

Abstract
Drug resistance is a serious problem for treatment of the HIV/AIDS pandemic. Potent clinical inhibitors of HIV-1 protease show several orders of magnitude worse inhibition of highly drug resistant variants. Hence, the structure and enzyme activity were analyzed for HIV protease mutant PRS5B from a clinical isolate that was selected by machine learning to represent high level drug resistance. PRS5B has 22 mutations including only one (I84V) in the inhibitor-binding site, however, clinical inhibitors had poor inhibition of PRS5B activity with Ki values of 4–1000 nM or 18–8000-fold worse than for wild-type PR. High resolution crystal structures of PRS5B complexes with the best inhibitors, amprenavir and darunavir (Ki ~4 nM), revealed only minor changes in protease-inhibitor interactions. Instead, two distinct clusters of mutations in distal regions induce coordinated conformational changes that decrease favorable internal interactions across the entire protein subunit. The largest structural rearrangements are described and compared to other characterized resistant mutants. In the protease hinge region, the N83D mutation eliminates a hydrogen bond connecting the hinge and core of the protease and increases disorder compared to highly resistant mutants PRS17 and PR20 with similar hinge mutations. In a distal β-sheet, mutations G73T and A71V coordinate with accessory mutations to bring about shifts that propagate throughout the subunit. Molecular dynamics simulations of ligand-free dimers show differences consistent with loss of interactions in mutant compared to wild-type PR. Clusters of mutations exhibit both coordinated and antagonistic effects suggesting PRS5B may represent an intermediate stage in the evolution of more highly resistant variants.
Keywords: HIV/AIDS, HIV Protease, Drug resistance, X-ray crystallography, Evolution of resistance
Introduction
Globally, human immunodeficiency virus (HIV) is estimated to infect 36.7 million people [1]. Treatments using a combination of inhibitors targeting HIV entry/fusion steps and the viral enzymes, reverse transcriptase (RT), integrase, and protease (PR), have greatly improved patient outcomes. However, HIV resistance to all current drugs is a serious problem due to the presence of numerous genomic polymorphisms and rapid evolution of virus[2]. Moreover, expansion of antiretroviral therapy has resulted in infection with virus resistant to RT inhibitors in greater than 10% of untreated individuals in some countries [3]. Thus, recent WHO guidelines promote second-line PR inhibitor (PI) treatments.
HIV evolves resistance to PIs primarily by mutations in the PR gene[4]. HIV-1 PR is required for proteolytic cleavage of Gag and Gag-Pol precursor polyproteins into individual proteins during viral maturation, and is essential for producing infectious virus[5]. The enzyme forms a dimer of 99-residue subunits. Mutations in over 50% of the PR residues are associated with resistance to all current clinical PIs and lead to decreased effectiveness and treatment failure [6]. Major mutations produce drug resistance by three general molecular mechanisms: mutations of residues in the inhibitor binding site can directly alter PR-PI interactions; some distal mutations confer drug resistance by altering dimerization via changes in inter-subunit interactions; and other distal mutations, such as L76V, can destabilize the dimer and decrease PI potency by creating structural rearrangements in key locations of the protease[7–11].
During drug treatment, the virus can evolve increasingly higher levels of resistance by combining major mutations with additional accessory or compensatory mutations[12] which can persist even after termination of treatment[13]. Several well characterized PR mutants include about 20 different mutations and show several orders of magnitude decreased susceptibility to clinical inhibitors [14]. Two such variants, PR20 and PRS17, form stable dimers with 2–4-fold less efficient Gag processing compared to wild-type PR while maintaining the same order of cleavage [15–17]. Mutations in PR20 coordinate to expand the binding cavity for inhibitors, consistent with dramatically decreased binding affinity. Analysis of ligand-free and darunavir (DRV)-bound X-ray crystal structures of PR20 and PRS17 showed clusters of mutations in the hinge and flap regions conferring changes in conformation of the flaps[18,19]. Nuclear Magnetic Resonance (NMR) spectroscopic analysis of ligand-free PR20 and PRS17 demonstrated that these mutants tend to exist in an open-flap conformation in the absence of inhibitor, while wild-type enzyme preferentially occurs in the closed conformation, even in the absence of inhibitor[18,20]. These changes in PR structure and dynamics are proposed to be important for acquiring high level resistance to PIs[18].
Here, we have investigated the enzyme kinetics and inhibitor-bound structures of an HIV protease mutant with 22 substitutions from a resistant clinical isolate, dubbed PRS5B. Like PRS17, the mutant was selected as a representative of highly drug-resistant PR by mean-shift clustering on genotype-phenotype data in Stanford HIVdb [21,22] using a unified encoding of the sequence and 3-D structure[23–25]. In this procedure, PRS17 was the sole mutant common to the clusters with highest level resistance for each of six PIs, whereas PRS5B mutant was the second most common mutant predicted to represent high-level resistance to only five PIs: atazanavir (ATV), nelfinavir (NFV), indinavir (IDV), tipranavir (TPV), and saquinavir (SQV). Two mutations in PRS5B have a major association with drug-resistance (M46L and I84V), while nine are classified as minor drug resistance mutations (L10I, V11I, M36I, I54V, I62V, I63P, I64V, A71V, and G73T)[6]. Mutation N83D is classified solely as a major resistance mutation for TPV[6]. Here, we confirmed by enzyme kinetics experiments that these drugs, in addition to lopinavir (LPV), amprenavir (APV), and DRV, have significantly worse inhibition for PRS5B relative to wild-type PR. Analysis of X-ray crystal structures of PRS5B in complexes with DRV and APV revealed how mutation clusters in distal regions of the protease may contribute to drug resistance via long-range indirect effects. The PRS5B structures are compared to PRS17 and PR20 complexes with the same inhibitors. Conformational variation of PRS5B and wild-type PR in the absence and presence of inhibitors is explored by molecular dynamics (MD) simulations. These studies illuminate how HIV PR can evolve drug resistance through synergistic effects of clusters of mutations in regions far from the inhibitor binding site. This information will be advantageous for improving inhibitor design against highly drug resistant virus.
Results and discussion
Enzyme kinetic parameters
Kinetic parameters for PRS5B were determined using a FRET-based assay that employed a substrate analog of the p2/NC cleavage site. The measured kcat of 109 ± 20 min−1 for PRS5B is ~2-fold lower than for wild-type PR (194 min−1), and PRS5B exhibits 5-fold higher Km of 160 ± 45 μM relative to PR (30 μM)[26]. The catalytic efficiency of PRS5B (kcat/Km = 0.69 ± 0.07 μM−1min−1) is comparable to that of PRS17 (0.49 μM−1min−1) under identical conditions[27] and close to that of PR20 (0.35 μM−1min−1) measured under different conditions with chromogenic substrate[16]. All three of these highly resistant mutants show about 10 to 20-fold reduced catalytic efficiency compared to PR (6.5 μM−1min−1 for FRET-substrate at 26 °C).
Inhibition constants of 8 PIs measured for PRS5B are compared with previously reported values for wild-type enzyme [15,28] in Table 1. Dose-response curves for a replicate of each PI are reported in Figure 1. The PIs showed Ki values of 4–1000 nM for PRS5B, or 18–8,000-fold worse than for wild-type enzyme. Machine-learning predictions suggesting resistance to inhibition by TPV, ATV, NFV, IDV, and SQV for PRS5B were confirmed by these enzyme inhibition results. PRS5B was poorly inhibited by SQV (2,500-fold relative to wild-type PR) and LPV (8,000-fold), while significantly worse inhibition of 40 to 800-fold was measured for DRV, ATV, TPV, NFV, and IDV. The lowest Ki value (3.6 nM) and relative inhibition (18-fold) for PRS5B was shown for APV, however, DRV also had a Ki of 4.0 nM. LPV showed significant resistance, although not the highest level, in the Stanford HIV database PhenoSense® assay for the isolate with PRS5B, which explains why resistance to this inhibitor was not identified in the mean-shift clustering analysis. DRV and APV PhenoSense® data were absent from the Stanford database at the time of the machine learning training. The lower resistance reported from the cell-based PhenoSense® assay compared to that observed by kinetics with the purified enzyme reflects differences between the two assays. Such discrepancies reinforce the importance of not over-training machine learning models for drug resistance.
Table 1: Ki values (nM) of clinical PIs for PRS5B.
PIs are listed in order of increasing Ki value for PRS5B. Ki fold change calculated as Ki for mutant/Ki for wild-type.
| Inhibitor | Ki for PRS5B | Ki for PRa | PRS5B Ki fold change | PRS17 Ki fold changea | PR20 Ki fold changec |
|---|---|---|---|---|---|
| APV | 3.6 ± 0.8 | 0.2 | 18 | 55 | 890 |
| DRV | 4.0 ± 1.3 | 0.005 | 800 | 10,000 | 8,200 |
| ATV | 8.4 ± 0.9 | 0.035 | 240 | 2,000 | - |
| TPV | 8.6 ± 1.2 | 0.019b | 453 | - | - |
| NFV | 15 ± 1.9 | 0.36 | 42 | 1,630 | - |
| IDV | 46 ± 7 | 0.25 | 184 | 3,200 | - |
| LPV | 160 ± 10 | 0.02 | 8,000 | 3,650 | - |
| SQV | 1000 ± 70 | 0.39 | 2,600 | 22,000 | 2,400 |
Figure 1: Kinetic measurement of clinical inhibitors.
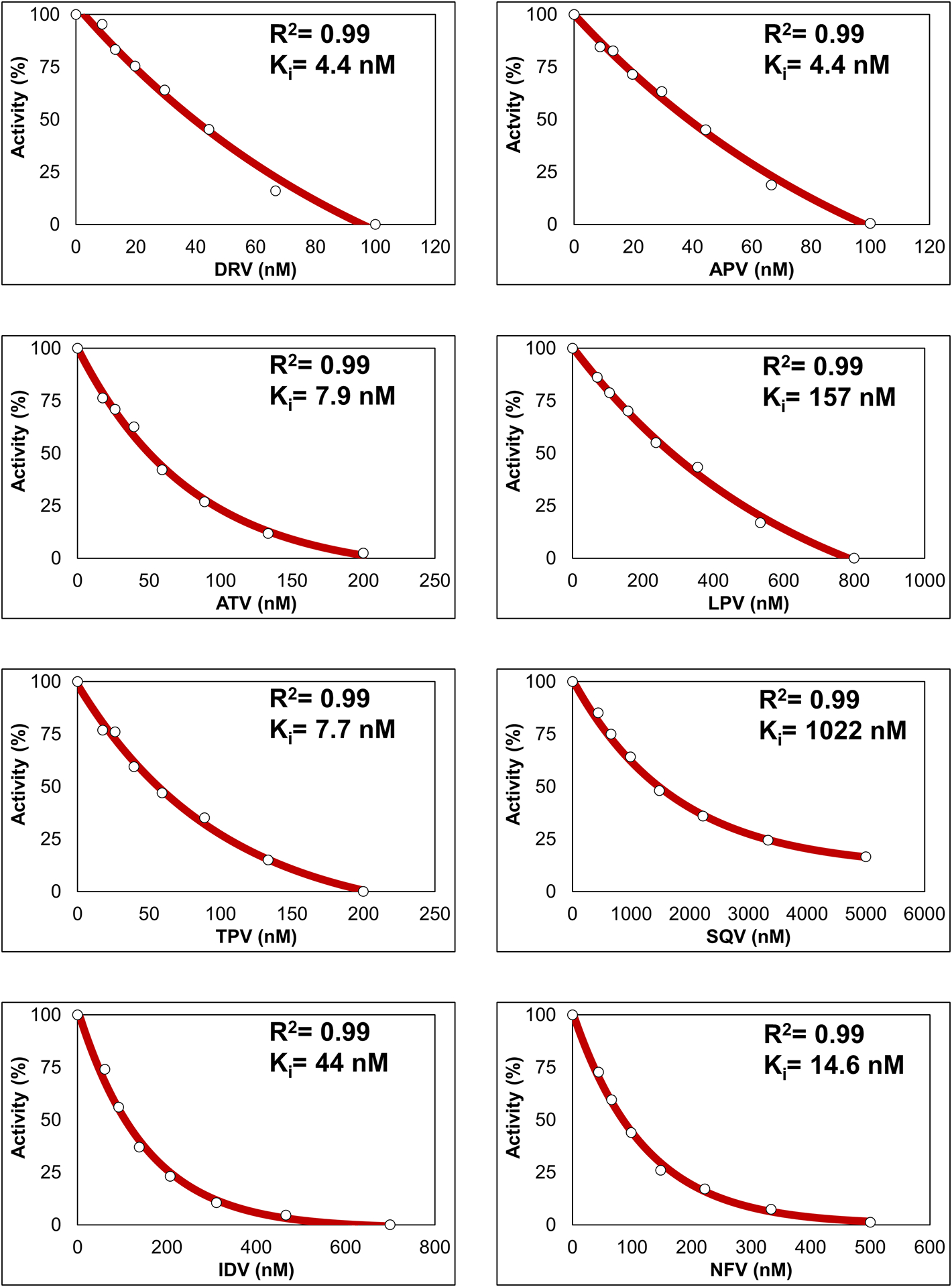
Kinetic inhibition dose-response curves of 8 clinical protease inhibitors for PRS5B proteolytic activity of FRET-based substrate. Reaction carried out in 50 mM MES pH 5.6, 200 mM sodium chloride, 0.5 mM EDTA, and 2.5% glycerol at 37°. Coefficient of determination for the regression (R2) and replicate Ki value are shown. Ki was calculated as Ki = (IC50 − [E]/2)/(1 + [S]/Km). Final Ki values are mean ± SD (n=3–5).
Drug-resistance mutations do not significantly affect protease sensitivity to urea
Proteolytic activity of PRS5B, PRS17, and PR20 was measured for a range of urea concentrations to assess sensitivity to denaturation. Loss of proteolytic activity in the presence of urea has been shown to reflect the unfolding process[29] and provides a chemical-based assay of enzyme stability. UC50 values for PRS5B, PRS17, and PR20 were measured to be 0.50 ± 0.21, 0.64 ± 0.18, and 0.73 ± 0.24 M, respectively, or very similar to the value measured for wild-type PR (0.7 M)[30].
For some mutants, increased sensitivity to urea denaturation is observed together with increased Kd for dimer dissociation relative to values for wild-type enzyme. For PR with single substitution of L76V (PRL76V), two-fold increased susceptibility to denaturation by urea was accompanied by a 7-fold higher dimer dissociation constant (Kd) relative to wild-type enzyme, although the catalytic efficiency was unchanged [9]. PR20 also showed 5-fold higher Kd relative to wild-type enzyme[17], although PR20 and PR have similar UC50 values indicating no association between the different measures of stability. PRS17 exhibited similar Kd[9] and UC50 values as wild-type enzyme, despite structural evidence for fewer intersubunit contacts[18]. Variants with multiple mutations can harbor substitutions conferring compensatory effects to restore dimer stability. Therefore, unlike PRL76V, the three drug resistant mutants with multiple substitutions and lower catalytic efficiency do not exhibit altered protein stability in the presence of urea.
Overall Structure and designation of clusters
X-ray crystal structures of PRS5B in complexes with DRV and APV were solved at 1.66 and 1.75 Å resolution, respectively. Data collection and refinement statistics are listed in Table 2. Both structures were solved in the tetragonal space group P43212 with one dimer per asymmetric unit. Structures of PRS5B/DRV and PRS5B/APV were refined to Rwork/Rfree of 0.18/0.21 and 0.18/0.22, respectively. Each dimer is numbered 1–99 for subunit A and 1′−99′ for subunit B corresponding to the orientation of the single conformation for the two peptidomimetic inhibitors. In both complexes, side chains of mutations were unambiguously modeled with three exceptions. Local side chain disorder was observed near the mutations E35N and S37D in subunit A, likely due to lack of crystal contacts as observed for subunit B and the clusters of mutations leading to loss of interactions in this region. The side chain atoms of mutated residue R41K show no electron density in both subunits. The basic side chain of residue 41 occurs at a polymorphic site on the protein surface. In the absence of crystal contacts, this side chain is typically disordered in PR structures. Alternate conformations were observed for main chain residues 34 and 35 in the hinge region as described in a later section.
Table 2: Crystallographic data collection and refinement statistics.
Parenthesis indicate highest resolution shell. The lowest resolution shells were from 50 Å to 3.58 and 3.77 for PRS5B/DRV and PRS5B/APV respectively.
| Structure | PRS5B/DRV | PRS5B/APV | |
|---|---|---|---|
| PDB entry | 6P9A | 6P9B | |
| Data Collection | Space group | P 43 21 2 | P 43 21 2 |
| a, b, c (Å) | 74.00, 74.00, 93.98 | 73.66, 73.66, 94.33 | |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | |
| Space group | P 43 21 2 | P 43 21 2 | |
| Resolution range (Å) | 50 – 1.66 (1.72–1.66) | 50 – 1.75 (1.81–1.75) | |
| Unique Reflections | 31,010 (3033) | 26,189 (2497) | |
| Completeness (%) | 99.9 (97.0) | 100 (95.0) | |
| Redundancy | 4.0 (4.1) | 4.1 (3.7) | |
| I/σ(I) | 18.1 (3.2) | 20.3 (2.8) | |
| Rmerge (%) | 6.6 (49.1) | 5.9 (48.9) | |
| CC1/2 (%) | 99.5 (88.8) | 100 (82.1) | |
| Refinement Statistics | Rwork (%) | 18.1 | 18.2 |
| Rfree (%) | 21.5 | 21.7 | |
| Solvent atoms | 157 | 141 | |
| Average B-factors (Å2) | |||
| Protein | 23.7 | 26.3 | |
| Inhibitor | 16.8 | 20.3 | |
| Solvent | 25.8 | 29.8 | |
| RMSD from ideality | |||
| Bond lengths (Å) | 0.013 | 0.011 | |
| 1.98 | 1.90 |
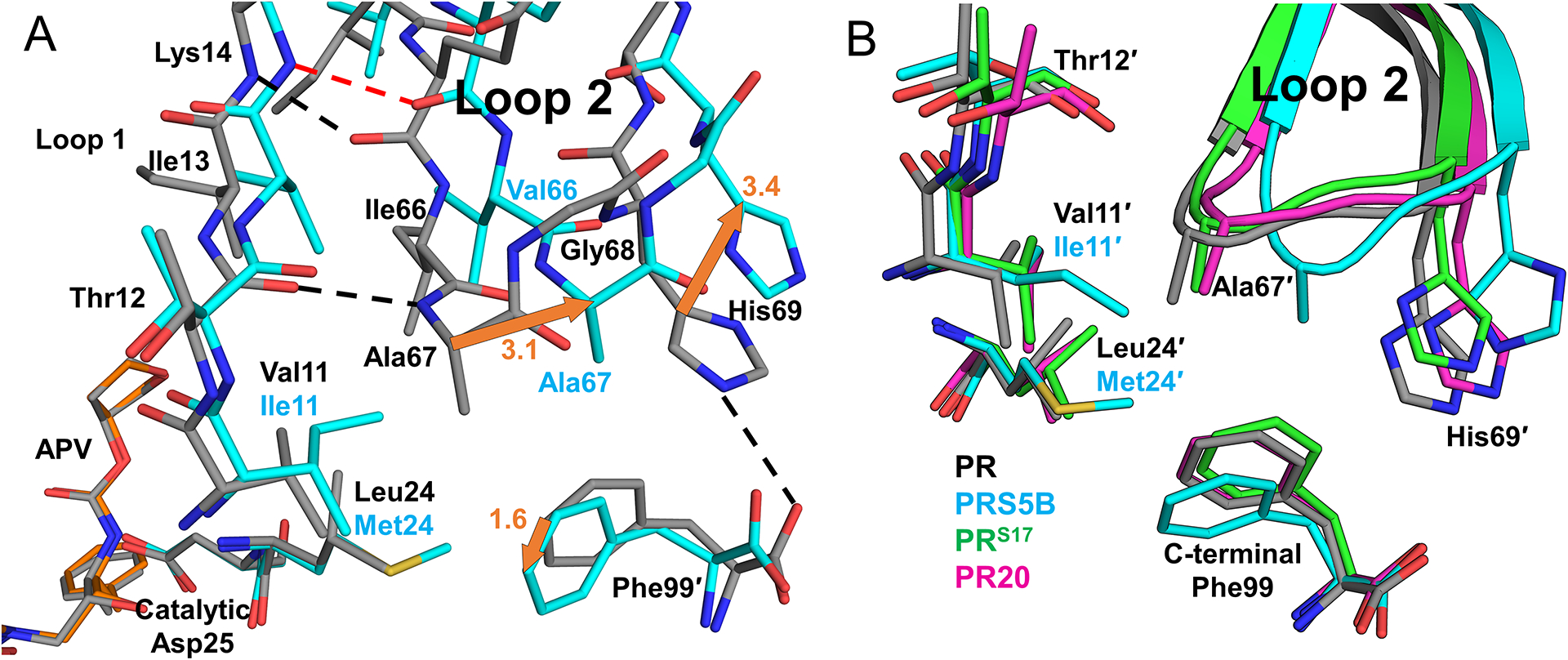
The dimer of PRS5B/DRV is shown in Figure 2 with key structural regions and mutation clusters labeled. Each protease monomer contains three major elements. The flaps (residues 42–56) control access to the active site and are important for binding of substrates and inhibitors. The dynamic flaps pivot about the hinge-loop (residues 34–41) which interacts with the main body of the protease at the 80’s loop (residues 78–85) and two other β-strands. The β-strand of the protease flap continues into two-stranded β-sheet referred to here as Loop 2 (residues 61–76). Loop 2 adjoins another two-strand antiparallel β-sheet, Loop 1 (residues 10–21), to form a 4-strand β-sheet that wraps around the sole α-helix (residues 87–93). Hydrophobic residues facing the center of this assembly create the hydrophobic core of the protein.
Figure 2: Distribution of mutations in PRS5B.
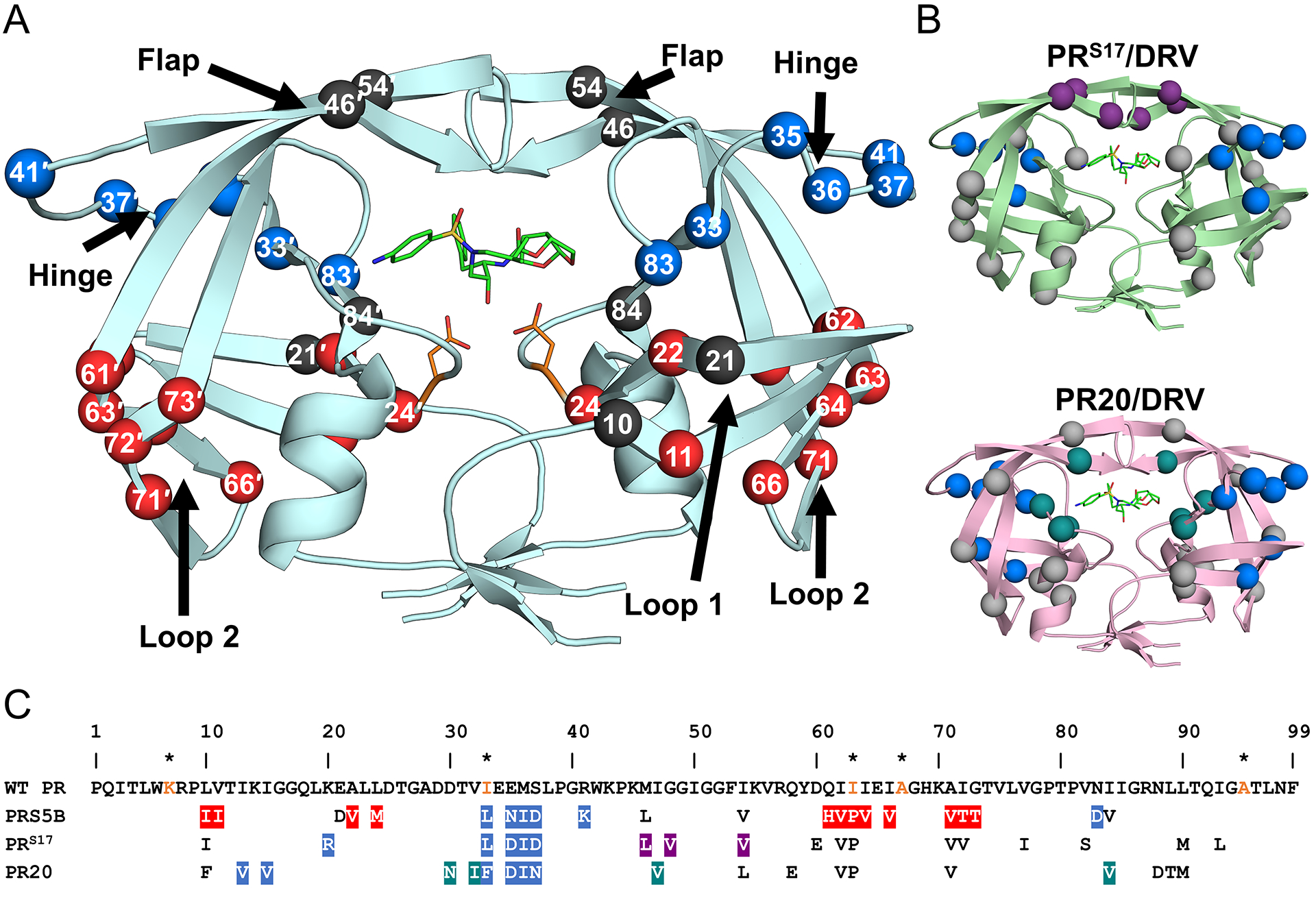
A. PRS5B dimer structure in ribbon representation. Major regions of the protease are indicated by arrows. Catalytic Asp25/25′ and protease inhibitor DRV are shown as orange and green sticks, respectively. Sites of mutation are shown as spheres at the Cα position. Mutation clusters affecting the hinge and Loop 2 regions are colored as blue and red spheres, respectively. Active site mutation I84V, flap mutations M46L and I54V, and Loop 1 mutations L10I and E21D are shown as black spheres. Spheres hidden from view in one subunit are labeled on the opposite subunit. B. Structures of drug resistant mutants PRS17 (light green) and PR20 (pink) complexed with DRV showing mutation clusters. A mutation cluster affecting the hinge is shared by all three mutants as shown by blue spheres. The flap cluster in PRS17 is indicated by purple spheres. Mutations in PR20 altering direct inhibitor interactions are shown in teal. All other mutations are shown in grey. C. Amino acid sequence alignment of wild-type HIV-1 PR with PRS5B, PRS17, and PR20. Mutated residues are colored as in figure 1A and 1B. Identical residues are omitted. HIV is a pseudo-species with polymorphic populations within an infected individual. This pseudo-wild-type PR contains mutations (orange) to restrict autoproteolysis (Q7K, L33I, and L63I) and cysteine-thiol oxidation (C67A and C95A). These optimizing mutations do not significantly alter the catalytic properties or structure of the enzyme[81,82]. For this study, substitutions from the reference wild-type are considered mutations in PRS5B. The sequences of PR, PRS17 and PR20 were obtained from PDB accession codes 3NU3, 5T2Z and 3UCB, respectively. The sequences were aligned manually. Structure figures were generated using PyMOL.
PRS5B contains 22 mutations, including only a single mutation (I84V) in the inhibitor binding site and two mutations (M46L and I54V) in the flaps. The other mutations fall into two main clusters, designated after the major protease regions they affect. Hinge and Loop 2 mutation clusters show coordinated effects on the hinge and Loop 2 regions of the protease respectively. Two additional mutations in Loop 1 bring about minor effects. The mutation clusters in PRS5B and in previously characterized drug resistant mutants PRS17 and PR20 are shown in Figure 2. The three PR mutants share a common cluster of mutations in the hinge region. Within each hinge mutation cluster, E35N/D and M36I are shared, although each mutant contains different accessory mutations. PRS17 contains a cluster of three flap mutations, M46L, G48V, and I54V, which primarily affect the conformation of the flaps[18,27]. PR20 shows an expanded inhibitor binding cavity due to a cluster of mutations (D30N, V32I, I47V, and I84V)[19,31]. Mutation L10I in Loop 1 of PRS5B was excluded from the designated clusters due to absence of significant structural changes as also seen for the same mutation in the PRS17/DRV structure. In PR20, however, a phenylalanine substitution at this position is associated with loss of an intersubunit ion pair between the nearby Arg8 and Asp29′ [16,31].
The 198 Cα atoms of PRS5B/DRV dimer superimpose on equivalent atoms of wild-type PR/DRV (PDB:2IEN[32]) with a root mean square deviation (RMSD) of 0.94 Å. The hinge and Loop 2 regions of PRS5B/DRV show the largest structural changes relative to PR/DRV suggesting an effect from the local mutation clusters in those regions. Likewise, the Cα atoms of PRS5B/APV superimpose onto PR/APV dimer (PDB: 3NU3[33]) with a RMSD of 1.02 Å. PRS5B/APV exhibits larger shifts in the hinge and Loop 2 relative to PR/APV compared to the corresponding DRV complexes. The two structures of PRS5B with DRV and APV are essentially identical and superimpose with a low RMSD of 0.16 Å.
PRS5B inhibitor-binding site and flaps
The inhibitors and their interactions with PRS5B are shown in Figure 3. The two inhibitors are chemically similar, apart from a bis-tetrahydrofuran (THF) moiety at P2 in DRV in place of the single THF ring of APV (Figures 3A and D). Both inhibitors bind to the active site of PRS5B in a single conformation, possibly due to crystallizing in the P43212 space group, unlike many crystal structures of PR-PI complexes that show inhibitor bound in two orientations related by 180°. The hydrogen bond interactions with inhibitors (Figures 3B and E) are conserved in PRS5B and wild-type PR complexes. The P2 THF group of APV accepts a hydrogen bond from the backbone amine of Asp30. In DRV, the P2 bis-THF group conserves this interaction in addition to forming hydrogen bonds with the amine of Asp 29. However, these additional interactions of DRV relative to APV do not alter the inhibition of PRS5B (Table 1).
Figure 3: PRS5B interactions with clinical inhibitors APV and DRV.
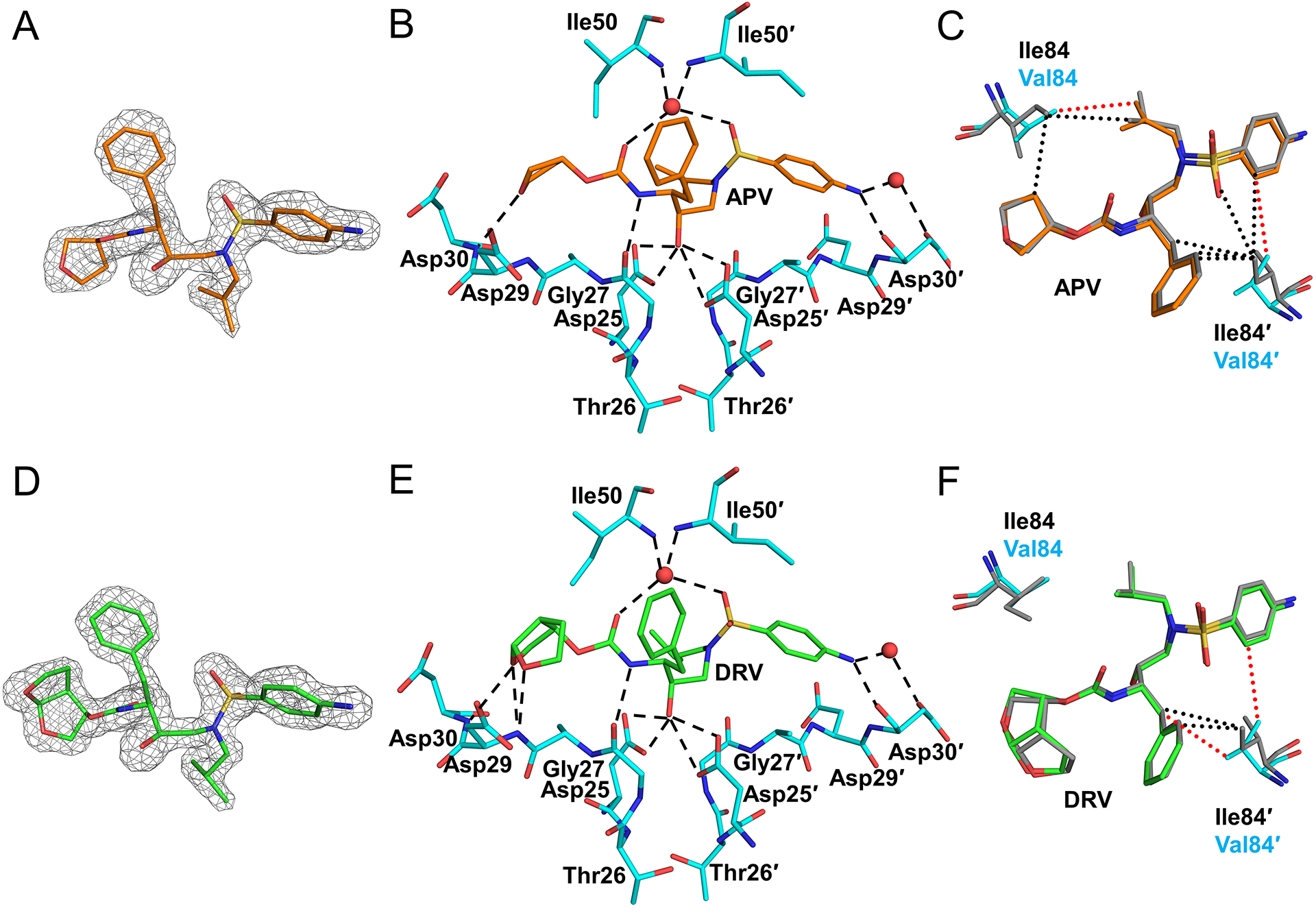
A. Omit map (grey mesh) for APV (orange sticks) B. APV hydrogen bonds with PRS5B. C. Comparison of van der Waals contacts of I84V with APV in PR and PRS5B. D. Omit map (grey mesh) for DRV (green sticks) E. DRV hydrogen bonds with PRS5B. F. Comparison of van der Waals contacts of I84V with DRV in PR and PRS5B. Omit maps are Fo-Fc maps contoured at 3σ (grey mesh) for APV and DRV. PRS5B residues are shown as cyan sticks. PR/APV and PR/DRV are shown as grey sticks. Water molecules are red spheres. Van der Waals interactions are shown as dotted lines in red and black for PRS5B and PR, respectively. Structure figures were generated using PyMOL.
I84V is the sole mutation in PRS5B with a side chain that faces the active site cavity. This mutation contributes resistance to all PIs[6]. Substitution of the smaller valine side chain at residue 84 increases the volume of the S2/S2′ pockets and results in fewer van der Waals contacts with APV compared to the PR complex (Figures 3C and F).
The side chains of flap mutations M46I and I54V do not form van der Waals contacts with these inhibitors. The single substitution of I54V in PR has been described previously to alter flap conformation and effect inhibitor binding [26]. This mutation is associated with resistance for all PIs except DRV [6]. In PRS5B structures, the shorter sidechain of I54V decreases hydrophobic interactions with the Ile50 side chain on the opposite subunit. PRS5B contains a second flap mutation M46I, which is a drug resistance mutation for all clinical PIs except DRV, SQV and TPV[6]. The side chain of flap residue 46 is adjacent to Phe53 at the protein surface, however, the shorter hydrophobic side chain of M46I shows no significant effects on the flap structure of PRS5B inhibitor complexes. M46I and I54V mutations co-occur frequently in drug resistance [28,34,35]. Adding M46I and I54V mutations to a RTV-resistant mutant was shown to improve catalytic efficiency while sustaining resistance [36]. Molecular dynamics simulation suggests these mutations may act by altering the flexibility of the flaps [37].
Hinge cluster mutations E35N and M36I coordinate with N83D to disengage flaps
Comparison of the new PRS5B structures with equivalent PR, PRS17, and PR20 complexes illustrates how the additional mutation of N83D replaces direct hydrogen bonds with water-mediated interactions at the hinge-body interface. The structures used for comparison are as follows: PR/APV (PDB:3NU3), PR/DRV (PDB:2IEN), PRS17/DRV (PDB:5T2Z), PR20/DRV (PDB:3UCB), and PR20/APV (PDB:4J5J). Consistent changes were observed in both PRS5B/DRV and PRS5B/APV structures. Structurally, the hinge (residues 34–41) is an anchoring point for the base of the flexible flap (residues 42–56). Mutations in the hinge region are known to increase flap flexibility and contribute to drug resistance in PR20, PRS17, and other mutants[18,19,28,31,38,39].
PRS5B harbors 4 mutations in the hinge, E35N, M36I, S37D, and R41K. Only M36I is associated with resistance to ATV, IDV, NFV and TPV, while the other hinge mutations have no significant association with resistance [6]. The side chain of residue 34 shows altered interactions with mutated residue N83D in the 80’s loop in the core of the dimer. With the exception of subunit A of PRS5B/DRV, hinge residues 34 and 35 display alternate conformations for the main chain, while the side chains of Glu34 and E35N show alternate conformations or disorder. Intermolecular crystal contacts partially mitigate the disorder in subunit A. Therefore, the extent of changes arising from hinge-loop mutations is described for subunit B and compared to PR (Figure 4). The loss of charge due to the E35N mutation in PRS5B eliminates the ion pair in wild-type enzyme between the side chains of Glu35′ and flap residue Arg57′ (Figure 4A). In PRS5B/APV, loss of this ionic interaction leads to a 2.8 Å shift in positions of Cα atoms for residue 35 and 7 Å shift in side chain position compared to wild-type PR. Furthermore, this change allows the Arg57′ side chain to move to a conformation which eliminates its hinge-flap hydrogen bond interaction with the carbonyl oxygen of Met36′. The new conformation of Arg57′ also retains a hydrogen bond with Tyr59′ and introduces new intra-flap π-interaction with the side chain of Trp42′. In the absence of stabilizing crystal contacts, the side chain of E35′N shows little electron density suggesting the mutated side chain does not form any significant new interactions compared to PR.
Figure 4: Conformational changes as a result of hinge cluster mutations E35N, M36I, and N83D.
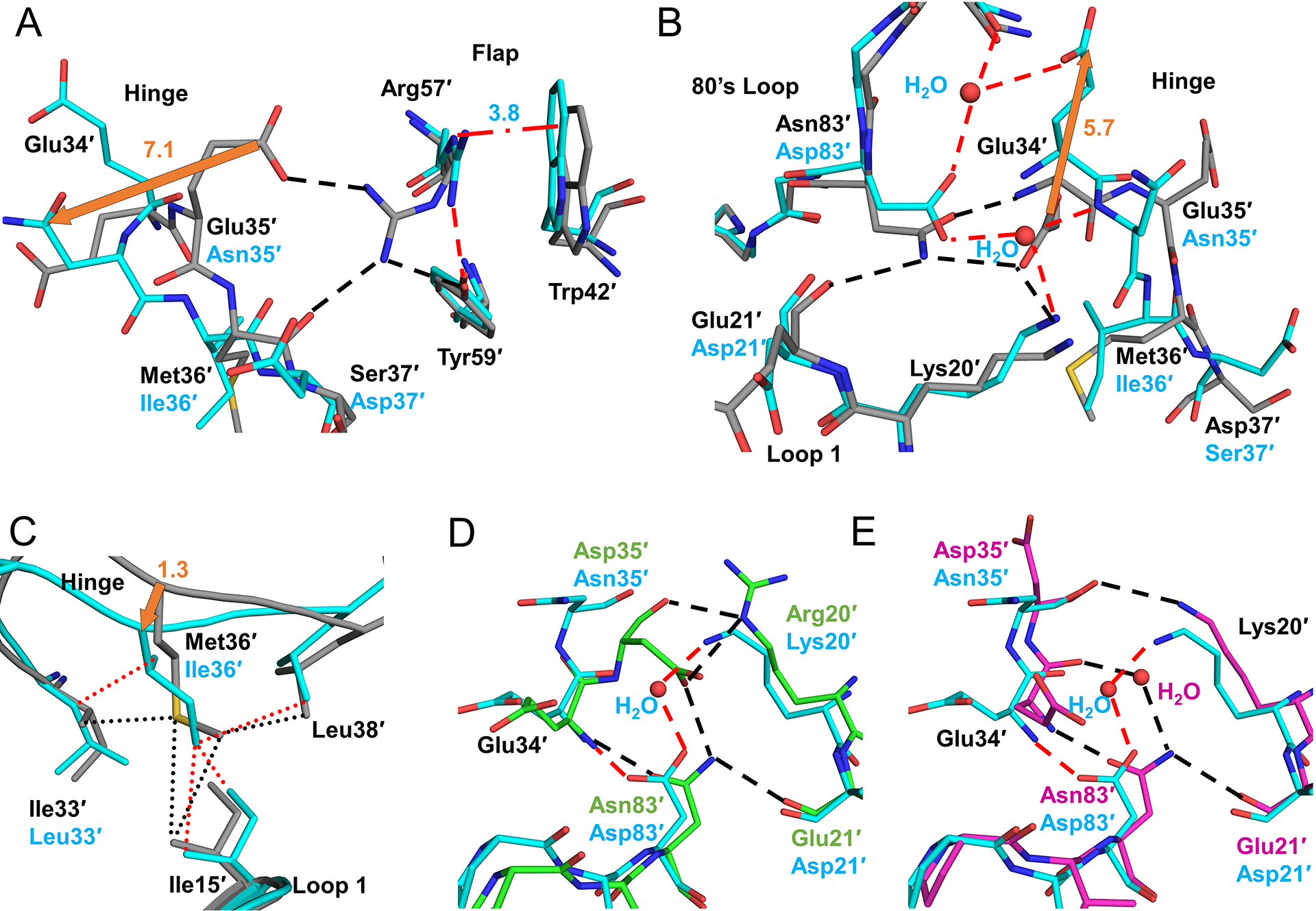
A. Hinge interactions for APV structures of PR (grey) and PRS5B (cyan) and conformational change due to E35′N. E35′N mutation eliminates ion pair with Arg57′. In PRS5B, altered conformation of Arg57′ forms coplanar π-interactions with Trp42′ (red dash-dots) and a hydrogen bond with Tyr59′. B. Rearrangements in the PRS5B hinge. Shifts due to mutations N83′D and E21′D alter the distribution of hydrogen bonds and introduce partial occupancy water molecules. C. M36′I mutation retains hydrophobic contacts (dots) with sidechains of I33′L, Ile′15, and Leu38′ due to shift in hinge main chain confirmation. D. Hinge of PRS5B/DRV (cyan) compared to PRS17/DRV (green). E. Hinge of PRS5B/DRV (cyan) compared to PR20/DRV (magenta). PR/APV (grey) and PRS5B/APV (cyan) carbons are shown as sticks. Hydrogen bonds are shown as dashes colored red for PRS5B and black for PR, PRS17, and PR20 structures. Waters are shown as red spheres. Orange arrows indicate major shifts with distances in Å. Alternate conformations were omitted for clarity. Structure figures were generated using PyMOL.
Glu34′ is the first residue in the hinge of PR. Its carboxylate side chain faces in the opposite direction to Glu35′ to form a bifurcated hydrogen bond with the side chains of Asn83′ in the body of the protease and Lys20′ in Loop 1 (Figure 4B). In this way, Glu34′ and Glu35′ of the hinge link the flap to the rest of the protease. These interactions are disrupted in PRS5B due to the N83D mutation. The pH for the crystallization and enzyme assay conditions (7.9 and 5.6) likely mean Asp83 is monoprotonated. N83D introduces a negatively charged carboxylate group and eliminates the hydrogen bonds with Glu34′ side chain and with the backbone carbonyl oxygen of Glu21′. Instead, the carboxylate side chain of Glu34′ shifts by ~6 Å away from the hinge-body interface to the protein surface of PRS5B/APV structure. The void left by the shifted Glu34′ side chain is filled by a single partial-occupancy water molecule that bridges the N83′D and Lys20′ side chains.
Met36′ in PR/DRV and PR/APV structures interacts with nearby side chains of Leu33′, Leu38′, and with Ile15′ in Loop 1. The hinge mutation M36I substitutes a shorter hydrophobic side chain. As shown in Figure 4C, the smaller M36I residue in PRS5B structures retains significant van der Waals contacts with Ile15′ and Leu38′ and the mutated I33L sidechain due to a shift in its main chain. The Cα of Ile36 is closer to Loop 1 by 1.3 and 1.9 Å in mutant complexes with DRV and APV structures, respectively, compared to PR structures. Mutations E35N and N83D drive two alternate conformations for the main chain of the hinge-loop at residues 34′ and 35′ through decreased interactions with 80’s loop and flap. By allowing a continuation of van der Waals contacts at the hinge-Loop 1 interface using a shorter side chain, M36I counterbalances the effects of E35N and N83D and stabilizing the hinge-loop. Coordinated substitutions of M36I and I33L maintain van der Waals contacts between the hinge and Loop 1, thus counteracting the loss of interactions of hinge with main chain of residue 21′ and flaps in mutant.
Comparison of the PRS5B/DRV hinge-region mutations with those in the DRV complexes with PRS17 and PR20 (Figures 4D and E) highlights the influence of the N83D mutation. Despite the presence of several mutations, introduction of a partially occupied (0.5) water molecule in PRS5B preserves many interactions of residues 20, 21, 34, 35 and 83 seen in the other two resistant mutants. All three resistant proteases have mutations for Glu35 (E35N or E35D) that break the ion pair with flap residue Arg57′ and a compensating M36I. However, PR20/DRV and PRS17/DRV lack the alternate main chain conformation of hinge-loop and side chain disorder exhibited by PRS5B/DRV. In PRS17/DRV, the E35D side chain forms an ion pair with K20R and a hydrogen bond with the side chain of Asn83′. PR20 does not contain a substitution for Asn83′ and thus retains the stabilizing interactions between the protease body and the hinge-loop. PR20 utilizes a full-occupancy water molecule interacting between Asn83′ and the main chain amide of residue 35′. Unlike PRS5B, both PRS17 and PR20 preserve the hydrogen bond between Asn83′ and the main chain carbonyl of Glu21′ that helps stabilize the body of the protease and Loop 2 with the hinge. PRS17 and PR20 also have additional compensating mutations that act synergistically with M36I (K20R in PRS17 and I33F, I13V and I15V in PR20) to further stabilize the hinge.
Mutations A71V and G73T coordinate with accessory mutations L11I, A22V, L24M, and I62/64/66V to induce structural shifts in Loop 2
Mutation A71V is associated with resistance to all PIs except for DRV, APV and TPV, while G73T only shows significant resistance to ATV. Comparison of both PRS5B/PI structures with analogous wild-type complexes reveals how G73T and A71V mutations act in synergy with other accessory mutations to drive structural shifts of almost 3 Å in Loop 2 in both subunits. This section describes the PRS5B/APV complex specifically unless noted. Subunit B of PRS5B/APV is compared with wild-type structure in Figure 5.
Figure 5: G73T and A71L drive distal movement of Loop 2 through flip in Leu89 sidechain.
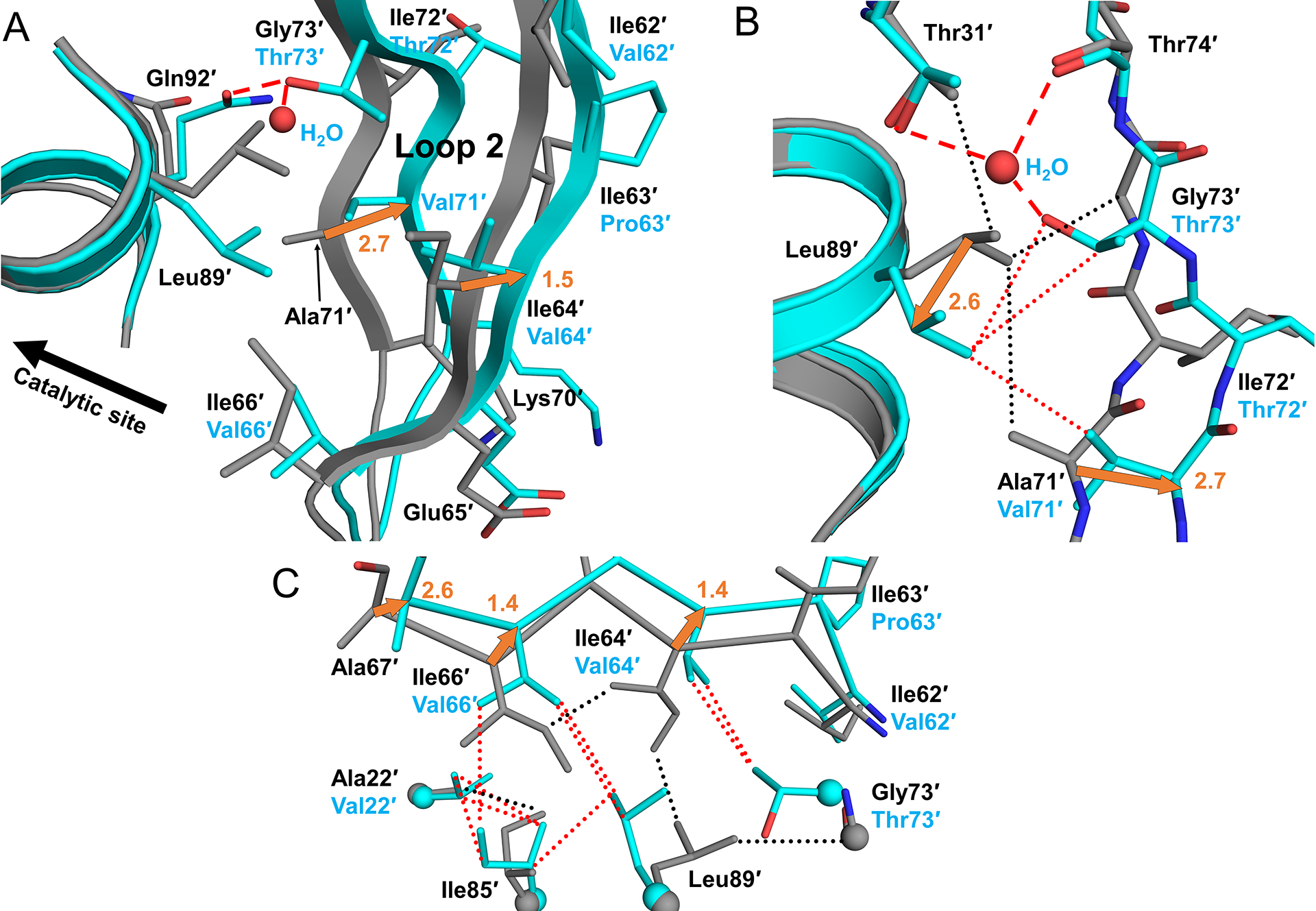
A. B-subunit Loop 2 of PR/APV (grey cartoon) compared to PRS5B/APV (cyan cartoon) with G73′T stabilizing H-bonds shown as red dashed lines. G73′T induces flipped conformation of Leu89′ which combines with larger A71′V side chain to rearrange Loop 2. B. New water molecule coordinates with Thr31′, Thr74′ and mutated G73′T. Flipped conformation of Leu89′ faces the valine side chain of A71′V mutation. Hydrogen bonds shown as dashed lines and van der Waals contacts as dotted lines. Side chains of Asn92′ and Asn88′ were omitted for clarity. C. Smaller hydrophobic side chains for residues 62′, 64′, and 66′ dampen distal movement of Loop 2. Distances in Å are indicated as orange arrows. Structure figures were generated using PyMOL.
Gly73 is situated at the top of Loop 2 where the Cα forms van der Waals contacts with the side chain of Leu89 (Figure 5A). In the wild-type PR, the large Leu89 side chain fits snugly into the hydrophobic core making contacts with Ile64, Thr31, Ile66, and Ala71. In PRS5B, Gly73 and Ala71 are replaced with larger residues, threonine and valine, respectively. The polar side chain of G73T is stabilized by hydrogen bonds with the side chain of Glu92 in a shifted conformation. In addition, subunit B of both PRS5B structures features a water molecule amidst this formerly non-polar core. This new water molecule forms a set of hydrogen bond interactions with the carbonyl oxygen of Thr74, the side chain hydroxyl groups of Thr31 and mutated Thr73, and the side chain of Asn88 (Figure 5B). More importantly, the larger side chain of G73T induces a 180° flip in the bulky Leu89 side chain where the Cγ atom is ~2.5 Å from the position in PR. The flipped Leu89 side chain is directed toward the new bulkier side chain of the A71V mutation and retains hydrophobic contacts with the larger side chains of mutated Val71 and Thr73. Together, these substituted residues combine with other compensatory mutations to initiate a 1.2–3.0 Å outward shift in all Cα atoms of Loop 2 from mutated I64V through G73T.
The coordinated effects of G73T, A71V, and flipped Leu89 conformation combine with clustered accessory mutations to bring about rearrangements in the Loop 2. Larger hydrophobic residues from mutations L11I, V22I, and L24M are located deep within the hydrophobic core of the protease and near the main chain of catalytic Asp25/25′. These three mutations help to fill voids in the hydrophobic core created by the shift of Loop 2 and propagate changes from Loop 2 to the catalytic site.
The bulge in Loop 2 induced by G73T and A71V is offset by ~1 Å due to smaller side chains in the I62V, I64V and I66V mutations, which have a compensatory effect (Figure 5C). Three other mutations in this cluster, Q61H, I63P, and I72V, project to the protein surface and do not appear to induce changes in the interior. Thus, the large distal movements for Loop 2 stemming from A71V and G73T mutations are compensated by repacking of the hydrophobic core from other mutations in the cluster.
Consequences of Loop 2 movement are best observed at the β-turn (residues 67–69) shown in Figure 6A. Large deviations in Cα atoms between PRS5B and PR structures (2.6–3.1 Å) occur at Ala67. In PR structures, the β-turn of Loop 2 is secured to the rest of the protease by two interactions: an anti-parallel β-sheet hydrogen bond from the backbone amine of Ala67 to the backbone carbonyl oxygen of Thr12 (Loop 1) and an intersubunit ion pair from the side chain of His69 to the C-terminal carboxylate of Phe99′. Both interactions are absent in PRS5B structures. The phenylalanine side chain of the C-terminal Phe99 interacts with Leu24, the amino acid adjacent to the catalytic aspartates 25/25′. The larger Met24 side chain in PRS5B displaces the side chain of Phe99′ by ~1.5 Å. The loss of interactions is significant as these two interactions contribute half of all polar contacts between Loop 2 from residues 62–73 and the rest of the protease present in the wild-type structures. It should be noted that the PRS5B structures possess no significant crystal contacts along the Loop 2 region. PRS5B structures feature mutations producing significant alternations of main chain conformation and repacking of hydrophobic side chains compared to wild-type PR structures. These changes likely result in altered dynamics that propagate to the active site and contribute to the poorer inhibition measured for clinical inhibitors.
Figure 6: Interactions of Loop 2 tip and comparison of three highly resistant mutants PRS5B, PRS17, and PR20 with wild-type PR.
A. B-subunit beta-loop of Loop 2 (residues 67–69) interacting with Loop 1 (residues 24 and 11–14 shown) and the C-terminus of A subunit. PRS5B/APV is shown in cyan sticks. PR/APV is grey sticks. Hydrogen bonds and ion-pair are shown as dashed lines. Distances in Å are indicated as orange arrows. B. PRS5B Loop 2 tip comparison with drug resistant mutants PRS17 and PR20. Protease/DRV cartoon complexes colored as follows: Wild-type PR (grey), PRS5B (cyan), PRS17 (green), and PR20 (magenta). Side chains for tip residues and interacting residues, including PRS5B mutations L24M and V11I are shown as sticks. Structure figures were generated using PyMOL.
PRS17 and PR20 also share the A71V mutation. However, the addition of G73T combined with the other mutations in the cluster produces a more dramatic expansion of Loop 2 in PRS5B than seen in the two other drug-resistant mutants. The tips of Loop 2 and adjacent residues are compared for DRV complexes with PRS5B, PRS17, PR20 and wild-type PR in Figure 6B. In PRS5B, the Cα atom of Ala67′ is 2.4 and 1.8 Å farther from Loop 2 at Cα atoms of Thr12′ and residue 11′ than in PR, whereas there is no significant difference in these distances for PRS17 and PR20. Similarly, the distance between the C-terminus of Phe99 to the His69′ imidazole ring is ~0.5 Å farther from the PR location in PRS5B compared to PRS17 and PR20.
Mutation clusters decrease polar interactions across the entire monomer
Mutation clusters at the hinge and Loop 2 are linked via mutation E21D in Loop 1. In subunit B of PR/APV structure, the large side chain of Glu21′ forms a hydrogen bond with the hydroxyl side chain of Thr12′ linking the two β-strands of Loop 1. The shorter E21D side chain in PRS5B mutant cannot form this hydrogen bond with Thr12′ side chain. Loop 1 residues 21 and 12 provide an interface between the corresponding mutation clusters in the hinge and Loop 2 (Figure 7). PRS5B has a total of six fewer polar interactions than the wild-type complex in this region spanning nearly the entire monomer from the C-terminus and tip of Loop 2 through the protease body (Loop 1 and residue 83′) to the hinge-flap interface (residues 34′−35′ to 57′).
Figure 7: Mutations in PRS5B eliminate polar interactions spanning the length of the PRS5B monomer from the terminus to the flap at Arg57.
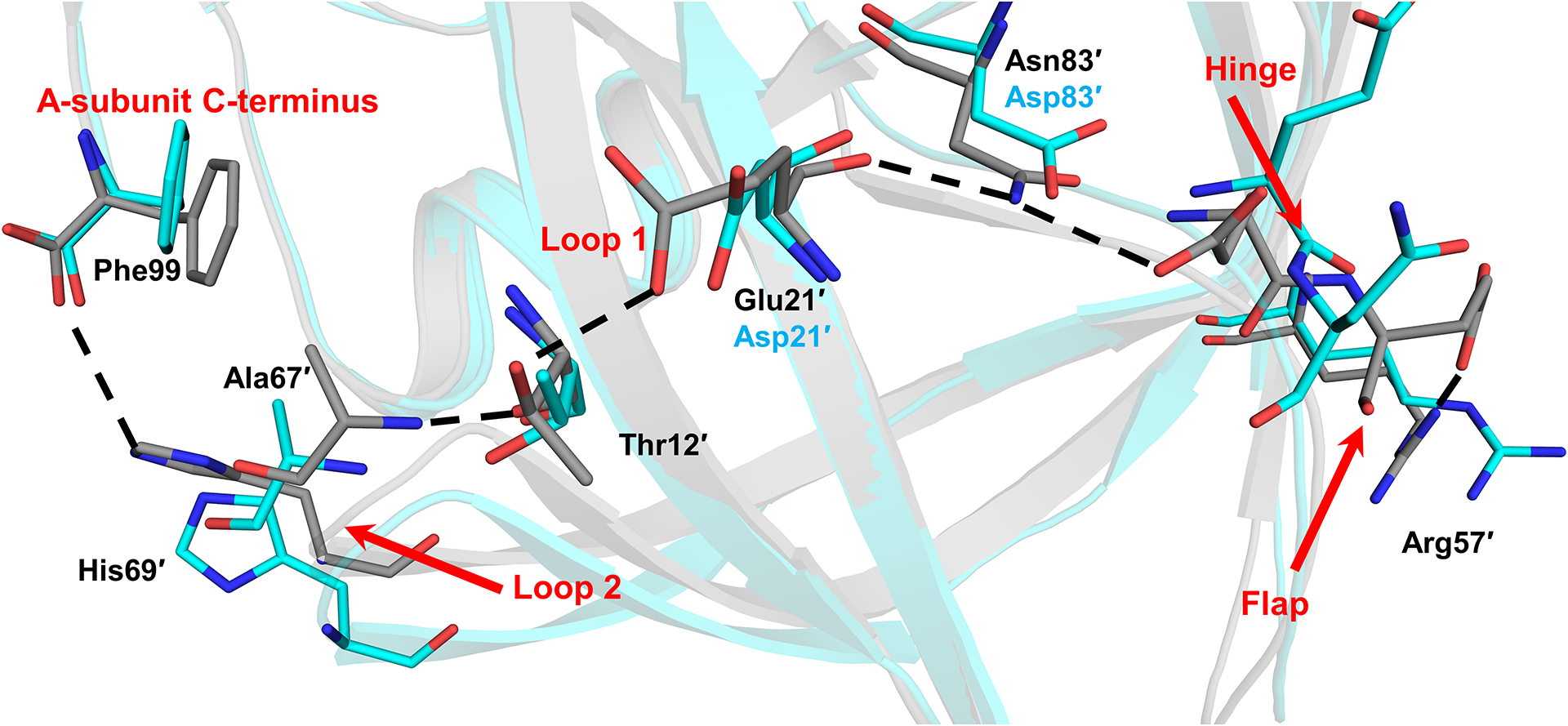
Hydrogen bonds present in PR/APV structure and absent in the PRS5B/APV structure are shown as black dashes. Notable regions of the protein are labeled in red. Alternate conformations omitted for clarity. PR/APV (3NU3) and PRS5B/APV are shown as grey and cyan sticks, respectively. Structure figures were generated using PyMOL.
Molecular dynamics simulations explore conformational variation in PRS5B and wild-type PR
Since the PRS5B structures showed only minor changes in the interactions with inhibitors compared to wild-type complexes, MD simulations were performed to evaluate the conformational variation and calculate the protease-inhibitor interaction energies. Simulations of the ligand-free enzymes were used to assess the variation in the absence of inhibitor. Inhibitor APV was removed from the starting structures prior to 20 ns MD simulations of the ligand-free enzymes. As shown in the trajectories (Figure 8A), the ligand-free structures equilibrate rapidly to about 2.2 Å RMSD on Cα atoms. The mutant has slightly higher RMSD values than the wild-type PR. In comparison, earlier MD simulations starting from open conformation crystal structures showed RMSDs rising to 4.5 Å for PR20 mutant and about 2 Å for wild-type PR compared to about 1.5 Å for simulations on closed conformation dimers after removing inhibitor[40–42].
Figure 8: Molecular dynamics simulations of ligand-free PR and PRS5B show conformational variation in the flaps, loops 1 and 2, and termini.
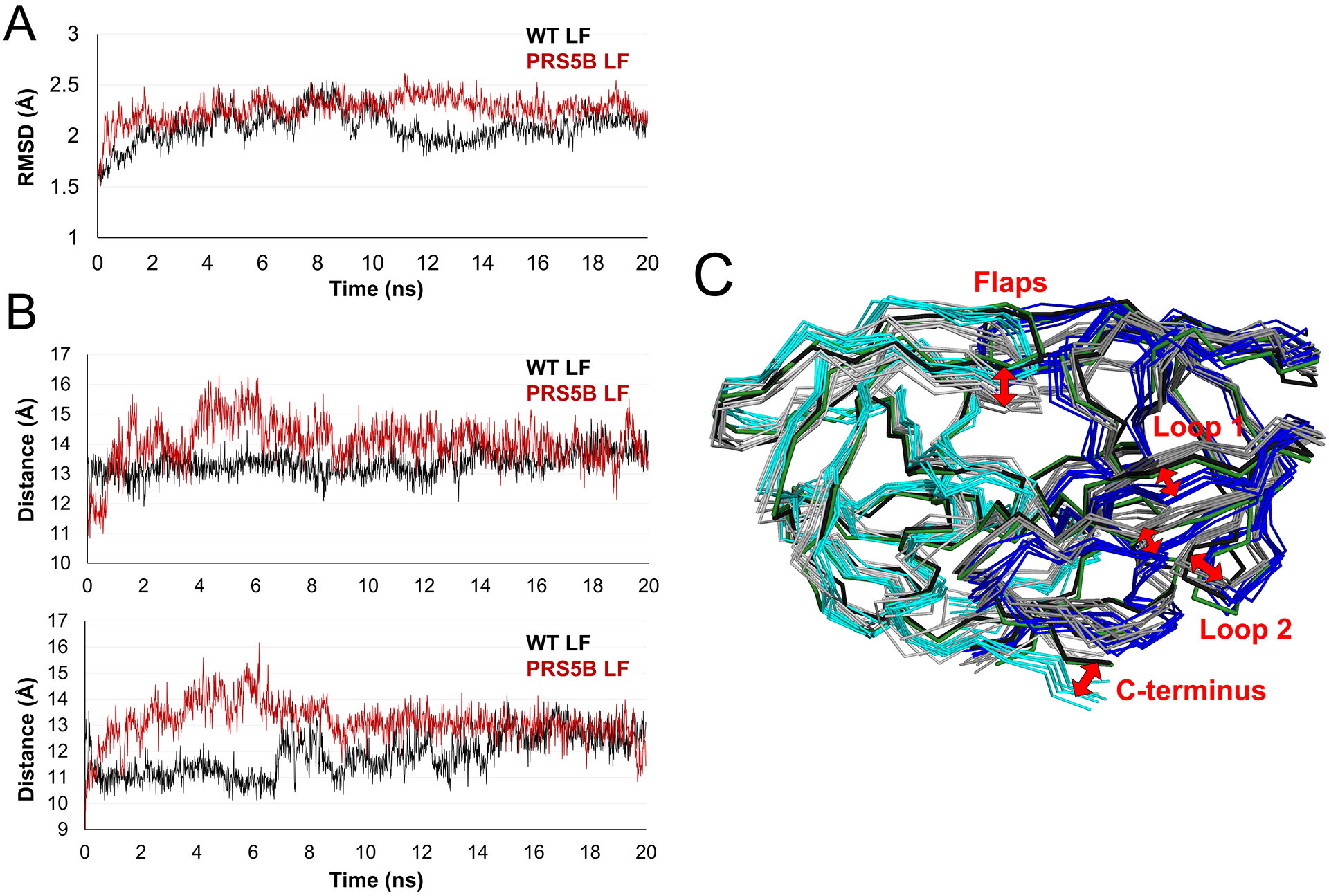
A. Trajectories of the simulations of ligand-free dimers. RMSD of Cα atoms after superposition on the starting crystal structure plotted for the 20 ns time course. PRS5B LF and PR LF simulations are colored in red and black respectively.
B. Distance between Cα atoms of the flap tip (Ile50) and the catalytic Asp25 for subunit A (top) and subunit B (bottom) for the time course of the simulation. Plots are colored as in A.
C. Comparison of PR and PRS5B ligand-free simulations. Snapshots of PR (grey shades) and PRS5B (cyan and blue) models for every 3 ns of the simulation shown as superimposed ribbons. The starting crystal structures of PR (black) and PRS5B (green) are shown. Labeled regions and red arrows indicate large conformational differences between wild-type and mutant protease. Structure figures were generated using PyMOL.
The variation in flap conformation was assessed by calculating the distances between Cα atoms of Ile50 and 50’ at the flap tips and the catalytic Asp 25 and 25’ (Figure 8B). These distances fluctuate during the first 15 ns and then stabilize at about 13–14 Å for mutant and wild-type PR. The two flaps in each dimer remain closer together with the separation between Cα atoms of Ile50 and 50’ at the flap tips varying around 5.8 Å for both mutant and wild-type protease.
The variation in Cα positions of ligand-free dimers during the simulations is illustrated by the superimposed structures in Figure 8C. PRS5B mutant and wild-type dimers exhibit larger displacements from the crystal structures of 2 to 5 Å in the flaps, both strands of loop 1, the tip of loop 2 and the C-terminus. The mutant and wild-type PR converge into separate conformations in these regions. Large variations in loop 2 and the C-terminus were also described in a recent MD simulation by another group [43]. The flaps of the wild-type dimer shift into the active site cavity in this orientation, whereas the mutant flaps remain closer to the conformation in the crystal structure. Loop 2 varies about two separate positions observed in the two crystal structures. Several shifts, especially those encompassing the two strands of loop 1 and the C-terminus, are consistent with the loss of polar interactions across the subunit of PRS5B relative to wild-type PR observed in the crystal structures (Figure 7).
MD simulations of the inhibitor-bound complexes of PRS5B/DRV, PRS5B/APV and the equivalent complexes of wild-type protease were run for 10 ns to assess the effect of inhibitor. The simulations had similar trajectories reaching about 2.2 Å RMSD from the starting structures, except for a slower divergence of PR/DRV to about 2.6 Å as shown in Figure 9A.
Figure 9: Molecular dynamics simulations of inhibitor-bound PR and PRS5B and calculated non-bonded interaction energies of inhibitors.
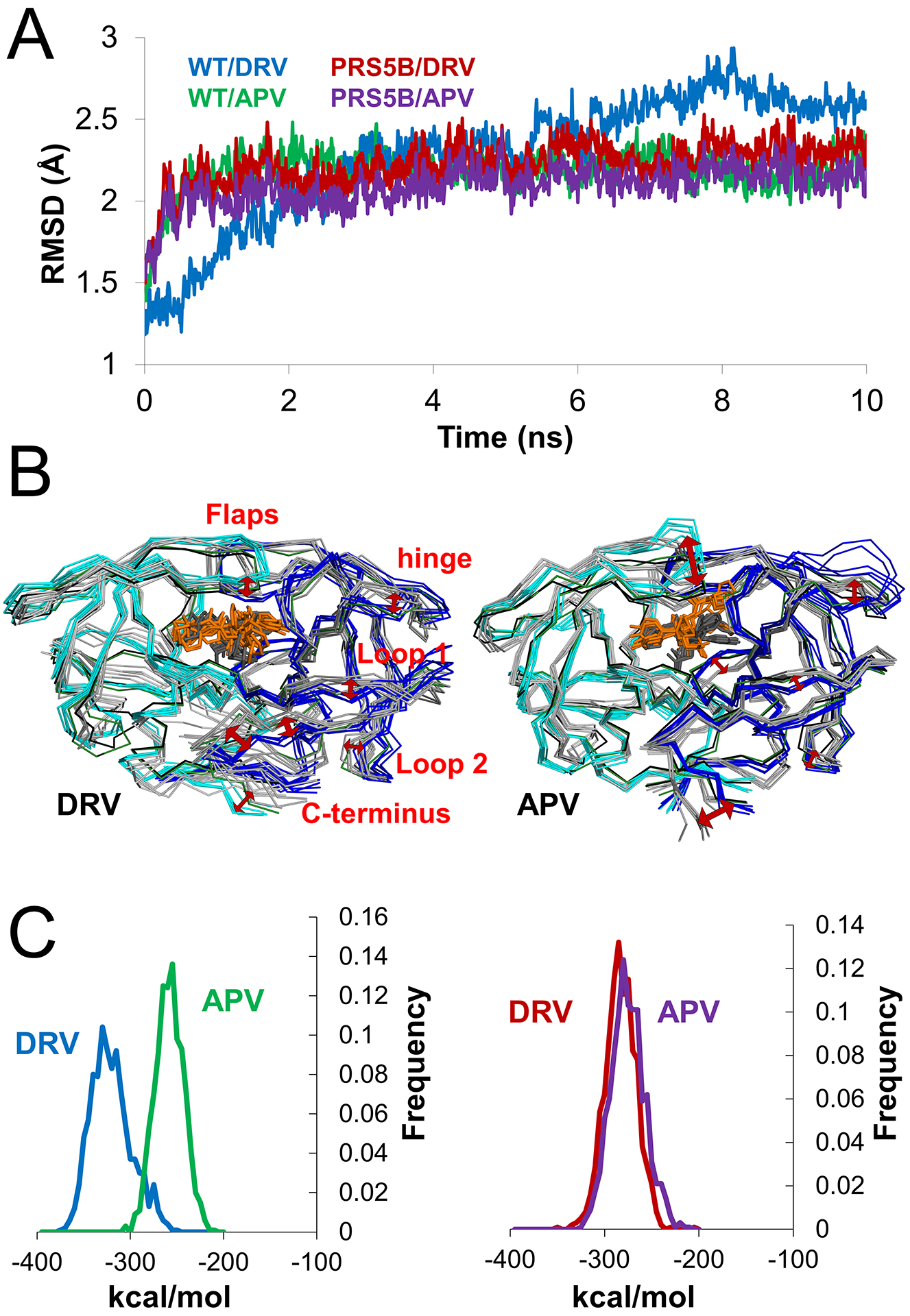
A. Trajectories of the 10 ns simulations of APV and DRV complexes of PR and PRS5B. RMSD of Cα atoms after superposition with the starting crystal structure plotted for the 10 ns time course. Simulations are colored as follows: PR/DRV (blue), PR/APV (green), PRS5B/DRV (red), and PRS5B/APV (purple).
B. Frequency plot of total non-bonded interaction energies of inhibitors in PR (left) and PRS5B (right) simulations. Simulations are colored as in A.
C. Comparison of PR with PRS5B for DRV (left) and APV (right) simulations. Snapshots of PR (grey shades, grey inhibitor as sticks) and PRS5B (cyan and blue, orange inhibitor as sticks) models for every 2 ns of the simulation are shown as superimposed ribbons. The starting crystal structures PR (black) and PRS5B (green) are shown. Red arrows indicate large conformational differences between wild-type and mutant protease. Structure figures were generated using PyMOL.
The structural variation during the simulations is illustrated in Figure 9B. Similar to the ligand-free dimers, the inhibitor complexes show higher variation in the flaps, loops 1 and 2, and termini. In addition, the hinge shows conformational changes, especially in the APV complexes, while the two DRV complexes have larger differences in the strand from residues 6–10. The flaps of the mutant move away from the catalytic site compared to their conformation in the wild-type PR. Moreover, the inhibitors show greater variation in the simulations for mutant (RMSD on non-hydrogen atoms of 2.3 and 1.7 Å for APV and DRV) than for wild-type PR (RMSD of 0.6 and 1.0 Å for APV and DRV). The greater fluctuation of inhibitors in the mutant is consistent with greater flexibility of residues in the binding cavity and thereby poorer inhibition. Increased fluctuation of inhibitor was also reported in a recent MD simulation by another group[44].
The non-bonded protease-inhibitor interaction energies were calculated over the 10 ns simulations (Figure 9C). For wild-type PR, the calculated interaction energy had a mean value of −323.0 ± 21.6 kcal/mol for DRV and −260.4 ± 15.4 for APV. The differences are in good qualitative agreement with the experimental values of 40-fold higher affinity for DRV relative to APV. Previously, another group reported close agreement with experimental values in more extensive 500 ns simulations of wild-type PR with DRV and APV[45]. In contrast, the interaction energies calculated for the mutant gave a mean of −285.3 ± 16.4 for DRV and −277.9 ± 18.0 kcal/mol for APV consistent with similar experimental inhibition values for the two inhibitors (Table 1). The calculations for mutant and wild-type complexes cannot be compared directly, however, since they do not account for contribution of the entropic differences in the two ligand-free enzymes and free inhibitors in solution. These differences are likely increased due to the large number of mutations (22) in PRS5B, including four changes that alter the charge of the side chain.
Implications for drug resistance
PRS5B is an HIV protease mutant from a clinical isolate that is poorly inhibited by all FDA-approved PIs as shown by enzyme kinetics experiments. Extremely poor inhibition was measured for LPV (Ki=160 nM) and SQV (Ki=1 μM). The best inhibitors, APV and DRV, displayed Ki values of approximately 4 nM for PRS5B. Structural analysis of PRS5B in complexes with APV and DRV demonstrates minor alteration in the inhibitor binding site, consistent with the presence of only I84V mutation. The distance between closest atoms of Ile47 and Val84 on opposite sides of the cavity increases by about 1 Å compared to the corresponding separation of Ile47 and Ile84 in the wild-type complexes. PR20 also contains the active site mutation I84V, which works synergistically with 3 other mutations to substantially increase the size of the inhibitor binding cavity as shown by 2 Å bigger distance between the closest atoms of Val47 and Val84 and thus decrease the affinity for PIs. Like PRS5B, PRS17 contains only one active site mutation, V82S, which does not significantly alter the size of the inhibitor binding site or interactions with DRV relative to those of wild-type PR/DRV but may contribute to substrate recognition when combined with the G48V flap mutation[27]. These three HIV protease variants, PR20, PRS17 and PRS5B, display decreased affinity for DRV despite retaining all hydrogen bond interactions with inhibitor observed in wild-type PR crystal structures.
In the case of PRS5B, mutations in the hinge and Loop 2 regions induce major conformational changes in regions distal to the active site that eliminate interactions observed in wild-type PR structures. Perturbations in PRS5B did not alter interactions with its two most effective inhibitors. Instead, significant conformational changes occurred far from the active site suggesting hinge and Loop 2 rearrangements lead to loss of inhibitor affinity through PI-independent mechanisms. MD simulations also show distinct conformations for these regions in the mutant and wild-type PR.
Unlike other studied examples of highly-resistant mutants, PRS5B contains a cluster of mutations protruding into and extending along two strands of Loop 2 (Figure 2). Non-active-site mutations in this region, such as A71V and G73S, have been shown to propagate changes to the catalytic site and provide cross-resistance for inhibitors[18,35,36,46–49]. Another study on single mutants V32I, L33F, L76V, and L90M showed how remote mutations can impact inhibitor binding by altering protein ensemble dynamics through a rearranged network of residues circulating to the active site[50]. Several molecular dynamics studies of proteins have shown that the effects of distal mutations can propagate to the active site and influence protein function[51,52]. These mechanisms combined with hydrophobic sliding described for HIV protease[11,33,53] are likely occurring in PRS5B. The dramatic perturbations resulting from the Loop 2 mutation cluster in PRS5B work synergistically with the hinge cluster and other mutations to decrease inhibitor affinity and catalytic activity.
N83D has long been identified as a major resistance associated mutation for TPV [54–56]. G73T is a rare minor drug resistance mutation for ATV [6,57–59]. This study is the first to describe the structural impact of these two mutations. L10I and L63P did not show structural changes in PRS5B but have been shown to help maintain thermal stability when combined with I84V[60].
NMR measurements of inhibitor-free PRS17 and PR20, supported by molecular dynamics simulations and crystal structures, demonstrate that these drug resistant proteases possess dynamic flaps that exist preferentially in an open conformation in the absence of inhibitor. This is consistent with Electronic Spin Resonance experiments on PR with D30N, M36I, and A71V mutations[61]. In contrast, wild-type PR tends to adopt a closed flap conformation even in the absence of ligand[17,18,62]. PRS5B shares similar hinge-loop mutations with PRS17 and PR20. In addition, PRS5B bears the unique N83D mutation which leads to fewer interactions between flap and hinge than observed in both PRS17 and PR20. MD simulations on PRS5B show larger variations in the surface features of the flaps, hinge, loop 1, the tip of loop 2 and the C-terminal residues. Overall, the conformational differences in the MD simulations are consistent with the loss of hydrogen bond interactions observed across the entire subunit in the crystal structures of the mutant compared to the wild-type PR. These conformational changes are expected to contribute to the changes in affinity for inhibitor. Protease-inhibitor interaction energies calculated in MD simulations agree qualitatively with the experimental difference in binding affinity of wild-type enzyme and mutant for DRV compared to APV.
Comparison of the three well-characterized examples of highly resistant mutants PR20, PRS17 and PRS5B reveals a trajectory of how clusters of mutations might evolve to promote drug-resistance as illustrated in Figure 10. All three mutants utilize mutations in the hinge region at 35 and compensating M36I mutation to drive rearrangements of the hinge and alter its separation from the flap. PRS5B, with its unique N83D mutation, features two backbone conformations for a section of the hinge-loop, whereas PRS17 and PR20 contain additional compensating mutations in Loop 1 to stabilize the hinge. Compared to PRS5B, PRS17 and PR20 have increasingly more effective mechanisms to compensate for loss of hinge-flap anchor point (Glu35-Arg57 ion pair) and show increasingly worse inhibition by PIs. PRS5B’s Loop 2 cluster facilitates more dramatic shifts compared to those in other mutants containing A71V, although three mutations serve to diminish these changes. Drug selection pressure may drive accumulation of mutations in protease initially towards the resistance-inducing changes in intermediate mutants like PRS5B, but over time, these viruses may be outcompeted by mutants with more synergistic mutations, such as PRS17. Other highly evolved mutants might resemble PR20 by introducing mutations that enhance monomer stability and expand the active site cavity or possess co-evolved mutations in Gag substrate cleavage sites [63].
Figure 10: Scheme for evolution of highly-drug resistant protease variants.
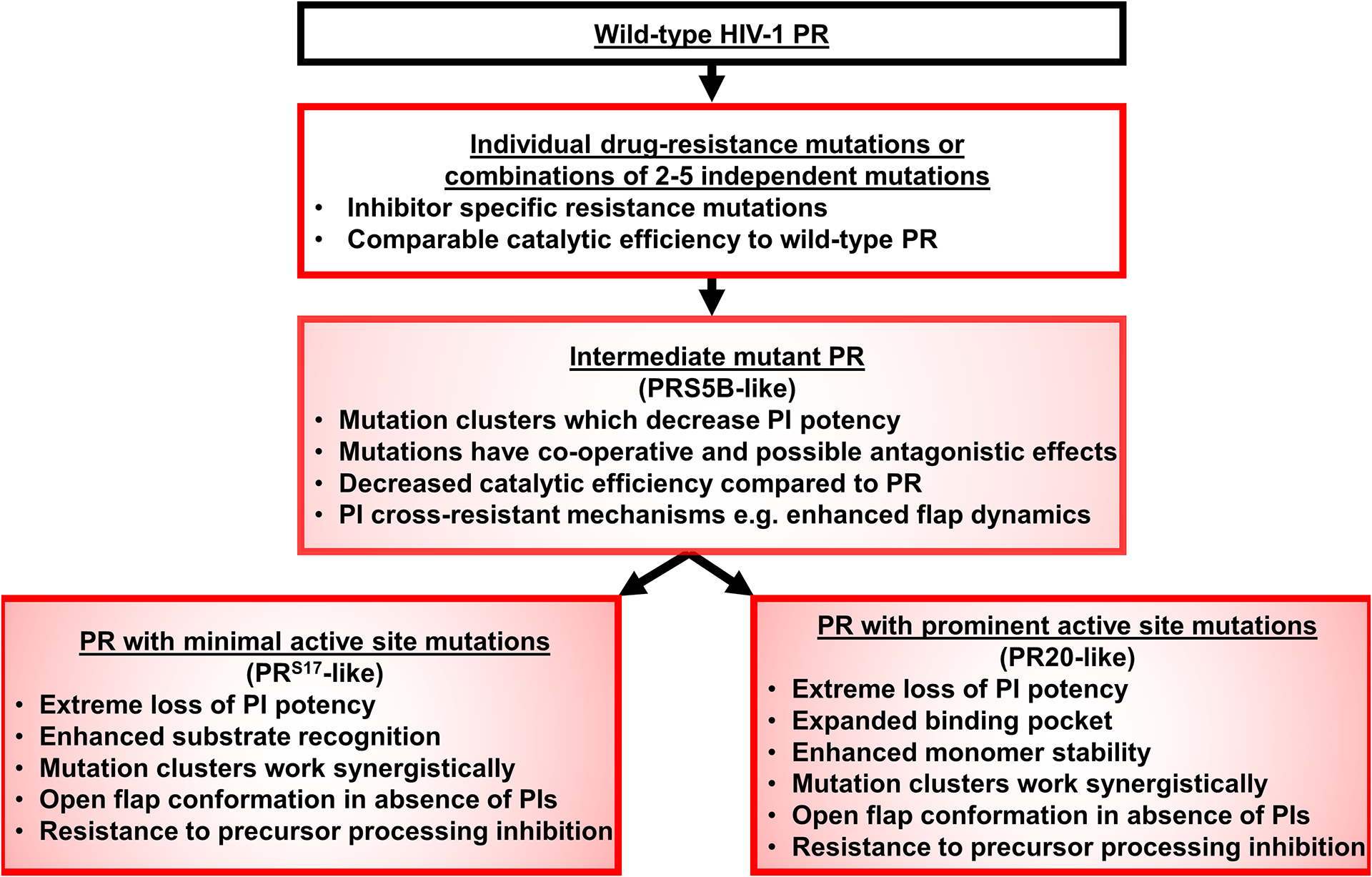
Antiretroviral treatment with PR inhibitors selects for PR variants able to process substrate in the presence of inhibitors. PRS5B represents an example of an intermediate mutant protease between the initial accumulation of resistance-associated mutations and more highly-evolved protease mutants.
The PRS5B mutant provides a prime example of how mutation clusters in distal regions can function synergistically to decrease inhibitor effectiveness while retaining viable catalytic efficiency and enzyme stability. Together, PRS5B, PRS17, and PR20 provide prototypes for the design of novel inhibitors to combat variations of cross-resistant mechanisms.
Materials and Methods
Selection of the PRS5B sequence
The protein sequence for PRS5B was obtained by the same machine learning procedure described for PRS17 [24]. Mutants representing sets of sequences with high levels resistance to each inhibitor were selected by mean shift clustering with regression analysis of genotype-phenotype data in the Stanford HIVdb (https://hivdb.stanford.edu) [24]. The procedure used a unified encoding of the sequence and 3D structure as described in [25]. Only a single mutant, PRS17, was observed in the highest resistance set for 6 inhibitors. PRS17 and PRS5B mutants were identified as representing high resistance to different combinations of 5 inhibitors.
Expression and purification of PRS5B
The gene coding for PRS5B was synthesized and cloned on a pJ414 plasmid with a T7 promoter [ATUM] and transformed into E. coli BL21 (DE3) (Invitrogen) cells. Mutation Q7K was introduced to reduce autoproteolysis and C67A and C95A to eliminate cysteine thiol crosslinking. Protein expression, purification, and folding were performed as described[27].
Enzyme kinetic assays
Kinetic experiments and enzyme dilutions were carried out in reaction buffer (50 mM MES pH 5.6, 200 mM sodium chloride, 0.5 mM EDTA, and 2.5% glycerol final reaction concentration). PIs obtained from the NIH AIDS Reagent program were dissolved in 100% DMSO. A spectroscopic fluorescence resonance energy-transfer (FRET) substrate analog derived from the HIV-1 Gag p2/NC cleavage site (H-2992 – BACHEM, Bubendorf, Switzerland) was used to monitor the initial rate of substrate hydrolysis by PRS5B at 37°C. The enzyme kinetic parameters (kcat, Km of FRET substrate, and kcat/Km) and inhibition constants (Ki) were determined as described in [27]. Enzyme concentration [E] was determined using active site titration with tight-binding inhibitor, APV. Ki was obtained from the equation Ki = (IC50 − [E]/2)/(1 + [S]/Km). Values are reported as mean ± SD (n=3–5).
UC50 for PRS5B, PRS17, and PR20 were measured using the same reaction buffer, substrate, enzyme, and temperature conditions as Ki measurements with the addition of 0–1.2 M urea as described in [30]. Values are reported as mean UC50 ± 3 standard deviations (n=3–4) where UC50 = urea concentration at 0.5 velocity at 0 M urea.
Crystallization and X-ray structure determination
Inhibitor dissolved in 100% DMSO was mixed at an 8:1 molar ratio with 4.2 mg/mL PRS5B solutions and incubated on ice for 30 minutes. Crystallization of the complexes were performed using hanging-drop vapor diffusion at room temperature using 1 μL protein/PI complex and 1 μL reservoir solution. Crystallization conditions for PRS5B/DRV were 2.2 M Ammonium Phosphate and 100 mM Tris buffer at pH 7.9. PRS5B/APV crystallized in 1.8 M Ammonium Phosphate and 100 mM Tris buffer at pH 7.9. Crystals grew after approximately 14 days and were cryo-protected stepwise in reservoir solution with 30% glycerol. Diffraction data were collected at 100°K on SER-CAT beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL, USA).
Diffraction data were integrated and scaled using HKL2000[64]. The crystal structure of PRS5B/DRV was solved by molecular replacement in Phaser using PR/DRV (2IEN) as a search model[65,66]. The refined PRS5B/DRV structure was used as a search model for PRS5B/APV. Integrative rounds of refinement using COOT[67] and REFMAC5[68] were used to construct the atomic models. In each structure, the inhibitor was fitted during refinement into unambiguous electron density in a single conformation. Statistical information was gathered using baverage and superpose[69]. Hydrogen bond interactions and van der Waals contacts were inferred from interatomic distances in the ranges of 2.4–3.4 Å and 3.0–4.2 Å, respectively. Structural figures were prepared using PyMOL[70].
Molecular dynamics simulations
Molecular dynamics (MD) simulations were performed for two ligand-free (PRS5B and PR) and four inhibitor-bound structures (PRS5B/APV, PRS5B/DRV, PR/APV and PR/DRV). The starting models were the wild-type PR complexes with DRV (PDB: 2IEN[32]) and APV (PDB: 3NU3[33]) and the corresponding complexes of the PRS5B mutant (PDB: 6P9A and 6P9B). Alternate conformations were deleted from the models, keeping the majority conformation. Inhibitors were removed for MD simulations of the ligand-free proteases. Solvent molecules from the crystal structures were included in the simulations and a mobile proton (H+) was introduced between the carboxylates of the catalytic Asp25 and 25’ to enable the protein to assume the variable protonation states observed in different neutron structures of HIV PR[71–73]. Each dimer was solvated with about five thousand water molecules generated randomly to fill the free space within a 30Å shell of the protein. Energy minimization and MD simulations used the program, AMMP[74] with the Tuna potential set[75], a modification of the SP4 set described in [76] with charges updated to those of Amber ff14SB[77]. In tests on small molecule benchmarks, the potential sets compare well to the CHARMM and AMBER sets[76]. AMMP is the molecular mechanics and dynamics engine of VegaZZ[78] and has undergone extensive professional code review as part of the SPEC2000 benchmark[79]. It is designed to be a computational backend that is plugged into other programs.
Positions were calculated for all hydrogen atoms and the geometry of the solvated protein system was minimized by conjugate gradients prior to starting the MD simulations. All-atom simulations were performed as described in [40,80]. The aperiodic amortized fast multipole algorithm was used for the electrostatic and non-bonded terms with parameters (mxdq = 0.75 Å; mmbox = 10.0 Å), 300K temperature constrained with Nose constraints, and a bounding volume constraint of 35 Å. Frames were saved for every 1 pc during the 10–20 ns simulations. The RMSD values and variance from the mean were calculated after superimposing each frame on the starting model. Selected interatomic distances were calculated using Python programs. The structural variation during the simulations was examined in a gif of the consecutive frames.
Acknowledgments
The research was supported in part by the National Institute of Health grant AI150461 (I.T.W., R.W.H.) and the Georgia State University Molecular Basis of Disease Fellowship (D.W.K.). We thank the staff at the Southeast Regional-Collaborative Access Team (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Abbreviations
- HIV
human immunodeficiency virus
- PR
HIV-1 protease (EC 3.4.23.16)
- PRS5B
PR with 22 mutations
- PRS17
PR with 17 mutations
- PR20
PR with 20 mutations
- DRV
Darunavir
- APV
Amprenavir
- Ki
kinetic inhibition value
- UC50
urea concentration at 50% maximum activity
- NMR
Nuclear magnetic resonance
- RMSD
root mean square deviation
- MD
Molecular dynamics
Footnotes
Databases: Structural data are available in Protein Data Bank under the accession codes 6P9A and 6P9B for PRS5B/DRV and PRS5B/APV respectively.
Conflicts of interests
The authors declare no conflicts of interest.
References
- 1.WHO | Data and statistics (2018) WHO. [Google Scholar]
- 2.Clutter DS, Jordan MR, Bertagnolio S & Shafer RW (2016) HIV-1 drug resistance and resistance testing. Infect. Genet. Evol 46, 292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO | HIV drug resistance report 2017 (2017) WHO. [Google Scholar]
- 4.Menéndez-Arias L (2013) Molecular basis of human immunodeficiency virus type 1 drug resistance: Overview and recent developments. Antiviral Res. 98, 93–120. [DOI] [PubMed] [Google Scholar]
- 5.Konvalinka J, Kräusslich H-G & Müller B (2015) Retroviral proteases and their roles in virion maturation. Virology 479, 403–417. [DOI] [PubMed] [Google Scholar]
- 6.Wensing AM, Calvez V, Günthard HF, Johnson VA, Paredes R, Pillay D, Shafer RW & Richman DD (2017) 2017 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med 24, 132–133. [PMC free article] [PubMed] [Google Scholar]
- 7.Wong-Sam A, Wang Y-F, Zhang Y, Ghosh AK, Harrison RW & Weber IT (2018) Drug Resistance Mutation L76V Alters Nonpolar Interactions at the Flap–Core Interface of HIV-1 Protease. ACS Omega 3, 12132–12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber IT & Agniswamy J (2009) HIV-1 Protease: Structural Perspectives on Drug Resistance. Viruses 1, 1110–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Louis JM, Zhang Y, Sayer JM, Wang Y, Robert W & Weber IT (2012) Drug Resistance Mutation L76V Decreases the Dimer Stability and Rate of Autoprocessing of HIV-1 Protease by Reducing Internal Hydrophobic Contacts. 50, 4786–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang Y-CE, Yu X, Zhang Y, Tie Y, Wang Y-F, Yashchuk S, Ghosh AK, Harrison RW & Weber IT (2012) Potent antiviral HIV-1 protease inhibitor GRL-02031 adapts to the structures of drug resistant mutants with its P1’-pyrrolidinone ring. J. Med. Chem 55, 3387–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldfarb NE, Ohanessian M, Biswas S, McGee TD Jr, Mahon BP, Ostrov DA, Garcia JP, Tang Y, McKenna R, Roitberg AE & Dunn BM (2014) Defective Hydrophobic Sliding Mechanism and Active Site Expansion in HIV-1 Protease Drug Resistant Variant Gly48Thr/Leu89Met: Mechanisms for the Loss of Saquinavir Binding Potency. Biochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nijhuis M, Schuurman R, de Jong D, Erickson J, Gustchina E, Albert J, Schipper P, Gulnik S & Boucher CA (1999) Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 13, 2349–59. [DOI] [PubMed] [Google Scholar]
- 13.van Maarseveen NM, Wensing AMJ, Jong D de, Taconis M, Borleffs JCC, Boucher CAB & Nijhuis M (2007) Persistence of HIV‐1 Variants with Multiple Protease Inhibitor (PI)–Resistance Mutations in the Absence of PI Therapy Can Be Explained by Compensatory Fixation. J. Infect. Dis 195, 399–409. [DOI] [PubMed] [Google Scholar]
- 14.Weber IT, Kneller DW & Wong-Sam A (2015) Highly resistant HIV-1 proteases and strategies for their inhibition. Future Med. Chem 7, 1023–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park JH, Sayer JM, Aniana A, Yu X, Weber IT, Harrison RW & Louis JM (2016) Binding of Clinical Inhibitors to a Model Precursor of a Rationally Selected Multidrug Resistant HIV-1 Protease Is Significantly Weaker Than That to the Released Mature Enzyme. Biochemistry 55, 2390–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louis JM, Aniana A, Weber IT & Sayer JM (2011) Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proc. Natl. Acad. Sci. U. S. A 108, 9072–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louis JM, Tözsér J, Roche J, Matúz K, Aniana A & Sayer JM (2013) Enhanced Stability of Monomer Fold Correlates with Extreme Drug Resistance of HIV-1 Protease. Biochemistry 52, 7678–7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agniswamy J, Louis JM, Roche J, Harrison RW & Weber IT (2016) Structural Studies of a Rationally Selected Multi-Drug Resistant HIV-1 Protease Reveal Synergistic Effect of Distal Mutations on Flap Dynamics. PLoS One 11, e0168616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agniswamy J, Shen CH, Aniana A, Sayer JM, Louis JM & Weber IT (2012) HIV-1 protease with 20 mutations exhibits extreme resistance to clinical inhibitors through coordinated structural rearrangements. Biochemistry 51, 2819–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roche J, Louis JM & Bax A (2014) Conformation of Inhibitor-Free HIV-1 Protease Derived from NMR Spectroscopy in a Weakly Oriented Solution. Chembiochem 16, 214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rhee S-Y, Gonzales MJ, Kantor R, Betts BJ, Ravela J & Shafer RW (2003) Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 31, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shafer RW (2006) Rationale and Uses of a Public HIV Drug‐Resistance Database. J. Infect. Dis 194, S51–S58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu X, Weber IT & Harrison RW (2013) Sparse Representation for Prediction of HIV-1 Protease Drug Resistance. Proc. SIAM Int. Conf. Data Min 2013, 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu X, Weber IT & Harrison RW (2015) Identifying representative drug resistant mutants of HIV. BMC Bioinformatics 16, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu X, Weber IT & Harrison RW (2014) Prediction of HIV drug resistance from genotype with encoded three-dimensional protein structure. BMC Genomics 15, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu F, Kovalevsky AY, Tie Y, Ghosh AK, Harrison RW & Weber IT (2008) Effect of Flap Mutations on Structure of HIV-1 Protease and Inhibition by Saquinavir and Darunavir. J. Mol. Biol 381, 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agniswamy J, Kneller DW, Brothers R, Wang Y-F, Harrison RW & Weber IT (2019) Highly Drug-Resistant HIV-1 Protease Mutant PRS17 Shows Enhanced Binding to Substrate Analogues. ACS Omega 4, 8707–8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muzammil S, Armstrong AA, Kang LW, Jakalian A, Bonneau PR, Schmelmer V, Amzel LM & Freire E (2007) Unique Thermodynamic Response of Tipranavir to Human Immunodeficiency Virus Type 1 Protease Drug Resistance Mutations. J. Virol 81, 5144–5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sayer JM, Agniswamy J, Weber IT & Louis JM (2010) Autocatalytic maturation, physical/chemical properties, and crystal structure of group N HIV-1 protease: Relevance to drug resistance. Protein Sci. 19, 2055–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pawar S, Wang Y-F, Wong-Sam A, Agniswamy J, Ghosh AK, Harrison RW & Weber IT (2019) Structural studies of antiviral inhibitor with HIV-1 protease bearing drug resistant substitutions of V32I, I47V and V82I. Biochem. Biophys. Res. Commun [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agniswamy J, Shen C-H, Wang Y-F, Ghosh AK, Rao KV, Xu C-X, Sayer JM, Louis JM & Weber IT (2013) Extreme multidrug resistant HIV-1 protease with 20 mutations is resistant to novel protease inhibitors with P1’-pyrrolidinone or P2-tris-tetrahydrofuran. J. Med. Chem 56, 4017–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tie Y, Boross PI, Wang Y-F, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW & Weber IT (2004) High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J. Mol. Biol 338, 341–52. [DOI] [PubMed] [Google Scholar]
- 33.Shen C-H, Wang Y-F, Kovalevsky AY, Harrison RW & Weber IT (2010) Amprenavir complexes with HIV-1 protease and its drug-resistant mutants altering hydrophobic clusters. FEBS J. 277, 3699–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohtaka H, Schön A & Freire E (2003) Multidrug Resistance to HIV-1 Protease Inhibition Requires Cooperative Coupling between Distal Mutations. Biochemistry 42, 13659–13666. [DOI] [PubMed] [Google Scholar]
- 35.Muzammil S, Ross P & Freire E (2003) A major role for a set of non-active site mutations in the development of HIV-1 protease drug resistance. Biochemistry 42, 631–638. [DOI] [PubMed] [Google Scholar]
- 36.Clemente JC, Moose RE, Hemrajani R, Whitford LRS, Govindasamy L, Reutzel R, McKenna R, Agbandje-McKenna M, Goodenow MM & Dunn BM (2004) Comparing the accumulation of active- and nonactive-site mutations in the HIV-1 protease. Biochemistry 43, 12141–12151. [DOI] [PubMed] [Google Scholar]
- 37.Meher BR & Wang Y (2015) Exploring the drug resistance of V32I and M46L mutant HIV-1 protease to inhibitor TMC114: Flap dynamics and binding mechanism. J. Mol. Graph. Model 56, 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Z, Huang X, Hu L, Pham L, Poole KM, Tang Y, Mahon BP, Tang W, Li K, Goldfarb NE, Dunn BM, McKenna R & Fanucci GE (2016) Effects of Hinge-region Natural Polymorphisms on Human Immunodeficiency Virus-Type 1 Protease Structure, Dynamics, and Drug Pressure Evolution. J. Biol. Chem 291, 22741–22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin P, Vickrey JF, Proteasa G, Jimenez YL, Wawrzak Z, Winters MA, Merigan TC & Kovari LC (2005) “Wide-open” 1.3 A structure of a multidrug-resistant HIV-1 protease as a drug target. Structure 13, 1887–95. [DOI] [PubMed] [Google Scholar]
- 40.Shen C-H, Chang Y-C, Agniswamy J, Harrison RW & Weber IT (2015) Conformational variation of an extreme drug resistant mutant of HIV protease. J. Mol. Graph. Model 62, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chetty S, Bhakat S, Martin AJM & Soliman MES (2016) Multi-drug resistance profile of PR20 HIV-1 protease is attributed to distorted conformational and drug binding landscape: Molecular dynamics insights. J. Biomol. Struct. Dyn 34. [DOI] [PubMed] [Google Scholar]
- 42.Henes M, Kosovrasti K, Lockbaum GJ, Leidner F, Nachum GS, Nalivaika EA, Bolon DNA, Kurt Yilmaz N, Schiffer CA & Whitfield TW (2019) Molecular Determinants of Epistasis in HIV-1 Protease: Elucidating the Interdependence of L89V and L90M Mutations in Resistance. Biochemistry 58, 3711–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gardner JM & Abrams CF (2019) Energetics of Flap Opening in HIV-1 Protease: String Method Calculations. J. Phys. Chem. B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henes M, Lockbaum GJ, Kosovrasti K, Leidner F, Nachum GS, Nalivaika EA, Lee S-K, Spielvogel E, Zhou S, Swanstrom R, Bolon DNA, Kurt Yilmaz N & Schiffer CA (2019) Picomolar to Micromolar: Elucidating the Role of Distal Mutations in HIV-1 Protease in Conferring Drug Resistance. ACS Chem. Biol., acschembio 9b00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu Y, Wang J, Shao Q, Shi J & Zhu W (2015) Effects of drug-resistant mutations on the dynamic properties of HIV-1 protease and inhibition by Amprenavir and Darunavir. Sci. Rep 5, 10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sasková KG, Kozísek M, Rezácová P, Brynda J, Yashina T, Kagan RM & Konvalinka J (2009) Molecular characterization of clinical isolates of human immunodeficiency virus resistant to the protease inhibitor darunavir. J. Virol 83, 8810–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clemente JC, Hemrajani R, Blum LE, Goodenow MM & Dunn BM (2003) Secondary Mutations M36I and A71V in the Human Immunodeficiency Virus Type 1 Protease Can Provide an Advantage for the Emergence of the Primary Mutation D30N †. Biochemistry 42, 15029–15035. [DOI] [PubMed] [Google Scholar]
- 48.Gonzalez LMF, Santos AF, Abecasis AB, Van Laethem K, Soares EA, Deforche K, Tanuri A, Camacho R, Vandamme A-M & Soares MA (2008) Impact of HIV-1 protease mutations A71V/T and T74S on M89I/V-mediated protease inhibitor resistance in subtype G isolates. J. Antimicrob. Chemother 61, 1201–1204. [DOI] [PubMed] [Google Scholar]
- 49.Liu F, Boross PI, Wang YF, Tozser J, Louis JM, Harrison RW & Weber IT (2005) Kinetic, stability, and structural changes in high-resolution crystal structures of HIV-1 protease with drug-resistant mutations L24I, I50V, and G73S. J. Mol. Biol 354, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ragland DA, Nalivaika EA, Nalam MNL, Prachanronarong KL, Cao H, Bandaranayake RM, Cai Y, Kurt-Yilmaz N & Schiffer CA (2014) Drug resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J. Am. Chem. Soc 136, 11956–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rajasekaran N, Suresh S, Gopi S, Raman K & Naganathan AN (2017) A General Mechanism for the Propagation of Mutational Effects in Proteins. Biochemistry 56, 294–305. [DOI] [PubMed] [Google Scholar]
- 52.Naganathan AN (2019) Modulation of allosteric coupling by mutations: from protein dynamics and packing to altered native ensembles and function. Curr. Opin. Struct. Biol 54, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foulkes-Murzycki JE, Scott WRP & Schiffer CA (2007) Hydrophobic sliding: a possible mechanism for drug resistance in human immunodeficiency virus type 1 protease. Structure 15, 225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baxter JD, Schapiro JM, Boucher CAB, Kohlbrenner VM, Hall DB, Scherer JR & Mayers DL (2006) Genotypic changes in human immunodeficiency virus type 1 protease associated with reduced susceptibility and virologic response to the protease inhibitor tipranavir. J. Virol 80, 10794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Llibre JM, Schapiro JM & Clotet B (2010) Clinical Implications of Genotypic Resistance to the Newer Antiretroviral Drugs in HIV‐1–Infected Patients with Virological Failure. Clin. Infect. Dis 50, 872–881. [DOI] [PubMed] [Google Scholar]
- 56.Bennett DE, Camacho RJ, Otelea D, Kuritzkes DR, Fleury H, Kiuchi M, Heneine W, Kantor R, Jordan MR, Schapiro JM, Vandamme A-M, Sandstrom P, Boucher CAB, van de Vijver D, Rhee S-Y, Liu TF, Pillay D & Shafer RW (2009) Drug Resistance Mutations for Surveillance of Transmitted HIV-1 Drug-Resistance: 2009 Update. PLoS One 4, e4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varghese V, Mitsuya Y, Fessel WJ, Liu TF, Melikian GL, Katzenstein DA, Schiffer CA, Holmes SP & Shafer RW (2013) Prototypical recombinant multi-protease-inhibitor-resistant infectious molecular clones of human immunodeficiency virus type 1. Antimicrob. Agents Chemother 57, 4290–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Le T, Chiarella J, Simen BB, Hanczaruk B, Egholm M, Landry ML, Dieckhaus K, Rosen MI & Kozal MJ (2009) Low-Abundance HIV Drug-Resistant Viral Variants in Treatment-Experienced Persons Correlate with Historical Antiretroviral Use. PLoS One 4, e6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rhee S-Y, Taylor J, Wadhera G, Ben-Hur A, Brutlag DL & Shafer RW (2006) Genotypic predictors of human immunodeficiency virus type 1 drug resistance. Proc. Natl. Acad. Sci. U. S. A 103, 17355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang MW & Torbett BE (2011) Accessory mutations maintain stability in drug-resistant HIV-1 protease. J. Mol. Biol 410, 756–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Vera IMS, Smith AN, Dancel MCA, Huang X, Dunn BM & Fanucci GE (2013) Elucidating a Relationship between Conformational Sampling and Drug Resistance in HIV-1 Protease. Biochemistry 52, 3278–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hornak V, Okur A, Rizzo RC & Simmerling C (2006) HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A 103, 915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kozísek M, Henke S, Sasková KG, Jacobs GB, Schuch A, Buchholz B, Müller V, Kräusslich H-GG, Rezácová P, Konvalinka J, Bodem J, Kožíšek M, Henke S, Šašková KG, Jacobs GB, Schuch A, Buchholz B, Müller V, Kräusslich H-GG, Řezáčová P, Konvalinka J & Bodem J (2012) Mutations in HIV-1 gag and pol compensate for the loss of viral fitness caused by a highly mutated protease. Antimicrob. Agents Chemother 56, 4320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Otwinowski Z & Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode Elsevier. [DOI] [PubMed] [Google Scholar]
- 65.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC & Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Storoni LC, McCoy AJ & Read RJ (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr. D. Biol. Crystallogr 60, 432–8. [DOI] [PubMed] [Google Scholar]
- 67.Emsley P & Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr 60, 2126–32. [DOI] [PubMed] [Google Scholar]
- 68.Murshudov GN, Vagin AA & Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- 69.Krissinel E, Henrick K & IUCr (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr 60, 2256–2268. [DOI] [PubMed] [Google Scholar]
- 70.DeLano WL (2002) Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 40, 82–92. [Google Scholar]
- 71.Gerlits O, Wymore T, Das A, Shen C-H, Parks JM, Smith JC, Weiss KL, Keen DA, Blakeley MP, Louis JM, Langan P, Weber IT & Kovalevsky A (2016) Long-Range Electrostatics-Induced Two-Proton Transfer Captured by Neutron Crystallography in an Enzyme Catalytic Site. Angew. Chemie Int. Ed 55, 4924–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gerlits O, Keen DA, Blakeley MP, Louis JM, Weber IT & Kovalevsky A (2017) Room Temperature Neutron Crystallography of Drug Resistant HIV-1 Protease Uncovers Limitations of X-ray Structural Analysis at 100 K. J. Med. Chem 60, 2018–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harrison RW & Weber IT (1994) Molecular dynamics simulations of HIV-1 protease with peptide substrate. “Protein Eng. Des. Sel 7, 1353–1363. [DOI] [PubMed] [Google Scholar]
- 74.Harrison RW (1993) Stiffness and energy conservation in molecular dynamics: An improved integrator. J. Comput. Chem 14, 1112–1122. [Google Scholar]
- 75.Fang B, Fu G, Agniswamy J, Harrison RW & Weber IT (2009) Caspase-3 binds diverse P4 residues in peptides as revealed by crystallography and structural modeling. Apoptosis 14, 741–752. [DOI] [PubMed] [Google Scholar]
- 76.Bagossi P, Zahuczky G, Tözsér J, Weber IT & Harrison RW (1999) Improved Parameters for Generating Partial Charges: Correlation with Observed Dipole Moments. J. Mol. Model 5, 143–152. [Google Scholar]
- 77.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE & Simmerling C (2015) ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stigliani J-L, Bernardes-Génisson V, Bernadou J & Pratviel G (2012) Cross-docking study on InhA inhibitors: a combination of Autodock Vina and PM6-DH2 simulations to retrieve bio-active conformations. Org. Biomol. Chem 10, 6341. [DOI] [PubMed] [Google Scholar]
- 79.188.ammp: SPEC CPU2000 Benchmark Description (2015)
- 80.Chen X, Weber IT & Harrison RW (2004) Molecular dynamics simulations of 14 HIV protease mutants in complexes with indinavir. J. Mol. Model 10, 373–381. [DOI] [PubMed] [Google Scholar]
- 81.Mildner AM, Rothrock DJ, Leone JW, Bannow CA, Lull JM, Reardon IM, Sarcich JL, Howe WJ, Tomich C-SC, Smith CW, Heinrickson RL & Tomasselli AG (1994) The HIV-1 Protease as Enzyme and Substrate: Mutagenesis of Autolysis Sites and Generation of a Stable Mutant with Retained Kinetic Properties. Biochemistry 33, 9405–9413. [DOI] [PubMed] [Google Scholar]
- 82.Szeltner Z & Polgár L (1996) Conformational stability and catalytic activity of HIV-1 protease are both enhanced at high salt concentration. J. Biol. Chem 271, 5458–63. [DOI] [PubMed] [Google Scholar]