

Abstract
Mechanisms of enzymatic epoxidation via oxygen atom transfer (OAT) to an olefin moiety is mainly derived from the studies on thiolate-heme containing epoxidases, such as cytochrome P450 epoxidases. The molecular basis of epoxidation catalyzed by non-heme-iron enzymes is much less explored. Herein, we present a detailed study on epoxidation catalyzed by the non-heme iron- and 2-oxoglutarate-dependent (Fe/2OG) oxygenase, AsqJ. The native substrate and analogs with different para substituents ranging from electron-donating groups (e.g. methoxy) to electron-withdrawing groups (e.g. trifluoromethyl) were used to probe the mechanism. The results derived from transient-state enzyme kinetics, Mössbauer spectroscopy, reaction product analysis, X-ray crystallography, density functional theory calculations and molecular dynamic simulations collectively revealed the following mechanistic insights: 1) The rapid O2 addition to the AsqJ Fe(II) center occurs with the iron-bound 2OG adopting an online-binding mode in which the C1 carboxylate group of 2OG is trans to the proximal histidine (His134) of the 2-His-1-carboxylate facial triad, instead of assuming the offline-binding mode with the C1 carboxylate group trans to the distal histidine (His211); 2) The decay rate constant of the ferryl intermediate is not strongly affected by the nature of the para substituents of the substrate during the OAT step, a reactivity behavior that is drastically different from non-heme Fe(IV)-oxo synthetic model complexes; 3) The OAT step most likely proceeds through a step-wise process with the initial formation of C(benzylic)-O bond to generate an Fe(III)-alkoxide species, which is observed in the AsqJ crystal structure. The subsequent C3-O bond formation completes the epoxide installation.
Introduction
The epoxide (oxirane) functional group is widely distributed in natural products with a broad spectrum of biological activities, including antibacterial, antifungal, antiviral, and antitumor activities.1 Furthermore, installation of epoxide moiety is an important and useful synthetic strategy to achieve structural and functional versatility in organic synthesis.2–6
In nature, two main strategies are utilized to install epoxide, namely formal dehydrogenation by cleaving a C-H and an O-H bond on two adjacent carbons, and an oxygen atom insertion, a.k.a. oxygen atom transfer (OAT), reaction onto an olefin moiety of the substrate (Scheme 1).1,7 For both strategies, molecular oxygen (O2) and in some cases H2O2, is utilized as an oxidant, a source of oxygen, or playing a dual role.1,8–17 Usually, the epoxidation is initiated by a highly reactive intermediate based on flavin,1 thiolate-heme,13–16 non-heme di-iron,8,10–12 or non-heme mononuclear iron.18–22 For non-heme mononuclear iron enzymes, epoxidation through the dehydrogenation pathway has been reported in the biosyntheses of scopolamine by hyoscyamine 6β-hydroxylase (H6H),18 fosfomycin by 2-hydroxyl propyl phosphonate epoxidase (HppE),19 clavulanic acid by clavamate synthase (CAS),23–26 and loline by LolO.27 Alternatively, epoxidation through the oxygen atom insertion is reported in the biosynthesis of Nβ-epoxy-succinamoyl-DAP-Val by DdaC,20 pentalenolactone by PenD (PntD) and PtlD,21,28 and quinolone alkaloid by AsqJ (Scheme 1).22 While both pathways require the formation of an iron(IV)-oxo (or ferryl) intermediate to initiate epoxidation, only recently such a species has been experimentally elucidated by us in the AsqJ catalyzed epoxide installation.28 AsqJ, together with H6H, CAS, LolO, and DdaC, belongs to the iron(II) and 2-oxoglutarate (Fe/2OG) dependent enzyme superfamily, whose members catalyze a broad array of transformations and are involved in many important natural product biosyntheses.29 Notably, AsqJ catalyzes stepwise desaturation and epoxidation in the biosynthesis of a quinolone-type fungal alkaloid, 4’-methoxy-viridicatin (Scheme 1).22
Scheme 1.
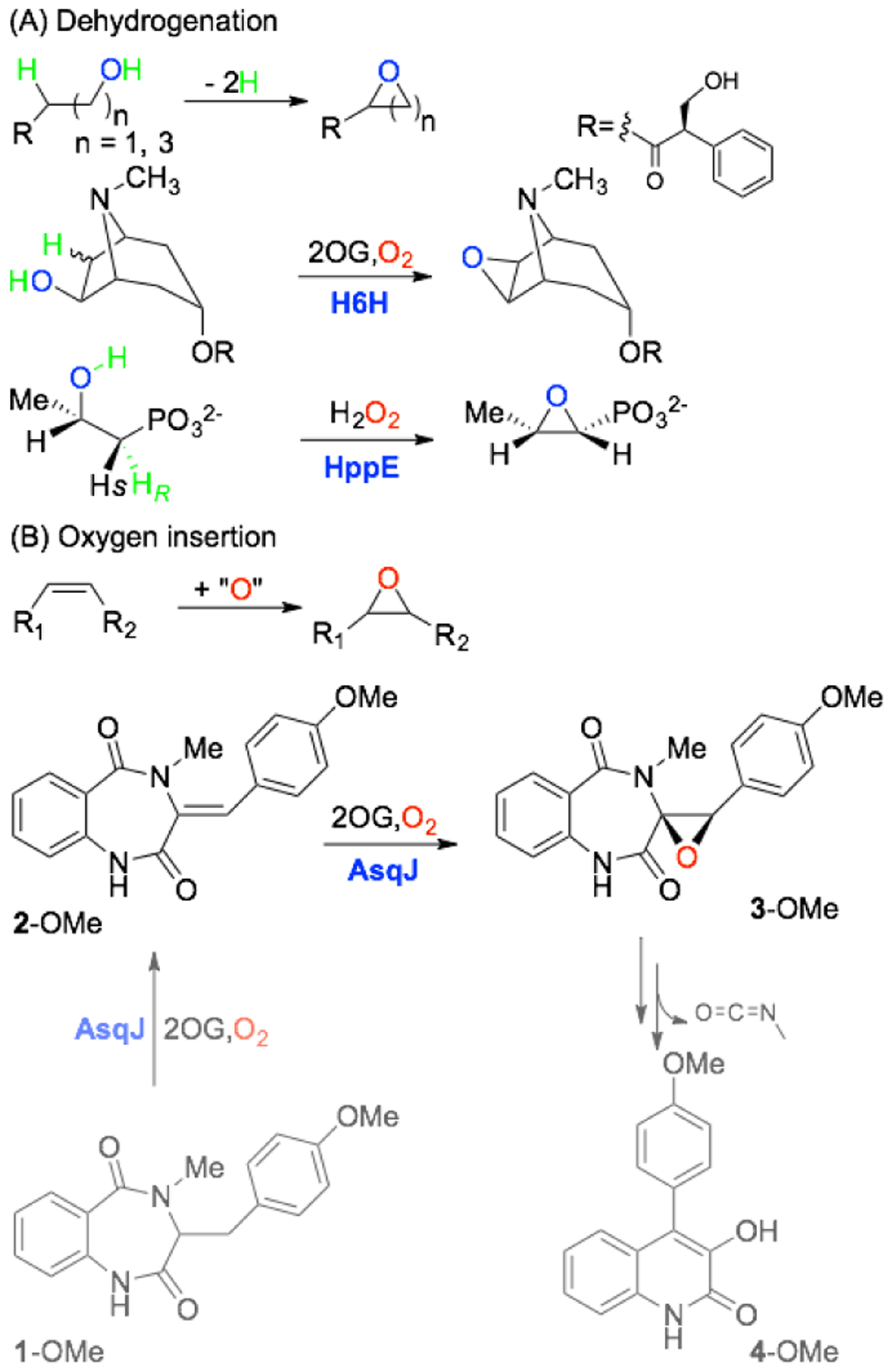
Examples of Epoxidation Catalyzed by Non-Heme Iron Enzymes via (A) Dehydrogenation and (B) Oxygen Atom Insertion.
Mechanistic understanding of enzyme-catalyzed epoxidation through the oxygen atom insertion onto an olefin moiety is mainly derived from the studies on thiolate-heme containing epoxidases, such as choloroperoxidases (CPOs) and cytochrome P450s.14 Briefly, the C-O bonds of an epoxide can be installed in a concerted fashion or a stepwise manner via a discrete radical or cation intermediate. A concerted mechanism is supported by the observation of retention of the olefin stereochemistry reported in P450 epoxidases. The stepwise mechanism with the involvement of a putative cation intermediate is implicated by the formation of the carbonyl product through a hydride or a chloride migration to the adjacent carbon.30–32 A cation-triggered phosphono group migration has also been reported for the non-heme iron enzyme HppE in the presence of a nonnative substrate.9 The possible involvement of a radical intermediate is suggested by the formation of alkylated porphyrin side product via alkylation of a pyrrole nitrogen of the porphyrin heme framework by the intermediate substrate radical species in some P450 epoxidases.33–35 It is generally accepted that the key intermediate in olefin epoxidation of CPOs and P450s is an iron(IV)-oxo porphyrin-cation-radical species, Compound I. To explain the energetic feasibility of these two competing mechanisms (concerted v.s. stepwise), computational studies on P450 Compound I catalyzed epoxidation suggest that the doublet- (S = 1/2) and quartet-spin (S = 3/2) states of Compound I are energetically close and both are suitable for epoxidation.34–39 The utilization of the doublet spin state of Compound I could result in a concerted epoxidation while the use of the quartet spin state intermediate is proposed to proceed through the stepwise mechanism.34,35 In addition, the Compound I related model complexes40–43 and the non-heme iron(IV)-oxo synthetic mimics44–46 generally exhibit strong electrophilic nature during epoxidation. Specifically, the studies conducted by using para-substituted styrenes revealed that the electron-donating groups (EDGs) could significantly increase the rate constant of the OAT reaction (by monitoring the decay rate constant of Compound I or iron(IV)-oxo species), while the electron-withdrawing groups (EWGs) exhibited the opposite effect, and the rate constant could be modulated by 2 order of magnitudes. However, Newcomb and coworkers showed that the OAT reaction rate constant was largely independent of para substitutions.47 More strikingly, a V-shaped Hammett plot was observed when a Mn(V)-oxo complex, [MnV(O)(TBP8Cz)(CN)]−, was examined, indicating a change of mechanism when going from EDGs to EWGs where the proposed high-valent metal-oxo species character shifts from electrophilic to weak nucleophilic, respectively.48
Compared to significant research efforts in elucidating plausible epoxidation mechanisms employed by thiolate-heme containing epoxidases, the molecular basis of non-heme-iron-enzyme-catalyzed epoxidation is much less studied. Inspired by the literature precedents, several possible reaction pathways (a step-wise mechanism via a cation intermediate, pathway i, or a radical intermediate, pathway ii, and a concerted mechanism, pathway iii) can be envisioned and are depicted in Scheme 2. Herein, we use AsqJ as an example to study detailed reaction mechanism of non-heme-iron-enzyme-catalyzed epoxidation by using a multi-faceted approach, including LC-MS analysis of the enzymatic reactions, transient-state kinetics, spectroscopic characterizations using Mössbauer and electron paramagnetic resonance (EPR), X-ray crystallography, density functional theory (DFT) calculations and molecular dynamics (MD) simulations. AsqJ native substrate (2-OMe) and several para-substituted analogs (2-H, 2-F and 2-CF3) were used in this study to probe the reactivity and plausible epoxidation mechanism. The transient-state kinetics and Mössbauer results showed that the the decay rate constant of the Fe(IV)-oxo intermediate is largely insensitive to the para substitutions on the enzyme substrate. This behavior is in contrast with the OAT reactivity exhibited by non-heme Fe(IV)-oxo synthetic complexes. In addition, the LC-MS analyses and x-ray crystallographic results strongly suggested that a step-wise mechanism is operative in AsqJ catalyzed epoxidation, where the Fe(IV)-oxo intermediate reacts with the olefin group of the substrates by first attacking the C10 (benzylic) position of the substrate. DFT calculations and MD simulations provide further support for the experimental observations.
Scheme 2.
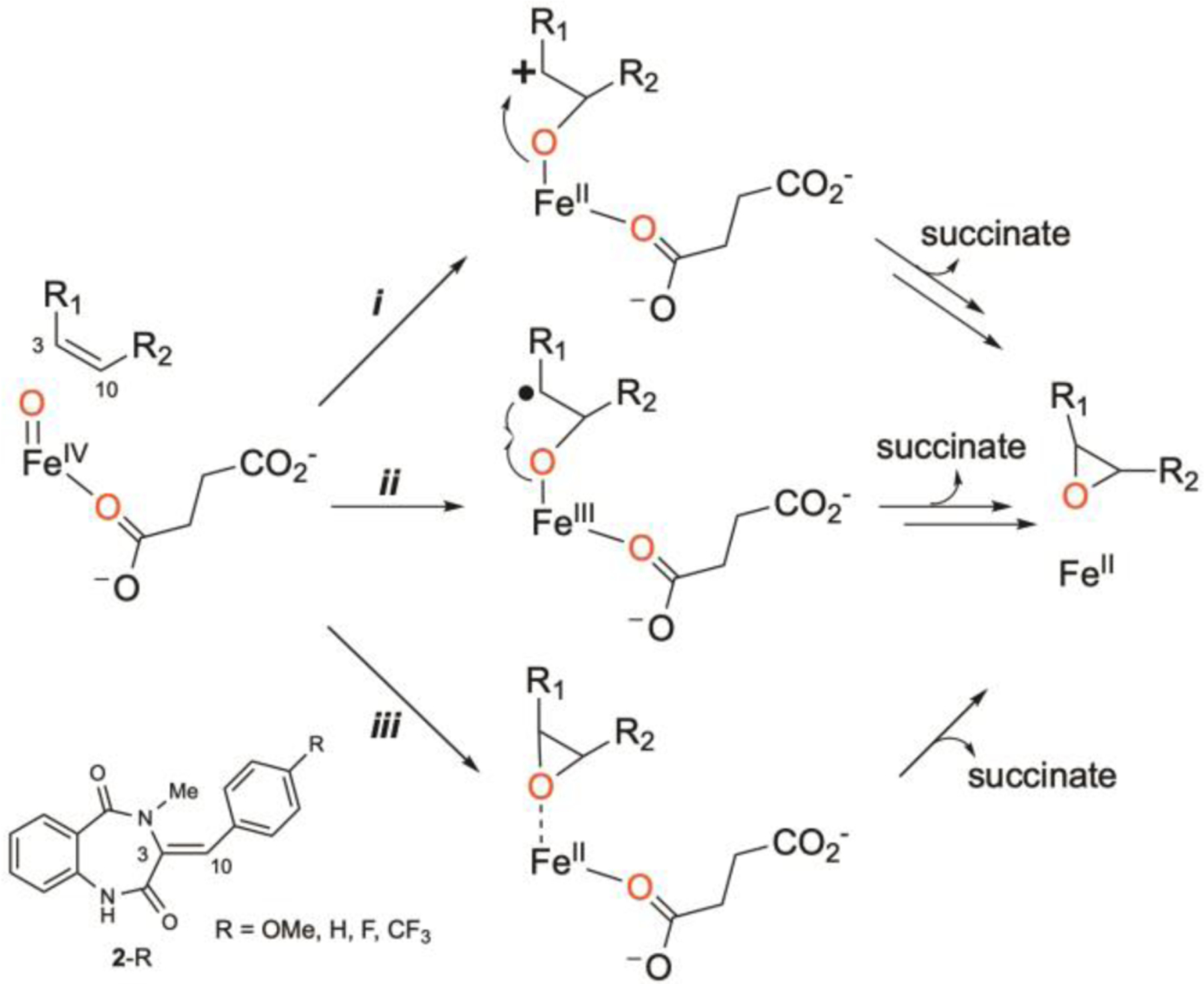
Proposed Reaction Mechanisms of Epoxidation via Oxygen Atom Transfer (OAT).
Results and Discussion.
Synthesis and Characterization of 2-R (R = OMe, H, F, CF3)
The synthesis of substrate analogs (2-R, R = OMe, H, F, and CF3) was carried out by using the published procedure with different para-substituted benzaldehyde as the starting material.49 The 1H NMR data of 2-OMe and 2-H in CDCl3 are consistent with the literature reported values. The 1H and 13C NMR data of 2-F and 2-CF3 are reported in the Supporting Information with spectra shown in Figures S1 and S2. Importantly, the 1H chemical shift at C10 of 2-R (~6.9 – 7.0 ppm) exhibits downfield shift in going from 2-OMe to 2-CF3 (Table S1) and is consistent with the expected para-substituent effect on the olefin moiety of 2-R in going from EDGs to EWGs.
Binding Affinity of 2-R to AsqJ.
To study AsqJ catalyzed epoxidation reaction mechanism, we first establish that the binding affinity of the analogs (2-H, 2-F, and 2-CF3) to AsqJ is not perturbed due to the para-substitution. The data indicate that the substrate and the analogs bind to AsqJ with similar affinity came from Mössbauer spectral analysis. With addition of only one equivalent (relative to the Fe2+-loaded enzyme) of 2-OMe or the analogs (2-H, 2-F, and 2-CF3), the Mössbauer spectrum (Figure S3) was completely converted from a broad quadrupole doublet (δ = 1.25 mm/s, |ΔEQ| = 2.26 mm/s, linewidth 0.51 mm/s), representing the AsqJ⋅Fe2+⋅2OG complex, to a sharp quadrupole doublet (δ = 1.25 mm/s, |ΔEQ| = 2.54 mm/s, linewidth, 0.30 mm/s). In addition, a Voigt line shape (a convolution of 50% Gaussian and 50% Lorentzian) had to be used to achieve satisfactory simulation for the AsqJ⋅Fe2+⋅2OG complex, as compared to a Lorentzian line shape used for the AsqJ⋅Fe2+⋅2OG⋅substrate quaternary complex, indicating substantial distribution of parameters in the former sample. This observation indicates that the binding of the substrate perturbs the electronic structure of the Fe(II) center, and the AsqJ⋅Fe2+⋅2OG⋅substrate quaternary complex was fully formed with the addition of stoichiometric amount of substrate. To further validate this result, Mn2+ loaded AsqJ was used to carry out substrate titration on 2-CF3. The ability of Mn2+ ion binding to the metal binding site of Fe/2OG enzymes has been reported for many crystal structures on Fe/2OG enzymes where Mn2+ was used as an alternative metal ion to reconstitute the enzymes. Indeed, when Mn2+ was incubated with apo AsqJ and 2OG (0.5 mM Mn2+, 0.75 mM AsqJ, and 5 mM 2OG), the resulted EPR signal was drastically different from the EPR signal of free Mn2+ (0.5 mM) in buffer solution (Figure 1). This type of spectroscopic change has been documented in the literature,50–53 and attributed to the change of the coordination environment of Mn2+ from a symmetric octahedral ligand field in the aqueous solution (with a close to zero axial zero field splitting parameter, D ~ 0) to a distorted octahedral environment in the protein metal binding site (with increased D values). Consistent with our experimental observation, the spectral simulations revealed that the AsqJ bound Mn2+ signal (in the presence of 2OG) originated from a single species with |D| ~ 0.08 cm−1, E/D ~ 0.20, and |A(Mn)| = 244 MHz. When an excess amount of 2-CF3 (2 eq. related to Mn2+ concentration) was introduced into the AsqJ-Mn2+−2OG complex, the Mn2+ EPR signal exhibited a subtle but clear change, suggesting that the presence of substrate further perturbs the metal site and indicates the formation of the AsqJ⋅Mn2+⋅2OG⋅2-CF3 quaternary complex. Spectral simulation showed that this new EPR signal originated from a single species with |D| ~ 0.08 cm−1, E/D ~ 0.16, and |A(Mn)| = 255 MHz. In addition, when the AsqJ⋅Mn2+⋅2OG⋅2-CF3 complex was exposed to air for 20 min, no obvious change was observed, indicating that the Mn2+ loaded quaternary complex does not readily react with O2. Based on the two EPR signals recorded under the conditions with and without substrate, we carried out the substrate titration on 2-CF3. The titration suggested that the dissociation constant (Kd) is ~ 0.005 mM for 2-CF3 (Figure S4). Thus taken together, both Mössbauer and EPR results confirm that all the substrate and analogs bind to AsqJ tightly.
Figure 1.
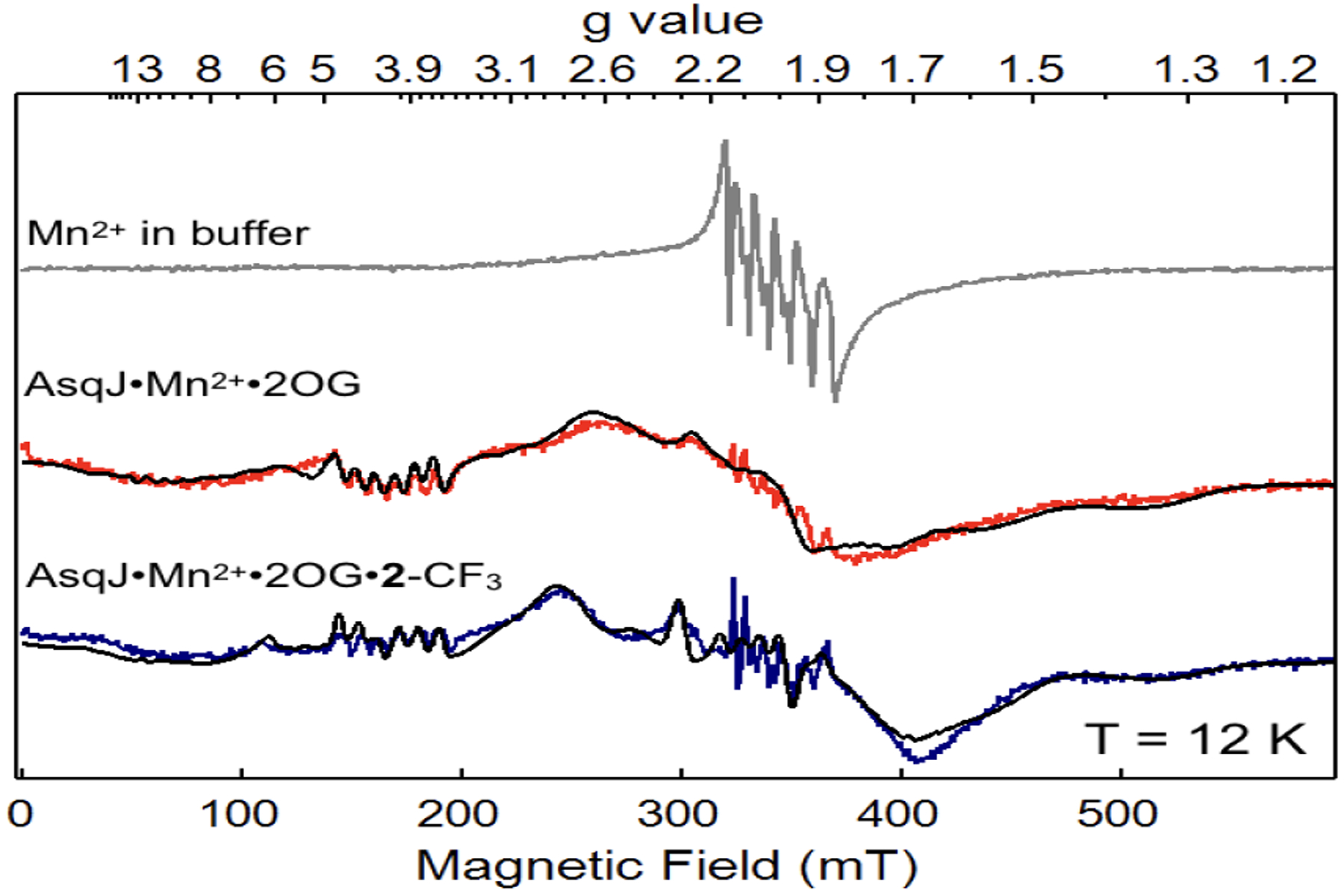
X-band EPR signals from samples containing Mn2+ in buffer solution (top, grey), AsqJ⋅Mn2+⋅2OG ternary complex (middle, red), and AsqJ⋅Mn2+⋅2OG⋅2-CF3 quaternary complex (bottom, blue). The corresponding spectral simulations are shown in black curves. The simulation parameters are for S = 5/2, I = 5/2. For the ternary complex (middle), g = 2.03, D = 0.08 cm−1, E/D = 0.20, |AMn| = 243 MHz; for the quaternary complex (bottom), g = 2.02, D = 0.08 cm−1, E/D = 0.13, |AMn| = 254 MHz. Measurement conditions: microwave frequency, 9.64 GHz; microwave power, 20 uW; modulation frequency, 100 kHz; modulation amplitude, 1 mT; temperature, 12 K.
Kinetic Analysis of AsqJ Catalyzed Epoxidation Reaction.
To further probe the mechanism, kinetics study of the AsqJ epoxidation reaction was carried out by rapidly mixing the anaerobic AsqJ⋅Fe2+⋅2OG⋅substrate complex with oxygenated buffer and recording time-dependent UV-Vis spectra using stopped-flow absorption spectroscopy (SF-Abs). We took advantage of the tight binding of the substrate analogs to AsqJ to conduct experiments under single-turnover conditions (0.12 mM enzyme, 0.1 mM Fe2+, 0.1 mM substrate (2-OMe, 2-H, 2-F, and 2-CF3), 4 mM α-KG and ~1.8 mM O2 before mixing) at 5 ∘C. When comparing SF-Abs derived kinetic traces, three major features were identified (Figure 2 and S5). 1) A decrease of the absorption band at 280–320 nm (for 2-OMe the band extends to > 330 nm) mainly corresponds to the consumption of the substrate. This assertion was derived from the difference of the absorption features for the substrate and the product in the near UV region.54 2) An initial decrease (up to ~ 0.4s) and subsequent recovery of a band centered at ~470 nm that corresponds to the Fe2+⋅2OG metal-to-ligand charge transfer (MLCT) band. 3) An initial increase (peaking at around 0.03 s) and subsequent decrease of a feature at 330–340 nm is also observed. We propose this latter feature corresponds to the formation and consumption of the Fe(IV)=O intermediate (see below). We next modeled the SF-Abs traces at 310, 330 and 470 nm with a 3-step kinetic model (Scheme 3). A similar kinetic model has been used to describe pre-steady state kinetics of several Fe/2OG enzymes.55–58 Under the current experimental conditions, the reaction likely proceeds through formation of the Fe(IV)=O intermediate via addition of O2 to the AsqJ⋅Fe2+⋅2OG⋅substrate quaternary complex with the rate constant k1. The Fe(IV)=O intermediate subsequently oxidizes the substrate to generate the AsqJ⋅Fe2+⋅succinate⋅product complex with the rate constant of k2, which in turn releases succinate and the product and regenerates the AsqJ⋅Fe2+⋅2OG ternary complex in the presence of excess 2OG with the rate constant of k3.
Figure 2.
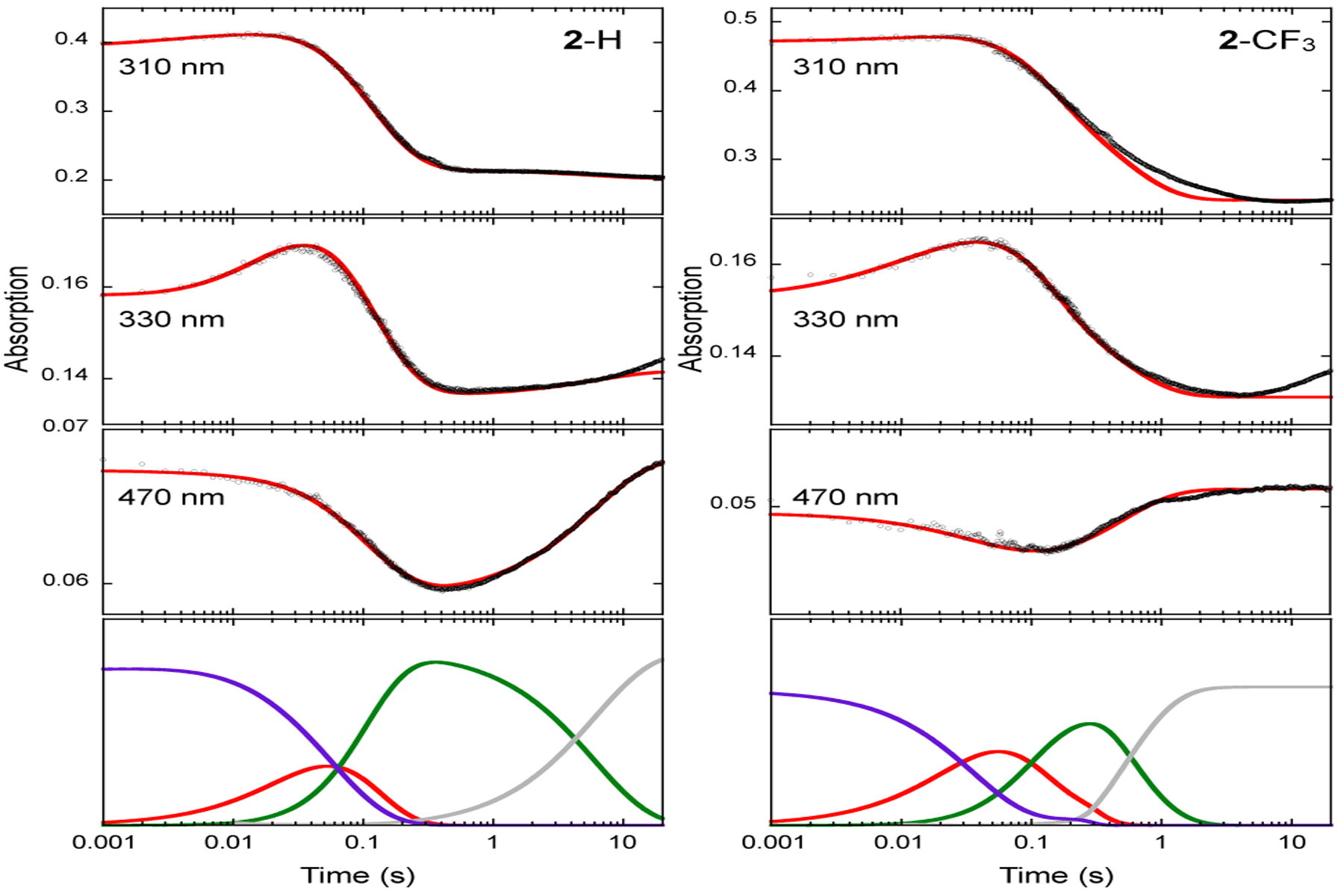
SF-Abs derived kinetic traces of the time-dependent absorption changes at various wavelengths in the AsqJ catalyzed epoxidation by using 2-H (left panel) and 2-CF3 (right panel) as the substrates. Experimental data are shown in black dots, the corresponding kinetic simulations are shown in red curves, and the corresponding rate constants are shown in Table 1. The kinetics of individual species are shown at the bottom of both panels: purple, the AsqJ•Fe2+•2OG•substrate complex; red, the Fe(IV)=O intermediate; green, the AsqJ-Fe2+-succinate-product complex; grey, the AsqJ•Fe2+•2OG complex. The results on the reactions with 2-OMe and 2-F are shown in Figure S5.
Scheme 3.
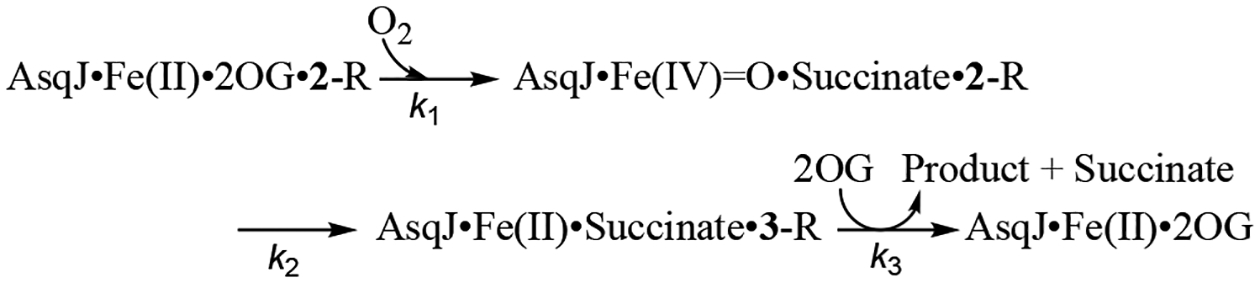
Three-Step Kinetic Model Used for Analyzing AsqJ Catalyzed Epoxidation.
The SF-Abs data and the kinetic simulations suggest that the pre-steady state kinetics are very similar when 2-OMe, 2-H, or 2-F are used as the substrate as shown in Figure 2 and Figure S5 (the simulation parameters are shown in Table 1). The O2 addition rate constant (k1) is further verified by SF-Abs data measured by using 2-H as the substrate and under varying O2 conditions (Figure S6). The formation and decay kinetics of the putative Fe(IV)=O intermediate is reflected by the 330 nm kinetic traces. The maximum accumulation of this intermediate is occurred at ~ 0.03 – 0.04 s, which is consistent with the previous published Mössbauer results.49 This observation further validates the assignment that the 330 nm trace represents mainly the Fe(IV)=O intermediate. However, for 2-CF3, the enzyme kinetics is altered. This is clearly shown by the kinetic trace at 470 nm, which mainly reflects the kinetics of the AsqJ quaternary and ternary complexes (Figure 2). Compared with the results obtained by using other analogs, both the decay and the reformation rates of 470 nm absorption are faster and the extent of absorption change in the reaction is only ~ 1/3 to that with 2-H. Simulation using a three-step kinetic model at three wavelengths (470nm, 330nm, and 310 nm) suggests that 2-CF3 has a faster formation rate constant (~ 2–3 times) and a slower decay rate constant (~ 2–3 times) of the Fe(IV)=O intermediate than those derived from the reaction with 2-H. The conversion rate constant of the enzyme product complex to the enzyme ternary complex is also faster (5–10 times) in the reaction with 2-CF3 (Table 1). These results indicate that the product dissociation from the enzyme binding pocket might be more rapid when 2-CF3 is used. In addition, an appreciable amount of the AsqJ⋅Fe2+⋅2OG⋅2-CF3 complex may not (or very slowly) react with O2 during the time period monitored by SF-Abs, which provides a possible explanation to the small absorbance changes at ~ 470nm in the reaction with 2-CF3.
Table 1.
Simulated Kinetic Constants Based on the 3-Step Kinetic Model.
| Substrate | k1 (mM−1 s−1) | k2 (s−1) | k3 (s−1) |
|---|---|---|---|
| 2-H | ~14 | 33 | 0.1 |
| 2-F | ~12 | 28 | 0.15 |
| 2-OMe | ~12 | 42 | 0.20 |
| 2-CF3 | ~30 | 12 | 1.2 |
Freeze-Quench-Coupled Mössbauer Analysis for the AsqJ Reaction with 2-CF3.
To further validate the kinetic model derived from the SF-Abs experiments, freeze-quench coupled Mössbauer analysis was carried out for the AsqJ reaction with 2-CF3. The anaerobic solution containing the AsqJ⋅Fe2+⋅2OG⋅2-CF3 complex (final concentration: 0.75 mM AsqJ, 0.6 mM Fe2+, 6 mM 2OG, and 0.7 mM 2-CF3) was rapidly mixed with O2 saturated buffer (final concentration: ~ 0.9 mM O2) and quenched at various time points. The samples were subject to Mössbauer measurements, and the resulted spectra are shown in Figure 3. Four time-dependent iron species were observed: (1) a quadrupole doublet representing the AsqJ quaternary complex (δ = 1.25 mm/s |ΔEQ| = 2.54 mm/s, linewidth 0.33 mm/s), (2) a second quadrupole doublet representing the Fe(IV)=O species (δ = 0.31 mm/s |ΔEQ| = 0.68 mm/s, linewidth 0.38 mm/s), (3) a third quadrupole doublet representing possibly the AsqJ enzyme product complex (δ = 1.21 mm/s |ΔEQ| = 2.96 mm/s, linewidth 0.32 mm/s), and (4) a fourth quadrupole doublet representing the AsqJ•Fe(II)•2OG ternary complex (δ = 1.25 mm/s |ΔEQ| = 2.26 mm/s, linewidth 0.55 mm/s with a Voigt line shape) Comparing with the previously published FQ Mössbauer results in the reactions with 2-OMe and 2-H,49 the AsqJ quaternary complex, the Fe(IV)=O species, and the AsqJ enzyme product complex observed here show identical Mössbauer parameters with those of the corresponding iron species in the reactions with 2-OMe and 2-H. However, the time dependent changes of these iron species are different. First, the kinetics of the Fe(IV)=O species and the amount accumulated (~ 20% of the total iron in the sample) are similar to the results from the reaction with 2-OMe and 2-H (in comparison, the maximum amount accumulated in the reactions with 2-OMe or 2-H was ~ 20–30%).49 However, a much smaller amount of the AsqJ quaternary complex was consumed. For example, at the time points (0.01, and 0.03 s) of the maximum accumulation of the Fe(IV)=O species, there are still ~ 60% (relative to the total iron in the sample) AsqJ quaternary complex left in the reaction with 2-CF3, but for 2-OMe and 2-H, the quaternary complex had diminished to ~ 30%.49 Even at a later time (1 s), the AsqJ quaternary complex only reduces to ~ 50% in the 2-CF3 reaction. This reduced consumption of the quaternary complex in the reaction with 2-CF3 is also correlated well with the amount of the enzyme product complex (δ = 1.21 mm/s |ΔEQ| = 2.96 mm/s, linewidth 0.32 mm/s) observed by Mössbauer, which is accumulated to a maximum of ~ 15% at 0.16 s. In addition, the enzyme product complex is rapidly converted to the AsqJ ternary complex (δ = 1.25 mm/s |ΔEQ| = 2.26 mm/s, linewidth 0.55 mm/s) at 1 s in the 2-CF3 reaction, while in the reactions with 2-OMe and 2-H, this enzyme product complex has a much longer lifetime so that at 1 s 70%−80% of the iron in the sample is still representing this species. Finally, ~ 10% O2 insensitive Fe(II) species was observed, which has δ = 1.29 mm/s |ΔEQ| = 3.45 mm/s. It has been attributed to the inactive portion of the enzyme. Thus far, combining the Mössbauer and the SF-Abs results, we can conclude that regardless of the nature of the para substitutions on the phenyl moiety of the substrate, the decay kinetics of the Fe(IV)=O intermediate is not strongly perturbed. Compare to strong electrophilicity demonstrated by synthetic Fe(IV)=O complexes in the OAT reactions, the OAT reactivity of the AsqJ Fe(IV)=O species does not exhibit a similar trend, suggesting that the electron richness of the olefin moiety is not the controlling factor in promoting the OAT reactivity of the ferryl intermediate in AsqJ. The small absorption change at ~ 470 nm in SF-Abs data from the 2-CF3 reaction is most likely the consequence that only a part of the AsqJ quaternary complex reacts rapidly with O2 as shown by the Mössbauer data.
Figure 3.
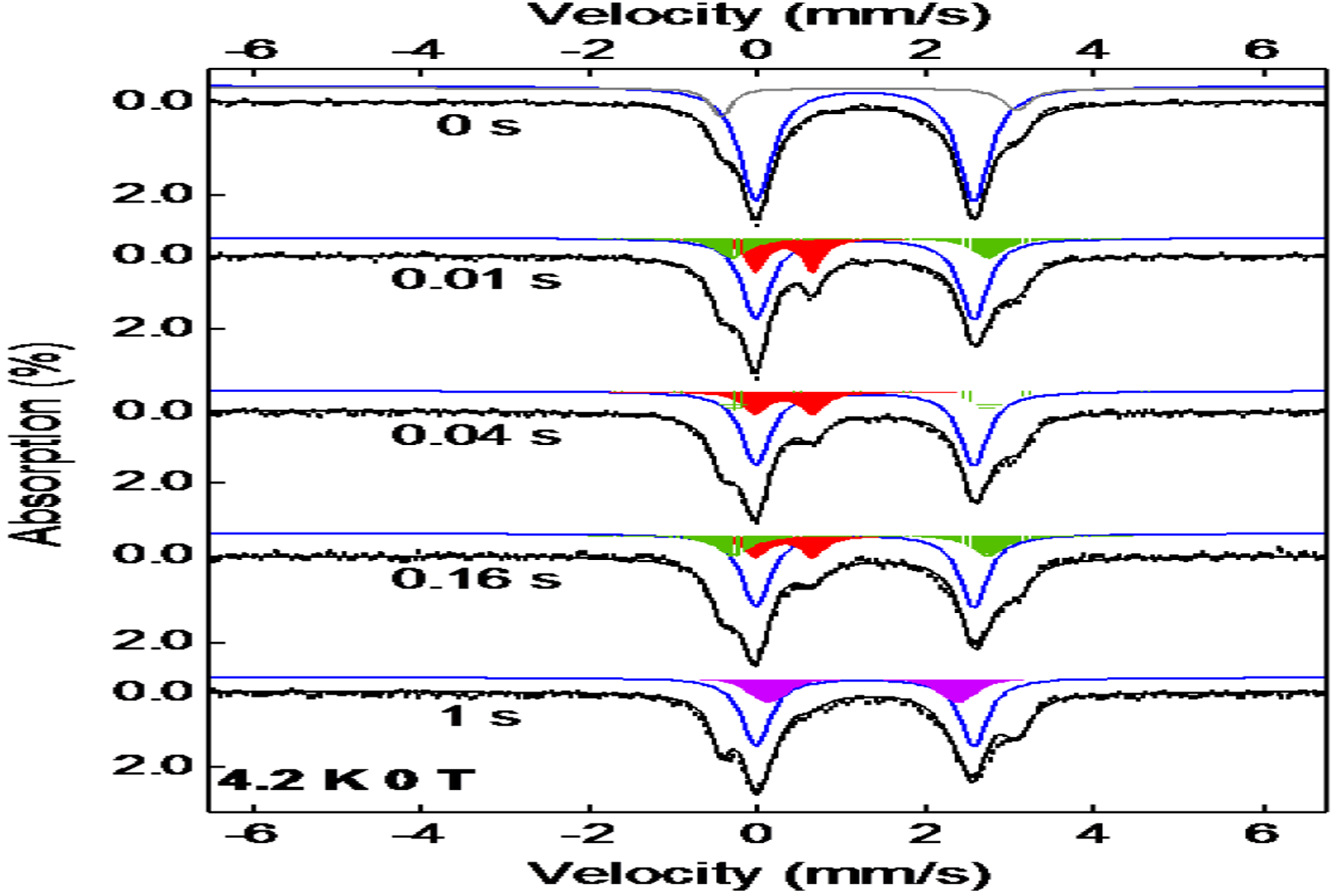
Zero-field Mössbauer spectra of samples obtained by freeze quenching the reaction of AsqJ with 2-CF3 at various time points. Black vertical bars: experimental data; Black lines: overall spectral simulation; the blue lines: the AsqJ⋅Fe2+⋅2OG⋅2-CF3 complex; the red areas: the Fe(IV)=O intermediate; the green areas: the AsqJ product complex; the purple areas: the AsqJ⋅Fe2+⋅2OG complex; the grey line: the inactive species. The simulation parameters and the relative amount of various species observed in different times are listed in the main text and in Table S2.
LC-MS Analysis of AsqJ Reactions with 2-R.
To further corroborate the spectroscopic results on AsqJ-catalyzed epoxidation, enzyme activity assay was carried out under multi-turnover conditions and the results were analyzed by liquid chromatography coupled mass spectrometry (LC-MS). As depicted in Figure 4, all analogs were converted to the corresponding epoxides (3-OMe, 3-H, 3-F and 3-CF3) respectively. Under the current experimental conditions, all analogs were consumed at a similar level. If a cation species centered at C10 is involved in the epoxidation, one anticipates the electron withdrawing groups, e.g. F or CF3, will disfavor the formation of such an intermediate, and likely result in decrease of product formation. However, the observation of a similar level product formation when replacing H with F and further by -CF3 (2-H vs. 2-F and 2-CF3) is not consistent with such a mechanism. In the literature, when a cation is involved in the reaction, the electron withdrawing group, such as F or CF3, usually diminishes the corresponding product formation drastically.59,60 Since no obvious perturbation was observed when different analogs were used, the LC-MS result is thus consistent with a mechanism where the AsqJ catalyzed epoxidation either initiates from the C10 position of the substrate or it starts from C3 center, but does not involve a cationic species at C10. In the latter scenario, the reaction likely proceeds via homolytic cleavage of the C=C bond of the substrate to form the C3-O bond and the C10 radical which is not sensitive to the para-substituent on the phenyl ring of the substrate.
Figure 4.
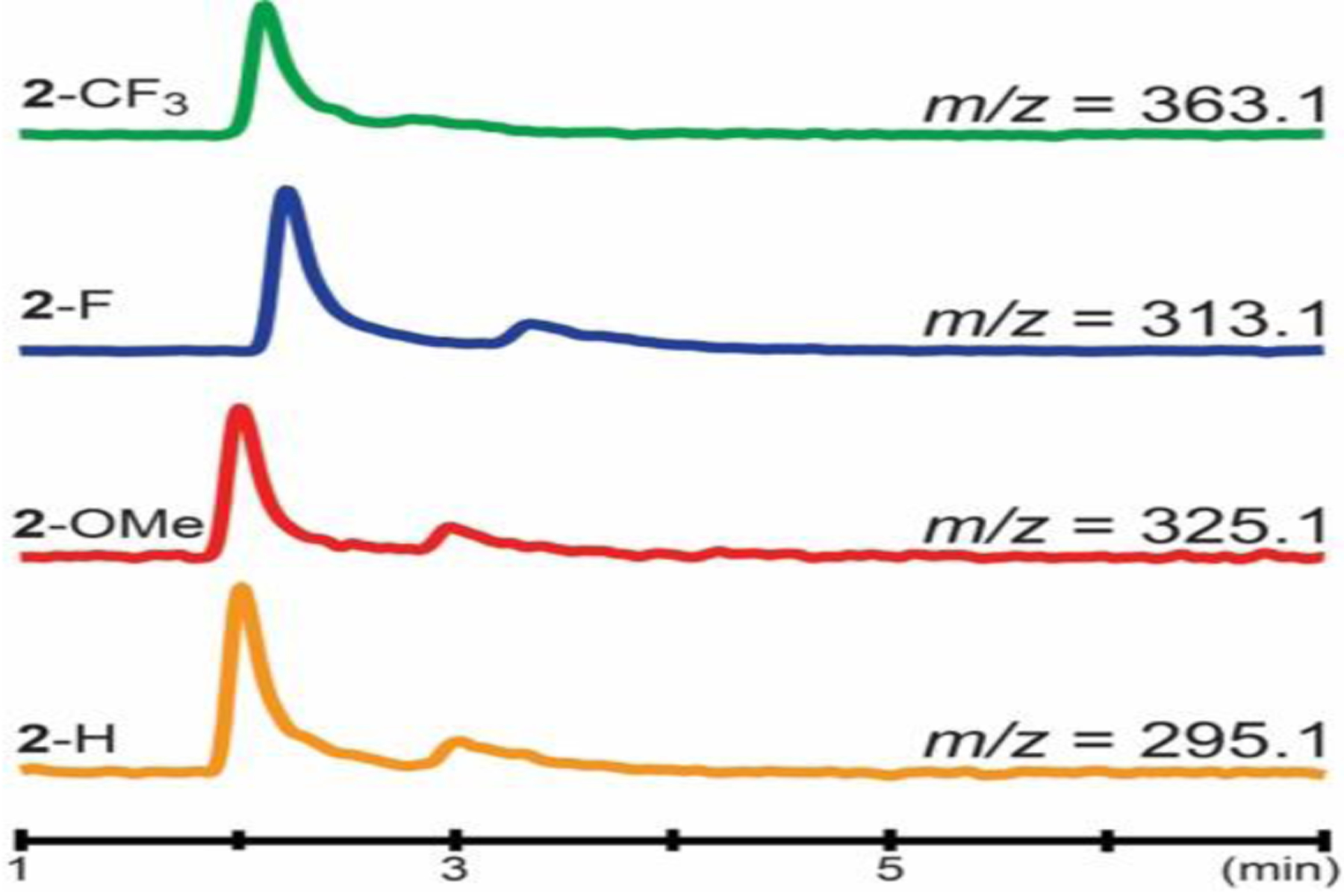
LC-MS chromatograms of epoxide products formed during AsqJ-catalyzed epoxidation when analogs (2-OMe, 2-H, 2-F and 2-CF3) were used. The MS traces are monitored and recorded using selected iron monitoring.
It is also worth noting that although only ~ 80% of 3-CF3 was produced (relative to the amount of 3-H), it is possibly due to substrate binding, rather than the substituent effect (see the crystallographic section).
Crystallographic Characterizations of the AsqJ Catalyzed Epoxidation.
To provide structural insights into the AsqJ-catalyzed epoxidation, we have determined the crystal structures of AsqJ at different stages of the catalytic cycle (Table S3). By using the published procedure,54 we first obtained crystals with 1-H (cyclopeptin) and acetate binding to the iron center. We then replaced 1-H and acetate with 2-H and 2OG respectively by post-crystallization soaking in the presence of O2. The resultant electron density map with a resolution of 1.7 Å showed unambiguously the presence of 2-H and 2OG (Figure. 5A), indicating that our ligand-exchange strategy was successful. In addition, an extra piece of electron density extending from the iron center and trans to His134 was identified. It could be fitted with a di-oxygen species or two mutually exclusive, closely spaced solvent molecules with partial occupancy. By allowing the occupancy of the proposed di-oxygen species to deviate from unity, the occupancy and O1-O2 distance converged at 0.6 and 1.5 Å, respectively, after the refinement. From the reported crystal structures on O2 activating mononuclear iron enzymes in the literature,61–63 the Fe-O2 adducts generally exhibit an O-O bond length of ~ 1.2 – 1.3 Å, and the Fe-(hydro)peroxo/Fe-alkyperoxo species generally show an O-O bond length of ~ 1.4 – 1.5 Å. Thus, the O1-O2 distance of 1.5 Å seen in our structure is close to those from the Fe-(hydro)peroxo/Fe-alkyperoxo species. In the O2 activation mechanism of Fe/2OG enzymes, two Fe-peroxy intermediates have been proposed.64–66 One is an Fe(II)-peroxysuccinate intermediate that is formed after the decarboxylation of 2OG while the O-O bond is not cleaved. The other is a bicyclic Fe(IV)-peroxyhemiketal complex where the peroxide bridges between the iron center and the C2 of 2OG. Several DFT studies suggested that the former intermediate is most likely involved in the O2 activation process based on relative energetics of these intermediates.65,66 Recently, the existence of the Fe(II)-peroxysuccinate intermediate has found some further support from a crystal structure of VioC, a Fe(II)/2OG dependent L-arginine hydroxylase, where the electron density map at the iron center was best fitted with an Fe(II)-peroxysuccinate moiety with O-O bond distance of 1.44 Å.67 In our case here, the existence of an intact 2OG molecule is well supported by the electron density map, thus it is possible that the extra electron density together with 2OG could represent the Fe(IV)-peroxyhemiketal complex. However, a close examination suggests it is unlikely because 1) the O2-C1(2OG) and the O2-C2(2OG) distances are almost the same (2.67 Å vs 2.66 Å), which rules out the possibility that a covalent bond is formed between O2 and C2 of 2OG; and 2) the Fe-O1 distance is 2.4 Å, which seems to be too long for an Fe(IV) center. Thus, this structure may represent a 2OG-bound Fe-(hydro)peroxo species. Alternatively, this extra piece of electron density may represent two closely spaced partially occupied solvent molecules. It should be noted that for all other reported AsqJ crystal structures, one solvent molecule is always present at the location occupied by O1.
Figure 5.
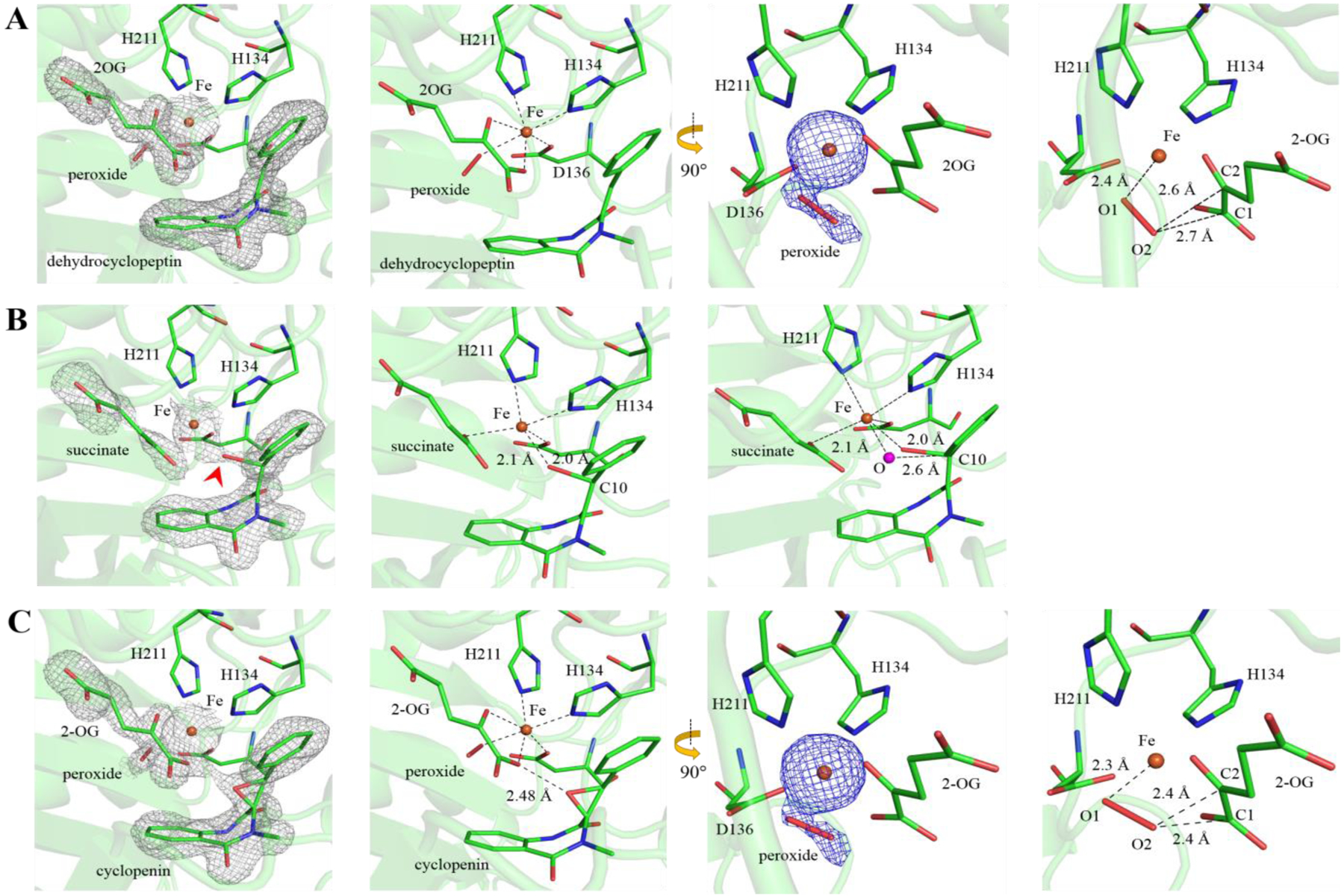
Crystallographic snapshots of AsqJ-catalyzed epoxidation. (A) The AsqJ·Fe·2OG·2-H·peroxide quinary complex (PDB code: 6K0E). (B) An intermediate structure show that the AsqJ-catalyzed epoxidation may start from the oxygenation of C10 (PDB code: 6JZM). The red arrowhead points to the position of the oxygen atom. The magenta sphere (right panel) shows the modeled oxygen atom in an idealized axial position. (C) The product-bound AsqJ·Fe·2OG·3-H·peroxide quinary complex (PDB code: 6K0F). The 2Fo-Fc maps (grey mesh) and unbiased Fo-Fc maps highlighting Fe and the extra piece of electron density (blue) are contoured at 1.0 σ and 3.4 σ, respectively. The bound ligands (cofactors and substrates) and peroxide are shown in stick form. The Fe is shown as orange sphere. The carbon atoms of ligands and selected amino acid side chains are colored green. The remaining atoms are colored according to the CPK scheme
When the same post-crystallization ligand-exchange was performed with a further addition of 0.5 mM ascorbate in the substitute mother liquor solution in the presence of O2, interestingly, difference electron density peaks (a resolution of 1.88 Å) appeared around the active center, suggesting that chemical reaction may have taken place in the crystal under this condition (Figure 5B). Reinterpretation of the electron density maps of the active site indicated that 2OG and 1-H had been converted to succinate and 2-H, respectively. Unexpectedly, a continuous electron density was observed between the Fe and the C10 of 2-H, which can be best fitted with an oxygen atom that is covalently linked to the Fe and the C10 of 2-H with the refined bond lengths of Fe-O and O-C10 are 2.1 and 2.0 Å, respectively (Figure 5B). The Fe center of this structure is penta-coordinated with a distorted trigonal bipyramidal geometry. Thus this Fe-alkoxide structure may represent an intermediate in AsqJ catalyzed epoxidation and implicates that the epoxidation of 2-H is initiated by the oxygenation of C10, which is fully consistent with the results obtained by SF-Abs, Mössbauer and LC-MS. A similar Fe-alkoxide species has recently been identified in a crystal structure of L-arginine-3-hydroxylase, VioC,67 and has also been proposed in taurine/2OG dioxygenase, TauD, based on resonance Raman data.68 Although the catalytic role of such an Fe-alkoxide species in aliphatic hydroxylation reaction, such as VioC catalyzed L-arginine hydroxylation, is still unclear, a similar species in the epoxidation reaction has long been proposed in experimental and computational studies on both heme dependent enzymes,30–36,39 and heme and non-heme biomimetic model complexes.37,38,45,69–74 Yet, it is the first time that such a species is experimentally identified in an enzymatic epoxidation reaction. In addition, the Fe-alkoxide species has also been proposed as the key intermediate for aromatic hydroxylation catalyzed by non-heme iron enzymes.75–77 Recently, an Fe-alkoxide species has been implicated in the reaction of a Rieske-type aromatic hydroxylase, Salicylate-5-Hydroxylase (S5H), based on the observed hyperfine broadening of the EPR resonance at g = 4.3 through a 17O2 experiment.78 Furthermore structure information for this type of species involved in aromatic hydroxylation has been reported on a non-heme iron model complex supported by a pyridine-based ligand that is involved in intramolecular aromatic hydroxylation.79,80 The existence of a similar Fe-alkoxide species in all these reaction types suggests strong similarities in their reaction mechanisms.
A further examination of this Fe-alkoxide structure revealed that C10 becomes sp3-hybridized and exhibits tetrahedral geometry. In contrast, C3 was found to adopt trigonal planar geometry in this epoxidation intermediate structure (Figure 6A), suggesting that C3 has remained in the sp2-hybridized state following the attack on C10. To further verify the observed bonding geometry, we deliberately modeled C3 as a tetrahedral carbon, and the hypothetical structural model was subjected to additional refinement cycles by Phenix. While planar C3 is fully enclosed within the electron density map, the tetrahedral C3 model was not justifiable due to a poor fit to both omitted and refined electron density maps (Figure 6B–D). Moreover, the refined bond length of C3-C10 is 1.47 Å (Figure 6B), which is shorter than the expected value (1.54 Å) for a C-C single bond. Taken together, these findings support the interpretation that C3 is likely in a sp2-hybridized state, which may imply a carbocation species at C3 (but see more discussion in the DFT section).
Figure. 6.
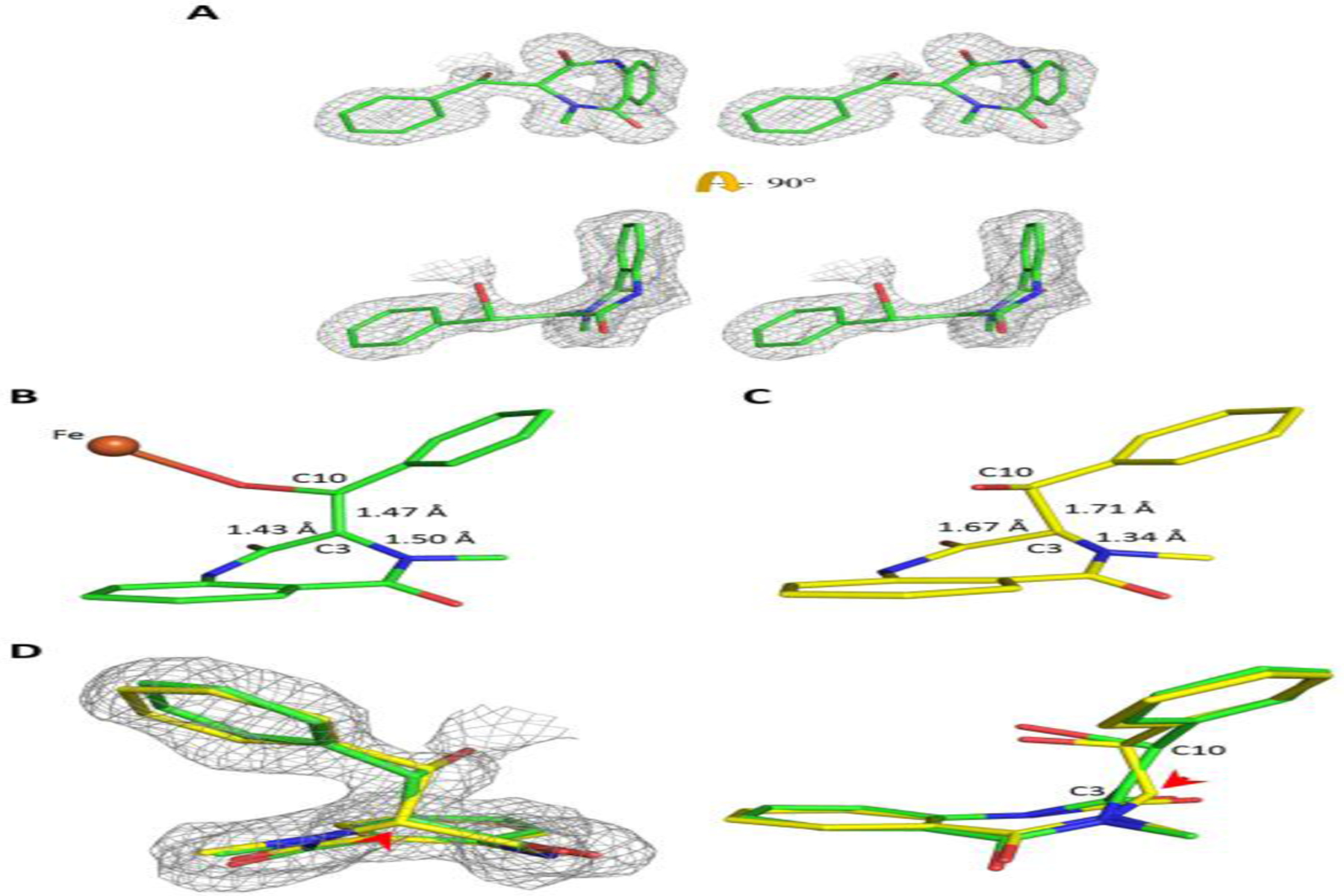
A sp2-hybridized C3 of the subtrate following the initial attack on C10 by the ferryl during epoxidation. (A) A wall-eye stereo diagram showing two orthogonal views of the observed epoxidation intermediate structure superimposed on the 2Fo-Fc electron density. (B, C) C3 of the epoxidation intermediate was modeled as sp2 (B, green) or sp3 (C, yellow) for structural refinement. (D) 2Fo-Fc electron density superimposed on the two refined structural models of the epoxidation intermediate with C3 being constrained to either adopt a planar (sp2, green) or tetrahedral (sp3, yellow) geometry. The red arrowhead indicates the position of C3. The 2Fo-Fc electron density maps (contoured at 1.0 σ) are shown in grey mesh. The Fe is shown as an orange sphere. The bound ligands are in stick form with carbon atoms colored either green and yellow, the remaining atoms are colored according to the CPK scheme.
Consistent with the occurrence of in-crystal epoxidation, the electron density maps (a resolution of 1.63 Å) derived from a 1-H-bound crystal exposed to excess amount of 2OG, 1.0 mM ascorbate, and O2 revealed the presence of 3-H, and 2OG in the active site (Figure 5C). This structure represents a product-bound, AsqJ·Fe·2OG·3-H complex. The presence of 2OG instead of succinate in the active site indicates that AsqJ likely binds 2OG with higher affinity than that of succinate. Collectively, these structures derived from our crystallographic studies provide new insights for the epoxidation steps of AsqJ and are fully consistent with other experimental data presented in the previous sections.
Furthermore, we determined the 2-CF3 bound structures by cocrystalization of 2-CF3 with iron bound AsqJ protein. The crystal structures of the complex were determined in different space groups C2221 and P212121 at 2.68 and 2.45 Å resolution (Table S4) with one and two AsqJ molecules per asymmetric unit, respectively. For both crystal forms, the electron density maps covering the benzyl moiety of 2-CF3 become bulkier, consistent with the presence of CF3 substituent (Figure 7A and 7B). Superimposition analysis showed that, while the spatial positions of active site residues are largely invariant between these two analog-bound structures, the plane defined by N1, C2 carbonyl (C2=O), and C3 of 2-CF3 exhibits a conformational change (Figure 7C). This finding indicates a greater flexibility in the binding of 2-CF3 to AsqJ than that of other substrates, and that the heptameric ring of 2-CF3 may undergo a ring-puckering change in the AsqJ active site. Moreover, compared to the 2-H-bound structure (Figure 7D), the trifluoromethylbenzene moiety seen in the P212121 structure displays a lateral movement with respect to the Fe and Fe-coordinating H134 (Figure 7D), likely caused by the steric repulsion between P132 and trifluoromethyl group of the analog. Due to the positional and ring-puckering change, the Fe-C3 distance seen in the P212121 structure (5.3 Å) is longer than that of the 2-H-bound structure (5.0 Å) (Figure 7D). We suspect the substrate binding mode seen in the P212121 structure is not optimal for catalysis, which may be a possible reason to explain why this analog is less reactive toward epoxidation.
Figure 7.
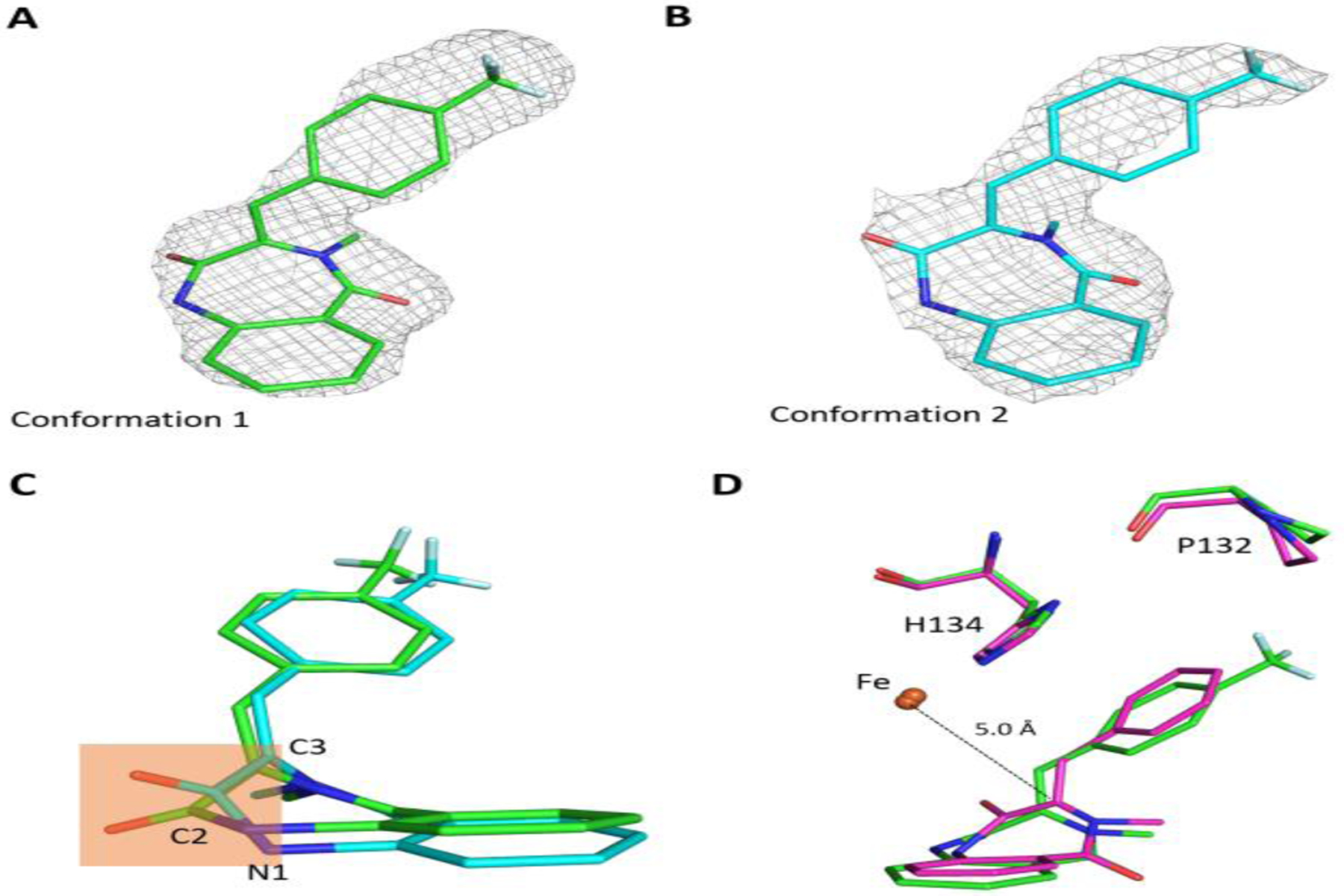
Crystal structures of AsqJ complexed with the 2-CF3 substrate analog. (A) and (B) Electron density maps and structures of the bound analog observed in space group P212121 (PDB code: 6KD9) and C2221 (PDB code: 6K3O), respectively. The 2Fo-Fc electron density maps (contoured at 1.0 σ) are shown in grey mesh. (C) Superimposition of the bound 2-CF3 analog. The conformations observed in space group P212121 and C2221, are in green and cyan, respectively. The orange-shaded region highlights the difference in ring-puckering seen in 2-CF3. (D) Structural comparison between AsqJ-bound 2-H (magenta) and 2-CF3 (green) in the orthorhombic crystal form (space group P212121) revealed the movement of 2-CF3 with respect to Fe, H134 and P132.
Density Functional Theory Calculations of the AsqJ Quaternary Complex and the Enzyme Product Complex.
DFT calculations were performed to provide structural models for the different experimentally observed iron species, in particular to help interpret the information about the iron sites and their coordination environment provided by Mössbauer spectra. Previous works on model complexes81–85 have demonstrated that quantum chemical calculations can estimate the Mössbauer parameters with good agreement to experiments. Recently, Lai and coworkers have also calculated parameters for the possible structures of the Fe(IV)-oxo intermediate of AsqJ.86 We performed DFT calculations using possible structures of the mononuclear iron active sites of AsqJ quaternary and product complexes. Of particular interest is the quadrupole splitting, ΔEQ, which reflects the charge distribution around the iron nucleus, especially the Fe valence electrons and the first coordination sphere. It provides signatures for different Fe species even if they have the same (formal) valence and spin states. For high-spin Fe(II) species with O/N ligands, |ΔEQ| is typically in the range of 1.6–3.2 mm/s.87
Models of the AsqJ mononuclear Fe active site were constructed based on a crystal structure reported in our previous work (PDB ID 5Y7R).54 The model contains an octahedral, 6-coordinate Fe, with the facial triad, the bidentate 2OG (keto group trans to Asp136, and carboxylate group trans to His211), and a water as the ligands, as well as the substrate 2-H in close vicinity but not directly bonding to the Fe ion (Figure S7). The “core” of the active site is modified according to different possible configurations. The geometries were optimized with the coordinates of certain anchoring atoms on the periphery of the model fixed at their crystal structure positions, as detailed in the SI. Based on the optimized structures, the Mössbauer parameters δ and ΔEQ were further calculated using the method described in the SI, which was also validated against several high-spin Fe(II) model complexes with known experimental |ΔEQ| (Table S5).
For the AsqJ⋅Fe2+⋅2OG⋅2-H complex (δ = 1.25 mm/s, |ΔEQ| = 2.54 mm/s, Figure S3), we considered the following possibilities: the 2OG could be chelating the iron with the carboxylate trans to His211 as in the crystal structure (the offline mode), or its C2-C3 bond could be rotated so that the carboxylate is trans to His134 (the online mode); the 6th ligand position could be occupied by a water, or unoccupied leaving a 5-coordinate species. The models and the calculated Mössbauer parameters are shown in Scheme 4. The calculated isomer shifts are similar across all models and consistent with experimental value, although the 5-coordinate species generally have lower isomer shifts. Rotating the 2OG from the binding configuration seen in the crystal structures (the offline mode) to the online mode results in little change in either the isomer shift or the quadrupole splitting. However, the 6-coordinate complexes generally have a larger |ΔEQ| than 5-coordinate complexes by approximately 0.4 mm/s. This difference is generally observed in all the DFT models tested in this study, showing a good correlation between the coordination number (four-, five- and six-coordinate) and the calculated quadrupole splittings (the quadrupole splitting increases with increasing coordination number, Figure S8). Thus, the Mössbauer parameters for the AsqJ quaternary complex are in best agreement with a 6-coordinate iron center. In addition, although in all the published crystal structures of AsqJ in the presence of 2OG and the structures included in this study, 2OG always shows an offline binding mode, based on the current DFT results the Mössbauer parameters (δ and ΔEQ) cannot be used to readily distinguish the 2OG binding configuration to the iron center (offline vs. online). Yet, our current calculations suggest that the 2OG online mode in a 6-coordinated iron center is energetically more stable than that of the offline mode by 4.5 kcal/mol (including only the electronic energy). For a 5-coordinate iron center (without the additional water molecule), the structures with two different 2OG binding modes are essentially isoenergetic, with the offline mode structure as the more stable structure by only ~1 kcal/mol. Therefore, two 2OG binding modes could both be present in aqueous solution in the AsqJ quaternary complex.
Scheme 4.
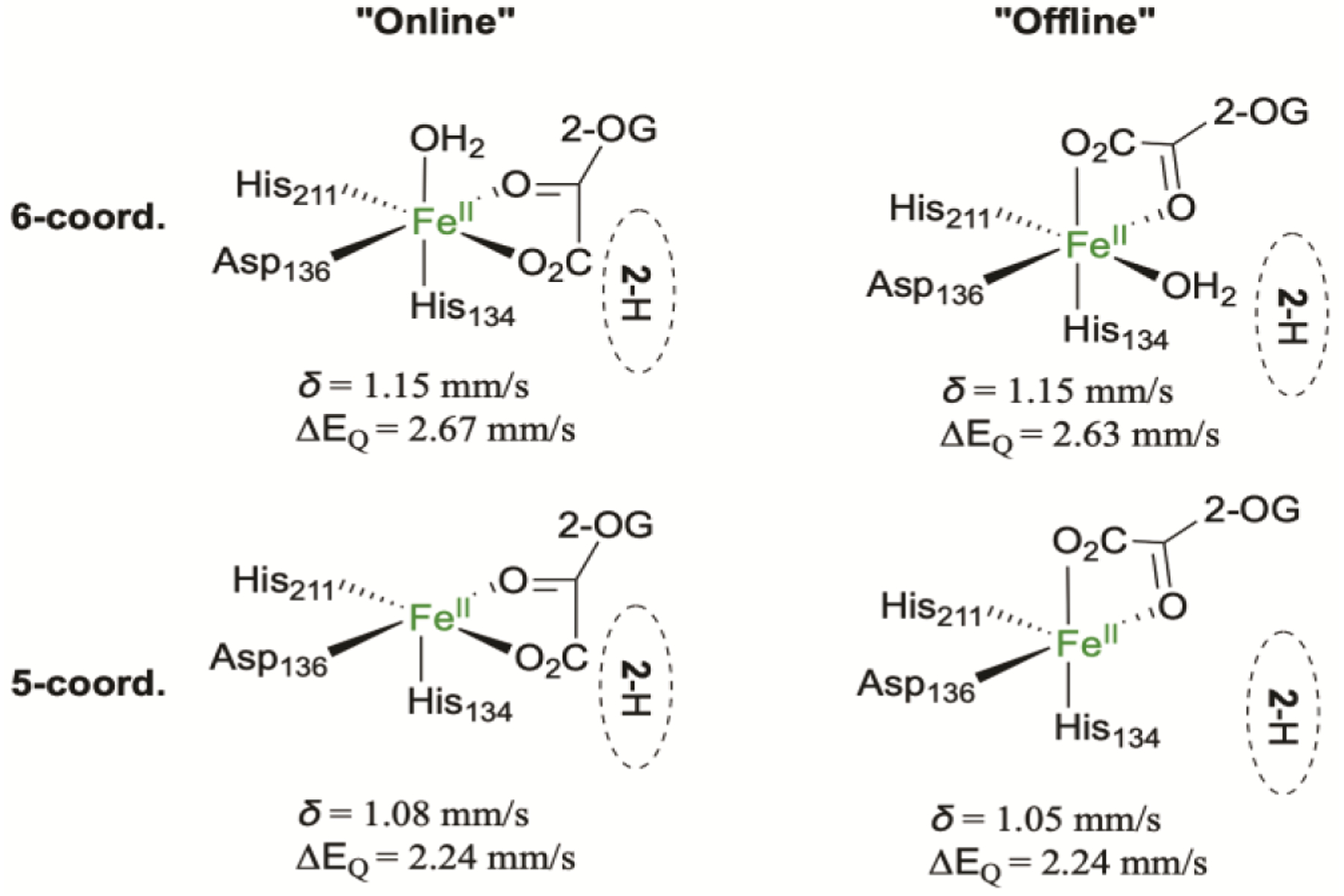
Schematic drawing of possible Fe center geometry of the AsqJ⋅Fe2+⋅2OG⋅2-H complex and their calculated Mössbauer parameters.
The intermediate Fe(II) species (δ = 1.21 mm/s, |ΔEQ| = 2.96 mm/s, See Figure 3 and our previous work49) appearing after the decay of the Fe(IV)=O intermediate is most likely a product complex with a bound succinate. To explore the structural models for such a product complex, we considered the following possibilities (Scheme 5): (i) a 4-coordinate complex without iron-bound water molecules; (ii) a 5-coordinate geometry with an iron bound water molecule either in the plane or out of the plane defined by the oxygens from succinate and Asp136 and nitrogen from His134; (iii) a 6-coordinate species with two water molecules, one of which interposes between the iron and the epoxide oxygen; (iv) an alternative 6-coordinate species with the epoxide oxygen directly coordinating to the iron. In addition, we considered the possibility of succinate carboxylate being a bidentate ligand to the iron center in the product complex, in light of various crystal structures as such included in88–90. However, geometry optimization attempts starting with bidentate binding invariably reverted back to a monodentate binding for succinate, likely dictated by a hydrogen bond interaction with Gln131, consistent with the arguments by Solomon and coworkers.91
Scheme 5.
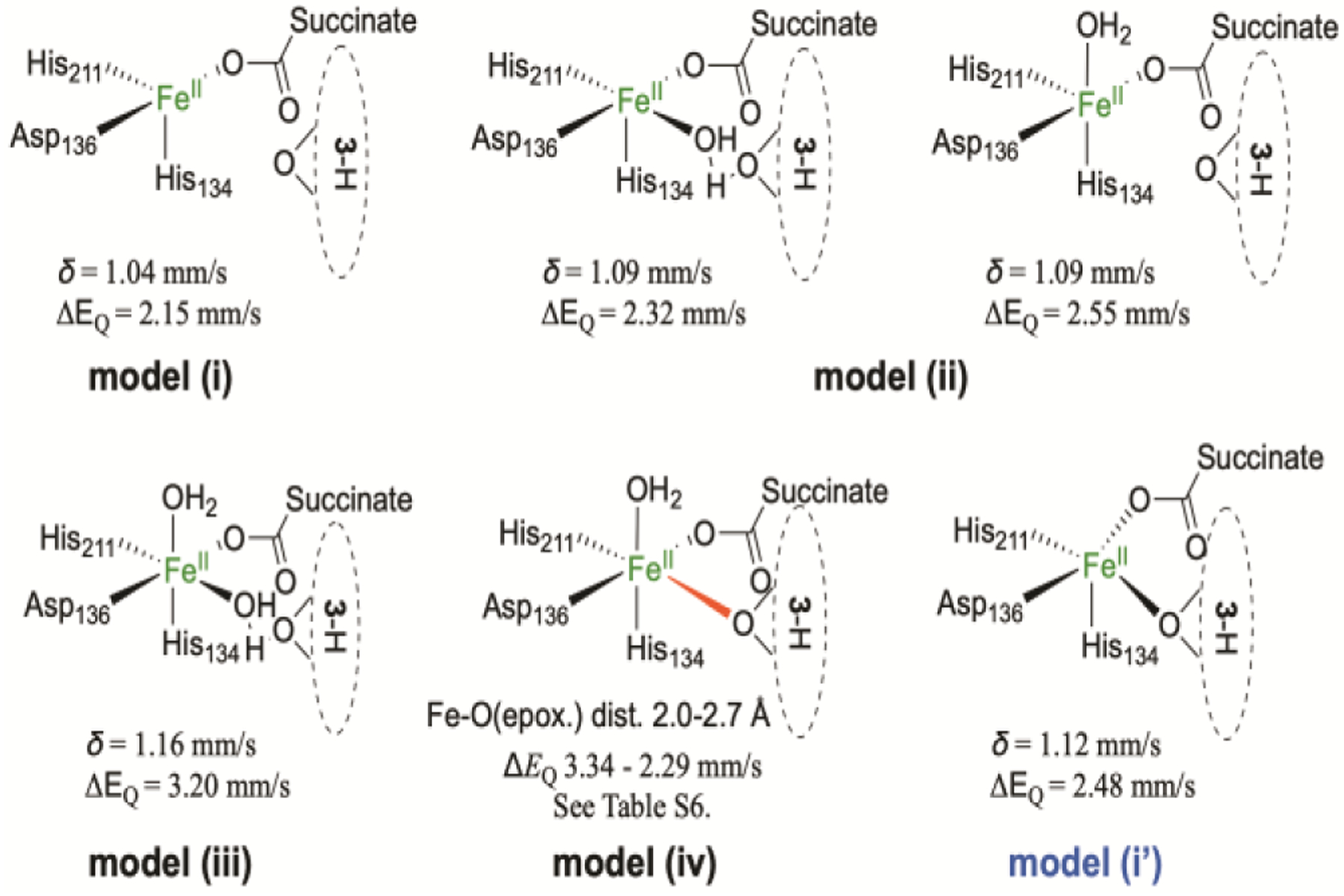
Possible Fe site structures of the AsqJ⋅Fe2+⋅succinate⋅3-H complex and the calculated Mössbauer parameters. For Fe-O distance scan in model (iv), see Table S6.
For the first two models listed above, the optimized structures exhibit Fe-O(epoxide) distances of 3.53, 3.46, and 4.03 Å respectively, and only the 6-coordinate species with two iron-bound waters reproduces the experimentally observed quadrupole splitting and isomer shift well (Scheme 5). For 5- and 4-coordinate species, the predicted quadruple splittings are generally much smaller than the experimentally determined one. It is also worth noting that the experimental and computational quadrupole splittings presented here for potentially 6-coordinate iron centers are also consistent with the observed |ΔEQ| for two 6-coordinate high-spin Fe(II) complexes supported by 2-pyrazinecarboxylate ligand (3.04 and 3.16 mm/s).92,93 The iron coordination environment of these complexes closely mimics that of the succinate-bound product complex, with two aromatic N ligands with neutral formal charge cis to each other, and two negatively charged carboxylate ligands trans to each other on an axis perpendicular to the N-Fe-N plane.
For the last model (model (iv), Scheme 5), geometry optimization did not result in the complex with directly coordinating epoxide oxygen in a 6-coordinate iron center. Rather, the structure relaxed into the 5-coordinate geometry of model (ii) above. This is possibly due to a hydrogen bond between the cyclopenin (3-H) C1 carbonyl and the acetamide group of Asn70 (See Figure S7), whose Cα is fixed in the DFT models. A close Fe-O (epoxide) distance would break this H-bond. We next brought the epoxide O close to the iron in a 6-coordinate geometry by freezing the Fe-O distance, then optimized a series of structures with fixed Fe-O distances ranging from 2.0 to 2.7 Å (which would break this H-bond) and calculated the Mössbauer parameters as well. The results are shown in Scheme 5 and Table S6. Again, the |ΔEQ| follows a decreasing trend with decreasing coordination number. The Fe-O distance scan results are also consistent with this trend, as the |ΔEQ| decreases with increasing Fe-O distance, essentially transitioning from 6- to 5-coordination. When the Fe-O distance is in the range of typical Fe(II)-O single bond (loosely defined to be 2.0 – 2.2 Å), |ΔEQ| is in the range of 2.98 – 3.34 mm/s, which is close to the experimentally observed value. However, as described above, the 6-coordinate complex with two iron-bound waters is also calculated to have a similar |ΔEQ| = 3.2 mm/s. Therefore, although the exact iron coordination environment cannot be exclusively established for the Fe(II) product complex based on these DFT calculations, it is clear that the Mössbauer parameters of the Fe(II) product complex support a 6-coordinate iron center with most likely 2 histidine nitrogen and 4 oxygen ligands. It should be noted that the Asn70 residue is located on a flexible loop near the surface of the protein (Figure S7).Therefore, it is possible that protein dynamics during the enzymatic reaction could modulate the H-bond between Asn70 and the substrate to allow the substrate to come close to the iron center during the epoxidation step, making it possible for an iron-bound epoxide to exist transiently.
We further investigated the possible electronic structure of the Fe-alkoxide species observed in one of the AsqJ crystal structures. Using the crystal structure of the Fe-alkoxide species (PDB code 6JZM) as the initial structure, the structural optimization cannot stabilize the Fe-alkoxide species, rather it causes the structure to relax into a fully formed epoxide with the C3-O and C10-O distances of 1.42 and 1.43 Å (Scheme 5, model (i’)). Interestingly, the optimized structure also features a 5-coordinate iron center with an Fe-O(epoxide) distance of 2.26 Å, suggesting that the formed epoxide is still bound to the iron center. In addition, in this structure the H-bond between Asn70 and the epoxide product is broken, with a distance of 5.70 Å between the C1 carbonyl oxygen of the 3-H and the nitrogen of Asn70, as compared to 3.10 Å in the model (i) discussed above. This optimized structure is essentially an alternative configuration of the product complex model with a 4-coordinate iron center (Scheme 5, model (i)). In that 4-coordinate iron center structure, the O(epoxide) does not coordinate to the iron center while the H-bond between Asn70 and the product is maintained. The electronic energy difference between these two optimized structures (model (i) vs. model (i’)) is only ~2 kcal/mol with the model (i) structure being a more stable structure. This further suggests that the modulation of the H-bond between Asn70 and the epoxide product may be important in determining the interaction between the iron center and the epoxide. Nonetheless, the quadrupole splitting predicted from this 5-coordinate Fe-epoxide structure (|ΔEQ| = 2.48 mm/s) is still smaller than that of the experimental value for the product complex ((|ΔEQ| ~ 3 mm/s).
Since the structural optimization by DFT cannot stabilize the Fe-alkoxide species, we performed a more constrained geometry optimization by only allowing the iron atom, the three protein ligands and the succinate (but not the alkoxide) to move. This optimization resulted in a structure with a shorter Fe-O bond length of 1.78 Å, compared to 2.1 Å in the crystal structure. Significant spin populations were found mostly on Fe (+4.02) and C3 (−0.57), while the population on O is only +0.13 (Figure S7). Calculation of Mössbauer parameters yielded δ = 0.54 mm/s and ΔEQ = 1.42 mm/s. The result signifies that the electronic structure of the iron center is essentially a high-spin Fe(III) (S = 5/2) antiferromagnetically coupled to a C3-radical. The spin density is visualized (contoured at 0.01 au−3) in Figure S9. <S2> was calculated to be 6.76, which were deviated from a <S2> = 6 for an isolated S = 2 iron center, but were consistent with a broken-symmetry solution having an S = 5/2 Fe(III) center antiferromagnetically coupled with a substrate radical.
In summary, DFT calculations suggest that the AsqJ quaternary complex with 2-H binding as well as the product complex (with the presence of the epoxide) most likely feature a 6-coordinate iron center based on the DFT predicted quadrupole splittings and isomer shifts on various models. The epoxide may directly coordinate to the iron center during the initial epoxide ring formation, and its dissociation from the iron center may rely on the hydrogen bond interaction between the epoxide product and the protein residues, most likely Asn70, as well as extra water molecules close to the iron center. The Fe-alkoxide species could not be stabilized during geometry optimization, suggesting that this species could only be captured in crystal due to the restraining crystal environment. In addition, its electron structure may be best described as a high-spin Fe(III) center antiferromagnetically coupled with an S = 1/2 radical localized at the C3 position of the substrate. This is a species that has long been proposed to be a potential intermediate in epoxidation reaction catalyzed by both heme and non-heme iron enzymes.33–38,86,94,95 Lastly, the offline and the online 2OG binding configurations to the iron center in the AsqJ quaternary complex cannot be readily distinguished by Mössbauer parameters and may coexist in aqueous solution based on close electronic energies of the two binding configurations. This is also predicted by a recent QM/MM study of an Fe/2OG-dependent histone demethylase.96
Molecular Dynamics Simulations on the Relevance of the Two 2OG Binding Configurations to the Initial O2 Reactivity towards the AsqJ Fe(II) center.
Two 2OG-binding configurations to the iron center have been observed in many published crystal structures of Fe/2OG enzymes.97 Although the C2-carbonyl oxygen of 2OG invariantly trans to the carboxylate of the 2-His-1-carboxylate facial triad, the position of the C1 carboxylate of 2OG adopts two orientations. In one orientation, the C1 carboxylate group is located trans to the proximal (closer to the N-terminus of the protein) histidine of the 2-His-1-carboxylate facial triad (the online configuration). In the other orientation, the C1 carboxylate group is located trans to the distal histidine (the offline configuration). For AsqJ, all the available crystal structures show an offline configuration for 2OG. As a result, the potential O2 binding site, which is normally occupied by a solvent molecule in the crystal structures, is pointed away from the substrate. Then the subsequently formed Fe(IV)=O intermediate followed by the O2 addition and the oxidative decarboxylation of 2OG would result in an offline mode ferryl intermediate where the Fe(IV)=O unit points away from the substrate, thus creating a problem for the subsequent chemical reaction between the Fe(IV)=O intermediate with the enzyme substrate. In our previous study on AsqJ catalyzed epoxidation reaction, a possible oxo-hydroxo/water tautomerism of the Fe(IV)=O intermediate has been proposed. Based on QM/MM calculations, Lai and coworkers suggested that this tautomerism could be the key mechanism to effectively rotate the Fe(IV)=O unit and bring the oxo close to the olefin moiety of 2-H for the subsequent steps of the reaction.86 The different orientations of the Fe(IV)=O moiety of the ferryl intermediate (online vs. offline) in the Fe/2OG dependent halogenase, SyrB2, have also been proposed to explain why SyrB2 favors halogenation than hydroxylation. Here, we investigate another possibility. Namely, which 2OG configuration (the online mode vs. the offline mode) is thermodynamically preferred for the initial O2 addition to the iron center of AsqJ.
For non-heme O2 activating mononuclear iron enzymes, it is generally accepted that the initial O2 addition to the Fe(II) center is largely facilitated by the generation of a five-coordinated iron center via the removal of the solvent molecule.98,99 Therefore, to answer the aforementioned question, we used thermodynamic integration (TI) to calculated free energy change in the process of replacing a coordinated water with an O2 at the iron site of AsqJ in the two 2OG configurations (online vs. offline). The TI simulations were performed repeatedly in presence and in absence of the enzyme substrate, 2-H. To fully account for the solvation effect and the dynamic nature of the enzyme, we used molecular dynamics (MD) simulations for these TI calculations (Scheme S2). The initial protein structures for both the online and offline modes of the 2OG binding were prepared from the crystal structure (See the SI for the details). The MD-equilibrated protein remained stable in the simulations in both the 2OG online and the 2OG offline modes. Simulated structures maintained conformations of the protein secondary structures and of the active site pocket, which can be overlapped with the initial crystal structure (Figure S10, S11). For calculating the free energy changes, the coordinated water in the initial structure is gradually transformed to an O2 for both the online and the off-line mode structures in the presence or the absence of 2-H (total four configurations were modeled, see the SI for the simulation details). The Gibbs free energy of the transformation was calculated by using the multistate Bennett acceptance ratio (MBAR) algorithm. Figure 8a shows free energies of transformation of a bound water to an O2 computed for four system configurations cumulatively averaged over 0.5ns MD intervals to demonstrate the statistical convergence of the simulated results. Computed ΔΔG are reported with respect to the free energy of the water to O2 substitution in the offline 2OG configuration with no substrate in the protein active site.
Figure 8.
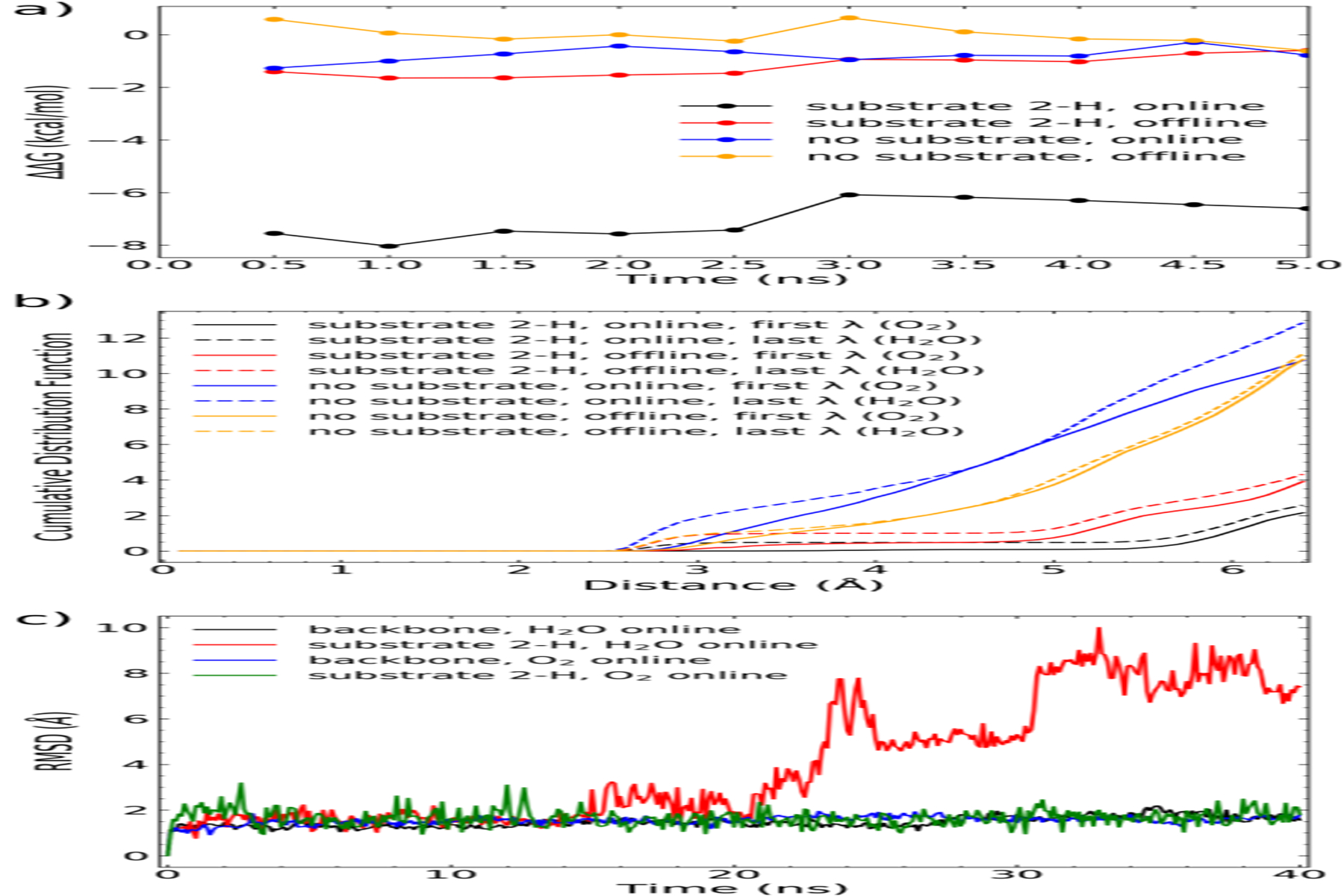
Free energy difference (ΔΔG) of O2 addition to the Fe(II) center of AsqJ in two different 2OG binding configurations derived from MD simulations. (a) Cumulative ΔΔG for four system configurations averaged over 0.5ns MD simulation intervals by using the the 2OG offline configuration without substrate as the reference state. (b) Cumulative distribution function of water molecules around iron-bound water or O2 in presence or absence of a substrate in two 2OG binding configurations. (c) Root-mean-squared deviation (RMSD) of the protein backbond and the substrate 2-H derived from MD simulations with either water or O2 bound to the iron and the 2OG in the online configuration. The simulations were performed with no positional restraints on either the substrate 2-H or the protein.
The results suggest that the presence of 2-H greatly facilitates the replacement of a bound water to an O2 only when 2OG is in the online mode (computed ΔΔG = −6.5 kcal/mol relative to the 2OG-online and no substrate configuration). At the same time, the 2OG offline mode is not affected significantly (computed ΔΔGs are within 1 kcal/mol for configurations with and without the substrate). Given that all other factors in the structure are similar this result suggests that the O2 addition rate should be much more rapid (>10000 times faster at 4 °C) in the 2OG online mode structure than in the offline mode once the substrate is present. This is likely because the O2 binding pocket created by the substrate binding is more hydrophobic in the online mode structure than in the offline mode. Indeed, Figure 8b shows the cumulative distribution function of water molecules around iron-bound water or O2 and suggests that the substrate binding squeezes water more strongly from the active site in the 2OG online configuration that results in a stronger destabilization of iron-bound water molecule. Figure 8c shows results of the 40 ns MD simulations of AsqJ in the 2OG online mode with unrestrained 2-H, with water or O2 bound to Fe. It shows that in the 2OG online mode the substrate binding is stable when oxygen is bound to iron but unstable when water is bound to iron. This may partially explain the difficulty to experimentally observe the online OG configuration of AsqJ/substrate complex in crystal structure.
In summary, although the 2OG-binding configuration in all the available crystal structures of AsqJ is in the offline mode, the rapid O2 addition to the Fe(II) center in solution could likely occur in the online binding mode of 2OG. Thus the subsequently formed ferryl intermediate will have the Fe(IV)=O moiety pointing towards the substrate to carry out rapid chemical transformations.
Conclusions
In this study, the AsqJ-catalyzed epoxidation was studied using the native substrate, 2-OMe, and several para-substituted substrate analogs (2-H, 2-F, and 2-CF3). All compounds triggered a rapid formation of the iron(IV)-oxo intermediate under pre-steady state conditions. Importantly, the decay kinetics of the ferryl species was largely insensitive to the para-substituents on the phenyl moiety bearing electron withdrawing or electron donating group (only 2–3 times of variation on the rate constant are observed). Thus, the OAT reactivity of the Fe(IV)-oxo intermediate in AsqJ does not depend on the electron richness of the olefin moiety of the substrate. This catalytic behavior is very different from non-heme Fe(IV)-oxo model complexes. In all the reported model complexes, the OAT reactivity for Fe(IV)-oxo species generally demonstrated strong electrophilic nature so that the para substitution on the phenyl moiety of the substrate (e.g. styrene) dramatically modulates the life-time of such a species (up to 100 fold in going from electron donating substituents to electron withdrawing ones). The lack of such a correlation in the AsqJ-catalyzed epoxidation also suggests that there is no significant charge separation (e.g. cationic species) in the transition state of the rate-determining step of the reaction. These experimental observations were further supported by the crystallographic data and the reaction product analysis obtained via LC-MS. The LC-MS analyses showed a similar level of substrate consumption and product formation when the enzymatic reactions were carried out in the steady-state turnover conditions regardless of the compound used. The observation of an Fe-alkoxide species in one of the crystal structures provided important support for a step-wise epoxidation mechanism where an initial attack from the Fe(IV)-oxo intermediate to the olefin moiety happens at the benzylic (C10) position of the substrate. This observation is also consistent with the substrate-bound crystal structures, which showed a shorter distance between the Fe and the C10 position than that between the Fe and the C3 position (4.4 Å vs. 5.0 Å).
Furthermore, the DFT calculations suggested that the Fe-alkoxide species observed in the crystallographic data may be best described as an Fe(III) center antiferromagnetically coupled with a substrate radical located at the C3 position of the substrate. However, this species was not observed in solution, suggesting that a restraining crystal environment is necessary to stabilize such an intermediate. In addition, the calculations also suggested that AsqJ quaternary complex as well as the product complex with the presence of the epoxide product most likely feature a 6-coordinate iron center. The epoxide may directly coordinate to the iron center during the initial epoxide ring formation, and its dissociation from the iron center may rely on the hydrogen bond interaction between the epoxide product and the protein residues, most likely Asn70, as well as extra water molecules close to the iron center. Finally, the MD simulations supports a scenario where the initial O2 addition to the iron center could happen in the online-binding mode of 2OG to the iron center, not the offline mode seen in the crystal structure. This is most likely due to the substrate binding, which provides a hydrophobic environment to facilitate the O2 addition to the iron center. Such a hydrophobic environment is more effectively formed when 2OG binds to the iron center in the online mode.
In summary, all the experimental and computational results strongly suggest a step-wise mechanism for AsqJ-catalyzed epoxidation reaction. The initial oxygen addition from the Fe(IV)-oxo species to the olefin moiety of the substrate happens at the benzylic position, which is most likely followed by the generation of a Fe(III)-alkoxide species with a carbon radical located at the C3 position of the substrate. The subsequent C3-O bond formation complete the epoxide ring formation.
EXPERIMENTAL SECTION
General Procedures.
All reagents were used directly as obtained from the commercial sources. Analytical thin layer chromatography (TLC) was carried out on pre-coated TLC aluminum plate (silica gel, grade 60, F254, 0.25 mm layer thickness) acquired from EMD Chemicals (Gibbstown, NJ). Flash column chromatography was performed using silica gel (230–400 mesh, grade 60) obtained from Sorbent Technologies. NMR spectra were recorded on a Bruker 500 MHz spectrometers. Chemical shifts (in ppm) are referenced using solvent (CHCl3, DMSO, water) peaks, with coupling constants reported in Hertz (Hz). The relative molecular mass and purity of enzyme samples were determined using SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The protein molecular weight marker used was purchased from Bio-Rad Laboratories (Precision Plus ProteinTM All Blue Standards).
Protein Overexpression and Purification.
Plasmids (pET-28a) carrying the Aspergillus nidulans AsqJ gene were synthesized by the commercial GeneArt Gene Synthesis service (Invitrogen). The plasmids were transformed into BL21 (DE3) E. coli cells (New England Biolabs) and cultured in LB medium. Overexpression was induced by Isopropyl β-d-1-thiogalactopyranoside (GeminiBio) at 27 °C for 4 hours before cells were harvested. The enzyme was purified using a Ni-NTA agarose column, and excess salt and protein-bound metal ions were removed by dialysis. Oxygen was removed from the concentrated protein solution by degassing on a Schlenk line and equilibrating in a glove box (MBraun). The protein stock was aliquoted and stored at −80 °C. Details of the experimental conditions are provided in the Supporting Information.
LC-MS experiments.
High performance liquid chromatography (HPLC) with detection by mass spectrometry (MS) was conducted on an Agilent Technologies (Santa Clara, CA) 1260 infinity series system coupled to an Agilent Technologies 6120 single quadruple mass spectrometer. The associated Agilent MassHunter software package was also used for data collection and analysis. Assay mixtures were separated on an Agilent ZORBAX Extended-C18 column (4.6 × 50 mm, 1.8 μm particle size) with an isocratic system of 40% of solvent A (25 mM aqueous ammonium formate, pH = 6.0) and 60% solvent B (methanol). Detection was performed using electrospray ionization in positive mode (ESI+). The corresponding products were extracted at the corresponding M+1 m/z values.
Reactions associated with Figure 4 was performed as follows: Each reaction contained a mixture of AsqJ, Fe(II), 2OG, substrate in 0.1 M tris buffer (pH = 7.5) with the final concentrations of 0.11 mM AsqJ, 0.1 mM Fe(II), 0.1 mM substrate, and 0.2 mM 2OG. Once all reaction samples were prepared in the glove box, samples were exposed to air by opening the reaction vials to allow oxygen infusion. After 20 mins under ambient environment exposure, proteins were removed using centrifugal filtering tubes using Nanosep® centrifugal filters.
Stopped-Flow Absorption Spectroscopy.
Stopped-flow absorption spectroscopy (SF-Abs) experiments were performed on an Applied Photophysics SX20 stopped-flow spectrometer operating in an MBraun Unilab glove box filled with nitrogen gas and maintained at < 0.5ppm O2 level. Sample and reaction chambers were kept at 5 °C for the entire duration of the experiment. Time-resolved absorbance scans from 300 to 700 nm were collected with a diode-array detector. The resulting data were processed using KaleidaGraph software and shown in Figure S5. The kinetic analysis was carried out by using a 3-step kinetic model depicted in Scheme 3, and the simulations were performed by Kintek Explorer software.100,101 Details of sample preparation are provided in the Supporting Information.
Freeze-quench Mössbauer Sample Preparation.
Freeze-quench experiments were performed using a KinTek quench-flow instrument. An oxygen-saturated buffer solution (~1.8 mM O2 in a 50 mM Tris-HCl, pH 7.5) buffer solution prepared at ~ 4 °C was rapidly mixed with an equal volume of an oxygen-free solution containing AsqJ (1.5 mM), 57Fe(II) (1.25 mM), 2OG (12.5 mM), and substrate (2-H or 2-CF3, 1.5 mM) to initiate the reaction at 5 °C. The resulting reaction was terminated by injection of the solution into liquid ethane maintained at 90 K at various time points. The resulting samples were first pumped to remove liquid ethane. Then the dry frozen solution powder was packed into in-house designed freeze quench Mössbauer sample cups cooled at liquid nitrogen temperature to generate Mössbauer samples. The reaction time of a freeze-quenched sample is the sum of the aging time and the quench time. The aging time was the transit time for the reaction mixture through the aging hose. The quench time corresponded to the time required after injection into the cryosolvent for the reaction mixture to be cooled sufficiently to prevent further reaction and was estimated to be ~ 5 ms.
Mössbauer analysis.
Mössbauer spectra were recorded with home-built spectrometers using Janis Research SuperVaritemp dewars, which allowed studies in the temperature range from 1.5 to 200 K, and applied magnetic fields up to 8.0 T. Mössbauer spectral simulations were performed using the WMOSS software package (SEE Co., Edina, MN). Isomer shifts are quoted relative to Fe metal at 298 K. All Mössbauer figures were prepared using SpinCount software.102
EPR analysis.
All EPR samples were produced in a N2 glove box with a sample volume of 250 uL and flash-frozen in liquid nitrogen with 10% (v/v) glucerol added as a glassing agent. X-band EPR spectra were recorded on a Bruker Elexsys spectrometer equipped with an Oxford ESR 910 cryostat and a Bruker bimodal cavity for generation of microwave fields parallel and transverse to the applied magnetic field. The modulation amplitude and frequency were 1mT and 100 kHz for all spectra. First-derivative spectra were recorded at 1024point with an integration time of 150 millliseconds per point. The microwave frequency was calibrated with a frequency counter and the magnetic field was calibrated with a calibrated Hall probe. The temperature was calibrated with a carbon glass resistor (Lakeshore CGR-1–1000). EPR signals were quantified by relative to a 1 mM Cu(II)EDTA standard in 10% glycerol. The spectral simulations were performed by using the SpinCount software.102
Protein Crystallization.
Crystallization of Fe(II)-bound complexes AsqJ•Fe•succinate•1-H and AsqJ•Fe•succinate•2-CF3 complexes was accomplished using the sitting-drop method as reported in previous work and detailed in the Supporting Information.54 Crystals of the peroxide-bound AsqJ•Fe•2OG•2-H were prepared by soaking the AsqJ•Fe•acetate•1-H crystals with a substitute mother liquor containing 25% PEG 1000, 100 mM imidazole/HCl (pH 8.0), 5 mM 2OG and 1 mM of dehydrocyclopeptin under aerobic condition for 24 h at 4°C. Crystals of AsqJ•Fe•succinate•2-H (with the O-C10 bond formation being observed) and AsqJ•Fe•2OG•3-H were obtained by soaking the pre-grown crystals of AsqJ•Fe•acetate•1-H in a substitute mother liquor containing 25% PEG 1000, 100 mM imidazole/HCl (pH 8.0) and 5 mM 2OG supplemented with 0.5 and 1 mM of ascorbate, respectively, for 24 h at 4°C.
Crystal Structure Determination.
The X-ray diffraction data of the crystals were collected at the BL15A1 and TPS beamline 05A at NSRRC (National Synchrotron Radiation Research Center, Taiwan). The data were indexed, integrated and scaled with the HKL-2000 program suite,103 while phasing and model building were accomplished with various components of the CCP4 suite,104 as detailed in the Supporting Information. The data collection and refinement parameters are listed in Tables S3 and S4. Atomic coordinates and structure factors have been deposited in the PDB with accession codes 6K0E (AsqJ·Fe·2OG·2-H·peroxide), 6JZM (O-C10 bond formation), 6K0F (AsqJ·Fe·2OG·3-H·peroxide), and 6KD9 and 6K3O (AsqJ•Fe•succinate•2-CF3 in space group P212121 and C2221, respectively).
Density Functional Theory Calculations.
Models for DFT calculations were built based on previously reported crystal structure (PDB ID 5Y7R)54 with truncation to keep only residues close to the active center. Various ligands are edited in and models are subject to geometry optimization using the Gaussian 09 software, revision D01.105 The Mössbauer parameters of the models were calculated using the ORCA software package, version 4.0.1.2.106 The calculated electron density was converted to isomer shift using a set of calibration formula from Pápai et al.84 The quadrupole splitting was calculated from the electric field gradient tensor at the Fe nucleus. The method for quadrupole splitting was also validated with a small set of model complexes, as detailed in Table S5. Further details of models, geometry optimization, and calculations were provided in the Supporting Information.
Molecular Dynamics Simulations.
MD simulations were carried out using the GROMACS software package.107 We performed thermodynamic integration for the free energy changes for the replacement of Fe-coordination water molecule by O2. The calculation was accomplished using the MBAR (Multistate Bennett Acceptance Ratio) method108 as implemented in gmx bar utility of GROMACS, applying an alchemical transformations of the Fe ligand O2 into H2O. 10 steps (11 λ points) have been used in the O2 to H2O transformation. Calculations have been carried out for 4 system configurations, namely with and without the bound 2-H substrate for the online and offline orientations of 2OG in the coordination complex. Each structure has been solvated and equilibrated for 20 ns before production MD runs. The length of production MDs was 5 ns for each of 11 intermediate (λ) states (vide infra). Mixed topology files have been prepared where the initial state is O2 coordination and the final state is H2O coordination. Details of model building, force field parameters, and restraints are provided in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by grants from the National Institutes of Health (GM125924 to Y.G., W.-c. C, and M. K.), National Taiwan University, and Ministry of Science and Technology grant 106-2113-M-002-021-MY3, R.O.C., Taiwan. We thank Prof. Michael P. Hendrich at CMU for the use of the FQ apparatus. The resource for computational work is partly provided by Extreme Science and Engineering Discovery Environment (XSEDE) and Pittsburgh Supercomputing Center (allocation number TG-CHE180042).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Additional experimental and computational details and data, including Schemes S1, Figures S1–S11, Tables S1 – S6 (PDF)
Cartesian coordinates of all DFT structural models (Zip)
The authors declare no competing financial interests.
REFERENCES
- (1).Thibodeaux CJ; Chang WC; Liu HW Enzymatic Chemistry of Cyclopropane, Epoxide, and Aziridine Biosynthesis. Chem. Rev 2012, 112, 1681–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sienel G; Rieth R; Rowbottom KT Epoxides In Ullmann’s Encyclopedia of Industrial Chemistry; 6th Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000; Vol. 13; pp 139–154. [Google Scholar]
- (3).Aziridines and Epoxides in Organic Synthesis; Yudin AK, Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- (4).Jacobsen EN Asymmetric Catalysis of Epoxide Ring-Opening Reactions. Acc. Chem. Res 2000, 33, 421–431. [DOI] [PubMed] [Google Scholar]
- (5).Ueberbacher BT; Hall M; Faber K Electrophilic and Nucleophilic Enzymatic Cascade Reactions in Biosynthesis. Nat. Prod. Rep 2012, 29, 337. [DOI] [PubMed] [Google Scholar]
- (6).Domínguez de María P; van Gemert RW; Straathof AJJ; Hanefeld U Biosynthesis of Ethers: Unusual or Common Natural Events? Nat. Prod. Rep 2010, 27, 370. [DOI] [PubMed] [Google Scholar]
- (7).Grüschow S; Sherman DH The Biosynthesis of Epoxides In Aziridines and Epoxides in Organic Synthesis; Yudin AK Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp 349–398. [Google Scholar]
- (8).Gallagher SC; Cammack R; Dalton H Alkene Monooxygenase from Nocardia Corallina B-276 Is a Member of the Class of Dinuclear Iron Proteins Capable of Stereospecific Epoxygenation Reactions. Eur. J. Biochem 1997, 247, 635–641. [DOI] [PubMed] [Google Scholar]
- (9).Chang W; Dey M; Liu P; Mansoorabadi SO; Moon S-J; Zhao ZK; Drennan CL; Liu H Mechanistic Studies of an Unprecedented Enzyme-Catalysed 1,2-Phosphono-Migration Reaction. Nature 2013, 496, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rather LJ; Weinert T; Demmer U; Bill E; Ismail W; Fuchs G; Ermler U Structure and Mechanism of the Diiron Benzoyl-Coenzyme A Epoxidase BoxB. J. Biol. Chem 2011, 286, 29241–29248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Grishin AM; Ajamian E; Tao L; Zhang L; Menard R; Cygler M Structural and Functional Studies of the Escherichia Coli Phenylacetyl-CoA Monooxygenase Complex. J. Biol. Chem 2011, 286, 10735–10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Teufel R; Friedrich T; Fuchs G An Oxygenase That Forms and Deoxygenates Toxic Epoxide. Nature 2012, 483, 359–362. [DOI] [PubMed] [Google Scholar]
- (13).Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cytochrome P450 - Structure, Mechanism, and Biochemistry, fourth.; Ortiz de Montellano PR, Ed.; Springer International Publishing: Cham, 2015. [Google Scholar]
- (15).Allain EJ; Hager LP; Deng L; Jacobsen EN Highly Enantioselective Epoxidation of Disubstituted Alkenes with Hydrogen Peroxide Catalyzed by Chloroperoxidase. J. Am. Chem. Soc 1993, 115, 4415–4416. [Google Scholar]
- (16).Zaks A; Dodds DR Chloroperoxidase-Catalyzed Asymmetric Oxidations: Substrate Specificity and Mechanistic Study. J. Am. Chem. Soc 1995, 117, 10419–10424. [Google Scholar]
- (17).Wang C; Chang W-C; Guo Y; Huang H; Peck SC; Pandelia ME; Lin G-M; Liu H-W; Krebs C; Bollinger JM Evidence That the Fosfomycin-Producing Epoxidase, HppE, Is a Non-Heme-Iron Peroxidase. Science 2013, 342, 991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hashimoto T; Hayashi A; Amano Y; Kohno J; Iwanari H; Usuda S; Yamada Y Hyoscyamine 6β-Hydroxylase, an Enzyme Involved in Tropane Alkaloid Biosynthesis, Is Localized at the Pericycle of the Root. J. Biol. Chem 1991, 266 (7), 4648–4653. [PubMed] [Google Scholar]
- (19).Liu P; Murakami K; Seki T; He X; Yeung S-M; Kuzuyama T; Seto H; Liu H Protein Purification and Function Assignment of the Epoxidase Catalyzing the Formation of Fosfomycin. J. Am. Chem. Soc 2001, 123, 4619–4620. [DOI] [PubMed] [Google Scholar]
- (20).Hollenhorst MA; Bumpus SB; Matthews ML; Bollinger JM; Kelleher NL; Walsh CT The Nonribosomal Peptide Synthetase Enzyme DdaD Tethers Nβ-Fumaramoyl-L-2,3-Diaminopropionate for Fe(II)/α-Ketoglutarate-Dependent Epoxidation by DdaC during Dapdiamide Antibiotic Biosynthesis. J. Am. Chem. Soc 2010, 132, 15773–15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Seo M-J; Zhu D; Endo S; Ikeda H; Cane DE Genome Mining in Streptomyces. Elucidation of the Role of Baeyer−Villiger Monooxygenases and Non-Heme Iron-Dependent Dehydrogenase/Oxygenases in the Final Steps of the Biosynthesis of Pentalenolactone and Neopentalenolactone. Biochemistry 2011, 50, 1739–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ishikawa N; Tanaka H; Koyama F; Noguchi H; Wang CCC; Hotta K; Watanabe K Non-Heme Dioxygenase Catalyzes Atypical Oxidations of 6,7-Bicyclic Systems To Form the 6,6-Quinolone Core of Viridicatin-Type Fungal Alkaloids. Angew. Chemie Int. Ed. 2014, 53, 12880–12884. [DOI] [PubMed] [Google Scholar]
- (23).Elson SW; Baggaley KH; Gillett J; Holland S; Nicholson NH; Sime JT; Woroniecki SR Isolation of Two Novel Intracellular β-Lactams and a Novel Dioxygenase Cyclising Enzyme from Streptomyces Clavuligerus. J. Chem. Soc., Chem. Commun 1987, 1736–1738. [Google Scholar]
- (24).Busby RW; Townsend CA A Single Monomeric Iron Center in Clavaminate Synthase Catalyzes Three Nonsuccessive Oxidative Transformations. Bioorg. Med. Chem 1996, 4, 1059–1064. [DOI] [PubMed] [Google Scholar]
- (25).Baldwin JE; Lloyd MD; Wha-Son B; Schofield CJ; Elson SW; Baggaleyb KH; Nicholson NH A Substrate Analogue Study on Clavaminic Acid Synthase: Possible Clues to the Biosynthetic Origin of Proclavamic Acid. J. Chem. Soc. Chem. Commun 1993, 500–502. [Google Scholar]
- (26).Salowe SP; Krol WJ; Iwata-Reuyl D; Townsend CA Elucidation of the Order of Oxidations and Identification of an Intermediate in the Multistep Clavaminate Synthase Reaction. Biochemistry 1991, 30, 2281–2292. [DOI] [PubMed] [Google Scholar]
- (27).Pan J; Bhardwaj M; Faulkner JR; Nagabhyru P; Charlton ND; Higashi RM; Miller A-F; Young CA; Grossman RB; Schardl CL Ether Bridge Formation in Loline Alkaloid Biosynthesis. Phytochemistry 2014, 98, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Deng Q; Liu Y; Chen L; Xu M; Naowarojna N; Lee N; Chen L; Zhu D; Hong X; Deng Z; Liu P; Zhao C Biochemical Characterization of a Multifunctional Mononuclear Nonheme Iron Enzyme (PtlD) in Neopentalenoketolactone Biosynthesis. Org. Lett 2019, 21, 7592–7596. [DOI] [PubMed] [Google Scholar]
- (29).2-Oxoglutarate-Dependent Oxygenases; Schofield C, Hausinger R, Eds.; Metallobiology; Royal Society of Chemistry: Cambridge, 2015. [Google Scholar]
- (30).Henschler D; Hoos WR; Fetz H; Dallmeier E; Metzler M Reactions of Trichloroethylene Epoxide in Aqueous Systems. Biochem. Pharmacol 1979, 28, 543–548. [DOI] [PubMed] [Google Scholar]
- (31).Miller RE; Guengerich FP Oxidation of Trichloroethylene by Liver Microsomal Cytochrome P-450: Evidence for Chlorine Migration in a Transition State Not Involving Trichloroethylene Oxide. Biochemistry 1982, 21, 1090–1097. [DOI] [PubMed] [Google Scholar]
- (32).Shinkyo R; Xu L; Tallman KA; Cheng Q; Porter NA; Guengerich FP Conversion of 7-Dehydrocholesterol to 7-Ketocholesterol Is Catalyzed by Human Cytochrome P450 7A1 and Occurs by Direct Oxidation without an Epoxide Intermediate. J. Biol. Chem 2011, 286, 33021–33028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Correia MA; Ortiz de Montellano PR Inhibition of Cytochrome P450 Enzymes In Cytochrome P450; Ortiz de Montellano PR, Ed.; Springer US: Boston, MA, 2005; pp 247–322. [Google Scholar]
- (34).De Visser SP; Ogliaro F; Shaik S How Does Ethene Inactivate Cytochrome P450 En Route to Its Epoxidation? A Density Functional Study. Angew. Chemie - Int. Ed. 2001, 40, 2871–2874. [DOI] [PubMed] [Google Scholar]
- (35).De Visser SP; Kumar D; Shaik S How Do Aldehyde Side Products Occur during Alkene Epoxidation by Cytochrome P450? Theory Reveals a State-Specific Multi-State Scenario Where the High-Spin Component Leads to All Side Products. J. Inorg. Biochem 2004, 98, 1183–1193. [DOI] [PubMed] [Google Scholar]
- (36).De Visser SP; Ogliaro F; Harris N; Shaik S Multi-State Epoxidation of Ethene by Cytochrome P450: A Quantum Chemical Study. J. Am. Chem. Soc 2001, 123, 3037–3047. [DOI] [PubMed] [Google Scholar]
- (37).Kumar D; Karamzadeh B; Sastry GN; De Visser SP What Factors Influence the Rate Constant of Substrate Epoxidation by Compound i of Cytochrome P450 and Analogous Iron(IV)-Oxo Oxidants? J. Am. Chem. Soc 2010, 132, 7656–7667. [DOI] [PubMed] [Google Scholar]
- (38).Kumar D; Latifi R; Kumar S; Rybak-Akimova EV; Sainna M. a.; de Visser SP Rationalization of the Barrier Height for p -Z-Styrene Epoxidation by Iron(IV)-Oxo Porphyrin Cation Radicals with Variable Axial Ligands. Inorg. Chem 2013, 52, 7968–7979. [DOI] [PubMed] [Google Scholar]
- (39).De Visser SP; Ogliaro F; Shaik S Stereospecific Oxidation by Compound I of Cytochrome P450 Does Not Proceed in a Concerted Synchronous Manner. Chem. Commun 2001, 3, 2322–2323. [DOI] [PubMed] [Google Scholar]
- (40).Groves JT; Watanabe Y The Mechanism of Olefin Epoxidation by Oxo-Iron Porphyrins. Direct Observation of an Intermediate. J. Am. Chem. Soc 1986, 108, 507–508. [DOI] [PubMed] [Google Scholar]
- (41).Smith JRL; Sleath PR Model Systems for Cytochrome P450 Dependent Mono-Oxygenases. Part 1. Oxidation of Alkenes and Aromatic Compounds by Tetraphenylporphinatoiron(III) Chloride and Lodosylbenzene. J. Chem. Soc. Perkin Trans 2 1982, 1009. [Google Scholar]
- (42).Song WJ; Ryu YO; Song R; Nam W Oxoiron(IV) Porphyrin π-Cation Radical Complexes with a Chameleon Behavior in Cytochrome P450 Model Reactions. J. Biol. Inorg. Chem 2005, 10, 294–304. [DOI] [PubMed] [Google Scholar]
- (43).Pan Z; Zhang R; Newcomb M Kinetic Studies of Reactions of Iron(IV)-Oxo Porphyrin Radical Cations with Organic Reductants. J. Inorg. Biochem 2006, 100, 524–532. [DOI] [PubMed] [Google Scholar]
- (44).Sastri CV; Sook Seo M; Joo Park M; Mook Kim K; Nam W Formation, Stability, and Reactivity of a Mononuclear Nonheme Oxoiron(Iv) Complex in Aqueous Solution. Chem. Commun 2005, 1405. [DOI] [PubMed] [Google Scholar]
- (45).Singh KK; Tiwari MK; Dhar BB; Vanka K; Sen Gupta S; Sen S Mechanism of Oxygen Atom Transfer from FeV(O) to Olefins at Room Temperature. Inorg. Chem 2015, 54, 6112–6121. [DOI] [PubMed] [Google Scholar]
- (46).Serrano-Plana J; Aguinaco A; Belda R; García-España E; Basallote MG; Company A; Costas M Exceedingly Fast Oxygen Atom Transfer to Olefins via a Catalytically Competent Nonheme Iron Species. Angew. Chemie Int. Ed. 2016, 55, 6310–6314. [DOI] [PubMed] [Google Scholar]
- (47).Zhang R; Nagraj N; Lansakara-P. DSP; Hager LP; Newcomb M Kinetics of Two-Electron Oxidations by the Compound I Derivative of Chloroperoxidase, a Model for Cytochrome P450 Oxidants. Org. Lett 2006, 8, 2731–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Neu HM; Yang T; Baglia RA; Yosca TH; Green MT; Quesne MG; De Visser SP; Goldberg DP Oxygen-Atom Transfer Reactivity of Axially Ligated Mn(V)-Oxo Complexes: Evidence for Enhanced Electrophilic and Nucleophilic Pathways. J. Am. Chem. Soc 2014, 136, 13845–13852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Chang W; Li J; Lee JL; Cronican AA; Guo Y Mechanistic Investigation of a Non-Heme Iron Enzyme Catalyzed Epoxidation in (−)-4′-Methoxycyclopenin Biosynthesis. J. Am. Chem. Soc 2016, 138, 10390–10393. [DOI] [PubMed] [Google Scholar]
- (50).Un S; Dorlet P; Voyard G; Tabares LC; Cortez N High-Field EPR Characterization of Manganese Reconstituted Superoxide Dismutase from Rhodobacter Capsulatus. J. Am. Chem. Soc 2001, 123, 10123–10124. [DOI] [PubMed] [Google Scholar]
- (51).Golynskiy MV; Gunderson W. a; Hendrich MP; Cohen SM Metal Binding Studies and EPR Spectroscopy of the Manganese Transport Regulator MntR. Biochemistry 2006, 45, 15359–15372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Gunderson WA; Zatsman AI; Emerson JP; Farquhar ER; Que L; Lipscomb JD; Hendrich MP Electron Paramagnetic Resonance Detection of Intermediates in the Enzymatic Cycle of an Extradiol Dioxygenase. J. Am. Chem. Soc 2008, 130, 14465–14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hadley RC; Gagnon DM; Brophy MB; Gu Y; Nakashige TG; Britt RD; Nolan EM Biochemical and Spectroscopic Observation of Mn(II) Sequestration from Bacterial Mn(II) Transport Machinery by Calprotectin. J. Am. Chem. Soc 2018, 140, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Liao H-J; Li J; Huang J-L; Davidson M; Kurnikov I; Lin T-S; Lee JL; Kurnikova M; Guo Y; Chan N-L; Chang W.-c. Insights into the Desaturation of Cyclopeptin and Its C3 Epimer Catalyzed by a Non-Heme Iron Enzyme: Structural Characterization and Mechanism Elucidation. Angew. Chemie Int. Ed. 2018, 57, 1831–1835. [DOI] [PubMed] [Google Scholar]
- (55).Price JC; Barr EW; Glass TE; Krebs C; Bollinger JMJ Evidence for Hydrogen Abstraction from C1 of Taurine by the High-Spin Fe(IV) Intermediate Detected during Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase (TauD). J. Am. Chem. Soc 2003, 125, 13008–13009. [DOI] [PubMed] [Google Scholar]
- (56).Matthews ML; Krest CM; Barr EW; Vaillancourt FH; Walsh CT; Green MT; Krebs C; Bollinger JM Substrate-Triggered Formation and Remarkable Stability of the C−H Bond-Cleaving Chloroferryl Intermediate in the Aliphatic Halogenase, SyrB2. Biochemistry 2009, 48, 4331–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Grzyska PK; Ryle MJ; Monterosso GR; Liu J; Ballou DP; Hausinger RP Steady-State and Transient Kinetic Analyses of Taurine/α-Ketoglutarate Dioxygenase: Effects of Oxygen Concentration, Alternative Sulfonates, and Active-Site Variants on the Fe(IV)-Oxo Intermediate. Biochemistry 2005, 44, 3845–3855. [DOI] [PubMed] [Google Scholar]
- (58).Yu C-P; Tang Y; Cha L; Milikisiyants S; Smirnova TI; Smirnov AI; Guo Y; Chang W Elucidating the Reaction Pathway of Decarboxylation-Assisted Olefination Catalyzed by a Mononuclear Non-Heme Iron Enzyme. J. Am. Chem. Soc 2018, 140, 15190–15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Poulter CD; Rilling HC Prenyltransferase: The Mechanism of the Reaction. Biochemistry 1976, 15, 1079–1083. [DOI] [PubMed] [Google Scholar]
- (60).Blanksby SJ; Ellison GB Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- (61).Kovaleva EG; Lipscomb JD Crystal Structures of Fe2+ Dioxygenase Superoxo, Alkylperoxo, and Bound Product Intermediates. Science 2007, 316, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Emerson JP; Farquhar ER; Que L Structural “Snapshots” along Reaction Pathways of Non-Heme Iron Enzymes. Angew. Chemie Int. Ed. 2007, 46, 8553–8556. [DOI] [PubMed] [Google Scholar]
- (63).Jeoung J-H; Bommer M; Lin T-Y; Dobbek H Visualizing the Substrate-, Superoxo-, Alkylperoxo-, and Product-Bound States at the Nonheme Fe(II) Site of Homogentisate Dioxygenase. Proc. Natl. Acad. Sci 2013, 110, 12625–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Hanauske-Abel HM; Günzler V A Stereochemical Concept for the Catalytic Mechanism of Prolylhydroxylase. Applicability to Classification and Design of Inhibitors. J. Theor. Biol 1982, 94, 421–455. [DOI] [PubMed] [Google Scholar]
- (65).Borowski T; Bassan A; Siegbahn PEM Mechanism of Dioxygen Activation in 2-Oxoglutarate-Dependent Enzymes: A Hybrid DFT Study. Chem. - A Eur. J. 2004, 10, 1031–1041. [DOI] [PubMed] [Google Scholar]
- (66).Ye S; Riplinger C; Hansen A; Krebs C; Bollinger JM; Neese F Electronic Structure Analysis of the Oxygen-Activation Mechanism by FeII- and α-Ketoglutarate (AKG)-Dependent Dioxygenases. Chem. - A Eur. J. 2012, 18, 6555–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Mitchell AJ; Dunham NP; Martinie RJ; Bergman JA; Pollock CJ; Hu K; Allen BD; Chang W; Silakov A; Bollinger JM; Krebs C; Boal AK Visualizing the Reaction Cycle in an Iron(II)- and 2-(Oxo)-Glutarate-Dependent Hydroxylase. J. Am. Chem. Soc 2017, 139, 13830–13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Grzyska PK; Appelman EH; Hausinger RP; Proshlyakov DA Insight into the Mechanism of an Iron Dioxygenase by Resolution of Steps Following the FeIV=O Species. Proc. Natl. Acad. Sci 2010, 107, 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Terencio T; Andris E; Gamba I; Srnec M; Costas M; Roithová J Chemoselectivity in the Oxidation of Cycloalkenes with a Non-Heme Iron(IV)-Oxo-Chloride Complex: Epoxidation vs. Hydroxylation Selectivity. J. Am. Soc. Mass Spectrom 2019, 30, 1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Meena BI; Lakk-Bogáth D; Kripli B; Speier G; Kaizer J Kinetics and Mechanism of Epoxidation of Olefins by Chiral Tetrapyridyl Oxoiron(IV) Complex. Polyhedron 2018, 151, 141–145. [Google Scholar]
- (71).Cantú Reinhard FG; de Visser SP Oxygen Atom Transfer Using an Iron(IV)-Oxo Embedded in a Tetracyclic N-Heterocyclic Carbene System: How Does the Reactivity Compare to Cytochrome P450 Compound I? Chem. - A Eur. J. 2017, 23, 2935–2944. [DOI] [PubMed] [Google Scholar]
- (72).Sainna MA; Kumar S; Kumar D; Fornarini S; Crestoni ME; de Visser SP A Comprehensive Test Set of Epoxidation Rate Constants for Iron(IV)–Oxo Porphyrin Cation Radical Complexes. Chem. Sci 2015, 6, 1516–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Ye W; Staples RJ; Rybak-Akimova EV Oxygen Atom Transfer Mediated by an Iron(IV)/Iron(II) Macrocyclic Complex Containing Pyridine and Tertiary Amine Donors. J. Inorg. Biochem 2012, 115, 1–12. [DOI] [PubMed] [Google Scholar]
- (74).Balland V; Charlot M-F; Banse F; Girerd J-J; Mattioli TA; Bill E; Bartoli J-F; Battioni P; Mansuy D Spectroscopic Characterization of an FeIV Intermediate Generated by Reaction of XO− (X = Cl, Br) with an FeII Complex Bearing a Pentadentate Non-Porphyrinic Ligand−Hydroxylation and Epoxidation Activity. Eur. J. Inorg. Chem 2004, 301–308. [Google Scholar]
- (75).Costas M; Mehn MP; Jensen MP; Que L Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- (76).Fitzpatrick PF Mechanism of Aromatic Amino Acid Hydroxylation. Biochemistry 2003, 42, 14083–14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Kovaleva EG; Lipscomb JD Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Rogers MS; Lipscomb JD Salicylate 5-Hydroxylase: Intermediates in Aromatic Hydroxylation by a Rieske Monooxygenase. Biochemistry 2019, 58, 5305–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Sahu S; Quesne MG; Davies CG; Dürr M; Ivanović-Burmazović I; Siegler MA; Jameson GNL; De Visser SP; Goldberg DP Direct Observation of a Nonheme Iron(IV)–Oxo Complex That Mediates Aromatic C–F Hydroxylation. J. Am. Chem. Soc 2014, 136, 13542–13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Sahu S; Zhang B; Pollock CJ; Dürr M; Davies CG; Confer AM; Ivanović-Burmazović I; Siegler MA; Jameson GNL; Krebs C; Goldberg DP Aromatic C–F Hydroxylation by Nonheme Iron(IV)–Oxo Complexes: Structural, Spectroscopic, and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138, 12791–12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Godbout N; Havlin R; Salzmann R; Debrunner PG; Oldfield E Iron-57 NMR Chemical Shifts and Mössbauer Quadrupole Splittings in Metalloporphyrins, Ferrocytochrome c, and Myoglobins: A Density Functional Theory Investigation †. J. Phys. Chem. A 1998, 102, 2342–2350. [Google Scholar]
- (82).Zhang Y; Mao J; Oldfield E 57 Fe Mössbauer Isomer Shifts of Heme Protein Model Systems: Electronic Structure Calculations. J. Am. Chem. Soc 2002, 124, 7829–7839. [DOI] [PubMed] [Google Scholar]
- (83).Sinnecker S; Slep LD; Bill E; Neese F Performance of Nonrelativistic and Quasi-Relativistic Hybrid DFT for the Prediction of Electric and Magnetic Hyperfine Parameters in 57Fe Mössbauer Spectra. Inorg. Chem 2005, 44, 2245–2254. [DOI] [PubMed] [Google Scholar]
- (84).Pápai M; Vankó G On Predicting Mössbauer Parameters of Iron-Containing Molecules with Density-Functional Theory. J. Chem. Theory Comput 2013, 9, 5004–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).McWilliams SF; Brennan-Wydra E; MacLeod KC; Holland PL Density Functional Calculations for Prediction of 57 Fe Mössbauer Isomer Shifts and Quadrupole Splittings in β-Diketiminate Complexes. ACS Omega 2017, 2, 2594–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Song X; Lu J; Lai W Mechanistic Insights into Dioxygen Activation, Oxygen Atom Exchange and Substrate Epoxidation by AsqJ Dioxygenase from Quantum Mechanical/Molecular Mechanical Calculations. Phys. Chem. Chem. Phys 2017, 19, 20188–20197. [DOI] [PubMed] [Google Scholar]
- (87).Gütlich P; Bill E; Trautwein AX Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications; Springer Berlin Heidelberg: Berlin, Heidelberg, 2011. [Google Scholar]
- (88).Helmetag V; Samel SA; Thomas MG; Marahiel MA; Essen L-O Structural Basis for the Erythro-Stereospecificity of the l-Arginine Oxygenase VioC in Viomycin Biosynthesis. FEBS J 2009, 276, 3669–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Chekan JR; Ongpipattanakul C; Wright TR; Zhang B; Bollinger JM; Rajakovich LJ; Krebs C; Cicchillo RM; Nair SK Molecular Basis for Enantioselective Herbicide Degradation Imparted by Aryloxyalkanoate Dioxygenases in Transgenic Plants. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 13299–13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Davis KM; Altmyer M; Martinie RJ; Schaperdoth I; Krebs C; Bollinger JM; Boal AK Structure of a Ferryl Mimic in the Archetypal Iron(II)- and 2-(Oxo)-Glutarate-Dependent Dioxygenase, TauD. Biochemistry 2019, 58, 4218–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Light KM; Hangasky J. a.; Knapp MJ; Solomon EI Spectroscopic Studies of the Mononuclear Non-Heme FeII Enzyme FIH: Second-Sphere Contributions to Reactivity. J. Am. Chem. Soc 2013, 135, 9665–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Tanase S; Marqués Gallego P; Bouwman E; Long GJ; Rebbouh L; Grandjean F; de Gelder R; Mutikainen I; Turpeinen U; Reedijk J Versatility in the Binding of 2-Pyrazinecarboxylate with Iron. Synthesis, Structure and Magnetic Properties of Iron(II) and Iron(III) Complexes. Dalt. Trans 2006, 1675–1684. [DOI] [PubMed] [Google Scholar]
- (93).Long GJ; Tanase S; Remacle F; Periyasamy G; Grandjean F Combined Mössbauer Spectral and Density Functional Theory Determination of the Magnetic Easy-Axis in Two High-Spin Iron(II) 2-Pyrazinecarboxylate Complexes. Inorg. Chem 2009, 48, 8173–8179. [DOI] [PubMed] [Google Scholar]
- (94).Su H; Sheng X; Zhu W; Ma G; Liu Y Mechanistic Insights into the Decoupled Desaturation and Epoxidation Catalyzed by Dioxygenase AsqJ Involved in the Biosynthesis of Quinolone Alkaloids. ACS Catal 2017, 7, 5534–5543. [Google Scholar]
- (95).Mader SL; Bräuer A; Groll M; Kaila VRI Catalytic Mechanism and Molecular Engineering of Quinolone Biosynthesis in Dioxygenase AsqJ. Nat. Commun 2018, 9, 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Chaturvedi SS; Ramanan R; Lehnert N; Schofield CJ; Karabencheva-Christova TG; Christov CZ Catalysis by the Non-Heme Iron(II) Histone Demethylase PHF8 Involves Iron Center Rearrangement and Conformational Modulation of Substrate Orientation. ACS Catal. 2019, 1195–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Gao S; Naowarojna N; Cheng R; Liu X; Liu P Recent Examples of α-Ketoglutarate-Dependent Mononuclear Non-Haem Iron Enzymes in Natural Product Biosyntheses. Nat. Prod. Rep 2018, 35, 792–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Zhou J; Kelly WL; Bachmann BO; Gunsior M; Townsend CA; Solomon EI Spectroscopic Studies of Substrate Interactions with Clavaminate Synthase 2, a Multifunctional Alpha-KG-Dependent Non-Heme Iron Enzyme: Correlation with Mechanisms and Reactivities. J. Am. Chem. Soc 2001, 123, 7388–7398. [DOI] [PubMed] [Google Scholar]
- (99).Neidig ML; Brown CD; Light KM; Fujimori DG; Nolan EM; Price JC; Barr EW; Bollinger JM; Krebs C; Walsh CT; Solomon EI CD and MCD of CytC3 and Taurine Dioxygenase: Role of the Facial Triad in α-KG-Dependent Oxygenases. J. Am. Chem. Soc 2007, 129, 14224–14231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Johnson KA; Simpson ZB; Blom T FitSpace Explorer: An Algorithm to Evaluate Multidimensional Parameter Space in Fitting Kinetic Data. Anal. Biochem 2009, 387, 30–41. [DOI] [PubMed] [Google Scholar]
- (101).Johnson KA; Simpson ZB; Blom T Global Kinetic Explorer: A New Computer Program for Dynamic Simulation and Fitting of Kinetic Data. Anal. Biochem 2009, 387, 20–29. [DOI] [PubMed] [Google Scholar]
- (102).Petasis DT; Hendrich MP Quantitative Interpretation of Multifrequency Multimode EPR Spectra of Metal Containing Proteins, Enzymes and Biomimetic Complexes. Methods Enzymol. 2015, 563, 171–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Otwinowski Z; Minor W Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (104).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Frisch MJ; Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2016. Please see Reference #10 in the Supporting Information for the full citation. [Google Scholar]
- (106).Neese F The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci 2012, 2, 73–78. [Google Scholar]
- (107).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- (108).Bennett CH Efficient Estimation of Free Energy Differences from Monte Carlo Data. J. Comput. Phys 1976, 22, 245–268. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.