astrocytes after the 48 h incubation (Fig. 9E). Aβ42 levels were higher in the media of Lrp4 mutant astrocytes, compared with control astrocytes (Fig. 9F). These results indicate that LRP4 deficiency impairs uptake of Aβ42 by astrocytes.
Figure 9.

Impaired Aβ uptake in Lrp4 mutated primary astrocytes. A, Experimental design. Primary astrocytes derived from P3 Lrp4f/f and GFAP-Lrp4f/f mice brain were incubated with FAM-Aβ42 for imaging (B) and FACS (D). B, Reduced FAM-Aβ42 intensity in astrocytes. Scale bar, 25 µm. C, Quantitative analysis of data in B. n = 8 for Lrp4f/f, n = 6 for GFAP-Lrp4f/f, t(12) = 3.212, p = 0.0075, unpaired t test. D, Decreased cell-associated Aβ42 in astrocyte-derived from GFAP-Lrp4f/f mice compared with astrocyte-derived from Lrp4f/f mice by FACS. n = 4 for each group, t(6) = 8.869, p = 0.0001, unpaired t test. E, F, Increased Aβ42 level in the Lrp4 mutant astrocyte medium compared with Lrp4f/f astrocytes medium, n = 8 for each group, t(14) = 5.887, p < 0.0001, unpaired t test. G, H, Reduced LRP4 interaction with the ApoE4. The nitrocellulose membrane was spotted with 500 ng of Ecto-LRP4 and incubated with different human ApoEs. ApoE was produced by mouse astrocytes that express lipidated and secrete human ApoE isoforms. Bound ApoE was detected using a biotinylated anti-ApoE antibody. Representative dot blots (G) and quantitative data (H), F(2,6) = 16.84, p = 0.0035, ANOVA. Data are mean ± SEM. **p < 0.01. ***p < 0.0001.
Members of the LDLR family, in particular, LDLR and LRP1, have been shown to interact with ApoE and thus promote Aβ clearance. We determined whether LRP4 was able to interact with ApoE and, if so, whether the interaction varies among different alleles. ApoE protein was produced by astrocyte lines that express different alleles of human ApoE. We found that LRP4 interaction with ApoE3 was better than that with ApoE4 (Fig. 9G,H). Together, these results support a working model where LRP4 in astrocytes promotes Aβ uptake by serving as a receptor for ApoE.
Discussion
Aβ clearance is regulated by LDLR family members in particular LDLR and LRP1 (LDLR-related protein 1) (Lane-Donovan et al., 2014; Lane-Donovan and Herz, 2017). Lack of Ldlr aggravates Aβ deposits in AD transgenic mice (Cao et al., 2006), whereas Ldlr overexpression reduces Aβ deposition and increases Aβ clearance (J. Kim et al., 2009). Mutation of RAP (receptor-associated protein), an antagonist of the LDLR family, increases Aβ deposit (Van Uden et al., 2002). Astrocytic Aβ uptake could be blocked by astrocyte-specific KO of Ldlr and Lrp1 (Koistinaho et al., 2004; Basak et al., 2012; C. C. Liu et al., 2017). Considering the abundance of astrocytes in the brain and their modulatory functions on multiple brain cells, they may represent a major mechanism for Aβ clearance. Here, we demonstrated that astrocytic LRP4 serves as a receptor for ApoE to uptake Aβ for degradation in a mouse model of amyloid. Genetically, deletion Lrp4 in astrocytes in 5xFAD mice exacerbated amyloid pathology. In vitro, primary culture astrocytes demonstrated that this effect due to without LRP4 in astrocyte impairing the astrocytic LRP4-mediated Aβ uptake and degradation. Receptor-mediated astrocyte clearance is an efficient way of reducing brain Aβ, due to internalizing by the ApoE receptor are delivered to lysosomes for degradation and the abundance of astrocytes (De Strooper and Karran, 2016; Yamazaki et al., 2019). Together with reduced levels of LRP4 in the brain of AD patients, these observations suggest a new pathologic mechanism of AD development.
LRP4 has a large ECD consisting of an LDLa domain and four β propeller domains that is capable of interacting with various proteins, including ApoE and agrin (Herz and Bock, 2002; May and Herz, 2003; Shen et al., 2015). Loss of LRP4 function in astrocytes impairs Aβ uptake may due to insufficient receptors for ApoE. ApoE is present in the amyloid plaque (Rebeck et al., 1993; Strittmatter et al., 1993a) and binds to Aβ via the C-terminus, which is also responsible for lipid binding (Strittmatter et al., 1993b; Tamamizu-Kato et al., 2008). LRP4 could interact with ApoE in vivo (Lu et al., 2007). Astrocyte-mediated degradation of Aβ occurs through a mechanism that depends on both ApoE and LRP1 (Koistinaho et al., 2004). In accord, Ldlr downregulation reduced Aβ uptake, whereas the upregulation enhanced both the uptake and clearance of Aβ in astrocytes (Basak et al., 2012). Because of the interaction between ApoE and Aβ occurs under physiological conditions, ApoE may be required for astrocyte clearance of Aβ (Verghese et al., 2013). ApoE isoforms have different binding affinities for Aβ, with ApoE3 with higher affinity than ApoE4, for example (LaDu et al., 1994). On the other hand, ApoE4 could impair Aβ clearance without altering Aβ production (Castellano et al., 2011). ApoE3-expressing mice develop fewer Aβ plaques than ApoE4-expressing mice (Holtzman et al., 1999, 2000). Human iPSC-derived astrocytes homozygous for ApoE4 are impaired in Aβ42 uptake, compared with ApoE3 controls (Y. T. Lin et al., 2018). Interestingly, LRP4 has a lower affinity for ApoE4 than for ApoE3, consistent with the notion that ApoE4 is less efficient in Aβ clearance.
LRP4 is a receptor for many ligands, including agrin and APP (Herz and Bock, 2002; May and Herz, 2003; Shen et al., 2015). Agrin is expressed in the brain (Ksiazek et al., 2007), in particular, the neuronal isoform, which contains a B/Z+ insert at the C-terminus that is required for binding to LRP4 and MuSK activation (Bezakova and Ruegg, 2003; B. Zhang et al., 2008; Zong et al., 2012). In addition, crystal structural analysis indicates the essential role of β1 in LRP4, which is required and sufficient to interact with agrin (W. Zhang et al., 2011). Agrin stimulation activates MuSK in astrocytes in a manner dependent on LRP4 (Sun et al., 2016). Moreover, agrin was shown to interact with Aβ (Cotman et al., 2000); and agrin mutation increases Aβ deposition, and overexpression of agrin reduces Aβ accumulation in APP/PS1 mice (Rauch et al., 2011). Whether Aβ uptake by astrocytes is regulated by agrin signaling warrants further investigation.
It is worth pointing out that, at the moment, there are no clinic data to link LRP4 to AD. However, LRP4 has been shown to interact with proteins whose genes are strongly associated with AD. For example, it interacts with APP, whose Swedish mutations cause early-onset AD (Haass et al., 1995) and ApoE, whose ε4 allele is implicated late-onset AD (Corder et al., 1993; Strittmatter et al., 1993a). Both APP's Swedish mutations and ApoE's ε4 allele are thought to increase AD risk (Hardy and Selkoe, 2002). APP interaction with LRP4 has been shown to promote agrin signaling, although its role in AD pathogenesis remains unclear. Here we provide evidence that the LRP4-ApoE interaction is compromised by ApoE4, compared with ApoE3. Members of the LDLR family, in particular LDLR and LRP1, have been shown to interact with ApoE and thus promote Aβ uptake (Kanekiyo et al., 2014). Because Lrp4 levels are 4- to 10-fold higher than those of Ldlr and Lrp1, we propose that LRP4 may play a key role in Aβ uptake by astrocytes. Indeed, Aβ uptake was reduced by Lrp4-deficient astrocytes. This notion is in agreement with the findings that Lrp4 mutation had little effect on APP expression or processing.
The mechanism for LRP4 reduction in postmortem brains of AD patients remains unclear. The mRNA of Lrp4 was in the brain between mice at ages of 2.5 and 24 months are increased (Clarke et al., 2018). Therefore, the reduction likely results from a post-translational mechanism. At the neuromuscular junction, LRP4 reduction in aged mice associates with reduced protein stability (Zhao et al., 2018). In ApoE4-expressing astrocytes, LRP1 surface expression was reduced because of altered endosomal pH (Prasad and Rao, 2018). We showed recently that LRP4 interacts with PRR (prorenin receptor), an auxiliary protein of v-ATPase that is critical to maintaining lysosomal/endosomal pH (Xiong et al., 2017). This interaction is required for LRP4 downregulation of PRR in osteoblasts (Xiong et al., 2017). Exactly how the stability of LRP4 is regulated in astrocytes warrants future study.
Pathologic mechanisms of cognitive impairments in Lrp4-deficient 5xFAD mice could be complex. Although Lrp4 expression in pyramidal neurons and microglial cells is minimal, functions of these cells could be altered indirectly by Lrp4 mutationin astrocytes. Furthermore, the deficits could involve Aβ-dependent as well as -independent mechanisms. Impaired astrocytes, because of LRP4 deficiency, may release cytokines or other molecules to alter the function of pyramidal neurons and microglia, and thus contribute to AD development. Indeed, we showed earlier that LRP4 deficiency increases ATP release and adenosine production in mutant mice (Sun et al., 2016). Purinergic and adenosine signaling has been implicated in AD pathogenesis. Damaged neurons as well as astrocytes can release ATP in response to Aβ peptides (Orellana et al., 2011; Riteau et al., 2012; Fiebich et al., 2014). ATP and its metabolite adenosine are key agonists for purinergic receptors (P1 and P2) or adenosine receptors (A1R, A2AR, A2BR, and A3R), respectively (Cieślak and Wojtczak, 2018). A1R at presynaptic terminals mediates adenosine inhibition of glutamate release (Kimura et al., 2003); however, its level is reduced in AD brains (Jaarsma et al., 1991). Interestingly, A2AR expression is increased in brains of aged animals, AD mice model, and AD patients (Canas et al., 2009; Viana da Silva et al., 2016). A2AR inhibition or genetic silencing could reduce Aβ in cultured neurons and protect against learning and memory deficits in AD animal models or during aging (Canas et al., 2009; Orr et al., 2015, 2018; Viana da Silva et al., 2016).These results suggest the involvement of A2AR activation in AD development. Among P2 receptors, P2X7 receptor (P2X7R) activation plays a role in AD development. An SNP (489C>T) of P2X7R is associated with AD (Sanz et al., 2014). P2X7R is upregulated in astrocytes and microglia near Aβ plaques in AD humans and mice (Martin et al., 2019a,b). Additionally, P2X7R activation is implicated in AD pathogenesis by promoting superoxide production in microglia (Martin et al., 2019a), activating the NLRP3 inflammasome (Heneka et al., 2013), and participating in Aβ-triggered release of IL-1β from microglia (Rampe et al., 2004; Sanz et al., 2009). Furthermore, pharmacological blockade of P2X7R signaling decreases Aβ load in AD mice (Diaz-Hernandez et al., 2012); and P2X7R KO reduces Aβ lesions, rescues cognitive deficits, and improves synaptic plasticity in AD mice (Martin et al., 2019b). Therefore, increased ATP and adenosine could be a mechanism for enhanced cognitive deficits in 5xFAD mice lacking Lrp4 by activating relevant receptors in neurons and microglia.
Footnotes
Work in the L.M. laboratory was supported by AG051510, MH083317, NS090083, and NS082007. Work in the J.G.S. laboratory was supported by National Institutes of Health Grants RF1AG058267 and RF1AG061797. Human tissues were obtained from National Institutes of Health NeuroBioBank (University of Miami Brain Endowment Bank and Human Brain and Spinal Fluid Resource Center). We thank Dr. David Holtzman (Washington University) and Dr. Lance Johnson (University of Kentucky) for ApoE-expressing astrocyte cell lines; Dr. Lin Gan for generating Lrp4-CreERT2 mice; members of the L.M. and W.-C.X. laboratories for helpful discussion; and the patients' families, the CJD Foundation, referring clinicians, and all the members of the National Prion Disease Pathology Surveillance Center for invaluable technical help.
The authors declare no competing financial interests.
References
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH (2011) The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7:270–279. 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's Association (2019) 2019 Alzheimer's disease facts and figures. Alzheimers Dement 15:321–387. [Google Scholar]
- Appel JR, Ye S, Tang F, Sun D, Zhang H, Mei L, Xiong WC (2018) Increased microglial activity, impaired adult hippocampal neurogenesis, and depressive-like behavior in microglial VPS35-depleted mice. J Neurosci 38:5949–5968. 10.1523/JNEUROSCI.3621-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, Li X, Rademakers R, Kang SS, Xu H, Younkin S, Das P, Fryer JD, Bu G (2015) Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem 290:26043–26050. 10.1074/jbc.M115.679043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik A, Lu Y, Sathyamurthy A, Bowman A, Shen C, Li L, Xiong WC, Mei L (2014) LRP4 is critical for neuromuscular junction maintenance. J Neurosci 34:13892–13905. 10.1523/JNEUROSCI.1733-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM (2012) Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. J Biol Chem 287:13959–13971. 10.1074/jbc.M111.288746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Aumont N, Dea D, Lussier-Cacan S, Davignon J, Poirier J (1999) Apolipoprotein E isoform-specific reduction of extracellular amyloid in neuronal cultures. Brain Res Mol Brain Res 68:181–185. 10.1016/s0169-328x(99)00073-x [DOI] [PubMed] [Google Scholar]
- Bezakova G, Ruegg MA (2003) New insights into the roles of agrin. Nat Rev Mol Cell Biol 4:295–308. 10.1038/nrm1074 [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- Bussiere T, Bard F, Barbour R, Grajeda H, Guido T, Khan K, Schenk D, Games D, Seubert P, Buttini M (2004) Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am J Pathol 165:987–995. 10.1016/s0002-9440(10)63360-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canas PM, Porciuncula LO, Cunha GM, Silva CG, Machado NJ, Oliveira JM, Oliveira CR, Cunha RA (2009) Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci 29:14741–14751. 10.1523/JNEUROSCI.3728-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D, Fukuchi K, Wan H, Kim H, Li L (2006) Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice. Neurobiol Aging 27:1632–1643. 10.1016/j.neurobiolaging.2005.09.011 [DOI] [PubMed] [Google Scholar]
- Casper KB, Jones K, McCarthy KD (2007) Characterization of astrocyte-specific conditional knockouts. Genesis 45:292–299. 10.1002/dvg.20287 [DOI] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM (2011) Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med 3:89ra57. 10.1126/scitranslmed.3002156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Zhang M, Yin DM, Wen L, Ting A, Wang P, Lu YS, Zhu XH, Li SJ, Wu CY, Wang XM, Lai C, Xiong WC, Mei L, Gao TM (2010) ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc Natl Acad Sci USA 107:21818–21823. 10.1073/pnas.1010669107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, Barres BA (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504:394–400. 10.1038/nature12776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieślak M, Wojtczak A (2018) Role of purinergic receptors in the Alzheimer's disease. Purinergic Signal 14:331–344. 10.1007/s11302-018-9629-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA (2018) Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA 115:E1896–E1905. 10.1073/pnas.1800165115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ML, Kim C, Haldiman T, ElHag M, Mehndiratta P, Pichet T, Lissemore F, Shea M, Cohen Y, Chen W, Blevins J, Appleby BS, Surewicz K, Surewicz WK, Sajatovic M, Tatsuoka C, Zhang S, Mayo P, Butkiewicz M, Haines JL, et al. (2015) Rapidly progressive Alzheimer's disease features distinct structures of amyloid-beta. Brain 138:1009–1022. 10.1093/brain/awv006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261:921–923. 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- Cotman SL, Halfter W, Cole GJ (2000) Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer's disease brain. Mol Cell Neurosci 15:183–198. 10.1006/mcne.1999.0816 [DOI] [PubMed] [Google Scholar]
- Crouzin N, Baranger K, Cavalier M, Marchalant Y, Cohen-Solal C, Roman FS, Khrestchatisky M, Rivera S, Féron F, Vignes M (2013) Area-specific alterations of synaptic plasticity in the 5XFAD mouse model of Alzheimer's disease: dissociation between somatosensory cortex and hippocampus. PLoS One 8:e74667. 10.1371/journal.pone.0074667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Karran E (2016) The cellular phase of Alzheimer's disease. Cell 164:603–615. 10.1016/j.cell.2015.12.056 [DOI] [PubMed] [Google Scholar]
- Diaz-Hernandez JI, Gomez-Villafuertes R, Leon-Otegui M, Hontecillas-Prieto L, Del Puerto A, Trejo JL, Lucas JJ, Garrido JJ, Gualix J, Miras-Portugal MT, Diaz-Hernandez M (2012) In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer's disease through GSK3beta and secretases. Neurobiol Aging 33:1816–1828. 10.1016/j.neurobiolaging.2011.09.040 [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51. 10.1016/s0896-6273(00)00084-2 [DOI] [PubMed] [Google Scholar]
- Fiebich BL, Akter S, Akundi RS (2014) The two-hit hypothesis for neuroinflammation: role of exogenous ATP in modulating inflammation in the brain. Front Cell Neurosci 8:260. 10.3389/fncel.2014.00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato H, Yoshimura M, Yamazaki T, Saido TC, Ito Y, Yokofujita J, Okeda R, Ihara Y (1998) Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am J Pathol 152:983–992. [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA (2006) Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44:611–621. 10.1002/dvg.20256 [DOI] [PubMed] [Google Scholar]
- Gomez AM, Froemke RC, Burden SJ (2014) Synaptic plasticity and cognitive function are disrupted in the absence of Lrp4. Elife 3:e04287. 10.7554/eLife.04287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N (2003) A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425:917–925. 10.1038/nature02033 [DOI] [PubMed] [Google Scholar]
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ (1995) The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med 1:1291–1296. 10.1038/nm1295-1291 [DOI] [PubMed] [Google Scholar]
- Handara G, Hetsch FJ, Juttner R, Schick A, Haupt C, Rathjen FG, Kroger S (2019) The role of agrin, Lrp4 and MuSK during dendritic arborization and synaptogenesis in cultured embryonic CNS neurons. Dev Biol 445:54–67. 10.1016/j.ydbio.2018.10.017 [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678. 10.1038/nature11729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Bock HH (2002) Lipoprotein receptors in the nervous system. Annu Rev Biochem 71:405–434. 10.1146/annurev.biochem.71.110601.135342 [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Herz J, Bu G (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2:a006312. 10.1101/cshperspect.a006312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM (1999) Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. J Clin Invest 103:R15–R21. 10.1172/JCI6179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM (2000) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 97:2892–2897. 10.1073/pnas.050004797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Das B, Hou H, He W, Yan R (2018) BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J Exp Med 215:927–940. 10.1084/jem.20171831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iram T, Trudler D, Kain D, Kanner S, Galron R, Vassar R, Barzilai A, Blinder P, Fishelson Z, Frenkel D (2016) Astrocytes from old Alzheimer's disease mice are impaired in Abeta uptake and in neuroprotection. Neurobiol Dis 96:84–94. 10.1016/j.nbd.2016.08.001 [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Sebens JB, Korf J (1991) Reduction of adenosine A1-receptors in the perforant pathway terminal zone in Alzheimer hippocampus. Neurosci Lett 121:111–114. 10.1016/0304-3940(91)90661-c [DOI] [PubMed] [Google Scholar]
- Jay TM, Glowinski J, Thierry AM (1989) Selectivity of the hippocampal projection to the prelimbic area of the prefrontal cortex in the rat. Brain Res 505:337–340. 10.1016/0006-8993(89)91464-9 [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE (2008) ApoE promotes the proteolytic degradation of Abeta. Neuron 58:681–693. 10.1016/j.neuron.2008.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RS, Minogue AM, Connor TJ, Lynch MA (2013) Amyloid-beta-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J Neuroimmune Pharmacol 8:301–311. 10.1007/s11481-012-9427-3 [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H, Bu G (2014) ApoE and Abeta in Alzheimer's disease: accidental encounters or partners? Neuron 81:740–754. 10.1016/j.neuron.2014.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Cirrito JR, Liu CC, Shinohara M, Li J, Schuler DR, Shinohara M, Holtzman DM, Bu G (2013) Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J Neurosci 33:19276–19283. 10.1523/JNEUROSCI.3487-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian ZS. (1985) Diagnosis of Alzheimer's disease. Arch Neurol 42:1097–1105. 10.1001/archneur.1985.04060100083029 [DOI] [PubMed] [Google Scholar]
- Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM (2009) Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron 64:632–644. 10.1016/j.neuron.2009.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ (2008) Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 135:334–342. 10.1016/j.cell.2008.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Saitoh N, Takahashi T (2003) Adenosine A(1) receptor-mediated presynaptic inhibition at the calyx of Held of immature rats. J Physiol 553:415–426. 10.1113/jphysiol.2003.048371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Ohno M (2009) Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis 33:229–235. 10.1016/j.nbd.2008.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10:719–726. 10.1038/nm1058 [DOI] [PubMed] [Google Scholar]
- Kounnas MZ, Moir RD, Rebeck GW, Bush AI, Argraves WS, Tanzi RE, Hyman BT, Strickland DK (1995) LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted beta-amyloid precursor protein and mediates its degradation. Cell 82:331–340. 10.1016/0092-8674(95)90320-8 [DOI] [PubMed] [Google Scholar]
- Ksiazek I, Burkhardt C, Lin S, Seddik R, Maj M, Bezakova G, Jucker M, Arber S, Caroni P, Sanes JR, Bettler B, Ruegg MA (2007) Synapse loss in cortex of agrin-deficient mice after genetic rescue of perinatal death. J Neurosci 27:7183–7195. 10.1523/JNEUROSCI.1609-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE (1994) Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem 269:23403–23406. [PubMed] [Google Scholar]
- Lane-Donovan C, Herz J (2017) ApoE, ApoE receptors, and the synapse in Alzheimer's disease. Trends Endocrinol Metab 28:273–284. 10.1016/j.tem.2016.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane-Donovan C, Philips GT, Herz J (2014) More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron 83:771–787. 10.1016/j.neuron.2014.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Tse W, Smith JD, Landreth GE (2012) Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem 287:2032–2044. 10.1074/jbc.M111.295451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Chen G, Xie K, Zaia KA, Zhang S, Tsien JZ (2006) Large-scale neural ensemble recording in the brains of freely behaving mice. J Neurosci Methods 155:28–38. 10.1016/j.jneumeth.2005.12.032 [DOI] [PubMed] [Google Scholar]
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng Z, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, et al. (2018) APOE4 causes widespread molecular and cellular alterations associated with Alzheimer's disease phenotypes in human iPSC-derived brain cell types. Neuron 98:1294. 10.1016/j.neuron.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Hu J, Zhao N, Wang J, Wang N, Cirrito JR, Kanekiyo T, Holtzman DM, Bu G (2017) Astrocytic LRP1 mediates brain Abeta clearance and impacts amyloid deposition. J Neurosci 37:4023–4031. 10.1523/JNEUROSCI.3442-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RX, Huang C, Bennett DA, Li H, Wang R (2016) The characteristics of astrocyte on Abeta clearance altered in Alzheimer's disease were reversed by anti-inflammatory agent (+)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeate. Am J Transl Res 8:4082–4094. [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Tian QB, Endo S, Suzuki T (2007) A role for LRP4 in neuronal cell viability is related to apoE-binding. Brain Res 1177:19–28. 10.1016/j.brainres.2007.08.035 [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44:5–21. 10.1016/j.neuron.2004.09.012 [DOI] [PubMed] [Google Scholar]
- Martin E, Amar M, Dalle C, Youssef I, Boucher C, Le Duigou C, Brückner M, Prigent A, Sazdovitch V, Halle A, Kanellopoulos JM, Fontaine B, Delatour B, Delarasse C (2019a) Expression of P2X7R mRNA in mouse astrocytes and microglia. Mol Psychiatry 24:1 10.1038/s41380-018-0331-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Amar M, Dalle C, Youssef I, Boucher C, Le Duigou C, Bruckner M, Prigent A, Sazdovitch V, Halle A, Kanellopoulos JM, Fontaine B, Delatour B, Delarasse C (2019b) New role of P2X7 receptor in an Alzheimer's disease mouse model. Mol Psychiatry 24:108–125. 10.1038/s41380-018-0108-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P, Herz J (2003) LDL receptor-related proteins in neurodevelopment. Traffic 4:291–301. 10.1034/j.1600-0854.2003.00086_4_5.x [DOI] [PubMed] [Google Scholar]
- Morikawa M, Fryer JD, Sullivan PM, Christopher EA, Wahrle SE, DeMattos RB, O'Dell MA, Fagan AM, Lashuel HA, Walz T, Asai K, Holtzman DM (2005) Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol Dis 19:66–76. 10.1016/j.nbd.2004.11.005 [DOI] [PubMed] [Google Scholar]
- Nielsen HM, Veerhuis R, Holmqvist B, Janciauskiene S (2009) Binding and uptake of A beta1-42 by primary human astrocytes in vitro. Glia 57:978–988. 10.1002/glia.20822 [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129–10140. 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana JA, Shoji KF, Abudara V, Ezan P, Amigou E, Saez PJ, Jiang JX, Naus CC, Saez JC, Giaume C (2011) Amyloid beta-induced death in neurons involves glial and neuronal hemichannels. J Neurosci 31:4962–4977. 10.1523/JNEUROSCI.6417-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr AG, Hsiao EC, Wang MM, Ho K, Kim DH, Wang X, Guo W, Kang J, Yu GQ, Adame A, Devidze N, Dubal DB, Masliah E, Conklin BR, Mucke L (2015) Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat Neurosci 18:423–434. 10.1038/nn.3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr AG, Lo I, Schumacher H, Ho K, Gill M, Guo W, Kim DH, Knox A, Saito T, Saido TC, Simms J, Toddes C, Wang X, Yu GQ, Mucke L (2018) Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol Dis 110:29–36. 10.1016/j.nbd.2017.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad H, Rao R (2018) Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc Natl Acad Sci USA 115:E6640–E6649. 10.1073/pnas.1801612115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampe D, Wang L, Ringheim GE (2004) P2X7 receptor modulation of beta-amyloid- and LPS-induced cytokine secretion from human macrophages and microglia. J Neuroimmunol 147:56–61. 10.1016/j.jneuroim.2003.10.014 [DOI] [PubMed] [Google Scholar]
- Rauch SM, Huen K, Miller MC, Chaudry H, Lau M, Sanes JR, Johanson CE, Stopa EG, Burgess RW (2011) Changes in brain beta-amyloid deposition and aquaporin 4 levels in response to altered agrin expression in mice. J Neuropathol Exp Neurol 70:1124–1137. 10.1097/NEN.0b013e31823b0b12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeck GW, Reiter JS, Strickland DK, Hyman BT (1993) Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron 11:575–580. 10.1016/0896-6273(93)90070-8 [DOI] [PubMed] [Google Scholar]
- Riteau N, Baron L, Villeret B, Guillou N, Savigny F, Ryffel B, Rassendren F, Le Bert M, Gombault A, Couillin I (2012) ATP release and purinergic signaling: a common pathway for particle-mediated inflammasome activation. Cell Death Dis 3:e403. 10.1038/cddis.2012.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath D, Sathyanesan M, Newton SS (2017) Cognitive dysfunction in major depression and Alzheimer's disease is associated with hippocampal-prefrontal cortex dysconnectivity. Neuropsychiatr Dis Treat 13:1509–1519. 10.2147/NDT.S136122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz JM, Falzoni S, Rizzo R, Cipollone F, Zuliani G, Di Virgilio F (2014) Possible protective role of the 489C>T P2X7R polymorphism in Alzheimer's disease. Exp Gerontol 60:117–119. 10.1016/j.exger.2014.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz JM, Chiozzi P, Ferrari D, Colaianna M, Idzko M, Falzoni S, Fellin R, Trabace L, Di Virgilio F (2009) Activation of microglia by amyloid {beta} requires P2X7 receptor expression. J Immunol 182:4378–4385. 10.4049/jimmunol.0803612 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8:595–608. 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LM, Dority MD, Gupta-Bansal R, Frederickson RC, Younkin SG, Brunden KR (1995) Amyloid beta protein (A beta) removal by neuroglial cells in culture. Neurobiol Aging 16:737–745. 10.1016/0197-4580(95)00055-j [DOI] [PubMed] [Google Scholar]
- Shen C, Xiong WC, Mei L (2015) LRP4 in neuromuscular junction and bone development and diseases. Bone 80:101–108. 10.1016/j.bone.2015.05.012 [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993a) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981. 10.1073/pnas.90.5.1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD (1993b) Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 90:8098–8102. 10.1073/pnas.90.17.8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stujenske JM, Likhtik E, Topiwala MA, Gordon JA (2014) Fear and safety engage competing patterns of theta-gamma coupling in the basolateral amygdala. Neuron 83:919–933. 10.1016/j.neuron.2014.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XD, Li L, Liu F, Huang ZH, Bean JC, Jiao HF, Barik A, Kim SM, Wu H, Shen C, Tian Y, Lin TW, Bates R, Sathyamurthy A, Chen YJ, Yin DM, Xiong L, Lin HP, Hu JX, Li BM, et al. (2016) Lrp4 in astrocytes modulates glutamatergic transmission. Nat Neurosci 19:1010–1018. 10.1038/nn.4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW. (1981) A direct projection from Ammon's horn to prefrontal cortex in the rat. Brain Res 217:150–154. 10.1016/0006-8993(81)90192-X [DOI] [PubMed] [Google Scholar]
- Tamamizu-Kato S, Cohen JK, Drake CB, Kosaraju MG, Drury J, Narayanaswami V (2008) Interaction with amyloid beta peptide compromises the lipid binding function of apolipoprotein E. Biochemistry 47:5225–5234. 10.1021/bi702097s [DOI] [PubMed] [Google Scholar]
- Tan Z, Robinson HL, Yin DM, Liu Y, Liu F, Wang H, Lin TW, Xing G, Gan L, Xiong WC, Mei L (2018) Dynamic ErbB4 activity in hippocampal-prefrontal synchrony and top-down attention in rodents. Neuron 98:380–393.e384. 10.1016/j.neuron.2018.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Schultz C, Dehghani F, Yamaguchi H, Braak H, Braak E (2000) Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol 100:608–617. 10.1007/s004010000242 [DOI] [PubMed] [Google Scholar]
- Van Uden E, Mallory M, Veinbergs I, Alford M, Rockenstein E, Masliah E (2002) Increased extracellular amyloid deposition and neurodegeneration in human amyloid precursor protein transgenic mice deficient in receptor-associated protein. J Neurosci 22:9298–9304. 10.1523/JNEUROSCI.22-21-09298.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM (2013) ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci USA 110:E1807–E1816. 10.1073/pnas.1220484110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana da Silva S, Haberl MG, Zhang P, Bethge P, Lemos C, Goncalves N, Gorlewicz A, Malezieux M, Goncalves FQ, Grosjean N, Blanchet C, Frick A, Nagerl UV, Cunha RA, Mulle C (2016) Early synaptic deficits in the APP/PS1 mouse model of Alzheimer's disease involve neuronal adenosine A2A receptors. Nat Commun 7:11915. 10.1038/ncomms11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu F, Chen W, Sun X, Cui W, Dong Z, Zhao K, Zhang H, Li H, Xing G, Fei E, Pan BX, Li BM, Xiong WC, Mei L (2018) Genetic recovery of ErbB4 in adulthood partially restores brain functions in null mice. Proc Natl Acad Sci USA 115:13105–13110. 10.1073/pnas.1811287115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Tang FL, Hong Y, Luo SW, Wang CL, He W, Shen C, Jung JU, Xiong F, Lee DH, Zhang QG, Brann D, Kim TW, Yan R, Mei L, Xiong WC (2011) VPS35 haploinsufficiency increases Alzheimer's disease neuropathology. J Cell Biol 195:765–779. 10.1083/jcb.201105109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J (1991) Spatial relationships between astrocytes and classical plaque components. Neurobiol Aging 12:593–600. 10.1016/0197-4580(91)90091-w [DOI] [PubMed] [Google Scholar]
- Wu H, Lu Y, Shen C, Patel N, Gan L, Xiong WC, Mei L (2012) Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron 75:94–107. 10.1016/j.neuron.2012.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9:453–457. 10.1038/nm838 [DOI] [PubMed] [Google Scholar]
- Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, Diwan A, Lee JM (2014) Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci 34:9607–9620. 10.1523/JNEUROSCI.3788-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L, Jung JU, Guo HH, Pan JX, Sun XD, Mei L, Xiong WC (2017) Osteoblastic Lrp4 promotes osteoclastogenesis by regulating ATP release and adenosine-A2AR signaling. J Cell Biol 216:761–778. 10.1083/jcb.201608002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G (2019) Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol 15:501–518. 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Leung S, Zhang J, Maus E, Baysac K, Bu G, Vassar R, Yu C, LaDu MJ (2011) Amyloid-beta42 alters apolipoprotein E solubility in brains of mice with five familial AD mutations. J Neurosci Methods 196:51–59. 10.1016/j.jneumeth.2010.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, Kim J, Eimer WA, Estus S, Rebeck GW, Weeber EJ, Bu G, Yu C, Ladu MJ (2012) APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem 287:41774–41786. 10.1074/jbc.M112.407957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L (2008) LRP4 serves as a coreceptor of agrin. Neuron 60:285–297. 10.1016/j.neuron.2008.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kang E, Wang Y, Yang C, Yu H, Wang Q, Chen Z, Zhang C, Christian KM, Song H, Ming GL, Xu Z (2016) Brain-specific Crmp2 deletion leads to neuronal development deficits and behavioural impairments in mice. Nat Commun 7:11773 10.1038/ncomms11773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sathyamurthy A, Liu F, Li L, Zhang L, Dong Z, Cui W, Sun X, Zhao K, Wang H, Ho HH, Xiong WC, Mei L (2019) Agrin-Lrp4-Ror2 signaling regulates adult hippocampal neurogenesis in mice. Elife 8:e45303 10.7554/eLife.45303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Coldefy AS, Hubbard SR, Burden SJ (2011) Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J Biol Chem 286:40624–40630. 10.1074/jbc.M111.279307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Shen C, Li L, Wu H, Xing G, Dong Z, Jing H, Chen W, Zhang H, Tan Z, Pan J, Xiong L, Wang H, Cui W, Sun XD, Li S, Huang X, Xiong WC, Mei L (2018) Sarcoglycan alpha mitigates neuromuscular junction decline in aged mice by stabilizing LRP4. J Neurosci 38:8860–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A (2001) hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31:85–94. 10.1002/gene.10008 [DOI] [PubMed] [Google Scholar]
- Zong Y, Zhang B, Gu S, Lee K, Zhou J, Yao G, Figueiredo D, Perry K, Mei L, Jin R (2012) Structural basis of agrin-LRP4-MuSK signaling. Genes Dev 26:247–258. 10.1101/gad.180885.111 [DOI] [PMC free article] [PubMed] [Google Scholar]