

Abstract
Missense variants in Kirrel3 are repeatedly identified as risk factors for autism spectrum disorder and intellectual disability, but it has not been reported if or how these variants disrupt Kirrel3 function. Previously, we studied Kirrel3 loss of function using KO mice and showed that Kirrel3 is a synaptic adhesion molecule necessary to form one specific type of hippocampal synapse in vivo. Here, we developed an in vitro, gain-of-function assay for Kirrel3 using neuron cultures prepared from male and female mice and rats. We find that WT Kirrel3 induces synapse formation selectively between Kirrel3-expressing neurons via homophilic, transcellular binding. We tested six disease-associated Kirrel3 missense variants and found that five attenuate this synaptogenic function. All variants tested traffic to the cell surface and localize to synapses similar to WT Kirrel3. Two tested variants lack homophilic transcellular binding, which likely accounts for their reduced synaptogenic function. Interestingly, we also identified variants that bind in trans but cannot induce synapses, indicating that Kirrel3 transcellular binding is necessary but not sufficient for its synaptogenic function. Collectively, these results suggest Kirrel3 functions as a synaptogenic, cell-recognition molecule, and this function is attenuated by missense variants associated with autism spectrum disorder and intellectual disability. Thus, we provide critical insight to the mechanism of Kirrel3 function and the consequences of missense variants associated with autism and intellectual disability.
SIGNIFICANCE STATEMENT Here, we advance our understanding of mechanisms mediating target-specific synapse formation by providing evidence that Kirrel3 transcellular interactions mediate target recognition and signaling to promote synapse development. Moreover, this study tests the effects of disease-associated Kirrel3 missense variants on synapse formation, and thereby, increases understanding of the complex etiology of neurodevelopmental disorders arising from rare missense variants in synaptic genes.
Keywords: cell adhesion, synapse specificity, target selection
Introduction
Autism spectrum disorder (ASD) has a strong genetic basis, but the mechanistic links between genetic risk variants and brain development remain poorly understood (Tick et al., 2016; Sandin et al., 2017; Bai et al., 2019). This is likely because ASD patients have a heterogeneous set of atypical behaviors presenting with a variety of comorbid conditions, and genetic variants in hundreds of genes are identified as ASD risk factors (American Psychiatric Association, 2013; De Rubeis et al., 2014; Iossifov et al., 2014; Constantino and Charman, 2016). As such, it is important to understand how different types of genetic risk variants functionally impact neurons to properly understand, diagnose, and treat the diverse etiologies of individual patients (Geschwind and State, 2015).
Kirrel3 is an immunoglobulin (Ig) domain-containing transmembrane protein (see Fig. 1A) that undergoes homophilic transcellular binding (Sellin et al., 2003; Gerke et al., 2005; Martin et al., 2015). Importantly, copy number and missense variants in Kirrel3 are repeatedly associated with neurodevelopmental disorders, including ASD and intellectual disability (ID) (for references, see Table 1). There are currently at least 17 missense variants identified from multiple, independent studies supporting the possibility that altering Kirrel3 function leads to ASD and ID. However, it remains unknown whether Kirrel3 missense variants affect protein function.
Figure 1.
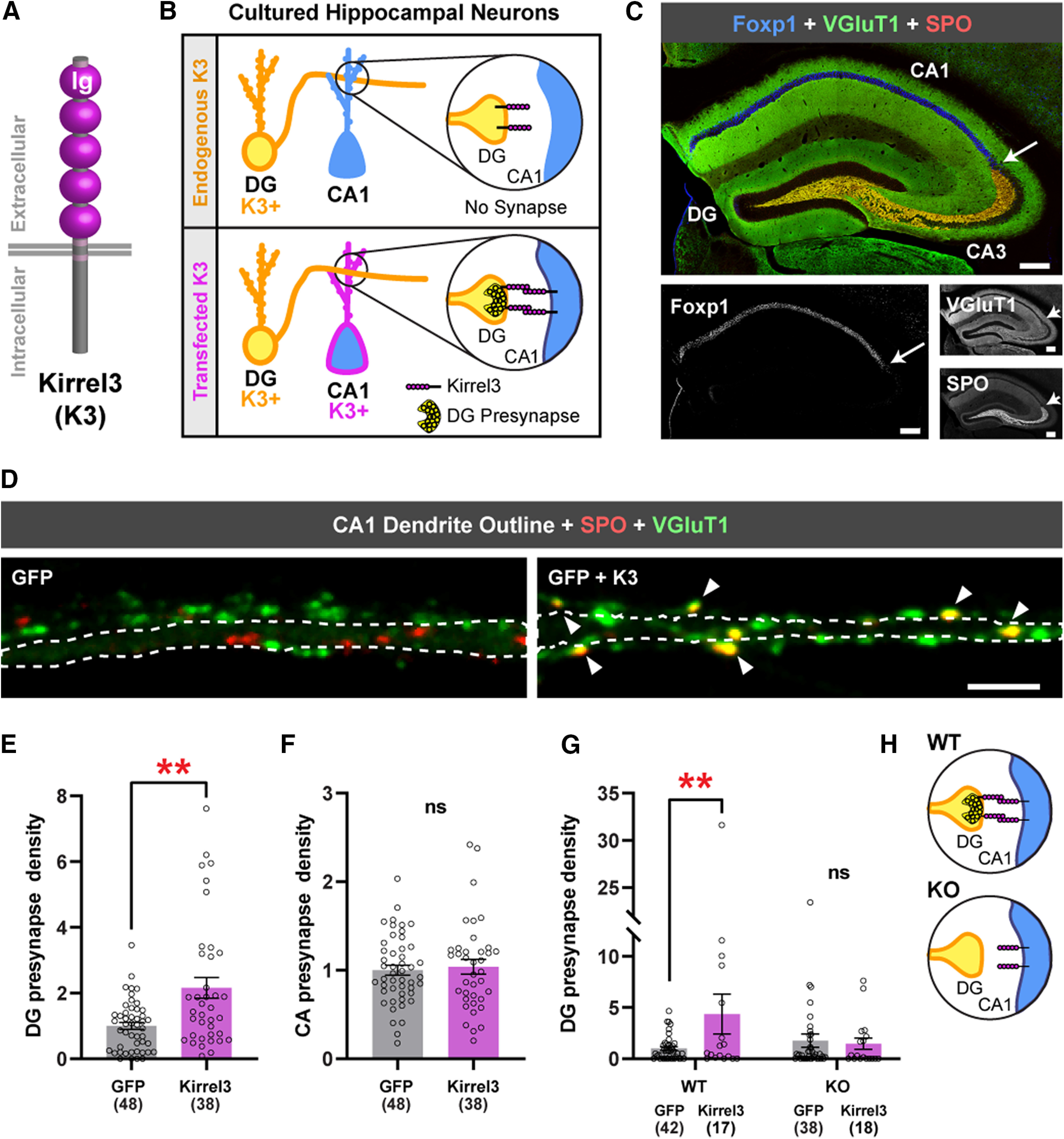
Ectopic Kirrel3 expression induces ectopic DG presynapses in cultured neurons. A, Schematic of Kirrel3 (K3) protein. Ig, Immunoglobulin domain. B, Schematic of the gain-of-function assay design. Top, In the control condition, a Kirrel3-positive DG neuron contacts a Kirrel3-negative CA1 dendrite and does not form a presynapse. Bottom, A Kirrel3-positive DG neuron contacts a Kirrel3-transfected CA1 neuron allowing a DG presynapse to form. C, Tiled 20× image of an adult mouse hippocampal section immunostained with the CA1 marker Foxp1 (blue) and the presynaptic markers VGluT1 (green) and SPO (red). Grayscale images below. In each image, the white arrow indicates approximate CA3 border. Scale bars, 200 μm. D, Dendrites from cultured rat CA1 dendrites transfected with GFP (left) or GFP + Kirrel3 (right). GFP is not shown but is outlined in white. DG presynapses (yellow) are identified by colabeling of SPO (red) and VGluT1 (green). Arrowheads point to DG presynapses. Scale bar, 5 μm. E, Quantification of DG presynapse density per length of CA1 dendrite and normalized to the GFP condition. n = 38-48 neurons (indicated under each bar) from three cultures. p = 0.0027 (Mann–Whitney test). F, Quantification of CA synapse density per length of CA1 dendrite and normalized to the GFP condition. n = 38-48 neurons (indicated under each bar) from three cultures. p = 0.9552 (Mann–Whitney test). G, Quantification of DG presynapse density per length of CA1 dendrite from Kirrel3 WT and KO cultures. All are normalized to the WT GFP condition. n = 17-42 neurons (indicated under each bar) from three cultures. WT: GFP versus GFP+Kirrel3, p = 0.0088; KO: GFP versus GFP+Kirrel3, p = 0.9560 (two-way ANOVA with Sidak's multiple comparisons, p = 0.0266; interaction). **p < 0.01. H, Schematic of WT versus KO assay results, suggesting that Kirrel3 must be present in both the presynaptic and postsynaptic neuron to induce DG synapse formation. E, F, G, Bar graphs show the mean ± SEM. Individual data points represented by open circles.
Table 1.
Kirrel3 Missense variants associated with neurodevelopmental disorders
| Variant ID | Amino acidchange | Associatedconditions | Reference |
|---|---|---|---|
| rs182260035 | Val30Met | ASD | De Rubeis et al., 2014 |
| rs119462978 | Arg40Trp | ID | Bhalla et al., 2008 |
| rs779979146 | Arg161His | ASD | Iossifov et al., 2014; Yuen et al., 2016 |
| rs376743687 | His171Gln | ASD | De Rubeis et al., 2014 |
| rs572991045 | Arg205Gln | ASD | De Rubeis et al., 2014 |
| Lys212Asn | ASD | De Rubeis et al., 2014 | |
| rs372786849 | Ser241Leu | ASD | Li et al., 2017 |
| rs201725914 | Val303Ala | ASD | De Rubeis et al., 2014 |
| rs114378922 | Arg336Gln | ID | Bhalla et al., 2008 |
| Trp358Cys | ASD | Li et al., 2017 | |
| rs201882059 | Ala393Thr | ASD | Wang et al., 2016 |
| rs369363937 | Ala393Val | ASD | Wang et al., 2016 |
| rs1053637755 | Gln428Lys | ASD | Li et al., 2017 |
| rs760499655 | Pro446Leu | ASD | Li et al., 2017 |
| Arg562Leu | ASD | Leblond, et al., 2019 | |
| rs773312950 | Arg650His | ASD | Guo et al., 2019 |
| rs1057519593 | M6731I | ID, ADHD | Present study |
| rs119462980 | Val731Phe | ID | Bhalla et al., 2008 |
We recently discovered that Kirrel3 is necessary to develop a specific type of synapse in the rodent hippocampus (Martin et al., 2015, 2017). Here, dentate granule (DG) neurons project axons, called mossy fibers, to area CA3 where they make unique excitatory synaptic complexes. These mossy fiber synapse complexes connect DG to CA3 neurons via giant presynaptic boutons containing dozens of active zones capable of vesicle release (Acsády et al., 1998; Bischofberger et al., 2006). Each bouton also projects several presynaptic filopodia that, in turn, build smaller excitatory synapses onto both CA3 and GABAergic neurons in area CA3 (Chicurel and Harris, 1992; Acsády et al., 1998; Martin et al., 2017). In the dorsal hippocampus, Kirrel3 is predominately detected in DG neurons and GABAergic neurons (Allen Institute for Brain Science, 2004; Lein et al., 2007; Martin et al., 2015). Consequently, Kirrel3 KO animals have a highly specific loss of DG to GABAergic neuron mossy fiber filopodial synapses, presumably via loss of transcellular contact recognition and downstream signaling mediated by Kirrel3 homophilic binding (Martin et al., 2015, 2017). Ultimately, Kirrel3 loss of function leads to a loss of feedforward inhibition and an excitation/inhibition imbalance in the developing hippocampus (Martin et al., 2015). In sum, previous work provides strong evidence that Kirrel3 is an important target-specific, pro-synaptogenic molecule in vivo.
Other studies also support a role for Kirrel3 in mammalian neurodevelopment. Specifically, DG neurons from Kirrel3 KO mice have more frequent miniature excitatory postsynaptic events in young animals, and Kirrel3 KO mice have behavioral phenotypes, including hyperactivity, social, and communication deficits (Choi et al., 2015; Roh et al., 2017; Hisaoka et al., 2018; Völker et al., 2018). Together, it is clear that lack of Kirrel3 function alters synapse development and adversely affects circuit function and behavior, but mechanisms underlying Kirrel3 function remain unknown.
Here, we sought to advance our understanding of Kirrel3 function and investigate how Kirrel3 variants could cause disease. To accomplish this, we developed a gain-of-function in vitro assay and discovered that ectopic Kirrel3 expression specifically induces ectopic DG presynapse formation via Kirrel3 homophilic interactions. We then tested the function of several disease-associated Kirrel3 missense variants identified in humans using the gain-of-function assay. We found that 5 of 6 disease-associated Kirrel3 variants tested have attenuated synaptogenic function. Intriguingly, we identified two classes of nonfunctional variants: one that can neither mediate homophilic transcellular interactions nor induce synapses, and one that can mediate homophilic transcellular interactions yet has impaired synaptogenesis. We conclude that transcellular binding is necessary but not sufficient for Kirrel3-mediated synapse formation, and provide strong support that most disease-associated Kirrel3 variants impair Kirrel3 function and are likely bona fide risk factors for neurodevelopmental disorders.
Materials and Methods
Kirrel3 variant identification and cloning
Single nucleotide polymorphism accession numbers for human disease-associated Kirrel3 missense variants are shown in Table 1 and were taken from the literature as indicated in the table and the SFARI gene database (https://gene.sfari.org) (Banerjee-Basu and Packer, 2010; Abrahams et al., 2013). The M673I variant is newly discovered and described below. Human Kirrel3 isoform 1 (NM_032531.4 nucleotide; NP_115920.1 protein) and mouse Kirrel3 Isoform A (NM_001190911.1 nucleotide; NP_001177840.1 protein) were aligned using Clustal Omega (Madeira et al., 2019) and found to share 97.94% amino acid identity with identical length. Thus, all variants are named and numbered according to these isoforms. However, all experiments in this study use a cDNA encoding mouse Kirrel3 isoform B (NM_026324.3 nucleotide; NP_080600.1) that is identical to isoform A, except that it lacks an alternatively spliced micro-exon encoding 12 amino acids near the transmembrane domain. Isoform B contains all the conserved missense variant sites and was previously cloned by our laboratory with an extracellular FLAG-tag in the vector pBOS (Martin et al., 2015). Variants were cloned using Q5 Site-Directed Mutagenesis (NEB catalog #E0554S) and QuikChangeII XL Site-Directed Mutagenesis (Agilent Technologies, catalog #200521-5) kits. Mouse cDNA codon changes were made as follows: R40W (CGG to TGG), R161H (CGG to CAT), R205Q (CGA to CAA), R336Q (CGA to CAA), M673I (ATG to ATT), and V731F (GTC to TTC).
Discovery of the KIRREL3 M673I variant
We evaluated a 12-year-old boy for ID, attention deficit hyperactivity disorder (ADHD), and obesity. He is the only child of healthy unrelated parents, born at term after an uneventful pregnancy; growth parameters at birth were normal (height −0.5 SD, weight −0.5 SD and head circumference 0.5 SD). Neonatal hypotonia was noted. The patient subsequently showed global developmental delay, with walking at 26 months and marked language delay (first words at 4 years old). Neuropsychological evaluation at age 12 years showed moderate ID. He is integrated in specialized schooling and cannot count or read. He has fine motricity deficits. Moreover, he suffers from marked ADHD and benefits from methylphenidate treatment. There are no autistic traits. Since 8 years of age, he has behavioral problems with frequent oppositional crises. In addition, the parents report hyperphagia and food craving. At 12 years of age, his height is at 0.5 SD, for a weight of + 3 SD (BMI 25, with truncal obesity) and head circumference at −1.5 SD. The patient also presents with mild dysmorphic features (i.e., exophthalmos, bulbous nose with slightly anteverted nares, thin upper lip, and wide-spaced teeth with two missing superior canines). Hands are normal, except for the presence of fetal pads; feet show second-third toe syndactyly. There are no transit or sleep issues. The boy had frequent otitis media during infancy, needing trans-tympanic drains; he is wearing glasses for mild hypermetropia and astigmatism. Cerebral MRI at age 2 showed mild temporal cortical atrophy and wide asymmetric aspect of posterior ventricular horns. Metabolic screening was normal as well as karyotype, FMR1 gene analysis, 15q11.2 DNA methylation, and array-CGH.
Exome sequencing followed by targeted analysis of a panel of 990 genes implicated in ID, identified a novel de novo missense variant in KIRREL3 c.2019G>A:p.(M673I), which was classified as likely pathogenic according to the ACMG guidelines (Richards et al., 2015). This variant has not been observed in gnomad version 2.1.1 (https://gnomad.broadinstitute.org/gene/ENSG00000149571?dataset=gnomad_r2_1; 248436 alleles), or gnomAD version 3 (https://gnomad.broadinstitute.org/gene/ENSG00000149571?dataset=gnomad_r3; 143348 alleles), or the Bravo databases (https://bravo.sph.umich.edu/freeze5/hg38/gene/ENSG00000149571; 125568 alleles) but a similar variant, M673V, has been observed once in gnomAD version 2.1.1, twice in gnomAD version 3, and twice in Bravo.
Animals
Experiments involving animals were conducted in accordance with National Institute of Health guidelines with approval from the University of Utah Institutional Animal Care and Use Committee. Timed pregnant Sprague Dawley dams were ordered from Charles River (Strain Code 400; RRID:RGD_737926). WT C57BL/6J mice (RRID:IMSR_JAX:000664) were originally obtained from The Jackson Laboratory. Generation of Kirrel3 KO mice was previously reported (Prince et al., 2013). KOs were maintained in a C57BL/6J background. Hippocampi from male and female newborn (P0) pups were combined for neuron cultures.
Immunostaining
Cells on glass coverslips were fixed at room temperature for 10-15 min in 4% PFA made in PBS. Cells were rinsed two times with PBS and blocked/permeabilized in PBS with 3% BSA and 0.1% Triton X for 30 min. Cells were incubated with primary antibodies diluted in PBS with 3% BSA for 1–2 h at room temperature with rocking, rinsed three times in PBS with 3% BSA, and incubated with secondary antibodies diluted in PBS with 3% BSA for 45 min. After rinsing, coverslips were mounted in Fluoromount-G (Southern Biotechnology, catalog #0100-01). For surface labeling, live neurons were incubated in serum-free media at 37°C in a 5% CO2 incubator with chicken-anti-FLAG for 20 min. After initial incubation, neurons were fixed, permeabilized, and stained as above.
Antibodies
Primary antibodies used include the following: rabbit-anti-Foxp1 (1:3600, Abcam, catalog #ab16645, RRID:AB_732428), guinea pig-anti-vesicular glutamate transporter 1 (VGluT1, 1:5000, Millipore, catalog #AB5905, RRID:AB_2301751), goat-anti-synaptoporin (SPO, 1:1000, Santa Cruz Biotechnology, catalog #sc-51 212, RRID:AB_1129844), goat-anti-GFP (1:3000, Abcam, catalog #ab6673, RRID:AB_305643), mouse-anti-PSD-95 (1:1000, Neuromab, catalog #75-348, RRID:AB_2315909), chicken-anti-FLAG (1:1000, Gallus Immunotech, catalog #AFLAG), mouse-anti-FLAG M2 (1:5000, Sigma Millipore, catalog #F1804, RRID:AB_262044), mouse anti-FLAG M2-Cy3 conjugated (1:1000, Sigma Millipore, catalog #A9594, RRID:AB_439700), and rabbit-anti-2A (1:1000, Millipore, catalog #ABS31, RRID:AB_11214282).
Secondary antibodies were all used at 1:1000 and include the following: donkey-anti-rabbit-Alexa-488 (Invitrogen, catalog #A21206, RRID:AB_141708), donkey-anti-rabbit-Cy3 (Jackson ImmunoResearch Laboratories, catalog #711-165-152, RRID:AB_2307443), donkey-anti-guinea pig-Alexa-647 (Jackson ImmunoResearch Laboratories, catalog #705-605-147, RRID:AB_2340437), donkey-anti-guinea pig-DyLight 405 (Jackson ImmunoResearch Laboratories, catalog #706-475-148, RRID:AB_2340470), donkey-anti-goat-Cy3 (Jackson ImmunoResearch Laboratories, catalog #705-165-147, RRID:AB_2307351), donkey-anti-goat-Alexa-647 (Jackson ImmunoResearch Laboratories, catalog #705-605-147, RRID:AB_2340437), donkey-anti-goat-Alexa-488 (Jackson ImmunoResearch Laboratories, catalog #705-545-147, RRID:AB_2336933), donkey-anti-mouse-Cy3 (Jackson ImmunoResearch Laboratories, catalog #715-165-150, RRID:AB_2340813), donkey-anti-mouse-Alexa-647 (Jackson ImmunoResearch Laboratories, catalog #715-605-150, RRID:AB_2340862), donkey-anti-mouse-Alexa-488 (Invitrogen, catalog #A21202, RRID:AB_141607), donkey-anti-chicken-Alexa-647 (Jackson ImmunoResearch Laboratories, catalog #703-605-155, RRID:AB_2340379), and donkey-anti-chicken-Cy3 (Jackson ImmunoResearch Laboratories, catalog #703-165-155, RRID:AB_2340363).
Neuron culture
Neuron cultures were prepared as previously described (Martin et al., 2015; Basu et al., 2017). Briefly, rat cortical astrocytes (combined from both sexes) were plated to glass coverslips coated with 0.03 mg/ml (∼7.5 μg/cm2) of PureCol (Advanced Biomatrix, catalog #5005) and grown to confluence in glia-feeding media: MEM (Invitrogen, catalog #11090-081) supplemented with 10% FBS (Atlas Biologicals, catalog #FP-0500-A), 30 mm glucose, and 1× penicillin-streptomycin solution (Invitrogen, catalog #15070-063). P0 mouse or rat hippocampi were dissected in ice-cold HBSS (Invitrogen, catalog #14185-052) supplemented with 2.5 mm HEPES, pH 7.4, 30 mm glucose, 1 mm CaCl2, 1 mm MgSO4, and 4 mm NaHCO3. Hippocampi were dissociated by incubating in papain solution (20 units/ml papain, Worthington, catalog #3126, 82 mm Na2SO4, 30 mm K2SO4, 5.8 mm MgCl2, 0.25 mm CaCl2, 1 mm HEPES, pH 7.4, 20 mm glucose, and 0.2 mm NaOH) for 30 min at 37°C followed by trituration through a polished glass pipette. Dissociated cells were rinsed and resuspended in neuron plating media: MEM supplemented with 10% horse serum (Invitrogen, catalog #26050-088), 20 mm glucose, 1× penicillin-streptomycin solution, 2 mm Glutamax (Invitrogen, catalog #35050-061), and 1 mm sodium pyruvate. Dissociated hippocampal cells (combined from both sexes) were electroporated with 1 pulse at 340 V for 900 μs pulse length using a square wave protocol in a cuvette with a 2 mm electrode (BTX, ECM830, Harvard Apparatus, catalog #45-0002) using the appropriate plasmids and plated at 1 × 105 cells/well onto glia-coated coverslips in a standard 12-well plate. Neurons were kept in a 37°C incubator maintained with 5% CO2. A few hours after plating, the neuron plating media was replaced with hippocampal feeding media: Neurobasal A (Invitrogen, catalog #10888-022) supplemented with 1× B-27 (Invitrogen, catalog #17504-044), 0.2× penicillin-streptomycin solution, 0.5 mm Glutamax, 12 mm glucose, and 1 μm AraC (Sigma Millipore, catalog #C6645).
Gain-of-function synapse induction assay
Neurons were transfected by electroporation at the time of plating with GFP-pBOS or cotransfected with GFP-pBOS + FLAG-Kirrel3-pBOS constructs. For the GFP-only negative control condition, 4 μg of plasmid was transfected per 5 × 104 cells. For the WT and missense variant transfections, 2.3 μg of GFP and 4 μg of Kirrel3 plasmid were cotransfected per 5 × 104 cells. Transfected neurons were mixed 50:50 with nontransfected neurons, and 1 × 105 total cells were plated per well. This results in sparse transfection of random neurons. Cultures were fixed at 14 DIV and stained with mouse-anti-GFP, rabbit-anti-Foxp1, guinea pig-anti-VGluT1, and goat-anti-SPO. GFP-positive CA1 neurons were identified after immunostaining by strong expression of the marker Foxp1 and pyramidal morphology. We scanned the entire coverslip and imaged all CA1 neurons that were not isolated near the outer edges of the coverslip or densely covered by nearby neurons. This approach ensured that synaptic puncta could be clearly assigned to individual dendrites and results in quantification of between 1 and 5 CA1 neurons per coverslip. Neurons were imaged at 63× magnification on an LSM-7 confocal microscope (Carl Zeiss) using identical settings for each condition per experiment. Maximum intensity projected images were analyzed in ImageJ. An ROI was made from the neuron area by thresholding the GFP channel. The soma and axons were removed, leaving only the dendritic area for analysis. The resulting dendrite area was expanded to include presynapses within 0.5 μm of dendrite border. VGluT1 and SPO puncta within the expanded dendrite area were counted using the ImageJ “analyze particles” tool on thresholded images. Only puncta > 0.2 μm2 were included. Synapse densities were calculated by dividing the number of VGluT1 only (originating predominantly from principle excitatory neurons of the CA3, CA2, and CA1 fields combined), SPO only (originating from interneurons), and VGluT1 + SPO colabeled puncta (originating from DG neurons) by the dendrite length per each cell. Image collection and analysis were performed blind to condition.
Neuron/HEK293 coculture assays
Neuron-HEK293 cocultures were prepared according to an established method (Biederer and Scheiffele, 2007). Briefly, P0 rat hippocampal neurons were plated to poly-D-lysine (10 μg/cm2; Millipore catalog #A-003-E)-coated coverslips at 5 × 104 neurons per well in a 24-well plate. HEK293T cells (ATCC, catalog #CRL-3216; RRID:CVCL_0063) were transfected with GFP-pBOS, GFP-pBOS + FLAG-Neuroligin-1 pCDNA, GFP-pBOS + FLAG-Neurexin-1β-AS4(-)-pCDNA, or GFP-pBOS + FLAG-Kirrel3-pBOS (WT) 24 h before seeding to neurons. In one condition for the presynaptic assay, the neurons were electroporated with FLAG-Kirrel3-pBOS before plating. HEK293 cells do not endogenously express significant levels of Kirrel3 or nephrin-1, a Kirrel3-interacting protein present in kidney and muscle (see Fig. 3A) (Khoshnoodi et al., 2003; Tanigawa et al., 2018).
Figure 3.

Kirrel3 in a non-neuronal cell is not sufficient to mediate synapse formation. A, Immunoblot of untransfected CHO and HEK293 cell lysates showing no detectable endogenous Kirrel3 expression. CHO cells transfected with FLAG-Kirrel3-pBOS were used as a positive control. B, Representative images of the presynaptic neuron/HEK293 coculture assay. Cultures were immunostained for GFP (blue) to label transfected HEK293 cells and VGluT1 (red) to label presynapses. Transfection conditions are indicated on each image. Nlgn1, Neuroligin-1; K3, Kirrel3. Bottom right, Arrow points to an outline of a FLAG-Kirrel3-positive axon. C, Quantification of presynaptic hemisynapse density from the coculture assay normalized to the GFP-only negative control (GFP). n = 32-61 cells (indicated under each bar) from 2 cultures. GFP versus Nlgn1, p = 0.0074; GFP versus Kirrel3, p = 0.3988; GFP versus K3+K3, p > 0.9999 (one-way ANOVA Kruskal–Wallis test, p = 0.0226, with Dunn's multiple comparisons). D, Images of the postsynaptic hemisynapse density from the coculture assay. Cultures were immunostained with GFP (blue), vGluT1 (red), and PSD-95 (green). Postsynaptic hemisynapses are labeled by PSD-95 alone with no VGluT1. Nrxn1, Neurexin-1. E, Quantification of postsynaptic hemisynapse density from coculture assay normalized to the GFP-only negative control. n = 19 or 20 cells (indicated under each bar) from one culture. GFP versus Nrxn1, p = 0.0252; GFP versus Kirrel3, p > 0.9999 (one-way ANOVA Kruskal–Wallis test, p = 0.0256, with Dunn's multiple comparisons). Scale bars: A, C, 20 μm. *p < 0.05. **p < 0.01. C, E, Bar graphs show the mean ± SEM with individual data points represented by open circles.
Test for presynapse induction
Transfected HEK293 cells were seeded to 6 DIV neurons at 3 × 104 cells per well. Twenty-four hours later, cocultures were fixed and immunostained with mouse-anti-FLAG, guinea pig-anti-VGluT1, and goat-anti-GFP. Transfected HEK293 cells in proximity to VGluT1-positive axons were imaged and analyzed. To determine the presynaptic hemisynapse density, images were thresholded and the total area of VGluT1 puncta on, or within 0.5 μm of the edge of each HEK293 cell, was quantified and normalized to the total area of the cell in ImageJ. Image collection and analysis were performed blind to condition.
Test for postsynapse induction
Transfected HEK293 cells were seeded to 14 DIV neurons at 3 × 104 cells per well. Twenty-four hours later, cocultures were fixed and immunostained with mouse-anti-FLAG, guinea pig-anti-VGluT1, mouse-anti-PSD-95, and goat-anti-GFP. HEK293 cells in proximity to PSD-95-positive dendrites were imaged and analyzed. To determine the postsynaptic hemisynapse density, images were thresholded and the total area of PSD-95-positive, VGluT1-negative puncta on, or within 0.5 μm of the edge of each HEK293, was quantified and normalized to the total area of the cell in ImageJ. Image collection and analysis were performed blind to condition.
Axon contact assay
P0 Kirrel3 KO neurons were dissociated and transfected for two conditions. For Condition 1 (Kirrel3 cells plus control cells): 2.5 × 104 neurons per well were cotransfected with 2.3 μg GFP-pBOS + 2.3 μg FLAG-Kirrel3-pBOS and another 2.5 × 104 neurons per well were transfected with 2.3 μg mCherry-pBOS. For Condition 2 (Kirrel3 cells plus Kirrel3 cells): 2.5 × 104 of the neurons per well were cotransfected with 2.3 μg GFP-pBOS + 2.3 μg FLAG-Kirrel3-pBOS and 2.5 × 104 neurons per well were cotransfected with 2.3 μg mCherry-pBOS + 2.3 μg FLAG-Kirrel3-pBOS. In both conditions, transfected neurons were plated with 5 × 104 untransfected neurons per well. Neurons were fixed at 7 DIV. At this neuron density and age, axon-dendrite contacts are only beginning to be made and thus simple enough to observe single axons crossing a neuron's dendritic field. Twenty-five GFP-positive neurons per coverslip were imaged at 63× blind to the mCherry channel. Neurons near the edge of the coverslip were avoided. Any images without mCherry-positive neurites in the FOV were omitted at time of analysis. Images were analyzed in ImageJ. To compare the relative amount of mCherry-positive axon contact with GFP-positive neurons between the conditions, we defined an axon contact index. We created a GFP-positive cell mask by thresholding the GFP channel and an axon mask by thresholding the mCherry channel. The area of mCherry axon mask within in the GFP cell mask was measured. Image collection and analysis were performed blind to condition.
Synapse localization assay
P0 rat hippocampi were dissociated and electroporated with WT mCherry-2A-FLAG-Kirrel3-pBOS, or the missense variants, and cultured as described above. At 21 DIV, neurons were live-labeled with chicken-anti-FLAG, fixed, and immunostained with rabbit-anti-2A, guinea pig-anti-VGluT1, and mouse-anti-PSD-95. mCherry-expressing neurons were imaged at 63× magnification and analyzed in ImageJ. The dendritic area was selected by thresholding the 2A signal and clearing the neuron soma and axons. The dendritic area was expanded to include synapses on or within 1 μm of the dendrite. The PSD-95-, VGluT1-, and Kirrel3 channels were thresholded, and puncta were counted within the expanded dendrite area. Synapses were counted by selecting thresholded PSD-95-positive puncta that had VGluT1-positive puncta within 1 μm. The number of FLAG-Kirrel3 puncta in proximity to synapses were determined by counting thresholded Kirrel3 puncta within 1 μm of the overlapping PSD-95 and VGluT1 puncta area. The Kirrel3 synapse proximity index was calculated by dividing the number of Kirrel3 puncta that were located in or near synapses by the total number of synapses within the dendrite area analyzed for each neuron. Image collection and analysis were performed blind to condition.
Surface expression assay
Chinese hamster ovarian-K1 (CHO) cells (ATCC, catalog #CCL-61, RRID:CVCL_0214) were plated on poly-D-lysine-coated (10 μg/cm2), 18 mm coverslips in a 12-well plate at 1 × 105 cells per well. At 24 h later, cells were transfected with GFP-pBOS or GFP-pBOS + FLAG-Kirrel3-pBOS constructs. Another 24 h later, cells were live-labeled with chicken-anti-FLAG before permeabilization to detect surface expressed FLAG-Kirrel3. Cells were then fixed, permeabilized, and stained with mouse-anti-FLAG to detect intracellular Kirrel3 along with goat-anti-GFP to label transfected cells. Transfected cells with healthy morphology that were not touching other transfected cells were imaged at 63× magnification, and maximum intensity projections were imaged in ImageJ. For each cell, the cell area was determined by GFP expression and was used to make a mask by thresholding. The integrated density of the chicken-anti-FLAG (extracellular) signal was quantified within the cell mask and divided by the integrated density of the mouse-anti-FLAG (intracellular) signal to determine the extracellular/intracellular ratio. General culture conditions and solutions for CHO cells were previously described (Martin et al., 2015; Basu et al., 2017). Image collection and analysis were performed blind to condition.
Cell aggregation assay
The aggregation assay was performed as previously described (Martin et al., 2015; Basu et al., 2017). CHO cells were transfected with mCherry-pBOS, WT mCherry-2A-FLAG-Kirrel3-pBOS, or the missense equivalents. Forty-eight hours later, cells were washed with HEPES Mg2+ free (HMF) buffer and detached from the dishes using 0.01% trypsin in HMF. Detached cells were spun down, resuspended in HMF, counted, and 100,000 cells were pipetted into single wells of 24-well plates precoated with 1% BSA in HMF. Subsequently, the plates were placed on a nutator for 90 min at 37°C. The cells were then fixed with PFA (4% final concentration in PBS), transferred to a 96-well glass-bottom plate, and imaged in an LSM 710 confocal microscope (Carl Zeiss). The aggregation index was calculated by dividing the mCherry integrated density in aggregated cell clumps by the total mCherry integrated density in the well. Image collection and analysis were performed blind to condition.
Immunoblotting
Immunoblotting was conducted by standard techniques. Briefly, CHO cells or HEK293 cells were transfected as needed in a 12-well plate. Cells were collected in 400 μl of Laemmli buffer, boiled, run on a 10% Tris-glycine gel, and transferred to a nitrocellulose membrane. The membrane was probed with mouse-anti-Kirrel3 (Neuromab, catalog #75-333, RRID:AB_2315857) and mouse-anti-GAPDH (Millipore, catalog #AB2302, RRID:AB_10615768). Secondary antibody was goat-anti-mouse-HRP (Jackson ImmunoResearch Laboratories, catalog #115-035-003, RRID:AB_10015289).
Experimental design and statistical analysis
Sample sizes were estimated a priori based on results from previous studies (Williams et al., 2011; Martin et al., 2015; Basu et al., 2017). Statistics were computed using GraphPad Prism (RRID:SCR_002798). All data were tested for normality using a D'Agostino and Pearson omnibus normality test, and then appropriate parametric or nonparametric tests were used and are reported in Results. For all multiple comparisons, we report the multiplicity adjusted p values. Graphs show mean ± SEM. A p value < 0.05 was considered significant for all tests. Data were normalized to the negative control for each culture. For k-means clustering, we used the mean rank values for each condition from the synapse induction assay and the cell aggregation assay, consistent with the nonparametric tests used to compare them. Mean ranks were normalized to the WT mean rank for ease of viewing.
Results
Exogenous Kirrel3 expression induces ectopic synapse formation between incorrect targets
Kirrel3 germline KO results in selective synapse loss in vivo (Martin et al., 2015, 2017), but it is unknown whether Kirrel3 plays an instructive role in selective synapse development. To complement our previous loss-of-function studies (Martin et al., 2015, 2017), we sought to test whether exogenous Kirrel3 expression induces synapse formation via a gain-of-function assay. To this end, we developed an in vitro assay based on the fact that target-specific synapse formation is largely maintained between cultured hippocampal neurons (Williams et al., 2011). Previously, it was shown that cultured DG neurons frequently synapse with CA3 neurons (an appropriate target in vivo) but only rarely synapse with CA1 neurons (an inappropriate target in vivo) despite the fact that DG axons similarly contact CA3 and CA1 dendrites in culture (Williams et al., 2011). Because most CA1 neurons do not express Kirrel3, we used this in vitro specificity to our advantage and tested whether exogenous Kirrel3 expression in CA1 neurons could induce ectopic DG-to-CA1 synapses (Fig. 1B).
To accomplish our goal, we needed to unambiguously identify CA1 neurons and DG synapses in culture. We demonstrate that CA1 neurons are identified by high expression of the transcription factor Foxp1, whereas presynapses arising from DG axons are identified by coexpression of the presynaptic proteins SPO and vesicular glutamate transporter 1 (VGluT1) (Fig. 1C) (Ferland et al., 2003; Williams et al., 2011). This combination of markers allows us to quantify the number of DG presynapses formed onto CA1 dendrites. We transfected dissociated rat hippocampal neurons with GFP alone or GFP plus FLAG-Kirrel3 and quantified the number of DG presynapses per unit length of CA1 dendrite to determine the DG presynapse density in GFP-positive neurons (Fig. 1D,E). Normally, cultured CA1 neurons have few DG presynapses, but exogenous expression of Kirrel3 in CA1 neurons significantly increases the density of DG presynapses onto CA1 neurons by more than twofold (Fig. 1E; Mann–Whitney U = 570, p = 0.0027). This effect is specific for DG synapses as we do not observe an increase in the number of synapses arising from other types of glutamatergic neurons in the culture. This includes CA3, CA2, and CA1 neurons, which are collectively marked by expression of VGluT1, but not SPO, and are referred to here as CA synapses (Fig. 1F; Mann–Whitney U = 905, p = 0.9552).
Kirrel3 mediates homophilic, transcellular adhesion in vitro, and in vivo loss-of-function studies suggest that Kirrel3 mediates synapse formation selectively between Kirrel3-expressing neurons (Gerke et al., 2005; Serizawa et al., 2006; Martin et al., 2015). Together, this suggests that Kirrel3's function requires transcellular interactions with other Kirrel3 molecules. Since all DG neurons normally express Kirrel3, we next tested whether Kirrel3's gain-of-function effect depends on the presence of endogenous Kirrel3 in DG axons. To do this, we performed the same assay in hippocampal neurons cultured from both Kirrel3 WT and KO mice, taking extra precaution to not quantify CA1 neurons that were contacted by transfected axons (marked by GFP). Similar to our results in rat neurons, we find that exogenous Kirrel3 expression potently induces ectopic DG-to-CA1 synapses in cultures prepared from WT mice, but Kirrel3 is not synaptogenic in cultures prepared from Kirrel3 KO mice (Fig. 1G; two-way ANOVA: F(1111) = 5.049, interaction, p = 0.0266; Sidak's multiple comparison WT-GFP vs WT-Kirrel3: p = 0.0088, KO-GFP vs KO-Kirrel3, p = 0.9560). This strongly suggests that, in our gain-of-function assay, postsynaptic Kirrel3 in the CA1 dendrite interacts with presynaptic Kirrel3 in DG axons to induce synapse formation via homophilic, transcellular interactions (Fig. 1H).
Kirrel3-induced synapses do not result from increased axon-dendrite contact
It is possible that Kirrel3 interactions directly instruct neurons to recruit synaptic components at existing axon-dendrite contact points. Alternatively, Kirrel3 expression may increase the amount of axon-dendrite contact between DG and CA1 neurons, and thereby increase the chance that synapses will form via other molecules. Thus, we next tested whether Kirrel3 expression increases axon-dendrite contacts. Tools to identify endogenously expressing Kirrel3-positive and -negative axons in culture are not available, so we cultured neurons from Kirrel3 KO mice and transfected Kirrel3 in a controlled manner. KO neurons were cotransfected with GFP and Kirrel3 and mixed with KO neurons transfected with either mCherry, or mCherry and Kirrel3. Thus, while in both conditions GFP-labeled neurons expressed Kirrel3: in one condition they were contacted by mCherry axons that were Kirrel3-negative, and in the other condition they were contacted by mCherry axons that were Kirrel3-positive. Quantification of the overlap between GFP dendrites and mCherry axons revealed a similar amount of axon-dendrite overlap regardless of whether the axons expressed Kirrel3 or not (Fig. 2A,B; Mann–Whitney U = 257, p = 0.2445). This suggests that Kirrel3 expression does not induce synapses simply by increasing axon-dendrite contact and supports a model in which Kirrel3 transcellular interactions actively recruit synaptic factors to existing points of axon-dendrite contact.
Figure 2.

Kirrel3 expression does not increase axon-dendrite contact in cultured neurons. A, Representative images of neurons transfected with GFP and Kirrel3 (green) contacted by mCherry (mCh) labeled axons with (left) or without (right) Kirrel3. Scale bar, 10 μm. B, Quantification of axon contact normalized to the Kirrel3-negative mCherry-labeled axon condition. Axon contact index is the area of mCherry-positive axon per area GFP-positive cell. n = 22–29 neurons (indicated under each bar) from 2 cultures; p = 0.2445 (Mann–Whitney test). dend = dendrite. Bar graph shows the mean ± SEM. Individual data points represented by open circles. ns = not significant.
Kirrel3-mediated synaptogenesis requires postsynaptic neurons
We next tested whether Kirrel3 is sufficient to induce synapses using a well-established coculture assay in which synaptogenic proteins are presented to neurons in non-neuronal cells (Scheiffele et al., 2000, Graf et al., 2004; Biederer and Scheiffele, 2007). In this assay, neurons abundantly contact HEK293 cells but do not recognize HEK293 cells as synaptic partners. However, if HEK293 cells express some postsynaptic molecules, such as Neuroligin-1, the neurons form presynapse structures, identified by VGlut1, on the HEK293 cells (Scheiffele et al., 2000). Conversely, when HEK293 cells express some presynaptic molecules, such as Neurexin-1, the neurons form postsynapse structures, identified by PSD-95, on the HEK293 cells (Graf et al., 2004). HEK293 cells do not express detectable levels of Kirrel3 (Fig. 3A), making them a suitable cell type to test Kirrel3 in the coculture assay. Interestingly, we find that when HEK293 cells are transfected with Kirrel3, cocultured neurons form neither presynapses nor postsynapses with HEK293 cells, although both of our positive controls (Neurexin-1 and Neuroligin-1) functioned as expected (Fig. 3B-E; presynapses: Kruskal–Wallis test with Dunn's multiple comparisons: GFP vs Kirrel3, p = 0.3988; GFP vs Neuroligin-1, p = 0.0074; Postsynapses: Kruskal–Wallis test with Dunn's multiple comparisons: GFP vs Kirrel3, p > 0.9999; GFP vs Neurexin-1, p = 0.0252). Because Kirrel3 expression specifically induces DG synapses, we also conducted this assay using the DG-specific marker SPO instead of the generic marker VGluT1. Unfortunately, we find that SPO is not reliably expressed in the 7 DIV neurons used for this assay. We circumvented this limitation by transfecting extra Kirrel3 into hippocampal neurons and then quantifying the density of presynaptic puncta on Kirrel3-expressing HEK293 cells contacting Kirrel3-transfected axons. We found that this still had no effect on the density of presynapses formed onto HEK293 cells (Fig. 3B,C; Kruskal–Wallis test with Dunn's multiple comparisons: GFP vs Kirrel3 neuron and HEK293 expression, p > 0.9999). In conclusion, we find that Kirrel3 is not sufficient to induce synapses when expressed in a non-neuronal cell. Together, our work indicates that Kirrel3's synaptogenic function requires both transcellular Kirrel3 binding and interactions with an additional cofactor(s) present in CA1 neurons but not in HEK293 cells.
Kirrel3 missense variants appropriately traffic to synapses and the cell surface
Thus far, we have used our in vitro gain-of-function assay to identify basic mechanistic principles of Kirrel3 signaling during synapse formation. Next, we tested whether and how disease-associated Kirrel3 missense variants affect its function. Accordingly, we cloned six missense variants identified as possible risk factors for neurodevelopmental disorders (Fig. 4A; Table 1) (Bhalla et al., 2008; De Rubeis et al., 2014; Iossifov et al., 2014; Yuen et al., 2016). These variants span the length of the Kirrel3 protein and are fully conserved between the mouse and human proteins, which share 98% amino acid identity (Gerke et al., 2005). Most variants are found in patients with both autism and ID. One variant, M673I, has not been reported previously. We identified this as a likely pathogenic variant in a male patient with developmental delay, ID, ADHD, and obesity (for complete description, see Materials and Methods).
Figure 4.

Kirrel3 missense variants show normal surface localization. A, Schematic of Kirrel3 protein with approximate positions of disease-associated missense variants. Magenta represents variants tested in this study. B, Representative images of CHO cells transfected with mCh-2A-FLAG-Kirrel3 variants. Cells were live-labeled with chicken-anti-FLAG (magenta) to show extracellular (EC) Kirrel3 protein and then permeabilized and stained with mouse-anti-FLAG (orange) to show intracellular (IC) Kirrel3 protein. Cell area was determined by GFP expression and is depicted by a white outline. Scale bar, 15 μm. C, Quantification of EC/IC Kirrel3 protein levels normalized to WT Kirrel3. n = 9 cells each from three cultures; p = 0.4424 (one-way ANOVA). D, Representative images showing a dendrite from an mCherry-2A-FLAG-Kirrel3 WT-transfected neuron. EC FLAG-Kirrel3 (yellow) localizes adjacent to juxtaposed synaptic markers VGluT1 (blue) and PSD-95 (red). mCherry-2A (white outline) served as a cell marker for dendrite area. Scale bar, 4 μm. E, Quantification of Kirrel3 synaptic localization. Shown is the synapse proximity index (a measure of juxtaposed VGluT1+, PSD-95+ puncta with adjacent FLAG-Kirrel3 puncta) for each variant normalized to WT Kirrel3. n = 9-19 neurons (indicated under each bar) from two cultures, p = 0.3197 (one-way ANOVA Kruskal–Wallis test). C, E, Bar graphs show the mean ± SEM with individual data points represented by open circles. ns = not significant.
Missense variants can impair the function of synaptic molecules by altering their trafficking or surface expression (Chih et al., 2004; Zhang et al., 2009; Jaco et al., 2010). Thus, we first tested whether Kirrel3 variants are expressed on the cell surface similar to WT Kirrel3. We transfected CHO cells, which do not express endogenous Kirrel3 (Fig. 3A), with Kirrel3 plasmids containing an extracellular FLAG tag that does not interfere with Kirrel3 homophilic binding (Martin et al., 2015). The cells were live-labeled with chicken-anti-FLAG antibodies to label Kirrel3 protein on the extracellular surface. Then, cells were fixed, permeabilized, and incubated with mouse-anti-FLAG antibodies to label intracellular Kirrel3 (Fig. 4B). The ratio of extracellular to intracellular Kirrel3 was quantified for individual cells. We find that the relative intensities of extracellular to intracellular FLAG-Kirrel3 are indistinguishable between groups, indicating that all six missense variants are appropriately trafficked to the cell surface similar to WT Kirrel3 (Fig. 4C; one-way ANOVA, F(6,56) = 0.9878, p = 0.4424).
WT Kirrel3 localizes at or near excitatory synapses (Martin et al., 2015; Roh et al., 2017). Thus, we tested whether Kirrel3 missense variants also localize to the cell surface near synapses in neurons. We expressed mCherry-2A-FLAG-Kirrel3 and missense variants in cultured neurons and costained for surface-expressed FLAG, VGluT1, and PSD-95 (Fig. 4D). We compared each variant's relative synaptic recruitment to WT Kirrel3 by creating a synaptic proximity index that counts Kirrel3 puncta near juxtaposed VGluT1 and PSD-95 puncta. All Kirrel3 variants display a punctate expression pattern, and their synaptic proximity indexes are similar to that of WT Kirrel3 (Fig. 4E; Kruskal–Wallis test = 7.013; p = 0.3197). Collectively, we do not observe general trafficking defects in disease-associated Kirrel3 missense variants.
Disease-associated Kirrel3 variants attenuate Kirrel3 synaptogenic function
We next tested the ability of each variant to induce DG presynapses when ectopically expressed in CA1 neurons compared with WT (as shown Fig. 1D). Importantly, we found that 4 of the 6 disease-associated variants (R161H, R205Q, R336Q, and V731F) significantly attenuate Kirrel3-mediated presynapse formation compared with WT Kirrel3 (Fig. 5A,B; Kruskal–Wallis test with Dunn's multiple comparisons: WT vs GFP, p < 0.0001; WT vs R40W, p = 0.2495; WT vs R161H, p = 0.0472; WT vs R205Q, p = 0.0218; WT vs R336Q, p = 0.0135; WT vs M673I, p = 0.9844; WT vs V731F, p = 0.0032). Another variant, R40W, appears to attenuate synapse formation but does not achieve statistical significance in this comparison. We then applied statistical tests to determine the ability of the missense variants to induce presynapse formation relative to negative control neurons transfected with GFP alone using the same dataset. Consistent with our previous analysis, DG-presynapse densities on CA1s expressing R161H, R205Q, R336Q, or V731F are not significantly different from GFP-only transfected neurons, suggesting that these variants do not induce synapse formation (Fig. 5C; Kruskal–Wallis test with Dunn's multiple comparisons: GFP vs WT, p < 0.0001; GFP vs R161H, p > 0.9999; GFP vs R205Q, p > 0.9999; GFP vs R336Q, p > 0.9999; GFP vs M673I, p = 0.0382; GFP vs V731F, p > 0.9999). In addition, we found that the R40W variant is also not significantly different from GFP-transfected controls (Kruskal–Wallis test with Dunn's multiple comparisons: GFP vs R40W, p = 0.1299), suggesting that this variant also does not induce synapse formation. The different outcomes for this variant depending on the statistics applied suggest that the R40W variant likely attenuates Kirrel3 function very near the sensitivity threshold of this functional assay. Thus, we conclude that 5 of the 6 tested disease-associated Kirrel3 variants have impaired synaptogenic function. Our results provide functional evidence that Kirrel3 missense variants may cause disease through the selective loss of synapses.
Figure 5.

Disease-associated Kirrel3 variants attenuate Kirrel3 synaptogenic function. A, Representative images of CA1 dendrites transfected with GFP only (GFP), or GFP and indicated FLAG-Kirrel3 variants. Outlines indicate dendrite area as determined from GFP expression. DG presynapses (yellow) are identified by colabeling of SPO (red) and VGluT1 (green). Arrows point to DG presynapses. Scale bar, 5 μm. B, Quantification of DG presynapse density on CA1 dendrites normalized to WT Kirrel3 and with multiple comparisons with WT Kirrel3. n = 30-55 neurons (indicated under each bar) from 3-5 cultures (one-way ANOVA Kruskal–Wallis test with multiple comparisons with WT Kirrel3). C, Same data as presented in B, but shown normalized to GFP and with multiple comparisons with GFP. *p < 0.05. **p < 0.01. ****p < 0.0001. For specific p values, see Results. B, C, Bar graphs show the mean ±SEM with individual data points represented by open circles.
Synapse-defective R161H and R336Q variants retain transcellular binding
Missense variants affecting Kirrel3 function span the extracellular and intracellular domains, suggesting that they may impair Kirrel3 function in distinct ways (Fig. 4A; Table 1). We reasoned that variants in the extracellular domain may eliminate Kirrel3 homophilic binding, whereas variants in the intracellular domain may alter downstream signaling. It is not yet possible to test Kirrel3 intracellular signaling because very little is known about Kirrel3's intracellular interactions or signaling mechanisms. Therefore, we tested the ability of each missense variant to mediate homophilic binding in trans using an established CHO cell aggregation assay. CHO cells do not normally form cell aggregates when they are mixed in a cell suspension (Fig. 6A,B). In contrast, CHO cells transfected with WT Kirrel3 form multicellular aggregates consistent with Kirrel3 undergoing homophilic, transcellular binding (Fig. 6A,B) (Martin et al., 2015). After testing each variant, we find that cells transfected with R40W, R161H, R336Q, and M673I aggregate similarly to WT Kirrel3, but the R205Q and V731F missense variants do not induce cell aggregation (Fig. 6B; Kruskal–Wallis test with Dunn's multiple comparisons: WT vs Ctrl, p < 0.0001; WT vs R40W, p > 0.9999; WT vs R161H, p > 0.9999; WT vs R205Q, p < 0.0001; WT vs R336Q, p > 0.9999; WT vs M673I, p > 0.9999; WT vs V731F, p = 0.0042). Subsequent k-means clustering defines four groups of variants with respect to synaptogenic and aggregation ability (Fig. 6C). WT Kirrel3 and the M673I variant group together as they exhibit homophilic transcellular binding and induce synapse formation to a greater extent than the other variants. The R40W variant is isolated, reflecting its intermediate ability to induce synapse formation with normal transcellular binding. R161H and R336Q form a group characterized by attenuated synaptogenic ability with normal transcellular binding. Finally, R205Q and V731F group with the negative control as they are completely binding deficient and do not induce synapse formation. Collectively, these results indicate that missense variants impact Kirrel3 function in distinct ways: some attenuate Kirrel3-mediated transcellular interactions, whereas others maintain an ability to mediate contact recognition but still fail to induce synaptogenesis, strongly implicating additional unidentified signaling mechanisms.
Figure 6.

Synapse-defective R161H and R336Q variants retain transcellular binding. A, Representative images from CHO aggregation assay showing CHO cells transfected with mCherry alone as a negative control (Ctrl), mCherry-2A-Kirrel3-WT (WT K3), or mCherry-2A-Kirrel3 variants. Red represents mCherry (mCh) signal. Blue represents DAPI. Scale bar, 50 μm. B, Quantification of CHO cell aggregation assay normalized to WT Kirrel3. n = 8-18 wells (indicated under each bar) from three cultures. *p < 0.05. ***p < 0.001. ****p < 0.0001. For specific p values, see Results. Bar graph shows the mean ± SEM with individual data points represented by open circles. C, k-means clustering of WT Kirrel3, missense variants, and negative control based on their ability to aggregate CHO cells and induce DG presynapse formation using mean rank values normalized to WT Kirrel3 for each condition from the two assays presented in Figures 5B and 6C.
Discussion
Kirrel3 functions in synapse development as a homophiliccell-recognition tag
Most synapses do not form randomly between any neurons that come intocontact with each other. Rather, neurites coursing through a dense microenvironment often select specific partners among a milieu of incorrect choices. A long-standing hypothesis proposes that neuronalwiring requires matching affinity tags selectively expressed on presynaptic and postsynaptic neurons (Sperry, 1963; Benson et al., 2001; Sanes and Yamagata, 2009; Südhof, 2018). The identity of these tags and how they signal at specific synapses remain largely unresolved.
Here, we directly tested whether Kirrel3 acts as synaptogenic, cell-recognition tag using a novel gain-of-function assay. We demonstrate that ectopic Kirrel3 expression in CA1 neurons specifically induces ectopic DG synapse formation, providing direct evidence that Kirrel3 plays an instructive role in synapse development. Furthermore, we show that ectopic Kirrel3 has no function when expressed in cultures prepared from Kirrel3 KO mice. This indicates that ectopically expressed Kirrel3 function depends on the presence of endogenous Kirrel3, most likely because ectopic Kirrel3 in the CA1 dendrites interacts with endogenous Kirrel3 in DG axons to induce DG synapses. This strongly suggests that Kirrel3's transcellular, homophilic interactions are necessary for its function. This is significant because, although prior work indicates that Kirrel3 can undergo homophilic interactions, a functional role of Kirrel3 homophilic interactions at synapses had not been directly tested (Gerke et al., 2005; Serizawa et al., 2006; Prince et al., 2013; Martin et al., 2015; Brignall et al., 2018). Moreover, in the mouse neuromuscular junction, mouse kidney, and at Caenorhabditis elegans synapses, Kirrel3 functions via heterophilic interactions with another Ig-superfamily protein Nephrin-1 (C. elegans Syg-1 and Syg-2, respectively) (Shen and Bargmann, 2003; Shen et al., 2004; Gerke et al., 2005; Ding et al., 2007; Chao and Shen, 2008; Komori et al., 2008; Chia et al., 2014; Özkan et al., 2014). However, Nephrin-1 is not appreciably expressed in the hippocampus (Allen Institute for Brain Science, 2004; Lein et al., 2007; Cembrowski et al., 2016). Our study does not rule out the possibility that Kirrel3 binds in trans to other molecules, but we clearly show that Kirrel3-Kirrel3 trans-homophilic interactions are necessary for its synaptogenic function.
Kirrel3 has multifaceted roles as a recognition, adhesion, and signaling molecule
Cell adhesion proteins are important for contact recognition between neurons, but very little is known about downstream signaling events that couple contact recognition to synapse development. Therefore, a common misperception is that adhesive proteins expressed on the cell surface act as a “molecular glue” to hold juxtaposed membranes together long enough for other general synaptogenic factors to be recruited. Indeed, many cell adhesion proteins act as more than molecular glue. For example, Cadherins, Neuroligins, Neurexins, SynCAM 1, and leucine-rich repeat containing cell adhesion proteins are shown to regulate synaptic transmission and plasticity (Jüngling et al., 2006; Kim et al., 2008; Robbins et al., 2010; Shipman and Nicoll, 2012; Basu et al., 2017; Schroeder et al., 2018; Restrepo et al., 2019). Our findings support a model in which Kirrel3 likely mediates a combination of contact recognition, adhesion, and downstream synaptogenic signaling and directly implicate Kirrel3 as a bona fide signaling molecule. Several lines of evidence support this conclusion. First, in support that Kirrel3 likely mediates contact recognition and adhesion, we identified two variants, R205Q and V731F, that cannot bind in trans and have no synaptogenic activity. This supports the conclusion that transcellular binding is necessary for Kirrel3 function. Interestingly, the V731F variant resides in the Kirrel3 intracellular domain, and we speculate that this variant produces a conformational change that directly inhibits transcellular interactions by the extracellular domain. Nonetheless, our results strongly suggest that transcellular binding is absolutely critical for Kirrel3 synaptic function. Second, in support of the idea that Kirrel3 has a nonadhesive signaling function, we also identified variants, R161H and R336Q, which bind normally in trans yet have impaired synaptogenic activity. These results indicate that transcellular binding is not sufficient for Kirrel3 function and strongly suggest that Kirrel3 is involved in synaptic signaling in addition to its role in transcellular recognition or adhesion. Additional support for this conclusion comes from the fact that Kirrel3 is not synaptogenic if presented to neurons by a non-neuronal cell. This also strongly suggests that transcellular binding is not sufficient for its synaptogenic function and that Kirrel3 function requires a neuronal cofactor. In sum, we find that transcellular binding is necessary but not sufficient for Kirrel3 function and that Kirrel3 must interact with other neuronal proteins to induce a synapse. Thus, Kirrel3 does more than act as synaptic glue.
What proteins might mediate a Kirrel3 signal?
For the presynaptic DG axon to differentiate between a HEK293 cell and neuron, we postulate that Kirrel3 likely interacts with another extracellular or transmembrane molecule in the postsynaptic neuron and that molecule must be lacking in the HEK293 cell. Interestingly, the Kirrel3 extracellular domain was reported to interact with the cell adhesion molecule Neurofascin, but this remains to be tested under physiological conditions (Völker et al., 2018).
Kirrel3 intracellular interactions are also likely to be important for Kirrel3-mediated synapse development. Most interestingly, the Kirrel3 intracellular domain ends in a conserved PDZ-binding domain, which is a common protein motif of synaptic organizing molecules. It was suggested that mouse Kirrel3 binds to the PDZ domain proteins CASK, PSD95, and ZO-1 (Huber et al., 2003; Gerke et al., 2006; Roh et al., 2017). Other evidence for putative Kirrel3 binding partners comes from studies using Drosophila and C. elegans. Here, Kirrel3 orthologs are shown to physically or genetically interact with Rols7 (Tanc1/2, a synaptic scaffold), Loner (Brag2, an Arf-GEF), Lin10 (Mint, an active zone component), WVE-1 (WAVE-1, an actin regulator), and SKR-1 (Skp1, an EIII ubiquitin ligase) (Kreisköther et al., 2006; Vishnu et al., 2006; Ding et al., 2007; Bulchand et al., 2010; Chia et al., 2014). However, none of these Kirrel3 interacting proteins has yet been well validated under physiological conditions in the mammalian brain. Further work is needed to identify new Kirrel3 signal transduction candidates and screen existing candidates. The identity of Kirrel3 signaling molecules and whether or not they are shared by other synapse specificity factors will be an important question to address in future studies.
Kirrel3 variants associated with disease impair synapse formation
Kirrel3 is an emerging hotspot for rare variants linked to neurodevelopmental disorders (Bhalla et al., 2008; Kaminsky et al., 2011; Ben-David and Shifman, 2012; Guerin et al., 2012; Michaelson et al., 2012; Neale et al., 2012; Talkowski et al., 2012; De Rubeis et al., 2014; Iossifov et al., 2014; Wang et al., 2016; Yuen et al., 2016; Li et al., 2017; Guo et al., 2019; Leblond et al., 2019). Here we provide functional evidence demonstrating that Kirrel3 variants could cause disease.
Interestingly, not all of the missense variants studied present a phenotype. Our newly reported M673I variant performs similar to WT Kirrel3 in all of the assays presented here. While this negative result could mean that this particular variant is not disease-causing, it is also possible that the variant requires a second mutation in another unidentified gene to impair its function or that our current set of tests for Kirrel3 function have not yet revealed the cellular phenotypes caused by this variant. For example, because CA1 neurons do not normally express Kirrel3, we would not expect this assay to identify dominant-negative mutations (discussed further below), only loss-of-function mutations. Thus, M673I could function as a dominant-negative, and we are in the process of developing new assays to test this class of mutations. Nonetheless, 5 of 6 tested disease-associated Kirrel3 variants have impaired function in our assays, providing functional evidence that most disease-associated Kirrel3 variants could cause neurodevelopmental disorders.
Most disease-associated Kirrel3 variants are rare, spontaneous mutations for which patients are heterozygous. If these variants cause disease, then Kirrel3 is likely haploinsufficient and/or disease-associated variants act as dominant-negative proteins. In support of haploinsufficiency, an interstitial deletion on part of chromosome 11, which leads to a complete loss of one copy of Kirrel3 along with other genes, is associated with Jacobsen syndrome, a developmental disorder with frequent neurologic symptoms, including ASD, epilepsy, and ID (Guerin et al., 2012). Furthermore, Kirrel3 has a pLI score, a probability measure that a gene is intolerant to heterozygous loss-of-function variation, of 0.97 suggesting haploinsufficiency for loss-of-function variants (gnomAD database; https://gnomad.broadinstitute.org/) (Lek et al., 2016). On the other hand, we find that all missense variants tested localize to the cell surface at synapses similar to WT Kirrel3, suggesting that Kirrel3 variants could interact in a dominant-negative fashion by sequestering Kirrel3 binding partners. Future work is needed to determine the precise mechanism of Kirrel3 and each variant in vivo, but results from this study provide a critical foundation that Kirrel3 variants are likely bona fide disease risk factors.
Footnotes
The authors declare no competing financial interests.
This work was supported by National Institute of Mental Health R01 MH105426 to M.E.W., Brain Research Foundation Fay/Frank Seed Grant to M.E.W., Childcare Foundation to S.E.A., and Autism Speaks Dennis Weatherstone Predoctoral Fellowship 10116 to E.A.M. We thank the patients and families reported in this study; Serena Hawatmeh for technical assistance; the entire M.E.W. laboratory for experimental help and discussion; and Richard Dorsky, Jan Christian, and Michael Deans for comments on the manuscript.
References
- Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, Menashe I, Wadkins T, Banerjee-Basu S, Packer A (2013) SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism 4:36. 10.1186/2040-2392-4-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acsády L, Kamondi A, Sík A, Freund T, Buzsáki G (1998) GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci 18:3386–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen Institute for Brain Science (2004) Allen Mouse Brain Atlas. Experiment 71920516, Gene Kirrel3. Available at https://mouse.brain-map.org/experiment/show/71920516. Accessed June 16, 2020.
- American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders: DSM-5. Washington, DC: American Psychiatric Association. [Google Scholar]
- Bai D, Yip BH, Windham GC, Sourander A, Francis R, Yoffe R, Glasson E, Mahjani B, Suominen A, Leonard H, Gissler M, Buxbaum JD, Wong K, Schendel D, Kodesh A, Breshnahan M, Levine SZ, Parner ET, Hansen SN, Hultman C, et al. (2019) Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry 76:1035–1043. 10.1001/jamapsychiatry.2019.1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee-Basu S, Packer A (2010) SFARI Gene: an evolving database for the autism research community. Dis Model Mech 3:133–135. 10.1242/dmm.005439 [DOI] [PubMed] [Google Scholar]
- Basu R, Duan X, Taylor MR, Martin EA, Muralidhar S, Wang Y, Gangi-Wellman L, Das SC, Yamagata M, West PJ, Sanes JR, Williams ME (2017) Heterophilic type II cadherins are required for high-magnitude synaptic potentiation in the hippocampus. Neuron 96:160–176.e8. 10.1016/j.neuron.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David E, Shifman S (2012) Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PLoS Genet 8:e1002556. 10.1371/journal.pgen.1002556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DL, Colman DR, Huntley GW (2001) Molecules, maps and synapse specificity. Nat Rev Neurosci 2:899–909. 10.1038/35104078 [DOI] [PubMed] [Google Scholar]
- Bhalla K, Luo Y, Buchan T, Beachem MA, Guzauskas GF, Ladd S, Bratcher SJ, Schroer RJ, Balsamo J, DuPont BR, Lilien J, Srivastava AK (2008) Alterations in CDH15 and Kirrel3 in patients with mild to severe intellectual disability. Am J Hum Genet 83:703–713. 10.1016/j.ajhg.2008.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Scheiffele P (2007) Mixed-culture assays for analyzing neuronal synapse formation. Nat Protoc 2:670–676. 10.1038/nprot.2007.92 [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Frotscher M, Jonas P (2006) Timing and efficacy of transmitter release at mossy fiber synapses in the hippocampal network. Pflugers Arch 453:361–372. 10.1007/s00424-006-0093-2 [DOI] [PubMed] [Google Scholar]
- Brignall AC, Raja R, Phen A, Prince JE, Dumontier E, Cloutier JF (2018) Loss of Kirrel family members alters glomerular structure and synapse numbers in the accessory olfactory bulb. Brain Struct Funct 223:307–319. 10.1007/s00429-017-1485-0 [DOI] [PubMed] [Google Scholar]
- Bulchand S, Menon SD, George SE, Chia W (2010) The intracellular domain of Dumbfounded affects myoblast fusion efficiency and interacts with rolling pebbles and loner. PLoS One 5:e9374. 10.1371/journal.pone.0009374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembrowski MS, Wang L, Sugino K, Shields BC, Spruston N (2016) Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. eLife 5:e14997. 10.7554/eLife.14997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao DL, Shen K (2008) Functional dissection of SYG-1 and SYG-2, cell adhesion molecules required for selective synaptogenesis in C. elegans. Mol Cell Neurosci 39:248–257. 10.1016/j.mcn.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia PH, Chen B, Li P, Rosen MK, Shen K (2014) Local F-actin network links synapse formation and axon branching. Cell 156:208–220. 10.1016/j.cell.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicurel ME, Harris KM (1992) Three-dimensional analysis of the structure and composition of CA3 branched dendritic spines and their synaptic relationships with mossy fiber boutons in the rat hippocampus. J Comp Neurol 325:169–182. 10.1002/cne.903250204 [DOI] [PubMed] [Google Scholar]
- Chih B, Afridi SK, Clark L, Scheiffele P (2004) Disorder-associated mutations lead to functional inactivation of neuroligins. Hum Mol Genet 13:1471–1477. 10.1093/hmg/ddh158 [DOI] [PubMed] [Google Scholar]
- Choi SY, Han K, Cutforth T, Chung W, Park H, Lee D, Kim R, Kim MH, Choi Y, Shen K, Kim E (2015) Mice lacking the synaptic adhesion molecule Neph2/Kirrel3 display moderate hyperactivity and defective novel object preference. Front Cell Neurosci 9:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino JN, Charman T (2016) Diagnosis of autism spectrum disorder: reconciling the syndrome, its diverse origins, and variation in expression. Lancet Neurol 15:279–291. 10.1016/S1474-4422(15)00151-9 [DOI] [PubMed] [Google Scholar]
- De Rubeis SD, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfield JM, Cai J, Campbell NG, et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M, Chao D, Wang G, Shen K (2007) Spatial regulation of an EIII ubiquitin ligase directs selective synapse elimination. Science 317:947–951. 10.1126/science.1145727 [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA (2003) Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol 460:266–279. 10.1002/cne.10654 [DOI] [PubMed] [Google Scholar]
- Gerke P, Sellin L, Kretz O, Petraschka D, Zentgraf H, Benzing T, Walz G (2005) NEPH2 is located at the glomerular slit diaphragm, interacts with nephrin and is cleaved from podocytes by metalloproteinases. J Am Soc Nephrol 16:1693–1702. 10.1681/ASN.2004060439 [DOI] [PubMed] [Google Scholar]
- Gerke P, Benzing T, Höhne M, Kispert A, Frotscher M, Walz G, Kretz O (2006) Neuronal expression and interaction with the synaptic protein CASK suggest a role for Neph1 and Neph2 in synaptogenesis. J Comp Neurol 498:466–475. 10.1002/cne.21064 [DOI] [PubMed] [Google Scholar]
- Geschwind DH, State MW (2015) Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol 14:1109–1120. 10.1016/S1474-4422(15)00044-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM (2004) Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119:1013–1026. 10.1016/j.cell.2004.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerin A, Stavropoulos DJ, Diab Y, Chénier S, Christensen H, Kahr WH, Babul-Hirji R, Chitayat D (2012) Interstitial deletion of 11q-implicating the Kirrel3 gene in the neurocognitive delay associated with Jacobsen syndrome. Am J Med Genet 158A:2551–2556. 10.1002/ajmg.a.35621 [DOI] [PubMed] [Google Scholar]
- Guo H, Duyzend MH, Coe BP, Baker C, Hoekzema K, Gerdts J, Turner TN, Zody MC, Beighley JS, Murali SC, Nelson BJ, University of Washington Center for Mendelian Genomics; Bamshad MJ, Nickerson DA, Bernier RA, Eichler EE (2019) Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genet Med 21:1611–1620. 10.1038/s41436-018-0380-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisaoka T, Komori T, Kitamura T, Morikawa Y (2018) Abnormal behaviours relevant to neurodevelopmental disorders in Kirrel3-knockout mice. Sci Rep 8:12 10.1038/s41598-018-19844-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber TB, Schmidts M, Gerke P, Schermer B, Zahn A, Hartleben B, Sellin L, Walz G, Benzing T (2003) The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J Biol Chem 278:13417–13421. 10.1074/jbc.C200678200 [DOI] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, Smith JD, Paeper B, Nickerson DA, Dea J, Dong S, Gonzalez LE, Mandell JD, Mane SM, Murtha MT, Sullivan CA, et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515:216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaco AD, Lin MZ, Dubi N, Comoletti D, Miller MT, Camp S, Ellisman M, Butko MT, Tsien RY, Taylor P (2010) Neuroligin trafficking deficiencies arising from mutations in the α/β-hydrolase fold protein family. J Biol Chem 285:28674–28682. 10.1074/jbc.M110.139519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jüngling K, Eulenburg V, Moore R, Kemler R, Lessmann V, Gottmann K (2006) N-cadherin transsynaptically regulates short-term plasticity at glutamatergic synapses in embryonic stem cell-derived neurons. J Neurosci 26:6968–6978. 10.1523/JNEUROSCI.1013-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, Richard G, Compton JG, Fuller AE, Gliem TJ, Huang S, Collinson MN, Beal SJ, Ackley T, Pickering DL, Golden DM, et al. (2011) An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 13:777–784. 10.1097/GIM.0b013e31822c79f9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnoodi J, Sigmundsson K, Öfverstedt LG, Skoglund U, Öbrink B, Wartiovaara J, Tryggvason K (2003) Nephrin promotes cell-cell adhesion through homophilic interactions. Am J Pathol 163:2337–2346. 10.1016/S0002-9440(10)63590-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SY, Lee YK, Park S, Choi JS, Lee CJ, Kim HS, Choi YB, Scheiffele P, Bailey CH, Kandel ER, Kim JH (2008) Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proc Natl Acad Sci USA 105:9087–9092. 10.1073/pnas.0803448105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, Gyobu H, Ueno H, Kitamura T, Senba E, Morikawa Y (2008) Expression of kin of irregular chiasm-like 3/mKirre in proprioceptive neurons of the dorsal root ganglia and its interaction with nephrin in muscle spindles. J Comp Neurol 511:92–108. 10.1002/cne.21838 [DOI] [PubMed] [Google Scholar]
- Kreisköther N, Reichert N, Buttgereit D, Hertenstein A, Fischbach KF, Renkawitz-Pohl R (2006) Drosophila Rolling pebbles colocalises and putatively interacts with alpha-Actinin and the Sls isoform Zormin in the Z-discs of the sarcomere and with Dumbfounded/Kirre, alpha-Actinin and Zormin in the terminal Z-discs. J Muscle Res Cell Motil 27:93–106. 10.1007/s10974-006-9060-y [DOI] [PubMed] [Google Scholar]
- Leblond CS, Cliquet F, Carton C, Huguet G, Mathieu A, Kergrohen T, Buratti J, Lemière N, Cuisset L, Bienvenu T, Boland A, Deleuze JF, Stora T, Biskupstoe R, Halling J, Andorsdóttir G, Billstedt E, Gillberg C, Bourgeron T (2019) Both rare and common genetic variants contribute to autism in the Faroe Islands. NPJ Genom Med 4:1. 10.1038/s41525-018-0075-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176. 10.1038/nature05453 [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang L, Guo H, Shi L, Zhang K, Tang M, Hu S, Dong S, Liu Y, Wang T, Yu P, He X, Hu Z, Zhao J, Liu C, Sun ZS, Xia K (2017) Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol Psychiatry 22:1282–1290. 10.1038/mp.2017.140 [DOI] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey AR, Potter SC, Finn RD, Lopez R (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47:W636–W641. 10.1093/nar/gkz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EA, Muralidhar S, Wang Z, Cervantes DC, Basu R, Taylor MR, Hunter J, Cutforth T, Wilke SA, Ghosh A, Williams ME (2015) The intellectual disability gene Kirrel3 regulates target-specific mossy fiber synapse development in the hippocampus. eLife 4:e09395. 10.7554/eLife.09395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EA, Woodruff D, Rawson RL, Williams ME (2017) Examining hippocampal mossy fiber synapses by 3D electron microscopy in WT and Kirrel3 knockout mice. eNeuro 4:ENEURO.0088-17.2017 10.1523/ENEURO.0088-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, Wu W, Corominas R, Peoples A, Koren A, Gore A, Kang S, Lin GN, Estabillo J, Gadomski T, Singh B, et al. (2012) Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151:1431–1442. 10.1016/j.cell.2012.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485:242–245. 10.1038/nature11011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özkan E, Chia PH, Wang RR, Goriatcheva N, Borek D, Otwinowski Z, Walz T, Shen K, Garcia KC (2014) Extracellular architecture of the SYG-1/SYG-2 adhesion complex instructs synaptogenesis. Cell 156:482–494. 10.1016/j.cell.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince JE, Brignall AC, Cutforth T, Shen K, Cloutier JF (2013) Kirrel3 is required for the coalescence of vomeronasal sensory neuron axons into glomeruli and for male-male aggression. Development 140:2398–2408. 10.1242/dev.087262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo S, Langer NJ, Nelson KA, Aoto J (2019) Modeling a neurexin-3α human mutation in mouse neurons identifies a novel role in the regulation of transsynaptic signaling and neurotransmitter release at excitatory synapses. J Neurosci 39:9065–9082. 10.1523/JNEUROSCI.1261-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins EM, Krupp AJ, Perez de Arce K, Ghosh AK, Fogel AI, Boucard A, Südhof TC, Stein V, Biederer T (2010) SynCAM 1 adhesion dynamically regulates synapse number and impacts plasticity and learning. Neuron 68:894–906. 10.1016/j.neuron.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JD, Choi SY, Cho YS, Choi TY, Park JS, Cutforth T, Chung W, Park H, Lee D, Kim MH, Lee Y, Mo S, Rhee JS, Kim H, Ko J, Choi SY, Bae YC, Shen K, Kim E, Han K (2017) Increased excitatory synaptic transmission of dentate granule neurons in mice lacking PSD-95-interacting adhesion molecule Neph2/Kirrel3 during the early postnatal period. Front Mol Neurosci 10:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, Reichenberg A (2017) The heritability of autism spectrum disorder. JAMA 318:1182–1184. 10.1001/jama.2017.12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Yamagata M (2009) Many paths to synaptic specificity. Annu Rev Cell Dev Biol 25:161–195. 10.1146/annurev.cellbio.24.110707.175402 [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T (2000) Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101:657–669. 10.1016/s0092-8674(00)80877-6 [DOI] [PubMed] [Google Scholar]
- Schroeder A, Vanderlinden J, Vints K, Ribeiro LF, Vennekens KM, Gounko NV, Wierda KD, de Wit J (2018) A modular organization of LRR protein-mediated synaptic adhesion defines synapse identity. Neuron 99:329–344.e7. 10.1016/j.neuron.2018.06.026 [DOI] [PubMed] [Google Scholar]
- Sellin L, Huber TB, Gerke P, Quack I, Pavenstädt H, Walz G (2003) NEPH1 defines a novel family of podocin interacting proteins. FASEB J 17:115–117. 10.1096/fj.02-0242fje [DOI] [PubMed] [Google Scholar]
- Serizawa S, Miyamichi K, Takeuchi H, Yamagishi Y, Suzuki M, Sakano H (2006) A neuronal identity code for the odorant receptor-specific and activity-dependent axon sorting. Cell 127:1057–1069. 10.1016/j.cell.2006.10.031 [DOI] [PubMed] [Google Scholar]
- Shen K, Bargmann CI (2003) The immunoglobulin superfamily protein SYG-1 determines the location of specific synapses in C. elegans. Cell 112:619–630. 10.1016/S0092-8674(03)00113-2 [DOI] [PubMed] [Google Scholar]
- Shen K, Fetter RD, Bargmann CI (2004) Synaptic specificity is generated by the synaptic guidepost protein SYG-2 and its receptor, SYG-1. Cell 116:869–881. 10.1016/s0092-8674(04)00251-x [DOI] [PubMed] [Google Scholar]
- Shipman SL, Nicoll RA (2012) A subtype-specific function for the extracellular domain of neuroligin 1 in hippocampal LTP. Neuron 76:309–316. 10.1016/j.neuron.2012.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperry RW. (1963) Chemoaffinity in the orderly growth of nerve fiber patterns and connections. Proc Natl Acad Sci USA 50:703–710. 10.1073/pnas.50.4.703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. (2018) Towards an understanding of synapse formation. Neuron 100:276–293. 10.1016/j.neuron.2018.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM, Pereira S, Ruderfer D, Kirby A, Ripke S, Harris DJ, Lee JH, Ha K, Kim HG, Solomon BD, Gropman AL, et al. (2012) Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149:525–537. 10.1016/j.cell.2012.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanigawa S, Islam M, Sharmin S, Naganuma H, Yoshimura Y, Haque F, Era T, Nakazato H, Nakanishi K, Sakuma T, Yamamoto T, Kurihara H, Taguchi A, Nishinakamura R (2018) Organoids from nephrotic disease-derived iPSCs identify impaired NEPHRIN localization and slit diaphragm formation in kidney podocytes. Stem Cell Rep 11:727–740. 10.1016/j.stemcr.2018.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tick B, Bolton P, Happé F, Rutter M, Rijsdijk F (2016) Heritability of autism spectrum disorders: a meta-analysis of twin studies. J Child Psychol Psychiatry 57:585–595. 10.1111/jcpp.12499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnu S, Hertenstein A, Betschinger J, Knoblich JA, Gert de Couet H, Fischbach KF (2006) The adaptor protein X11Lα/Dmint1 interacts with the PDZ-binding domain of the cell recognition protein Rst in Drosophila. Dev Biol 289:296–307. 10.1016/j.ydbio.2005.09.016 [DOI] [PubMed] [Google Scholar]
- Völker LA, Maar BA, Guevara BA, Bilkei-Gorzo A, Zimmer A, Brönneke H, Dafinger C, Bertsch S, Wagener JR, Schweizer H, Schermer B, Benzing T, Hoehne M (2018) Neph2/Kirrel3 regulates sensory input, motor coordination, and home-cage activity in rodents. Genes Brain Behav 17:e12516. 10.1111/gbb.12516 [DOI] [PubMed] [Google Scholar]
- Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, Turner TN, Liu Y, Zhao W, Hoekzema K, Vives L, Xia L, Tang M, Ou J, Chen B, Shen Y, Xun G, Long M, Lin J, Kronenberg ZN, et al. (2016) De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 7:13316. 10.1038/ncomms13316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, Wilke SA, Daggett A, Davis E, Otto S, Ravi D, Ripley B, Bushong EA, Ellisman MH, Klein G, Ghosh A (2011) Cadherin-9 regulates synapse-specific differentiation in the developing hippocampus. Neuron 71:640–655. 10.1016/j.neuron.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Merico D, Cao H, Pellecchia G, Alipanahi B, Thiruvahindrapuram B, Tong X, Sun Y, Cao D, Zhang T, Wu X, Jin X, Zhou Z, Liu X, Nalpathamkalam T, Walker S, Howe JL, Wang Z, MacDonald JR, Chan A, et al. (2016) Genome-wide characteristics of de novo mutations in autism. NPJ Genom Med 1:16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Milunsky JM, Newton S, Ko J, Zhao G, Maher TA, Tager-Flusberg H, Bolliger MF, Carter AS, Boucard AA, Powell CM, Südhof TC (2009) A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J Neurosci 29:10843–10854. 10.1523/JNEUROSCI.1248-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]