

Summary
Active non-muscle myosin II (NMII) enables migratory cell polarization and controls dynamic cellular processes, such as focal adhesion formation and turnover, and cell division. Filament assembly and force generation depend on NMII activation through the phosphorylation of Ser19 of the regulatory light chain (RLC). Here, we identify amino acid Tyr (Y) 155 of the RLC as a novel regulatory site that spatially controls NMII function. We show that Y155 is phosphorylated in vitro by the Tyr kinase domain of EGF receptor. In cells, phosphorylation of Y155, or its phospho-mimetic mutation (Glu), prevents the interaction of RLC with the myosin heavy chain (MHCII) to form functional NMII units. Conversely, Y155 mutation to a structurally similar, but non-phosphorylatable amino acid (Phe), restores the more dynamic cellular functions of NMII, such as myosin filament formation and nascent adhesion assembly; but not those requiring stable actomyosin bundles, e.g. focal adhesion elongation or migratory front-back polarization. In live cells, phospho-Y155 RLC is prominently featured in protrusions, where it prevents NMII assembly. Our data indicate that Y155 phosphorylation constitutes a novel regulatory mechanism that contributes to the compartmentalization of NMII assembly and function in live cells.
In brief
Aguilar-Cuenca et al. determine the cellular function of myosin light chain (RLC) phosphorylation on tyrosine. Phospho-Tyr155 RLC mainly appears at lamellipodia, preventing the interaction of the regulatory light chain with myosin heavy chain II, impairing formation of functional myosin hexamers and limiting myosin assembly during protrusion.
Introduction
Non-muscle myosin II (NMII) is a motor protein that produces actin-associated intracellular forces in non-muscle cells [1]. It integrates converging biochemical and mechanical signals, converting them into mechanical work by displacing and crosslinking actin filaments. Its activation and assembly generate diverse actomyosin structures of different mechanical properties and stability, which control migratory polarization and cell division, etc. (reviewed in [2, 3]). These actomyosin structures organize asymmetrically inside cells. Asymmetry is generated by spatially restricted activation of NMII, which begins with phosphorylation of the RLC on S19 (reviewed in [4]). This event activates NMII and enables its self-association into bipolar filaments that may disassemble, or evolve into higher order structures [5].
Integrins, growth factors and chemoattractants elicit S19 phosphorylation with different stoichiometry and kinetic profiles [6–8]. Additional phosphorylation events on different residues of the RLC have diverse effects on the stability of the actomyosin bundles. Phosphorylation of RLC on T18 increases twofold the ATPase activity of NMII [9, 10], defining the most stable subgroup of actomyosin filaments that delineate the trailing edge of polarized cells [7]. Additional regulatory sites include S1 and S2, which negatively regulate NMII upon phosphorylation by PKC [11], inactivating NMII at the leading edge during mesenchymal chemotaxis [12].
The second mechanism of sorting of actomyosin bundles in polarized cells depends on the intrinsic properties of NMII paralogues as defined by different MHCII genes. Mammalian cells express up to three paralogues (NMII-A, -B and -C), and the corresponding MHCII isoforms are encoded in different genes: Myh9 encodes MHCII-A; Myh10 encodes MHCII-B; and Myh14 encodes MHCII-C [13]. NMII-B defines the more stable actomyosin assemblies, and deletion of MHCII-B impairs front-back migratory polarity [14, 15]. Conversely, NMII-A is more dynamic [16] and its deletion impairs adhesion formation and compromises cell coherence [15, 17]. The stability of the NMII assemblies is also regulated by phosphorylation of specific residues in the tail domain of the heavy chain, including S1943 in MHCII-A [18], S1935 in MHCII-B [19], and other residues in MHCII-B and II-C [20, 21].
There are additional regulatory mechanisms of NMII function. For example, Tyr phosphorylation of MHCII motor domain affects NMII function, although the mechanism is poorly defined [22, 23]. On the other hand, Tyr phosphorylation of RLC was originally reported by Krebs and co-workers over 30 years ago [24], but its function in live cells has remained unexplored. Here, we find that replacing one specific Tyr residue, Y155, with a phospho-mimetic residue (Y→E), abrogates the association of the RLC to NMII. Y155 mutation prevents the formation of large and stable actomyosin bundles that define migratory polarity. When Y155 is phosphorylated by growth factor receptors, RLC is not incorporated into NMII hexamers. Likewise, RLC does not become phosphorylated on Y155 when associated to the NMII hexamer. RLC phosphorylated on Y155 is mainly localized at the leading edge of migrating cells. Together, these data reveal the existence of a novel mechanism that regulates NMII assembly and is crucial for the inhibition of NMII at the leading edge, driving the generation and maintenance of the intracellular gradients of assembled actomyosin that define front-rear polarity and adhesion dynamics in migrating cells.
Results
Detection of RLC phosphorylation on Y142 and Y155 by mass spectrometry
Tyr phosphorylation of the RLC on residues Y142 and Y155 in response to EGF was described early [24]. These phosphorylations were confirmed in several phosphoproteomics studies (https://www.phosphosite.org/, genes Myl9 and Myl12), but their biological significance remained unaddressed. To investigate this, we first expressed low levels of FLAG-RLC wild type (10–20% the level of the endogenous protein [14]) in HEK-293 cells, then treated the cells with peroxovanadate and calyculin A to inhibit Tyr and Ser/Thr phosphatases [25]. Subsequent mass spectrometric analysis of immunoprecipitated FLAG-RLC revealed that a substantial amount of RLC (≤10% total) was phosphorylated on Y155 (Figure 1A–C), and on Y142 (Figure 1A–B,D). As expected, S19 phosphorylation was readily detected (≈ 70% of total RLC), as well as a smaller percentage of bis-phosphorylated peptides, T18+S19 and Y142+Y155 (Figure 1A–B). In untreated conditions, Tyr phosphorylation was detectable, but much lower (<1%).
Figure 1. Identification of novel RLC tyrosine phosphorylations sites on myosin RLC by mass spectrometry.
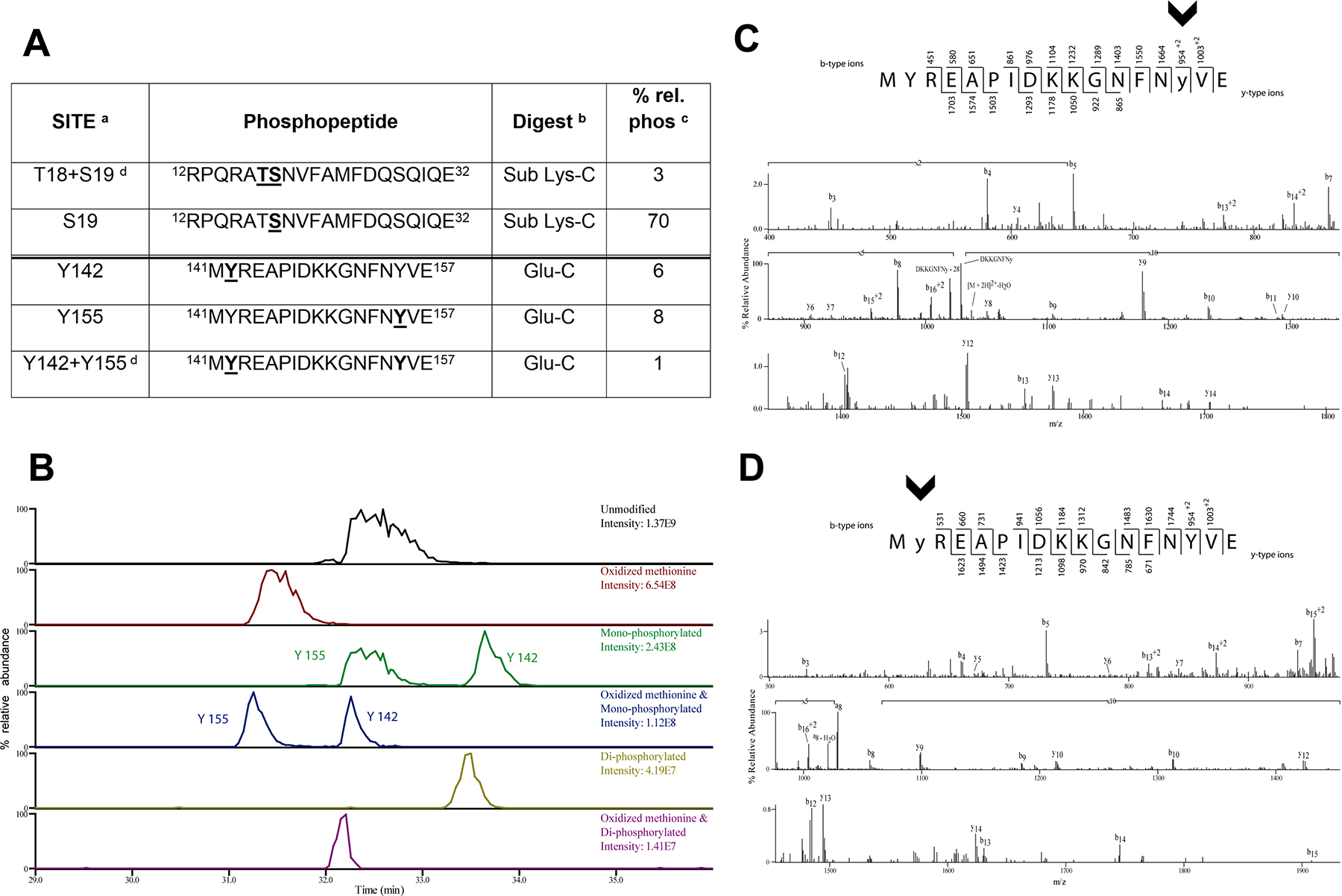
(A) RLC phosphorylated peptides identified by mass spectrometry. Phosphorylated residues are in underlined bold within the sequence. Footnotes: a, the sites of phosphorylation were manually confirmed; b, phosphorylation sites were determined from either peptides generated from the Glu-C digest or from the further digestion with Lys-C; c, relative percent phosphorylation is calculated by dividing the peak area of the phosphorylated form of the peptide over the total peak area of all forms of the peptide; d, there are two bisphosphorylated peptides identified.
(B) The extracted ion chromatograms of the RLC peptide 141-MYREAPIDKKGNFNYVE-157 unmodified, with an oxidized methionine, monophosphorylated with and without oxidized methionine, and bis-phosphorylated with and without oxidized methionine are displayed to show the relative abundance of these species.
(C,D) CAD MS/MS spectra acquired in the ion trap of [M+2H]2+ ion (m/z 1077.99) corresponding to the RLC GluC digest peptide MYREAPIDKKGNFNYVE phosphorylated on Y155 (C) and phosphorylated on Y142 (D). Identified product ions of the b2- and y2- types are listed above and below the peptide sequence, respectively. The identified ions are also indicated in the spectrum and are sufficient to localize the site of phosphorylation (lowercase and arrowheads).
Phosphomimetic and non-phosphorylatable mutants of RLC Y142 and Y155 display a different ability to associate to NMII filaments and mediate actomyosin-based contraction
To study the significance of RLC Tyr phosphorylation in live cells, we generated RLC-GFP carrying phosphomimetic (Y to E) and non-phosphorylatable (Y to F) mutations of Y142 and Y155. Low levels of mutant RLCs [14] were expressed in CHO.K1 cells, which were allowed to spread on fibronectin. None of the mutants affected the expression levels or localization of MHCII-A (Figure 2A). We found that RLC Y142F localized to myosin filaments similarly to wild type RLC, whereas RLC Y142E localized there less frequently, often appearing in nuclei (Figure 2A–B). RLC Y155F behaved similarly to RLC Y142E. Finally, RLC Y155E almost never appeared in myosin filaments (Figure 2A–B), suggesting a decreased ability of RLC Y142E, Y155F, and particularly Y155E, to compete with wild type RLC once contractile myosin filaments are formed. We next examined focal adhesion number, size and distribution, which are proxies of contractility [26]. Unexpectedly, cells expressing the RLC mutants displayed similar numbers of adhesions of comparable size (Figure 2A, C–D). Next, we serum-starved CHO.K1 cells expressing wild type RLC or the RLC Y155 mutants, which reduced the numbers of NMII filaments and focal adhesions (Figure 2E–G, compare to Figure 2A–D). Upon stimulation with IGF-I for 3h, cells expressing wild type RLC displayed a marked increase in the number and area of focal adhesions (Figure 2E–G). The number of adhesions also increased significantly in cells bearing the Y155F or Y155E mutant, although less than in wild type RLC-expressing cells (Figure 2F). However, the increase in total adhesive area in both mutants was very modest and not significant (Figure 2G). These experiments suggested that, in cells simultaneously expressing endogenous, wild type RLC and the RLC Y155 mutants, the latter prevented the downstream signals through which IGF-I triggers NMII-dependent contraction from reaching endogenous RLC, effectively inhibiting focal adhesion elongation and maturation.
Figure 2. Y155 is required for the correct localization of RLC to actomyosin filaments and adhesion elongation.
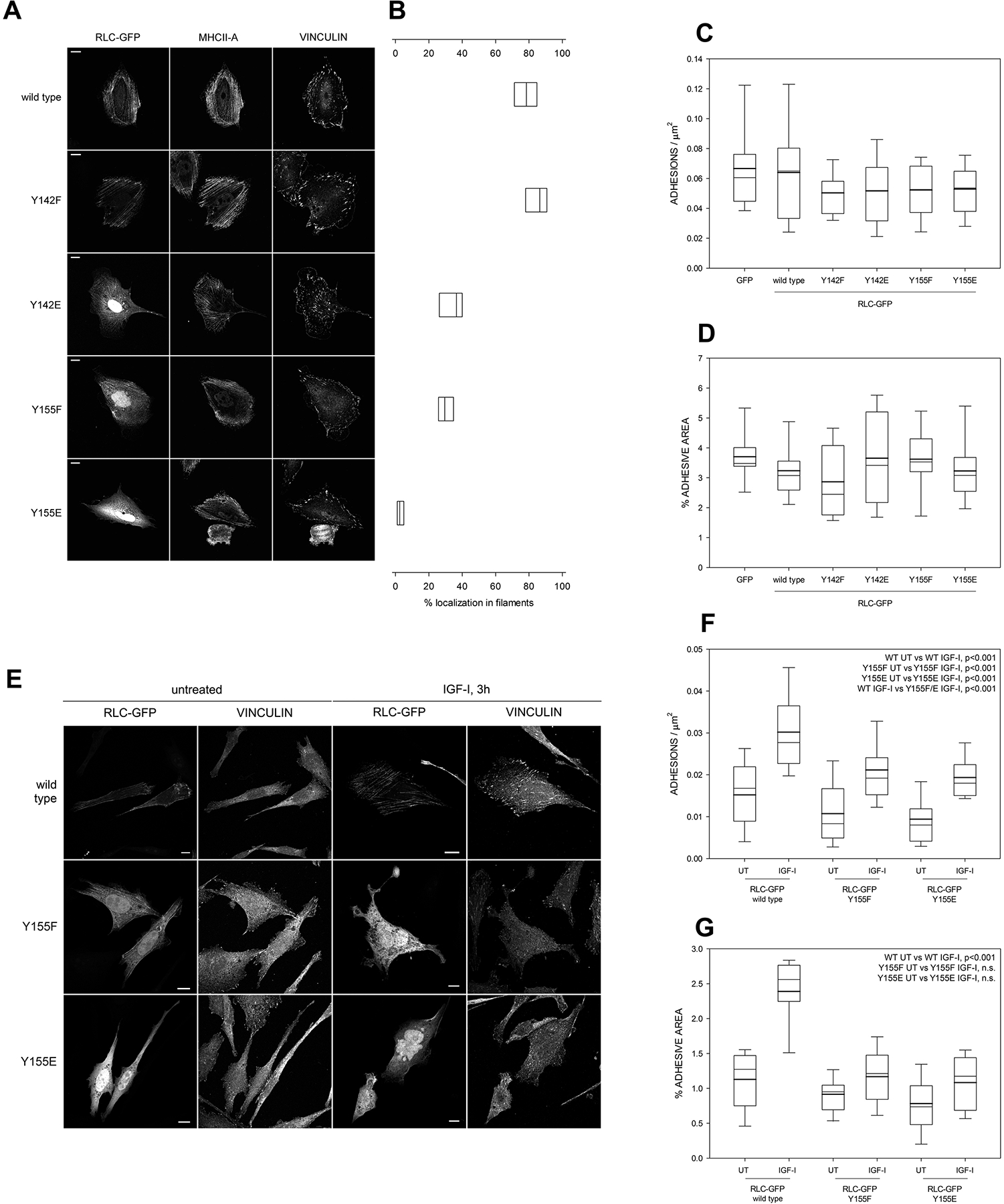
(A) Images of fibronectin-bound, CHO.K1 cells transfected with the indicated GFP-tagged versions of RLC. Bars=10 μm. Pictures are representative of >500 cells examined in three independent experiments.
(B) Quantification of the filamentous localization of the RLC mutants shown in (A). Boxes represent the 25th and 75th percentile of three independent experiments in which >500 cells were analyzed. p<0.01 between wild type/Y142F and the other conditions; also between Y155E and the other conditions.
(C-D) Quantification of the number of adhesions per μm2 (C) and total percentage of the area of the cell that corresponds to focal adhesions, that is, adhesive area (D). Boxes represent the 25th and 75th percentile and whiskers correspond to the 10th and 90th percentile of three independent experiments in which n ≥ 20 cells (>2000 adhesions) were analyzed. Throughout the study, thick line represents the mean and thin line the median. There are no statistically significant differences among conditions.
(E) Images of fibronectin-bound, CHO.K1 cells transfected with the indicated GFP-tagged versions of RLC. After 2h of adhesion, cells were starved overnight and then stimulated with IGF for 3h. Bars=10 μm. Pictures are representative of >200 cells from three independent experiments.
(F-G) Quantification of the number of adhesions per μm2 (F) and adhesive area (G) in the conditions represented in (E). n ≥ 20 cells from three separate experiments (>2000 adhesions per condition). The significances of the relevant comparisons between conditions are shown on the top right corner of each panel. WT stands for wild type throughout the study.
Phosphomimetic and non-phosphorylatable mutants of RLC Y142 and Y155 differentially affect the ability of NMII to promote adhesion maturation, front-back polarity, cell migration and cytokinesis
The previous experiments suggested that residue Y155 plays a fundamental role in controlling NMII function in live cells. To further study this, we first decreased the interference of endogenous RLC by using a specific shRNA (Figure 3A). As previously published [27], RLC depletion also decreased the expression of MHCII-A and MHCII-B (Figure 3A–B), causing a spiky, round phenotype (Figure 3B). Expression of wild type RLC and the Y142F, Y142E and Y155F mutants to levels comparable to those of endogenous RLC (see STAR Methods) restored expression of MHCII-A (Figure 3B) and MHCII-B (Figure S1A). Conversely, Y155E did not (Figure 3B and S1A). RLC depletion had no effect on Myo18A, another myosin that interacts with RLC [28, 29] (Figure S1A–B).
Figure 3. Replacement of endogenous RLC with phosphomimetic/non-phosphorylatable mutants reveals a key role for Y155 in NMII assembly and adhesion maturation.
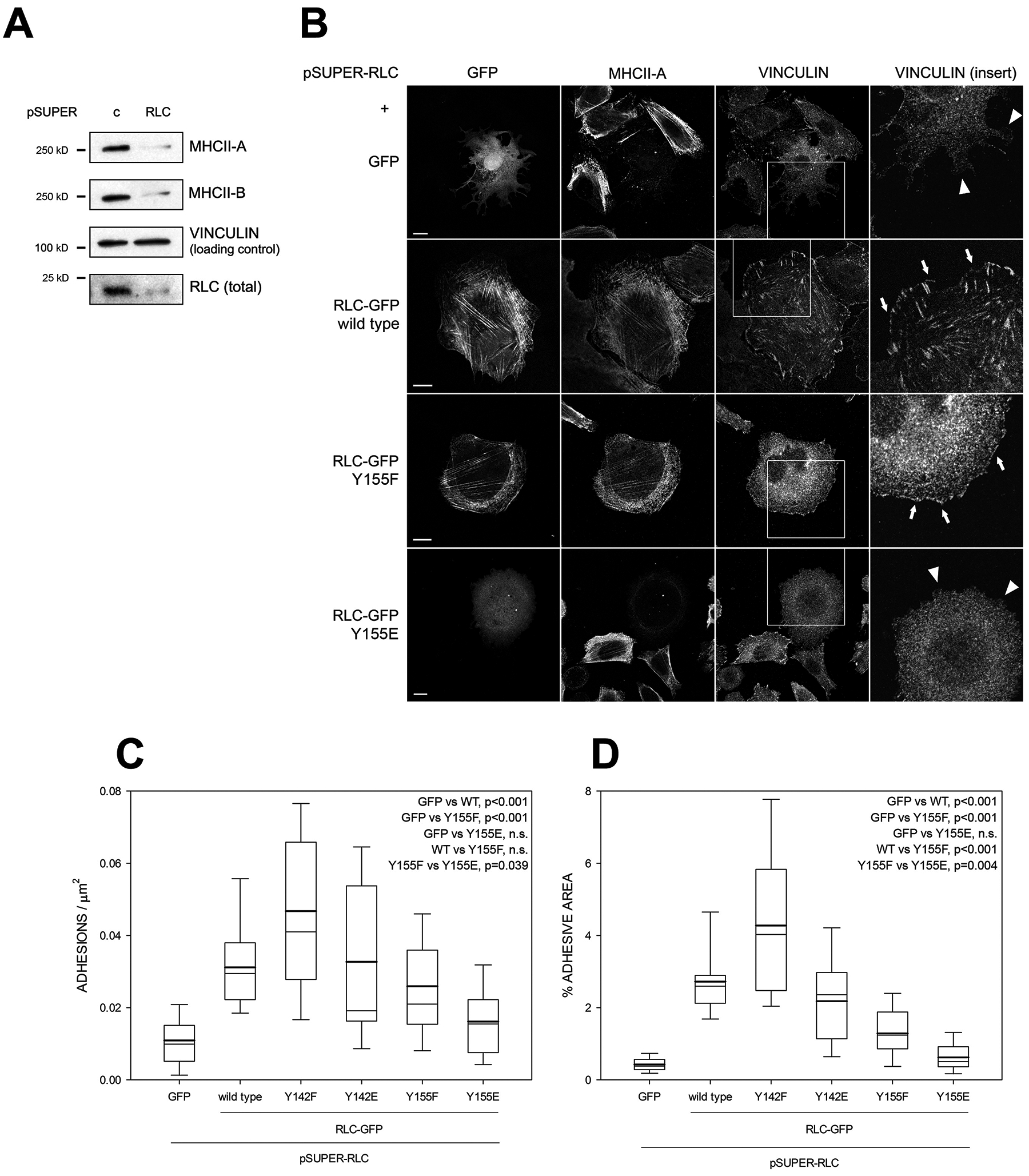
(A) CHO.K1 cells were transfected with pSUPER-RLC or a control sequence (pSUPER-C), cultured for 96h and lysed. Lysates were separated by PAGE/SDS and blotted against RLC, MHCII-A and MHCII-B. Vinculin is shown as a loading control. Experiment is representative of eight performed.
(B) CHO.K1 cells were co-transfected with pSUPER-RLC together with GFP or RLC-GFP, wild type, Y155F or Y155E (first column). After 96h, cells were allowed to spread on fibronectin for 2h, fixed, permeabilized and stained as indicated. Bars=10 μm. Fourth column, magnification of the boxed regions in the vinculin column. Arrows point to adhesions. Arrowheads indicate the lack of adhesions in comparable regions.
(C-D) Quantification of the number of adhesions per μm2 (C) and adhesive area (D) in the conditions represented in (B). n ≥ 20 cells from three independent experiments (>2000 adhesions per condition). Significances are shown in the figures.
See also Figures S1 and S2.
RLC depletion also abrogated focal adhesion formation (Figure 3B–D, compare to 2A,C–D). Surprisingly, this effect extended to nascent adhesions in lamellipodia (Figure 3B, arrowheads in top-right panel). RLC wild type restored adhesion elongation and distribution (Figure 3B, arrows). Both Y142 mutants did as well, Y142F even better than wild type RLC (Figure 3C–D). Importantly, Y155E-expressing, endogenous RLC-depleted cells remained devoid of elongated adhesions in the lamellum and cell body, and nascent adhesions in the lamellipodium (Figure 3B, arrowheads and C–D). Interestingly, RLC Y155F efficiently restored the number of adhesions (Figure 3C), including lamellipodial nascent adhesions (Figure 3B, arrows); but not their size (Figure 3D). We also observed that depletion of endogenous RLC improved the localization of RLC Y155F, but not Y155E, to myosin filaments (Figure 3B and 4A).
Figure 4. Y155 is required to generate NMII-dependent front-back polarity and enable cell migration.
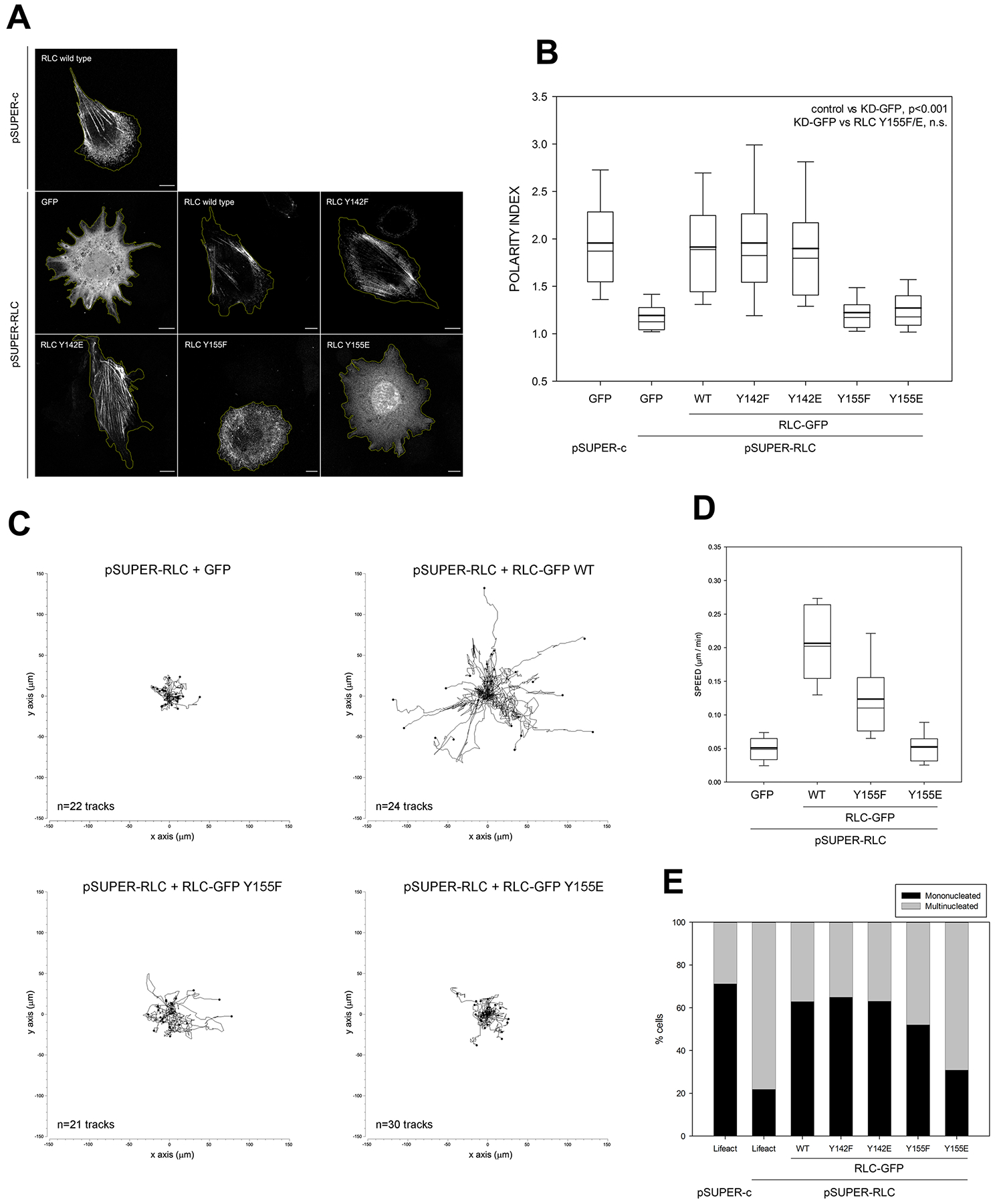
(A) CHO.K1 cells were co-transfected with pSUPER-C and pSUPER-RLC and GFP or the indicated GFP-coupled RLC mutants. After 96h, cells were allowed to adhere to fibronectin for 2h and fixed. Bars=10 μm. Images are representative of >500 examined per condition in three representative experiments.
(B) Quantification of the polarity index as indicated in STAR Methods from three independent experiments. Relevant significances are indicated in the figure.
(C) CHO.K1 cells were transfected with pSUPER-RLC and co-transfected with GFP (control) or GFP-coupled wild type (WT) RLC, Y155F or Y155E mutant as indicated. After 96h, cells were sorted by flow cytometry, adhered to fibronectin-coated coverslips and filmed as they migrate freely for sixteen hours. Data represent the indicated number of tracks from a representative experiment out of three performed.
(D) Speed measurements from the analysis of migratory tracks as shown in (C). All the conditions are significantly different with respect to each other except GFP vs. Y155E.
(E) CHO.K1 cells were transfected with pSUPER-c or pSUPER-RLC and co-transfected with GFP (control) or GFP-coupled wild type (WT) RLC, Y155F or Y155E mutant as indicated, adhered to fibronectin, fixed and stained for lamin A to determine multinucleation. Data include >200 cells/ condition from three independent experiments. All the rescues are statistically significant with respect to the non-rescued condition (second column), except Y155E.
RLC depletion also increased cellular spreading and produced a significant loss of front-back polarity (Figure 4A–B). Expression of near-endogenous levels of wild type RLC and both Y142 mutants also reversed this effect, but RLC Y155E did not. Interestingly, RLC Y155F did not restore polarity either, despite its ability to partially support NMII focal adhesion assembly and actomyosin filament formation (Figure 3C and 4A). RLC Y155F also displayed defective phosphorylation on S19 and T18+S19 in response to adhesion (Figure S2). Loss of front-rear polarity resulted in the inability of the cells to migrate properly, which was restored by expression of wild type RLC, but not RLC Y155E (Figure 4C–D). RLC Y155F restored cell migration, although only partially (Figure 4C–D).
NMII depletion or inhibition causes multinucleation [30]. We recapitulated this by removing RLC. Multinucleation was reversed by expression of wild type RLC and both Y142 mutants. Conversely, RLC Y155E did not reverse multinucleation. RLC Y155F reversed multinucleation, but again only partially (Figure 4E). Together, these data delineate a key role for Y155 in the control of the cellular functions of NMII.
Phosphorylation of the RLC on Y155 impairs its association to the myosin heavy chain to form functional NMII hexamers
The two most plausible mechanisms of action of Y155 phosphorylation are: (i) Y155 phosphorylation controls the conformation of NMII; (ii) it regulates the interaction of the RLC with MHCII as it incorporates into the NMII hexamer. To distinguish between the two possibilities, we first examined the interaction of the RLC Y155 mutants with MHCII (experiments targeting MHCII-B are shown; targeting MHCII-A yielded similar data) in control, live cells by co-immunoprecipitation. Immunoprecipitation of MHCII-B pulled down a reduced amount of RLC Y155F compared to wild type RLC, and a negligible amount of RLC Y155E (Figure 5A, second row). Reciprocal immunoprecipitation of RLC-GFP mutants revealed that MHCII-B was almost undetectable in RLC Y155E and reduced in RLC Y155F immunoprecipitates (Figure 5B, first row). These results suggested that Y155 phosphorylation negatively regulates the interaction of RLC with MHCII to form NMII hexamers. They also suggested that phospho-Y155 RLC does not interact properly with MHCII. However, Y155E RLC mimics phosphorylation, but it may not faithfully replicate all the effects of phosphorylation. To address this, we generated a polyclonal antibody (8471) that recognized phospho-Y155 RLC. The 8471 antibody recognized RLC (Mw ≈ 20 kD) in FLAG-transfected, peroxovanadate-treated CHO.K1 cells (Figure S3A). The antibody was deemed specific because it did recognize FLAG-tagged wild type and Y142F-RLC, but not Y155F or a double mutant, YYFF (Figure S3A). Its intensity also decreased in peroxovanadate-treated, RLC-depleted cells with respect to non-depleted cells (Figure S3B).
Figure 5. Y155 phosphorylation abrogates the association of RLC to NMII hexamers and filaments.
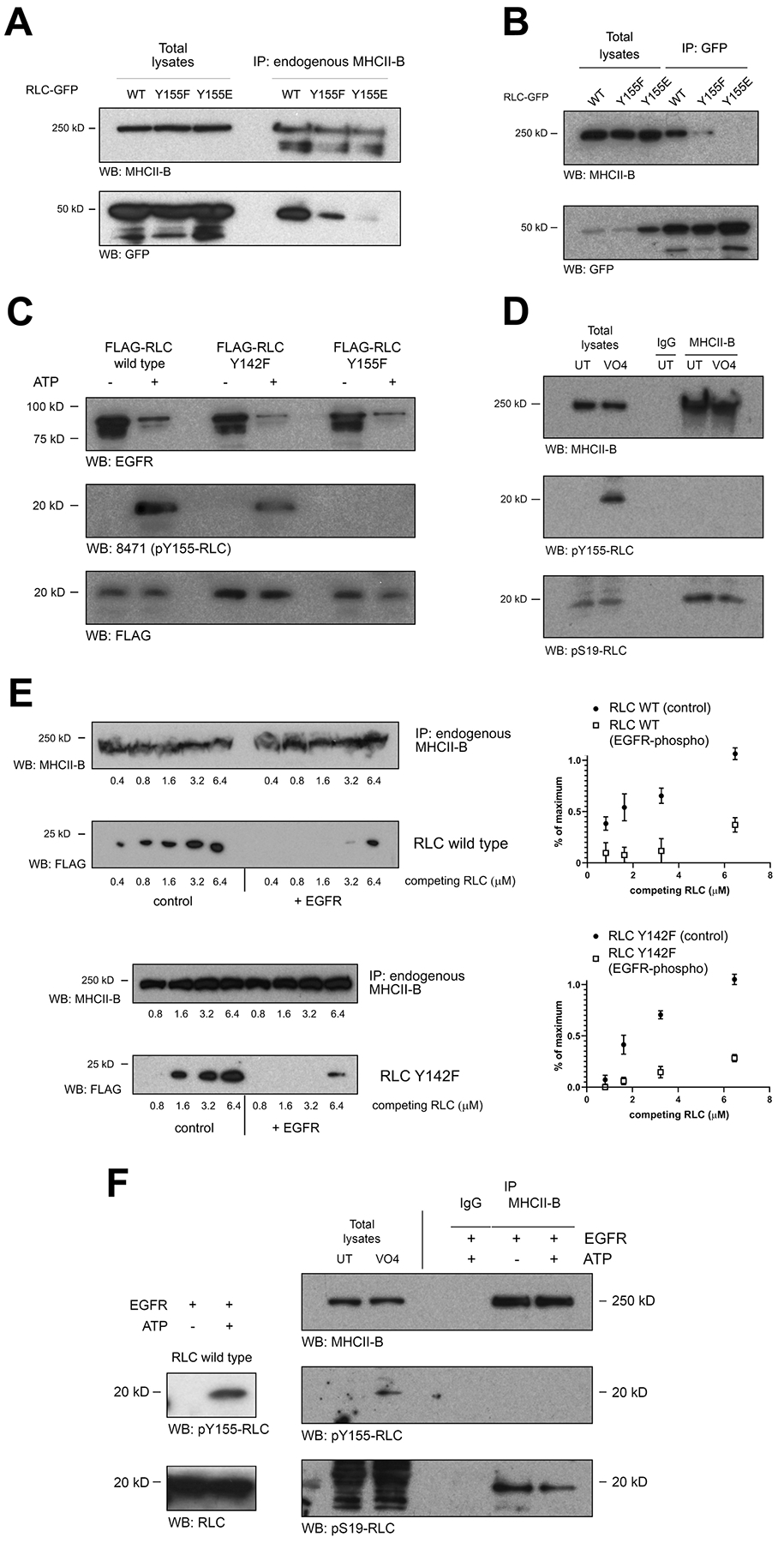
(A) Cells were transfected with wild type (WT) RLC-GFP or the indicated mutants. After 24h, endogenous MHCII-B was immunoprecipitated, separated by PAGE-SDS and blotted for MHCII-B (immunoprecipitation yield, top) and GFP (bottom). A representative experiment of three performed is shown.
(B) Cells were transfected and lysed as in (A). RLC-GFP variants were then immunoprecipitated with GFP-TRAP magnetic agarose, separated by PAGE-SDS and blotted for MHCII-B (top) and GFP (immunoprecipitation yield, bottom). A representative experiment of three performed is shown.
(C) In vitro kinase assay of recombinant FLAG-RLC, wild type or the indicated mutants (substrates) and recombinant active GST-EGFR (kinase). The in vitro kinase assay was carried out for 30 min at 30°C in the presence (+) or absence (−) of ATP, stopped with Laemmli buffer, separated by PAGE/SDS and blotted using antibodies against EGFR (top), pY155 (middle) and FLAG (bottom). A representative experiment out of five performed is shown.
(D) CHO.K1 cells were treated (VO4) or not (UT) with 1μM sodium peroxovanadate for 30 min, lysed and endogenous MHCII-B was immunoprecipitated with a specific antibody. Immunoprecipitated proteins were separated by PAGE-SDS and blotted for MHCII-B (immunoprecipitation yield, top) or NMII-bound RLC, phosphorylated in pY155 (middle) or pS19 (bottom). A representative experiment of three performed is shown.
(E) MHCII-B was immunoprecipitated from untreated CHO.K1 cells as in (D). Equal amounts of the immunoprecipitates were separated and incubated for 30 min with increasing amounts of competing FLAG-tagged RLC, wild type (top) or Y142F, non-phosphorylated (left) or phosphorylated with recombinant active GST- EGFR kinase + ATP (right). First and third box depict MHCII-B (immunoprecipitation yield); second and fourth box depict FLAG. A representative experiment is shown. Right panels represent the quantification of the densitometric analysis of three independent experiments.
(F) In vitro kinase assay of recombinant RLC (left) or NMII-B-bound RLC. Blots on right represent endogenous MHCII-B immunoprecipitated as in (A). Immunoprecipitated material was incubated with EGFR kinase (rEGFR) fragment in the presence (+) or absence (−) of ATP as indicated before. Total lysates of untreated and peroxovanadate-treated cells are also shown as reference. Samples were blotted with anti-MHCII-B (immunoprecipitation yield, top); anti-pY155 RLC (middle boxes) or pS19 RLC (bottom boxes). Panels in left depict an in vitro kinase assay done in parallel to the one shown in the panels on right, using the same reagents and conditions except the phosphorylation substrate, which was 1 μg of recombinant, wild type FLAG-RLC. A representative experiment of three performed is shown.
See also Figures S3, S4 and S5.
We next aimed at identifying the kinase(s) that could mediate Y155 phosphorylation. The Scansite algorithm [31] predicted that RTK receptors (PDGFR, EGFR) could phosphorylate RLC directly on Y155. To test this, we produced recombinant, FLAG-tagged RLC (Figure 5C, third row) and used it as a substrate for a GST-fusion protein containing the active kinase domain of EGFR (Figure 5C, first row). The kinase domain of EGFR robustly phosphorylated wild type and Y142F, but not Y155F, RLC (Figure 5C, second row). We also addressed whether Y155 phosphorylation altered the conformation of the RLC. Using trypsin to cleave in vitro phosphorylated recombinant RLC, we found that phospho-Y155 RLC (and the Y155E mutant) displayed a modestly increased accessibility to proteolytic digestion (Figure S4A–B), suggesting that the conformation of phospho-Y155 RLC is slightly, but not dramatically, more accessible than that of non-phosphorylated RLC.
We next immunoprecipitated MHCII-B from peroxovanadate-treated cells and examined the phosphorylation of NMII-B-bound RLC on Y155 and S19. As expected, we detected RLC phosphorylated on S19 (Figure 5D, third row). However, the immunoprecipitates did not contain phospho-Y155 RLC (Figure 5D, second row, right lanes), despite phospho-Y155 RLC being present in total lysates (Figure 5D, second row, left lanes). Together with the co-immunoprecipitation assays using RLC Y155E, these experiments suggest that, when phosphorylated on Y155, RLC interacts poorly with MHCII. To confirm this, we pulled down endogenous MHCII-B and incubated the immunoprecipitates with increasing amounts of FLAG-tagged wild type (or Y142F RLC to prevent interferences from the potential phosphorylation of Y142 by EGFR), non-phosphorylated or phosphorylated in Y155 by pre-incubation with active EGFR kinase and ATP. In both cases, non-phosphorylated RLC readily displaced endogenous RLC from immunoprecipitated MHCII-B; but Y155-phosphorylated RLC required a much higher concentration (Figure 5E, second and fourth rows and panels on right). This indicates that Y155-phosphorylated RLC displays lower affinity for MHCII-B than the non-phosphorylated form. These experiments prompted us to probe whether RLC could become phosphorylated on Y155 while bound to the NMII hexamer. In vitro phosphorylation experiments in which we used the entire NMII-B hexamer (immunoprecipitating MHCII-B) as a substrate for recombinant EGFR kinase revealed that, when in the context of the NMII-B hexamer, RLC does not become phosphorylated on Y155 (Figure 5F, second row, last lane), whereas it is readily phosphorylated in the NMII-unbound, free form (Figure 5F, left boxes). Also, experiments using recombinant subfragment-1 NMII-A produced in baculovirus transduced-Sf9 cells, or free recombinant His-tagged RLC, followed by in vitro phosphorylation with recombinant EGFR kinase and mass spectrometry analysis yielded a similar conclusion (Figure S5). These data show that RLC becomes phosphorylated in Y155 when free, that is, outside of the context of the NMII hexamer, and that Y155 phosphorylation effectively prevents its incorporation to the hexamer.
Preferential phosphorylation of the RLC on Y155 at the leading edge contributes to the inhibition of local NMII assembly in protrusions
Growth factors induce actin polymerization-driven protrusion. Others and we have shown that NMII is not actively assembling in the protruding region [32, 33]. Since growth factors promote Y155 phosphorylation in carcinoma cells [24], we sought to examine the potential role of phosphorylation of RLC on Y155 as a regulator of NMII assembly in the context of growth factor-driven protrusion. CHO.K1 cells respond strongly to IGF-I [34], eliciting many common downstream signals with EGF [35]. In these cells, we observed a modest and sustained increase phospho-Y155 RLC in response to IGF-I (Figure 6A–B). The kinetics of Y155 phosphorylation was different from those of S19, T18/S19 and S1, which reached a maximum after 30 min, declining thereafter (Figure 6A–B). Similar observations were made in A549 cells (Figure 6C) and in HEK-293 cells in a dose-dependent manner in response to EGF (Figure 6D). These experiments indicate that growth factors of the EGF/IGF-I family trigger phosphorylation of RLC on Y155 in live cells, albeit modestly. To investigate whether the increase in Y155 phosphorylation was spatially restricted, we examined the localization of phospho-Y155 RLC in fibronectin-bound CHO.K1 cells treated with IGF-I or PMA. The antibody was extensively validated for use in immunofluorescence (Figure S6). We observed that, in cells treated (10 min) with IGF-I or PMA, phospho-Y155 RLC appeared in lamellipodia, whereas it was largely absent from the lamellum (Figure 7A). These data indicate that phospho-Y155 RLC, which is unable to associate to NMII filaments, accumulates at protrusive regions of the cell. Together with the fact that RLC exchanges rapidly in and out of NMII filaments as shown by FRAP (up to 80% in 60s [36], which is higher than MHCII-A [15]), these experiments are consistent with a model in which growth factor-driven phosphorylation of RLC on Y155 at the leading edge locally abrogates the exchange of RLC in and out of NMII filaments, thereby preventing filament stabilization and assembly, and maintaining the edge of the protrusion free of assembled NMII (Figure 7B).
Figure 6. Growth factor-dependent phosphorylation of RLC on Y155 is kinetically different from its phosphorylation on S1, S19 and T18/S19.
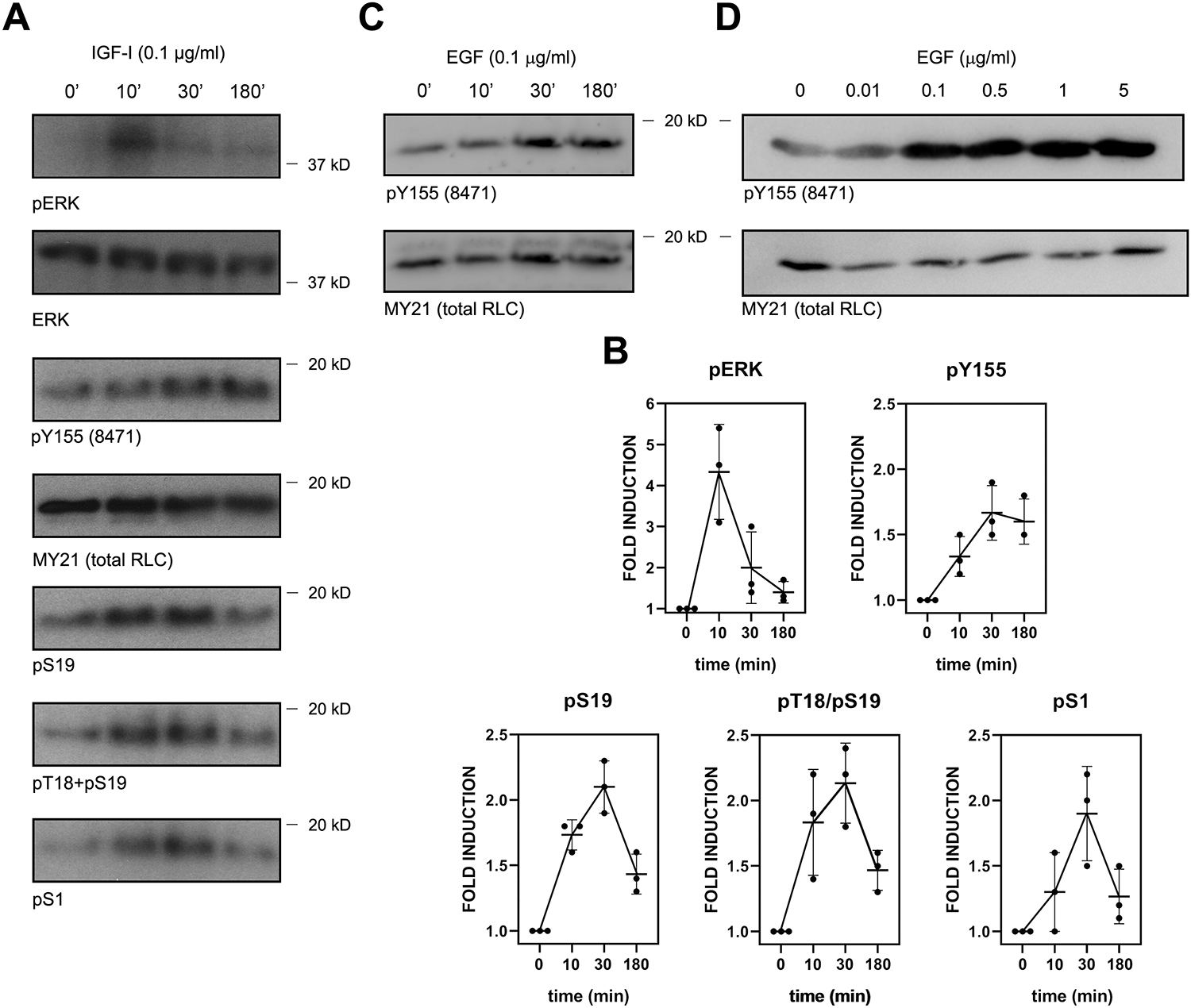
(A) CHO.K1 cells were serum-starved for 16h, then treated with IGF-I (0.1 μg/ml) for the indicated times, lysed with Laemmli buffer and blotted for the indicated antigens. Experiment is representative of three performed.
(B) Quantification of the densitometric profiles of the blots shown in (A) and its biological replicates.
(C) Serum-starved A549 cells were treated for the indicated times with 0.1 μg/ml EGF and processed as in (A). Experiment is representative of three performed.
(D) Serum-starved HEK-293 cells were treated for 30 min with the indicated dose of EGF. Experiment is representative of three performed.
Figure 7. Y155-phosphorylated RLC preferentially localizes to the leading edge of polarized cells.
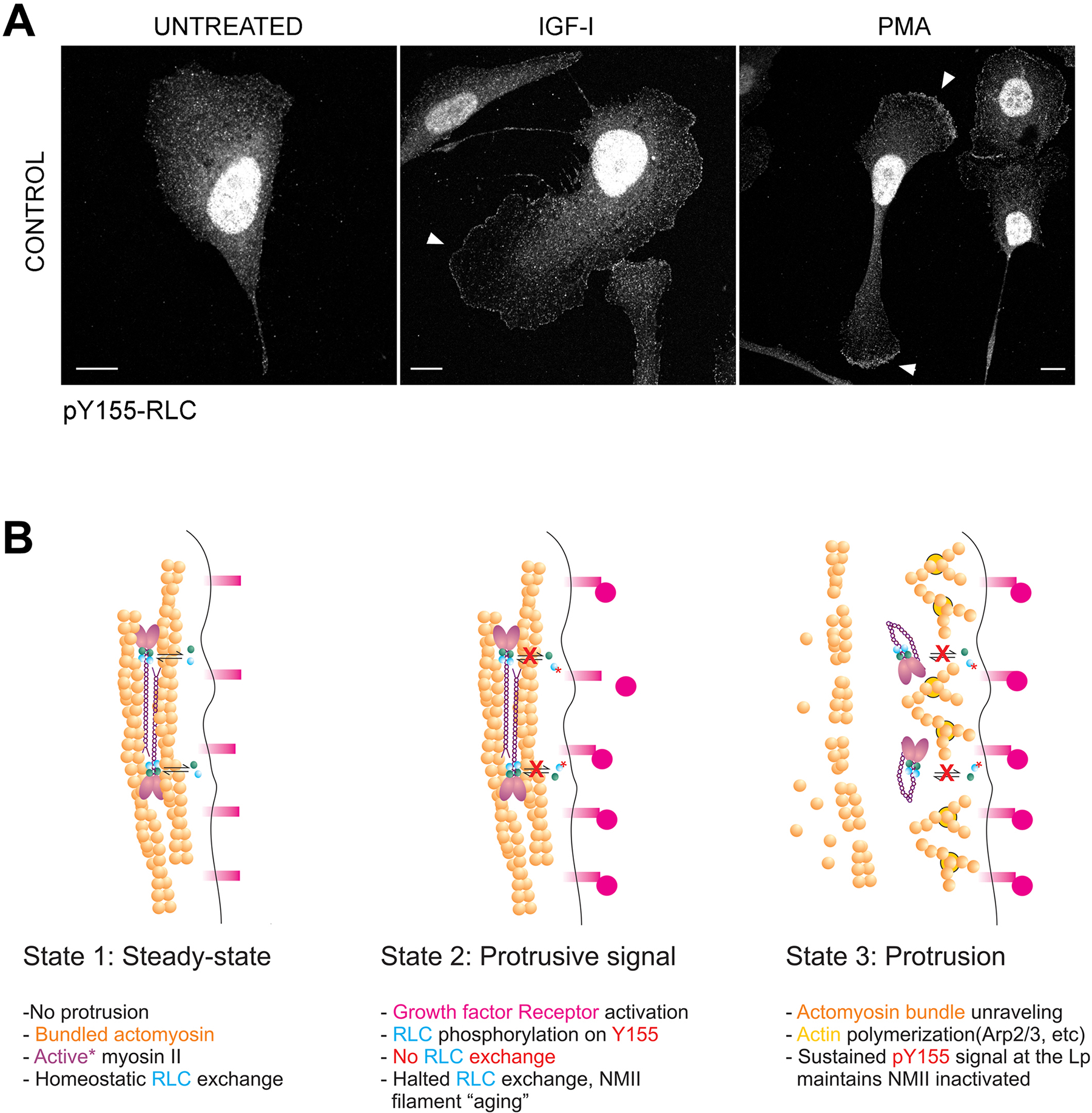
(A) CHO.K1 cells were spread on fibronectin, serum-starved for 16h, then treated with IGF-I (100 ng/ml) or PMA (20 ng/ml) for 10 min, fixed and stained with the pY155 antibody. Bars=10 μm.
(B) A model of regulation of NMII assembly by Y155 in the lamellipodium. Left panel represents an edge of the cell in steady-state, in the absence of stimulus and with fully formed actomyosin bundles that prevent spontaneous protrusion. A crucial assumption is that bundle maintenance requires bound RLC (cyan balls) exchanging with the free RLC fraction (indicated by bidirectional black arrows), which is inferred from published FRAP data [36]; upon growth factor stimulation (middle panel), RLC becomes phosphorylated on Y155 (red asterisk), effectively lowering the affinity of RLC for NMII and thus blocking the homeostatic exchange of RLC (red Xs); as a consequence, NMII filaments cannot exchange RLC and eventually disassemble (right panel). The region is now devoid of assembled NMII and can undergo protrusion.
See also Figure S6.
Discussion
In this study, we demonstrate that phosphorylation of residue Y155 of the RLC inhibits NMII assembly at the leading edge. In non-muscle cells, actin and NMII appear in two major states: non-filamentous and filamentous. Filamentous actomyosin can be structurally divided into small and dynamic bundles, and large and stable bundles [14, 32]. A large amount of stable actomyosin bundles increases the mechanical resistance of cells [37]. The intracellular distribution of actomyosin bundles of different stability controls the ability of the cells to become stably polarized. This hinges on the assembly competence of NMII, which depends on S19 (and T18) phosphorylation [4]. Here, Y155 emerges as a crucial residue that controls the incorporation of RLC into the NMII hexamer. When phosphorylated, or phospho-mimicked (Y-to-E), RLC interacts very poorly with MHCII. This is not likely due to improper folding of RLC Y155E since it Y155E is expressed at levels comparable to the functional forms, does not localize to the aggresome (not shown) and it displays similar in vitro accessibility to proteolysis as phospho-Y155 RLC. It also diverts IGF-I signaling, indicating its functionality as a downstream target of growth factor signaling; however, RLC Y155E cannot convert these signals into contraction. The inability of RLC Y155E to restore the loss of MHCII-A/B expression caused by depletion of endogenous RLC suggests that its affinity for MHCII-A/B is so low that it does not stabilize NMII hexamers even in the non-filamentous (10S) conformation. This confirms in live cells that RLC interaction with MHCII is necessary for the stabilization of NMII, as shown in vitro for smooth muscle myosin [38].
On the other hand, RLC Y155F becomes incorporated into NMII filaments in the absence of endogenous RLC. However, its activation (measured by S19 and T18/S19 phosphorylation) is very modest, and clearly insufficient to generate the required amount of large and stable actomyosin filaments that define front-rear migratory polarity. This suggests that myosin filaments containing RLC Y155F are less stable and smaller than those containing wild type RLC. However, RLC Y155F-containing myosin filaments are sufficient to drive nascent adhesion formation. Interestingly, blebbistatin does not deplete NMII hexamers and does not remove nascent adhesions [39]. This is in line with another study that suggested that NMII activation in the absence of bipolar filament formation underlies nascent adhesion formation [40].
Our data are consistent with a model in which Y155 would function as a switch that enables the cell to use RLC to form assembly-competent NMII only when Y155 is not phosphorylated. Y155 localizes to the C-terminal lobe of RLC. Taylor and colleagues used cryo-EM to show that, in smooth muscle myosin II, RLC Y155 is solvent-accessible [41]. Together with the observations described here, their data suggest that Y155 phosphorylation could prevent the RLC from adopting a conformation that allows RLC binding to MHCII to form NMII hexamers.
As for the possible role of Y155 phosphorylation in a physiological context, our experiments indicate that Y155 phosphorylation in response to growth factors is modest. However, phospho-Y155 RLC appears locally in lamellipodia and is largely absent from the lamellum. This suggests that growth factors prevent NMII assembly in the lamellipodium through this mechanism. This is particularly important in light of the fact that RTKs can also activate NMII via S19 phosphorylation, hence the same stimulus could trigger paradoxical, protrusive and anti-protrusive, signals. We propose that, in active, protruding lamellipodia, Y155 phosphorylation by growth factors is locally dominant, preventing NMII assembly. Later, lamellipodia stall because the effect of growth factors on S19 phosphorylation becomes dominant, promoting NMII filament assembly, traction stress and adhesion elongation. The kinetics of Y155 phosphorylation, which plateaus between 30 min and 3h; and S1/S19, which reach a maximum between 10–30 min and decline sharply afterwards; suggest that Y155 phosphorylation does not likely control NMII activation, but regulates the amount of NMII that can become activated at lamellipodial edges.
In addition to NMII, RLC also interacts with other myosins [42], for example Myo18A. Interestingly, Myo18A was unaffected by RLC depletion. Given that Myo18A lacks ATPase activity [28, 43, 44], it is feasible that the RLC-dependent mechanism that controls MHCII-A/B expression is tied to their ability to act as actin motors, or their ability to form filaments.
Our data support a model in which phosphorylation of Y155 inhibits the incorporation of RLC into the NMII hexamer in the lamellipodium (Figure 7B). In steady-state, filamentous NMII undergoes homeostatic RLC (cyan ball) exchange with the free fraction of RLC, as shown by FRAP [36]. This exchange is essential for the stability of the NMII filaments. Upon local growth factor stimulation, free RLC would become locally phosphorylated on Y155, greatly reducing its affinity for NMII hexamers and impeding its exchange. As a consequence, NMII filaments in this subcellular region would no longer be exchanging fresh RLC and would disassemble. If the region is initially devoid of actomyosin filaments, this mechanism would ensure that no actomyosin assemblies form during the protrusive phase of the cellular response to growth factors. It seems clear that the regulation of NMII function lies at the center of the ability of cells to maintain polarity and coherence during migration. Recent studies have shown that filamentous NMII localizes to the trailing edge through a mechanism that involves actin retrograde flow [45]. In keratocytes, NMII filaments control traction stress in a manner dependent on the distance to the center of mass of the cell [46], consistent with the aberrant elongation of the cell body observed when NMII-A is deleted [15, 17]. By maintaining RLC in a soluble, non-filamentous state, Y155 phosphorylation represents a mechanism to maintain a reservoir of exchangeable RLC that can be used by the cell to reassemble NMII filaments and generate traction in response to changes in their microenvironment.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Miguel Vicente-Manzanares (miguel.vicente@csic.es).
Materials availability
All unique reagents generated in this study are available without restriction. The pY155 polyclonal antibody was custom made, hence supplies are limited, and will be provided while supplies last.
Data and code availability
This study did not generate any unique code. The authors declare that all data supporting the findings of this study are available within the manuscript and its supplementary files or are available from the lead contact on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and transfection
HEK-293 (ATCC #CRL-1573) and A549 (ATCC #CCL-185) cell lines were cultured in high-glucose DMEM medium. CHO.K1 cells (ATCC #CCL-61) were cultured in lowglucose DMEM medium. Both media contained 10% FBS, 4 mM L-glutamine, 1% nonessential amino acids and 100U/mL penicillin and 100U/mL streptomycin. Cells were transfected as described [33]. Analysis was performed 24h (overexpression) or 96h (depletion and rescue) post-transfection. In co-transfection experiments, pSUPER plasmids containing the shRNA sequences were used in 10:1 excess to GFP-containing plasmids to ensure knockdown in fluorescence-positive cells. For immunofluorescence rescue experiments, cells were selected as follows: in an initial experiment, we transfected cells with pSUPER-C or pSUPER-RLC and RLC-GFP (wild type). After four days, we performed flow cytometry on both populations, staining for RLC with Rockland’s rabbit RLC antibody (# 600-401-938) and a goat anti-rabbit antibody coupled to AlexaFluor647. We compared the intensity of the RLC channel in the GFP-transfected cells to the intensity of the RLC in non-transfected cells. We selected a population of cells within a comparable range of intensities, examined the GFP channel and used this population to create a flow cytometry gate. Another population of these cells were then sorted and adhered to fibronectin-coated coverslips, stained for RLC as indicated above and examined by confocal microscopy. These cells were used to establish the desired intensity in the GFP channel that corresponds to normal RLC levels. These conditions were saved and applied in all subsequent experiments. Spodoptera frugiperda (Sf9) insect cells were from Thermo Fisher Scientific and cultured in Sf-900™ II SFM medium according to the manufacturer’s instructions. Infection with His-RLC and FLAG-tagged myosin II-A subfragment-1 was performed using standard procedures [16].
METHOD DETAILS
Plasmids
Wild type chicken myosin RLC coupled to GFP was a generous gift from Kathleen Kelly (NCI, Bethesda, MD). The gene encoded in this plasmid is Gallus gallus Myl9 (NIH accession number XM_015296240.1), which bears 97% protein homology with hamster non-muscle RLC, gene Myl12B). The Y142E/F, Y155E/F and YYFF (Figure S3) mutations were obtained by site-directed mutagenesis using the GeneTailor kit (Thermo Fisher Scientific). FLAG-tagged chicken RLC versions were obtained by routine subcloning into pCDNA3 with a built-in FLAG tag in the N-terminus [47]. His-tagged and tandem GST-FLAG-tagged RLC for in vitro kinase assays were obtained by in frame cloning of chicken RLC in pET302 or pGEX-6P1 (GE Healthcare). pSUPER-RLC and pSUPER-c (control) plasmids were obtained by inserting the sequences GGGATGGCTTCATTGACAA, specific for RLC (mouse, genes Myl12A and Myl12B, hamster, gene Myl12B) but absent in chicken RLC, gene Myl9 (2 mismatches, experimentally verified to be insensitive to the siRNA sequence, data not shown) and CGTACGCGGAATACTTCGA in pSUPER vector (Oligoengine) according to the manufacturer’s instructions. The expression construct for FLAG-tagged human MHCII-A subfragment-1 was described in [16]. Lifeact-GFP (used as control in Figure 4E) was obtained from R. Wedlich-Soldner (Max Planck Institute, Martinsried) [48].
Antibodies
Antibodies against the following antigens were used: MHCII-A and -B (#909801 and #909901, respectively) from Biolegend; Myo18A (#NB100-79839) from Novus Biologicals; myosin RLC pS19 (#3675) and RLC pT18/S19 (#3674) from Cell Signaling Technologies; RLC pS1 (#MP3461) from ECM Biosciences; total RLC (indicated as RLC in the figures, # 600-401-938) from Rockland; EGFR (#AHR5072), from Thermo Fisher Scientific; RLC (indicated as MY-21 in the figures, # M4401), vinculin (#V9131), α-tubulin (#T9026), FLAG (#F3165) and vimentin (#V6630) from Merck; and GFP (#9996) from Santa Cruz Biotechnology. Anti-GFP-conjugated magnetic beads (#gtma-20) were from Chromotek. The 8471 antiserum was custom developed specifically for us in a rabbit host by Thermo Fisher Scientific, using the following immunizing peptide conjugated to KLH: KGNFN(pY)VEFTRIC. Four routine immunizations were administered over 72 days and serum samples were collected and tested at days 24, 48 and 72. An additional boost was administered at day 73, and serum was collected at day 84 (final). The antiserum was affinity-purified and tested by ELISA at the Thermo Fisher Scientific facility, followed by our own affinity tests as shown in Figure S3 and S6. Before use, the antibody was preincubated for 1h at room temperature with a tenfold excess of the non-phosphorylated peptide, KGNFNYVEFTRIC, to prevent non-specific binding. Highly cross-adsorbed Alexa Fluor568 (mouse, A11031; rabbit, A11036), AlexaFluor647 (mouse, A32728; rabbit, A21245) or HRP (mouse, A16078; rabbit, A16110)-conjugated antibodies were from Thermo Fisher Scientific. Regarding antibodies for immunoprecipitation, the following were used: mouse IgG-agarose (A0919) and anti-FLAG M2 magnetic beads (M8823), from Merck; and anti-GFP-conjugated magnetic beads (gtma-20), from Chromotek. In some cases, Surebeads protein-A magnetic beads (1614013, BioRad) were used to conjugate the specific antibodies as indicated in the Immunoprecipitation section.
Immunofluorescence
Cells were allowed to adhere to 2 μg/ml fibronectin-coated coverslips for 60 min, fixed using 4% paraformaldehyde (methanol-free) in PBS, and permeabilized with 0.5% Triton X-100 for 5 min. Coverslips were incubated with primary antibodies diluted and a species-appropriate, highly crosslinked, secondary antibody coupled to either Alexa Fluor 568/647 and/or phalloidin-AlexaFluor647 where indicated, rinsed and mounted in coverslips using Prolong Glass antifade medium.
Production of recombinant proteins and in vitro kinase assay
FLAG-MHCII-A subfragment-1 was expressed in the Bac-to-Bac baculovirus/Sf9 system (Thermo Fisher Scientific) and purified by FLAG-affinity chromatography followed by size exclusion chromatography [49]. Briefly, pFastBac1-FLAG MHCII-A S1 (human) and vectors encoding the RLC and ELC were used to generate recombinant bacmids that were transfected into Sf9 cells to generate recombinant viruses. The protein was purified exactly as described [49], with the modification that the protein was further purified with a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) and concentrated with Vivaspin centrifugal concentrators (50 kDa MWCO, GE Healthcare). His-RLC in pET302 vector was produced in E.coli according to the protocol of the vector supplier (Thermo Fisher Scientific). For intact molecule mass spectrometry, 10 mM FLAG-MHCII-A subfragment-1 or 10 mM His-RLC was incubated with 0.003 mg/ml EGFR in 10mMMOPS pH 7.2, 50mMNaCl, 5mMMgCl2, 0.1mMEGTA, 2mMMnCl2, 1x phosphatase inhibitor cocktail PhosStop and 100 mMATP for 2 h at room temperature. For overnight incubation, samples were placed on ice after a 2 h incubation at room temperature. Kinase was omitted for the controls. Samples were mixed 1:1 with 5% acetonitrile+0.05% trifluoroacetic acid and subjected to HPLC-mass spectrometry [50].
For expression of recombinant RLC, pGEX-6P1 plasmids encoding tandem GST-FLAG-tagged RLC mutants were introduced in BL21(DE3)pLysS competent cells, which were further allowed to grow to plateau phase overnight in 40 ml LB medium at 37°C under constant shaking. Cultures were expanded to 400 ml and incubated for 2 more hours. 0.3 mM IPTG was added to the cultures, which were grown at 30°C for 3h. Cells were collected, lysed using 1 mg/ml lysozyme in ice-cold TBS and sonicated five times for 30s. Lysates were clarified by centrifugation (11,000 ×g, 30 min at 4°C) and incubated for 2h with 300 μl glutathione-agarose beads. The beads were rinsed five times with ice-cold TBS and incubated with 20 U of Prescission protease in 200 μl Prescission buffer at 4°C (to remove the GST tag, leaving the FLAG tag in) according to the manufacturer’s instructions. His-tagged RLC was prepared according to standard procedures. For IVK assays, 5 μg of recombinant RLC or 50 μl of MHCII-B immunoprecipitate (in experiments shown in Figure 5F) were mixed with 0.5 μg of human active EGFR in 30 μl kinase buffer containing 5 mM MOPS, pH 7.2, 100 μM ATP, 2 mM β-glycerol-phosphate, 5mM MgCl2, 2mM MnCl2, 1mM EGTA, 0.5mM EDTA and incubated for 30 min at 30°C. The reaction was stopped using 30 μl of 2× Laemmli buffer, separated using PAGE/SDS, transferred to PVDF membranes and blotted as indicated.
Immunoprecipitation
For initial detection of phosphorylated residues in RLC by mass spectrometry, cells (3× p100 dishes per condition) were lysed in 1ml ice-cold 50mM Tris-HCl buffer pH7.4 containing 1% Triton X-100, 50 mM NaCl, 5mM MgCl2, 5mM ATP, and protease and phosphatase inhibitor cocktails. Lysates were clarified by centrifugation (13000 ×g, 15 min at 4°C) and incubation with 50 μl mouse IgG-agarose for 2h at 4°C under continuous stirring. Clarified lysates were incubated with 30 μl anti-FLAG-conjugated magnetic beads for 90 min at 4°C under continuous stirring. The beads were rinsed four times with ice-cold TBS and immunoprecipitated proteins were removed from the beads using 80 μl Laemmli buffer. For the data shown in Figure 5, immunoprecipitations were carried out using protein-A-conjugated magnetic beads pre-coupled to rabbit anti- MHCII-B (5 μl of purified antibody + 50 μl beads in 500 μl of PBS + 1% BSA, incubated overnight at 4°C under continuous rocking); or anti-GFP-conjugated beads. Immunoprecipitates were then separated by PAGE/SDS, transferred to PVDF membranes, blocked with BSA and blotted with the antibodies indicated in each case. When the molecular weight coincided with that of the antibody light chains, protein A-HRP or protein G-HRP were used instead of HRP-conjugated antibodies, to avoid detection of the antibody bands. Blots were scanned in a Canon 9000F-II scanner and quantified by densitometry using ImageJ.
Mass spectrometry based phosphoproteomics
HEK-293 cells were transfected with FLAG-RLC wild type and maintained in DMEM medium for 24h. To inhibit phosphatase activity, cells were treated with 1μM peroxovanadate and 10 nM calyculin A 30 min prior to lysis. Subsequently, cells were lysed in a buffer containing 20 mM Tris, 300 mM NaCl, 10 mM MgCl2 and 5 mM ATP with protease and phosphatase inhibitors, and immunoprecipitated for 90 min using anti-FLAG-conjugated magnetic beads, rinsed three times with lysis buffer without protease inhibitors, and eluted using 100 μg/ml FLAG peptide. FLAG-RLC immunoprecipitates were reduced for 1 h at room temperature (RT) with dithiothreitol (DTT), at a final concentration of 0.05 mM and alkylation for 45 minutes at RT with iodoacetamide (IAD), at a final concentration of 0.12 mM. Bound protein was digested using endoproteinase Lys-C or endoproteinase Glu-C as indicated in Figure 1A, at a ratio of 1:20, enzyme to protein, for 6 hrs with agitation at RT. The peptide mixture was removed from the beads and acidified to pH 4.0 with glacial acetic acid. An aliquot of the peptide mixture was loaded onto a kasil fritted, polyimide coated, fused silica capillary “pre-column” (360 μm o.d. × 75 μm i.d.) packed with 8 cm of irregular C18 reverse phase (RP) packing material (ODS-A, 120 Ǻ pore size, 5 – 20 μm). The sample was washed with solvent A (100 mM acetic acid) for 5 column volumes. The pre-column was butt-connected to an analytical column (360 μm o.d. × 50 μm i.d.) containing 6–8 cm of C18 RP packing material (ODS-A, 120 Ǻ pore size, 5 μm diameter) using polytetrafluoroethylene (PTFE) tubing (0.06” o.d. × 0.012” i.d.). The analytical column was equipped with an integrated, laser-pulled (P-2000), emitter tip for ESI. Peptides were gradient eluted into a LTQ Orbitrap mass spectrometer modified for front-end, electron transfer dissociation (FETD) using an Agilent 1100 series binary liquid chromatography (LC) pump at a split flow rate of approximately 60 nL/min using a 65 min gradient from 0% to 60% solvent B (70% acetonitrile, 100 mM acetic acid). Solvent B was then ramped up to 100% and was held at 100% for 20minutes for column wash. High resolution full MS spectra were acquired with a resolution of 100,000, an AGC target of 5e5, a maximum ion time of 50 ms, and scan range of 300 to 2000 m/z. Following each full MS scan 10 data-dependent CAD MS/MS spectra were acquired in the ion trap with normal scan rate. Monoisotopic precursor selection (MIPS) was on and dynamic exclusion was set to 30 seconds exclusion time after two selections in 15 seconds. All MS/MS spectra were collected using the following instrument parameters: AGC target of 1e5, maximum ion time of 100 ms, one microscan, 2.0 m/z isolation window, and Normalized Collision Energy (NCE) of 35. All MS/MS spectra were searched against both a RLC protein database and the NCBI NR human and chicken protein database using the Open Mass Spectrometry Search Algorithm (OMSSA) [51]. All searches were completed using the GluC digest parameter, precursor and peptide mass tolerances were set to +/− 0.05 and +/− 0.35, respectively. Search parameters included the variable modifications; +56 Da for carbamidomethylation of cysteines, +16 Da for the oxidation of methionines, and +80 Da for the presence of a phosphate group on serine, threonine, and tyrosine residues. Precursor charge state determination was set to read input file. Results from the searches were subject to manual interpretation in order to determine sequence coverage and modification site identification.
Proteolytic protection assay
The protocol used was modified from [52]. Briefly, recombinant, FLAG-tagged wild type RLC was phosphorylated in vitro with recombinant GST-EGFR as indicated above. The kinase was then turned off irreversibly by adding 10 nM afatinib for 15 min. Then, 1 μg of phosphorylated RLC, non-phosphorylated RLC and the Y155E mutant were separately incubated with 1 ng trypsin for 5 or 10 min (total volume 20 μl). Reaction was stopped by placing the tubes on dry ice and adding an equal volume of 2× Laemmlii buffer. The reaction was then separated by PAGE/SDS and bands were visualized by Coomassie staining. Gels were scanned in a Canon 9000F-II scanner and quantified by densitometry using ImageJ.
Confocal microscopy
Images were obtained in Leica SP8 confocal spectral microscope at 63× magnification. As source of illumination, the microscope was fitted with a pulsed white light (WL) laser (80 MHz). GFP was excited at 491 nm; whereas AlexaFluor568 and Alexa Fluor647 probes were excited with at 568 and 647 nm, respectively. Photon collection was set using internal standards of the microscope for each wavelength in sequential mode.
Migration assay
Cells were plated on glass-bottomed dishes pre-incubated overnight with 2 μg/ml fibronectin in imaging medium for 1 h, and maintained at 37°C at pH 7.4. The transfected cells were identified using fluorescence for the first frame of the time-lapse video. The rest of the images were collected using very low levels of transmitted light, one image every 10 min for 16 h.
QUANTIFICATION AND STATISTICAL ANALYSIS
Polarity quantification and focal adhesion analysis
Polarity and focal adhesion number and size (represented as cellular adhesive area) were quantified as described elsewhere [33]. Briefly, to quantify cell polarity (axis ratio), cells were fixed, stained and images acquired in the Leica SP8 confocal microscope as described before. For each individual cell, the axis ratio was calculated dividing the length of the long, migration-defined axis by the perpendicular axis passing by the nucleus of the cell. For adhesion analysis, we obtained images as before. We then used ImageJ [53] to subtract background, applied the CLAHE (Contrast Limited Adaptive Histogram Equalization) algorithm to create a high-contrast image that was subject to the threshold command of ImageJ and quantified using the Analyze Particles ImageJ built-in feature. % of adhesive areas was obtained by dividing the adhesive area of the cell determined as described above divided by the total area of the cell determined manually.
For migration assays, the centroids of migrating cells were determined using the “Manual Tracking” plugin for ImageJ, transferred and plotted using Chemotaxis tool from Ibidi. Velocities were determined using the same software.
Statistics
When comparing number of adhesions, adhesive area or polarity, a normality test (Shapiro-Wilk) was first applied to the conditions under comparison. If both conditions passed this test (i.e., they were normal), Student’s t test was used to determine significance. However, when data failed Shapiro-Wilk’s test, Mann Whitney’s U test was used to determine the significance of the observed differences. Due to the importance of these comparisons for the interpretation of the data throughout the study, significance is indicated either in the figure itself or in its legend.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Non Muscle Myosin Heavy Chain II-A | Biolegend | Cat#909801 |
| Rabbit polyclonal anti-Non Muscle Myosin Heavy Chain II-B | Biolegend | Cat#909901 |
| Rabbit polyclonal anti-Myo18A | Novus Biologicals | Cat#NB100–79839 |
| Rabbit polyclonal anti-Phospho-Myosin Light Chain 2 (Ser19) | Cell Signaling Technologies | Cat#3675 |
| Rabbit polyclonal anti-Phospho-Myosin Light Chain 2 (Thr18/Ser19) | Cell Signaling Technologies | Cat#3674 |
| Rabbit polyclonal anti-Phospho-Myosin Light Chain (Ser1) | ECM Biosciences | Cat#MP3461 |
| Rabbit polyclonal anti-Myosin Regulatory Light Chain | Rockland | Cat#600-401-938 |
| Mouse monoclonal anti-Myosin Regulatory Light Chain (clone MY-21) | Merck | Cat#M4401 |
| Mouse monoclonal anti-EGFR | Thermo Fisher Scientific | Cat#AHR5072 |
| Mouse monoclonal anti-Vinculin | Merck | Cat#V9131 |
| Mouse monoclonal anti-α-Tubulin | Merck | Cat#T9026 |
| Mouse monoclonal anti-FLAG | Merck | Cat#F3165 |
| Mouse monoclonal anti-Vimentin | Merck | Cat#V6630 |
| Mouse monoclonal anti-GFP | Santa Cruz Biotechnology | Cat#9996 |
| Rabbit serum anti-Phospho-Myosin Light Chain 2 (Tyr155) – Antiserum 8471 | This paper, custom made by Thermo Fisher Scientific | N/A |
| Goat anti mouse AlexaFluor568, highly cross-adsorbed | Thermo Fisher Scientific | Cat#A11031 |
| Goat anti mouse AlexaFluor647, highly cross-adsorbed | Thermo Fisher Scientific | Cat#A32728 |
| Goat anti rabbit AlexaFluor568, highly cross-adsorbed | Thermo Fisher Scientific | Cat#A11036 |
| Goat anti rabbit AlexaFluor647, highly cross-adsorbed | Thermo Fisher Scientific | Cat#A21245 |
| Goat anti mouse-HRP | Thermo Fisher Scientific | Cat#A16078 |
| Goat anti rabbit-HRP | Thermo Fisher Scientific | Cat#A16110 |
| Mouse IgG−Agarose | Merck | Cat#A0919 |
| Anti-GFP-conjugated magnetic beads | Chromotek | Cat#gtma-20 |
| Anti-FLAG M2 Magnetic Beads | Merck | Cat#M8823 |
| Bacterial and Virus Strains | ||
| BL21(DE3)pLysS | EMD Millipore | Cat#70236 |
| Biological Samples | ||
| None | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sodium orthovanadate | Merck | Cat#S6508 |
| Calyculin A | Enzo Life sciences | Cat#BML-EI192–0025 |
| ATP | Merck | Cat#A7699 |
| Lysozime | Merck | Cat#L6876 |
| UltraCruz® PVDF Transfer membrane | Santa Cruz Biotech. | Cat#SC-3723 |
| Bovine serum albumin | NZY Tech | Cat#MB04603 |
| Lys-C Protease, MS-grade | Thermo Fisher Scientific | Cat#90307 |
| Glu-C endoproteinase | Merck | Cat# 11420399001 |
| Iodoacetamide | Merck | Cat#I1149 |
| Fibronectin | Merck | Cat#F2006 |
| rhIGF-1 | Thermo Fisher Scientific | Cat#PHG0071 |
| rhEGF | Thermo Fisher Scientific | Cat#PHG0311 |
| Active human EGFR | Merck | Cat#SRP0239 |
| PreScission™ Protease | GE Healthcare | Cat#GE27-0843-01 |
| IPTG | NZY Tech | Cat#MB026 |
| Triton X-100 | Merck | Cat#T8787 |
| Phosphatase Inhibitor Cocktail 2 | Merck | Cat#P5726 |
| Phosphatase Inhibitor Cocktail 3 | Merck | Cat#P0044 |
| SureBeads™ Protein A Magnetic Beads | BioRad | Cat#1614013 |
| Afatinib | SelleckChem | Cat#S1011 |
| Prolong Glass Antifade medium | Thermo Fisher Scientific | Cat# P36984 |
| Phalloidin-AlexaFluor 647 | Thermo Fisher Scientific | Cat#22287 |
| Pierce™ Recombinant Protein A, Peroxidase Conjugated | Thermo Fisher Scientific | Cat#32400 |
| Pierce™ Recombinant Protein G, Peroxidase Conjugated | Thermo Fisher Scientific | Cat#31499 |
| Pierce™ 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher Scientific | Cat# 28906 |
| BD BaculoGold Linearized Baculovirus DNA | BD Biosciences | Cat#554739 |
| Acetonitrile | Merck | Cat#900667 |
| Betamercaptoethanol | Merck | Cat#M6250 |
| Benzonase Nuclease | Merck | Cat#E1014 |
| CelLyticB | Merck | Cat#B7435 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Merck | 11873580001 |
| DL-Dithiothreitol (DTT) | Biomatik | A2452–5G |
| Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) | Merck | Cat# 3777 |
| Flag peptide | Biomatik | Custom synthesis |
| Imidazole | Merck | Cat#792527 |
| MagicMedia | Thermo Fisher Scientific | Cat# K6803 |
| 3-(N-Morpholino)propanesulfonic acid (MOPS) | Merck | Cat# RDD003 |
| Ni-NTA | Qiagen | Cat# 30230 |
| Magnesium chloride (MgCl2) | Quality Biological | Cat# 351-033-721 |
| Manganese chloride (MnCl2) | Merck | Cat# 805930 |
| PhosSTOP | Roche | Cat# 04906837001 |
| Trifluoroacetic acid (TFA) | Merck | Cat#302031 |
| Trypsin, from bovine pancreas | Merck | Cat#T1426 |
| Glutathione-agarose medium | ABT | Cat#4B-Glu-10 |
| NZY Speedy miniprep | NZY Tech | Cat#MB21002 |
| T4 ligase | Thermo Fisher Scientific | Cat#EL0011 |
| Critical Commercial Assays | ||
| GeneTailor Kit | Thermo Fisher Scientific | Cat#12397–014 |
| Deposited Data | ||
| None | ||
| Experimental Models: Cell Lines | ||
| Hamster: CHO.K1 cells | ATCC | Cat#CCL-61 |
| Human: HEK-293 cells | ATCC | Cat#CRL-1573 |
| Human: A549 cells | ATCC | Cat#CCL-185 |
| Insect: Sf9 cells | Thermo Fisher Scientific | Cat#12659017 |
| Experimental Models: Organisms/Strains | ||
| None | ||
| Oligonucleotides | ||
| Listed in Table S1 | This paper | Oligos IDs listed in Table S1 |
| Recombinant DNA | ||
| Plasmid: pCDNA3-FLAG | [47] | N/A |
| Plasmid: pCDNA3-FLAG-RLC wild type | This paper | N/A |
| Plasmid: pCDNA3-FLAG-RLC Y142F | This paper | N/A |
| Plasmid: pCDNA3-FLAG-RLC Y155F | This paper | N/A |
| Plasmid: pCDNA3-FLAG-RLC Y142F+Y155F | This paper | N/A |
| Plasmid: pEGFP-N1-RLC wild type | Kathleen Kelly (NCI, Bethesda, MD) | N/A |
| Plasmid: pEGFP-N1-RLC Y142F | This paper | N/A |
| Plasmid: pEGFP-N1-RLC Y142E | This paper | N/A |
| Plasmid: pEGFP-N1-RLC Y155F | This paper | N/A |
| Plasmid: pEGFP-N1-RLC Y155E | This paper | N/A |
| Plasmid: pSUPER-c | This paper | N/A |
| Plasmid: pSUPER-RLC | This paper | N/A |
| Plasmid: Lifeact-GFP | [48] | N/A |
| Plasmid: pGEX-6P-1-FLAG-RLC wild type | This paper | N/A |
| Plasmid: pGEX-6P-1-FLAG-RLC Y142F | This paper | N/A |
| Plasmid: pGEX-6P-1-FLAG-RLC Y155F | This paper | N/A |
| Plasmid: pGEX-6P-1-FLAG-RLC Y155E | This paper | N/A |
| Plasmid: pET302-His-FLAG-RLC wild type | This paper | N/A |
| Plasmid: pVL1392-FLAG MHCII-A S1 (human) | [16] | N/A |
| Software and Algorithms | ||
| ImageJ | [53] | https://imagej.nih.gov/ij/ |
| SigmaPlot | Systat Software, Inc. | N/A |
| Open Mass Spectrometry Search Algorithm | [51] | N/A |
| Chemotaxis and Migration Tool | Ibidi | https://ibidi.com/chemotaxis-analysis/171-chemotaxis-and-migration-tool.html |
| MassHunter version B.06.00 | Agilent Technologies | N/A |
| Origin | OriginLab | N/A |
| Other | ||
| Polyimide coated, fused silica capillary column, 360 μm o.d. × 75 μm i.d. | Polymicro Technologies | N/A |
| C18 reverse phase packing material (ODS-A, 120 Ǻ pore size, 5 – 20 μm) | YMC | N/A |
| C18 reverse phase packing material (ODS-A, 120 Ǻ pore size, 5 μm) | YMC | N/A |
| Laser-pulled (P-2000) emitter tip for ESI | Sutter Instruments | N/A |
| LTQ Orbitrap | Thermo Fisher Scientific Scientific | N/A |
| 16/600 Superdex 200 pg column | GE Healthcare Life Sciences | 28989335 |
| Agilent 1100 series HPLC | Agilent Technologies | N/A |
| Agilent 6224 mass spectrometer | Agilent Technologies | N/A |
| ÄKTA FPLC | GE Healthcare Life Sciences | N/A |
| Zorbax 300 SB-C18 (2.1×50 mm, 3.5 μm) column | Agilent Technologies | N/A |
| Vivaspin concentrators | GE Healthcare | 28-9323-60, 28-9323-62 |
Highlights.
Myosin light chain (RLC) is phosphorylated in tyrosine.
Aberrant cell function by RLC deletion is not corrected by phospho-mimetic RLC.
Phosphorylation of RLC in Y155 impairs formation of functional myosin hexamers.
Phospho-Y155 RLC mainly appears at lamellipodia.
Acknowledgments
The authors thank Sarah Conrad, Leanna Whitmore, and Rick Horwitz for their help during the initial stages of this project; Pekka Lappalainen and Laura Machesky for very helpful advice; Jose Maria de Pereda (IBMCC, Salamanca, Spain) for help with the proteolysis assay; and Victor López Huete for GFP-based cell sorting. We also thank the Biochemistry Core Facility of the National Heart, Lung, and Blood Institute and the Unit of Molecular Imaging of the IBMCC for support, advice, and use of facilities. C.L.-G. is supported by a predoctoral fellowship from the Junta de Castilla y León. M.M.-S. is supported by a predoctoral fellowhip from the AECC. This work was funded by the following grants: Programa de Apoyo a Planes Estrategicos de Investigación de Estructuras de Investigación de Excelencia (CLC–2017–01) from the Junta de Castilla-Leon with FEDER funds (Spain); SAF2014-54705-R and SAF2017-87408-R from MINECO (Spain); CIVP16A1831 from the Ramon Areces Foundation (Spain); 14-BBM-340 from the BBVA Foundation (Spain); and IDEAS-VICE18 from the Asociacion Española Contra el Cancer (AECC, Spain) to M.V.-M.; NIH GM 037537 (D.F.H.); and K22HL131869 (S.M.H.) and the Intramural Research Program (J.R.S.) of the National Heart, Lung, and Blood Institute, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare that they have no competing financial interests.
References
- 1.Heissler SM, and Sellers JR (2016). Various Themes of Myosin Regulation. J Mol Biol 428, 1927–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilar-Cuenca R, Juanes-Garcia A, and Vicente-Manzanares M (2014). Myosin II in mechanotransduction: master and commander of cell migration, morphogenesis, and cancer. Cell Mol Life Sci 71, 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma X, and Adelstein RS (2014). The role of vertebrate nonmuscle Myosin II in development and human disease. Bioarchitecture 4, 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ikebe M (2008). Regulation of the function of mammalian myosin and its conformational change. Biochem Biophys Res Commun 369, 157–164. [DOI] [PubMed] [Google Scholar]
- 5.Dasbiswas K, Hu S, Schnorrer F, Safran SA, and Bershadsky AD (2018). Ordering of myosin II filaments driven by mechanical forces: experiments and theory. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 373, 20170114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betapudi V, Rai V, Beach JR, and Egelhoff T (2010). Novel regulation and dynamics of myosin II activation during epidermal wound responses. Exp Cell Res 316, 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vicente-Manzanares M, and Horwitz AR (2010). Myosin light chain monoand di-phosphorylation differentially regulate adhesion and polarity in migrating cells. Biochem Biophys Res Commun 402, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vicente-Manzanares M, Cabrero JR, Rey M, Perez-Martinez M, Ursa A, Itoh K, and Sanchez-Madrid F (2002). A role for the Rho-p160 Rho coiled-coil kinase axis in the chemokine stromal cell-derived factor-1alpha-induced lymphocyte actomyosin and microtubular organization and chemotaxis. J Immunol 168, 400–410. [DOI] [PubMed] [Google Scholar]
- 9.Kamisoyama H, Araki Y, and Ikebe M (1994). Mutagenesis of the phosphorylation site (serine 19) of smooth muscle myosin regulatory light chain and its effects on the properties of myosin. Biochemistry 33, 840–847. [DOI] [PubMed] [Google Scholar]
- 10.Ikebe M, Hartshorne DJ, and Elzinga M (1986). Identification, phosphorylation, and dephosphorylation of a second site for myosin light chain kinase on the 20,000-dalton light chain of smooth muscle myosin. J Biol Chem 261, 36–39. [PubMed] [Google Scholar]
- 11.Nishikawa M, Sellers JR, Adelstein RS, and Hidaka H (1984). Protein kinase C modulates in vitro phosphorylation of the smooth muscle heavy meromyosin by myosin light chain kinase. J Biol Chem 259, 8808–8814. [PubMed] [Google Scholar]
- 12.Asokan SB, Johnson HE, Rahman A, King SJ, Rotty JD, Lebedeva IP, Haugh JM, and Bear JE (2014). Mesenchymal chemotaxis requires selective inactivation of myosin II at the leading edge via a noncanonical PLCgamma/PKCalpha pathway. Dev Cell 31, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vicente-Manzanares M, Ma X, Adelstein RS, and Horwitz AR (2009). Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vicente-Manzanares M, Koach MA, Whitmore L, Lamers ML, and Horwitz AF (2008). Segregation and activation of myosin IIB creates a rear in migrating cells. J Cell Biol 183, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, and Horwitz AF (2007). Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol 176, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovacs M, Wang F, Hu A, Zhang Y, and Sellers JR (2003). Functional divergence of human cytoplasmic myosin II: kinetic characterization of the non-muscle IIA isoform. J Biol Chem 278, 38132–38140. [DOI] [PubMed] [Google Scholar]
- 17.Cai Y, Biais N, Giannone G, Tanase M, Jiang G, Hofman JM, Wiggins CH, Silberzan P, Buguin A, Ladoux B, et al. (2006). Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys J 91, 3907–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dulyaninova NG, Malashkevich VN, Almo SC, and Bresnick AR (2005). Regulation of myosin-IIA assembly and Mts1 binding by heavy chain phosphorylation. Biochemistry 44, 6867–6876. [DOI] [PubMed] [Google Scholar]
- 19.Juanes-Garcia A, Chapman JR, Aguilar-Cuenca R, Delgado-Arevalo C, Hodges J, Whitmore LA, Shabanowitz J, Hunt DF, Horwitz AR, and Vicente-Manzanares M (2015). A regulatory motif in nonmuscle myosin II-B regulates its role in migratory front-back polarity. J Cell Biol 209, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Even-Faitelson L, and Ravid S (2006). PAK1 and aPKCzeta regulate myosin II-B phosphorylation: a novel signaling pathway regulating filament assembly. Mol Biol Cell 17, 2869–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark K, Middelbeek J, Dorovkov MV, Figdor CG, Ryazanov AG, Lasonder E, and van Leeuwen FN (2008). The alpha-kinases TRPM6 and TRPM7, but not eEF-2 kinase, phosphorylate the assembly domain of myosin IIA, IIB and IIC. FEBS Lett 582, 2993–2997. [DOI] [PubMed] [Google Scholar]
- 22.Almeida MT, Mesquita FS, Cruz R, Osorio H, Custodio R, Brito C, Vingadassalom D, Martins M, Leong JM, Holden DW, et al. (2015). Src-dependent tyrosine phosphorylation of non-muscle myosin heavy chain-IIA restricts Listeria monocytogenes cellular infection. J Biol Chem 290, 8383–8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai RK, and Discher DE (2008). Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol 180, 989–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallis B, Edelman AM, Casnellie JE, and Krebs EG (1983). Epidermal growth factor stimulates tyrosine phosphorylation of the myosin regulatory light chain from smooth muscle. J Biol Chem 258, 13089–13093. [PubMed] [Google Scholar]
- 25.Webb DJ, Schroeder MJ, Brame CJ, Whitmore L, Shabanowitz J, Hunt DF, and Horwitz AR (2005). Paxillin phosphorylation sites mapped by mass spectrometry. J Cell Sci 118, 4925–4929. [DOI] [PubMed] [Google Scholar]
- 26.Dumbauld DW, Shin H, Gallant ND, Michael KE, Radhakrishna H, and Garcia AJ (2010). Contractility modulates cell adhesion strengthening through focal adhesion kinase and assembly of vinculin-containing focal adhesions. Journal of cellular physiology 223, 746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park I, Han C, Jin S, Lee B, Choi H, Kwon JT, Kim D, Kim J, Lifirsu E, Park WJ, et al. (2011). Myosin regulatory light chains are required to maintain the stability of myosin II and cellular integrity. Biochem J 434, 171–180. [DOI] [PubMed] [Google Scholar]
- 28.Guzik-Lendrum S, Heissler SM, Billington N, Takagi Y, Yang Y, Knight PJ, Homsher E, and Sellers JR (2013). Mammalian myosin-18A, a highly divergent myosin. J Biol Chem 288, 9532–9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan I, Yong J, Dong JM, Lim L, and Leung T (2008). A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 135, 123–136. [DOI] [PubMed] [Google Scholar]
- 30.Bao J, Jana SS, and Adelstein RS (2005). Vertebrate nonmuscle myosin II isoforms rescue small interfering RNA-induced defects in COS-7 cell cytokinesis. J Biol Chem 280, 19594–19599. [DOI] [PubMed] [Google Scholar]
- 31.Obenauer JC, Cantley LC, and Yaffe MB (2003). Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic acids research 31, 3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cramer LP, Siebert M, and Mitchison TJ (1997). Identification of novel graded polarity actin filament bundles in locomoting heart fibroblasts: implications for the generation of motile force. J Cell Biol 136, 1287–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vicente-Manzanares M, Newell-Litwa K, Bachir AI, Whitmore LA, and Horwitz AR (2011). Myosin IIA/IIB restrict adhesive and protrusive signaling to generate front-back polarity in migrating cells. J Cell Biol 193, 381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sunstrom NA, Baig M, Cheng L, Payet Sugyiono D, and Gray P (1998). Recombinant insulin-like growth factor-I (IGF-I) production in Super-CHO results in the expression of IGF-I receptor and IGF binding protein 3. Cytotechnology 28, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voudouri K, Berdiaki A, Tzardi M, Tzanakakis GN, and Nikitovic D (2015). Insulin-like growth factor and epidermal growth factor signaling in breast cancer cell growth: focus on endocrine resistant disease. Analytical cellular pathology 2015, 975495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe T, Hosoya H, and Yonemura S (2007). Regulation of myosin II dynamics by phosphorylation and dephosphorylation of its light chain in epithelial cells. Mol Biol Cell 18, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kassianidou E, and Kumar S (2015). A biomechanical perspective on stress fiber structure and function. Biochim Biophys Acta 1853, 3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trybus KM, and Lowey S (1988). The regulatory light chain is required for folding of smooth muscle myosin. J Biol Chem 263, 16485–16492. [PubMed] [Google Scholar]
- 39.Choi CK, Vicente-Manzanares M, Zareno J, Whitmore LA, Mogilner A, and Horwitz AR (2008). Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat Cell Biol 10, 1039–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shutova M, Yang C, Vasiliev JM, and Svitkina T (2012). Functions of nonmuscle myosin II in assembly of the cellular contractile system. PLoS One 7, e40814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baumann BA, Taylor DW, Huang Z, Tama F, Fagnant PM, Trybus KM, and Taylor KA (2012). Phosphorylated smooth muscle heavy meromyosin shows an open conformation linked to activation. J Mol Biol 415, 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heissler SM, and Sellers JR (2015). Four things to know about myosin light chains as reporters for non-muscle myosin-2 dynamics in live cells. Cytoskeleton 72, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Billington N, Beach JR, Heissler SM, Remmert K, Guzik-Lendrum S, Nagy A, Takagi Y, Shao L, Li D, Yang Y, et al. (2015). Myosin 18A coassembles with nonmuscle myosin 2 to form mixed bipolar filaments. Curr Biol 25, 942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taft MH, Behrmann E, Munske-Weidemann LC, Thiel C, Raunser S, and Manstein DJ (2013). Functional characterization of human myosin-18A and its interaction with F-actin and GOLPH3. J Biol Chem 288, 30029–30041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maiuri P, Rupprecht JF, Wieser S, Ruprecht V, Benichou O, Carpi N, Coppey M, De Beco S, Gov N, Heisenberg CP, et al. (2015). Actin flows mediate a universal coupling between cell speed and cell persistence. Cell 161, 374–386. [DOI] [PubMed] [Google Scholar]
- 46.Messi Z, Bornert A, Raynaud F, and Verkhovsky AB (2020). Traction Forces Control Cell-Edge Dynamics and Mediate Distance Sensitivity during Cell Polarization. Curr Biol 30, 1–8. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H, Webb DJ, Asmussen H, and Horwitz AF (2003). Synapse formation is regulated by the signaling adaptor GIT1. J. Cell Biol 161, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nature methods 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heissler SM, Chinthalapudi K, and Sellers JR (2015). Kinetic characterization of the sole nonmuscle myosin-2 from the model organism Drosophila melanogaster. FASEB J 29, 1456–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vasquez CG, Heissler SM, Billington N, Sellers JR, and Martin AC (2016). Drosophila non-muscle myosin II motor activity determines the rate of tissue folding. eLife 5, e20828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, and Bryant SH (2004). Open mass spectrometry search algorithm. Journal of proteome research 3, 958–964. [DOI] [PubMed] [Google Scholar]
- 52.Manso JA, Gomez-Hernandez M, Carabias A, Alonso-Garcia N, Garcia-Rubio I, Kreft M, Sonnenberg A, and de Pereda JM (2019). Integrin alpha6beta4 Recognition of a Linear Motif of Bullous Pemphigoid Antigen BP230 Controls Its Recruitment to Hemidesmosomes. Structure 27, 952–964. [DOI] [PubMed] [Google Scholar]
- 53.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique code. The authors declare that all data supporting the findings of this study are available within the manuscript and its supplementary files or are available from the lead contact on request.