

Abstract
TCR signal strength is critical for CD8+ T cell clonal expansion after antigen stimulation. Levels of the transcription factor IRF4 control the magnitude of this process through induction of genes involved in proliferation and glycolytic metabolism. The signaling mechanism connecting graded TCR signaling to the generation of varying amounts of IRF4 is not well understood. Here we show that antigen potency regulates the kinetics, but not the magnitude of NFAT1 activation in single mouse CD8+ T cells. Consequently, T cells that transduce weaker TCR signals exhibit a marked delay in Irf4 mRNA induction resulting in decreased overall IRF4 expression in individual cells and increased heterogeneity within the clonal population. We further show that the activity of the tyrosine kinase ITK acts as a signaling catalyst that accelerates the rate of the cellular response to TCR stimulation, controlling the time to onset of Irf4 gene transcription.
These findings provide insight into the function of ITK in TCR signal transduction that ultimately regulates IRF4 expression levels in response to variations in TCR signal strength.
Introduction
A central process of the adaptive immune response to infection is the activation and expansion of cytotoxic T cells. Characterized by their expression of the co-receptor CD8, these lymphocytes are critical for identification and removal of intracellular pathogens. Activation of naïve CD8+ T cells is primarily driven by binding of MHC:peptide antigen to their T cell receptor (TCR). Upon receptor binding, a signaling cascade is triggered which directs the cell to respond by reorganizing its cytoskeleton, upregulating glycolytic pathways, and preparing for cell division and migration. The cell rapidly changes its transcriptional program and thousands of new genes are expressed to meet this new functional demand (1). While numerous studies have characterized the activated T cell transcriptome, detailed insight into pathways that link proximal TCR signaling dynamics to graded gene expression has remained elusive.
TCRs can bind to MHC:peptide complexes with wide variations of ligand binding kinetics. Antigen abundance can also vary greatly, as a result of differences in pathogen burden and differing efficiencies of antigen presentation. Graded or variable responses to changes in TCR signal strength are known to generate diverse functional outcomes in responding T cells, a phenomenon best illustrated by the selection processes that shape the repertoire of developing T cells in the thymus (2). The responses of peripheral T cells to infection or immunization are also modulated by variations in TCR signal strength. When antigen doses are held constant, CD8+ T cells with higher affinity TCRs for MHC:peptide show a greater magnitude of expansion, and exhibit enhanced production of IFNγ relative to low affinity clones (3). However, it remains unclear whether higher antigen doses are able to compensate for lower TCR affinity during peripheral T cell activation, or whether intrinsic differences in TCR interactions with MHC:peptide establish limits on the downstream outcomes of T cell activation.
One important component regulating TCR signal strength is the Tec family tyrosine kinase ITK. A key molecule in TCR signal transduction, ITK is activated by the Src kinase Lck and recruited to the LAT signaling complex along with its substrate, phospholipase C (PLCγ). Once activated, ITK phosphorylates and activates PLCγ. Activated PLCγ cleaves the membrane phospholipid PIP2 into the two secondary messengers diacylglycerol (DAG) and inositol triphosphate (IP3), which both help propagate downstream signaling pathways to regulate gene expression (4). However, despite this biochemical insight into ITK’s function in TCR signaling, there is limited understanding of how ITK activity contributes to overall TCR signal strength.
To gain insight into the regulation of TCR signal strength, and the role of ITK in this process, we focused on dissecting the TCR-induced upregulation of the transcription factor, Interferon Regulatory Factor 4 (IRF4). This choice was motivated by our previous studies demonstrating that IRF4 upregulation was highly dependent on ITK activity (5), an observation also verified by others (6, 7). IRF4 has been extensively characterized in lymphocytes (8). For CD8+ T cells, the levels of IRF4 expression following T cell activation determine the magnitude of terminal effector cell (TEC) expansion in vivo (9–11). IRF4 expression is nearly undetectable in naïve and memory T cells, but is rapidly induced following TCR stimulation. Along with its frequent binding partner BATF, IRF4 upregulates a host of genes in newly-activated CD8+ T cells, specifically genes critical for the metabolic switch to glycolysis (10, 12). In TH2 cells, strength of TCR stimulation correlated with BATF-IRF4 binding to different enhancer regions of the genome, leading to variations in gene expression patterns (13). While graded expression of IRF4, which changes the magnitude of the effector response, is dependent on TCR signal strength, the signaling mechanism regulating the control of IRF4 expression levels in CD8+ T cells is not well understood.
To address this issue, we utilized primary naïve CD8+ T cells, and varied both the MHC:peptide dose and the affinity of the TCR interaction with individual MHC:peptide complexes. Using this system to fine tune TCR signaling, we then employed several single-cell assays to measure the consequences of varying TCR signal activity, thereby providing insight into the pathways contributing to graded expression of IRF4. We found that CD8+ T cells are able to integrate the digital activation of NFAT into graded IRF4 protein expression by varying the kinetics of activation. Furthermore, we showed that ITK activity contributes to TCR signaling by accelerating the rate of this response. Thus, under conditions of weak TCR signaling and/or in the absence of ITK activation, delayed kinetics of NFAT activation and Irf4 mRNA expression create a population of CD8+ T cells that, as shown previously, has reduced proliferative capacity in response to infection (9, 14, 15).
Materials and Methods
Mice
Mice were bred and housed in specific pathogen-free conditions at the University of Massachusetts Medical School (UMMS) in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines. OTI Rag−/− (B6.129S7-Rag1tm1Mom Tg(TcraTcrb)1100Mjb N9+N1) were purchased from Taconic (Hudson, NY). CD45.1 (B6.SJL-Ptprca Pep3b/BoyJ) and CD4-Cre mice were purchased from The Jackson Laboratory (Bar Harbor, ME). OTI Rag−/− Itk−/− CD45.1 mice were generated by crossing Itk−/−, OTI Rag−/− and CD45.1 mice. Itk−/− mice have been described previously (16). Nur77-GFP reporter mice (a gift from Dr. S. Swain, UMMS) were crossed to OTI Rag−/− mice. Nur77-GFP reporter mice have been described previously (17). NFAT-AV mice were crossed to CD4-Cre and have been described previously (18). For experiments, male or female littermate mice at 8-12 weeks of age were used.
Cell culture
Splenocytes from OT-I Rag−/− mice (and the genetic variants) were stimulated ex-vivo with recombinant peptide (N4, T4, or G4) at the indicated doses for 24h. Antigen-presenting-cells (APCs) in the bulk splenocyte population were sufficient for stimulation. (Figures 1–2) Recombinant peptides were purchased from 21st Century Biochemicals (Marborough, MA). PRN694 was generously provided by Principia Pharmaceuticals (19). For stimulation readouts earlier than 24h, OT-I T cells were stimulated at a 5:1 ratio of APCs to T cells. The APCs used were bulk splenocytes isolated from WT C57BL/6 mice, then pulsed with peptide prior to the addition of T cells. (Fig. 4) APCs used in Fig. 5 were stimulated with LPS (1ug/mL) and pulsed with peptide for 1h prior to addition of T cells. LPS, FK506, PMA, and Ionomycin were purchased from Sigma-Aldrich (St. Louis, MO). Fractionated CD8 T cells used in Fig. S3C–D were stimulated with 1µg/mL of plate-bound αCD3ε antibody (Clone 1742) purchased from Biolegend for 12-36h.
Figure 1. CD69 and IRF4 respond differently to graded TCR signaling in activated CD8+ T cells.
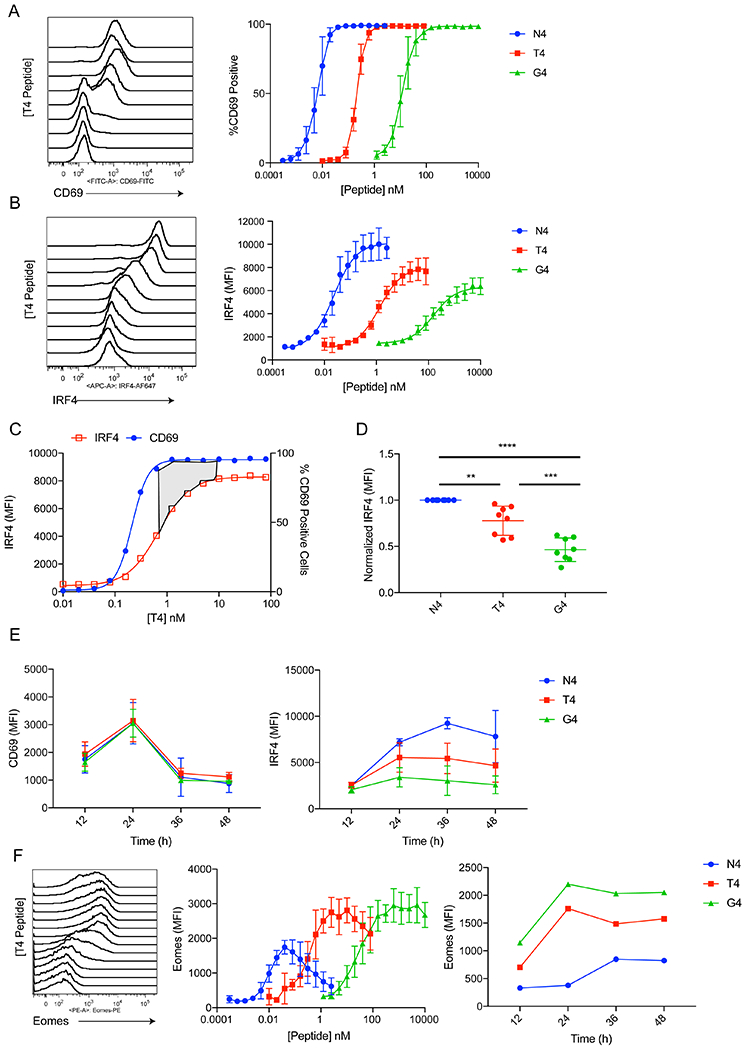
(A) Representative histogram plots of CD69 staining on OT-I cells stimulated with different doses of T4 peptide for 24h. Cells were gated on live CD8+ TCRβ+. Plots of %CD69+ values are shown for all three peptides at right.
(B) Representative histogram plots of IRF4 intracellular staining in OT-I cells stimulated with different doses of T4 peptide for 24h. Cells were gated on live CD8+ TCRβ+. Plots of IRF4 MFI values are shown for all three peptides at right.
(C) IRF4 MFI and %CD69+ values for the T4 peptide dose response were plotted on the same graph. The area shaded in gray emphasizes the concentration of antigen that yields maximum CD69 expression but is still on the upslope for IRF4 expression.
(D) IRF4 MFI values were normalized to N4 stimulation over multiple experiments. These data show results of stimulations with 1nM N4, 100nM T4, and 1μM G4, which are the relative concentrations eliciting maximum IRF4 expression for each peptide. ** p≤0.01, *** p≤0.001, **** p≤0.0001 (one-way ANOVA followed by Dunnett’s test for N4 comparisons. Unpaired student t test for T4 and G4 comparison)
(E) OT-I T cells were stimulated for 12-48h with 1nM N4, 100nM T4, or 1μM G4. MFI values for CD69 (left) and IRF4 (right) are plotted over time.
(F) OT-I cells were treated with varying doses of N4, T4, or G4 peptides for 24h. Representative histograms of Eomes intracellular staining are shown for the T4 peptide (left). Eomes MFI is plotted for each peptide dose (middle). Cells were treated with 1nM N4, 100nM T4, or 1μM G4 from 12-48h and MFI for each peptide is plotted over time.
Data are representative of three to five experiments.
Figure 2. ITK inhibition reduces maximum IRF4 expression in CD8+ T cells in a graded manner.
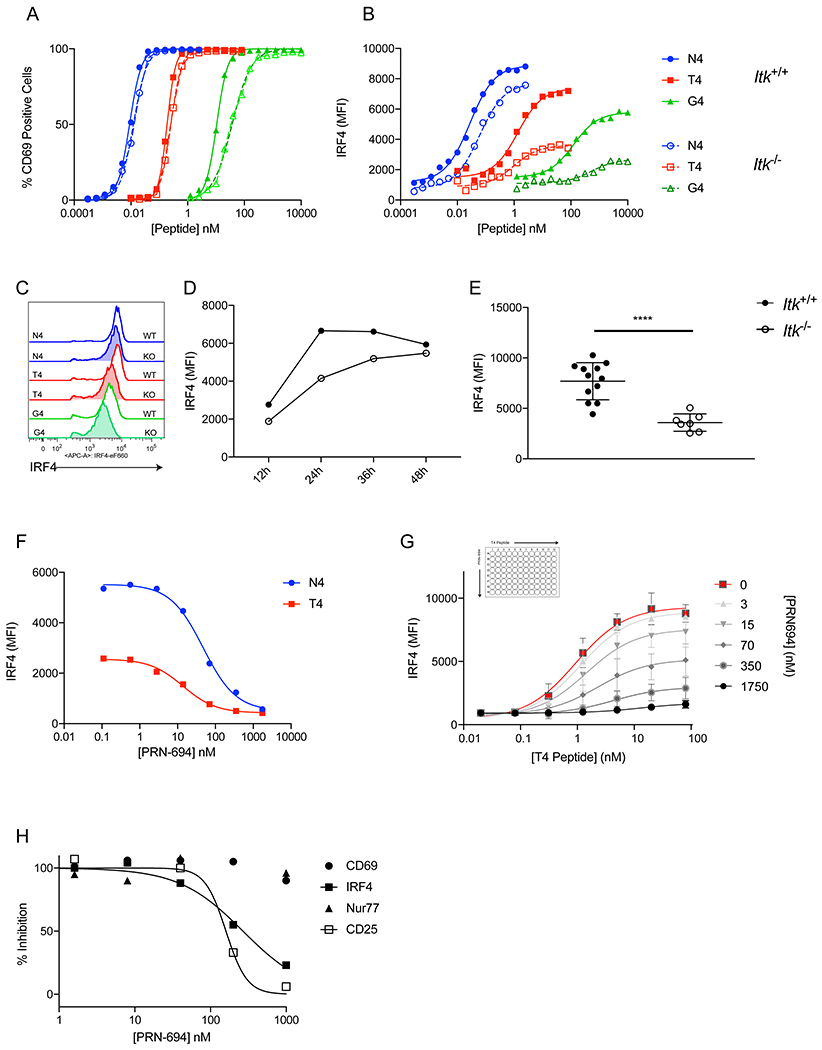
(A-B) OT-I WT (Itk+/+) or OT-I Itk−/− (Itk−/−) T cells were stimulated each peptide for 24h at multiple doses. %CD69+ (A) and IRF4 MFI (B) are plotted for each peptide concentration.
(C) Representative histograms of IRF4 intracellular staining for 1nM N4, 100nM T4, and 1μM G4 peptide stimulations of Itk+/+ or Itk−/− cells.
(D) Itk+/+ or Itk−/− OT-I cells were stimulated with 100nM T4 peptide at timepoints from 12-48h, and IRF4 MFI is plotted.
(E) IRF4 MFI values of Itk+/+ or Itk−/− cells stimulated with 100nM T4 for 24h. **** p≤0.0001 (unpaired student t test)
(F) OT-I T cells were stimulated with 1nM N4 and 100nM T4 peptide while simultaneously treated with varying doses of the ITK/RLK inhibitor PRN694 for 24h. Cells were stained for intracellular IRF4 and MFI of IRF4 staining under each condition is displayed.
(G) OT-I T cells were stimulated with a range of doses of T4 peptide in a matrix using varying doses of PRN694. After 24h the cells were stained for IRF4, and the IRF4 MFI for each condition is displayed. Each curve represents a dose response of T4 peptide at a single concentration of PRN694.
(H) OT-I T cells were stimulated with 100nM T4 peptide with varying doses of PRN694 for 24h. The cells were stained for IRF4, CD69, and CD25. For Nur77, the Nur77-GFP reporter was used. The MFI values were normalized to a positive and negative control to obtain % Inhibition.
Data are representative of three to five experiments.
Figure 4. TCR signal strength and ITK activity drives digital NFAT activation in CD8+ T cells.
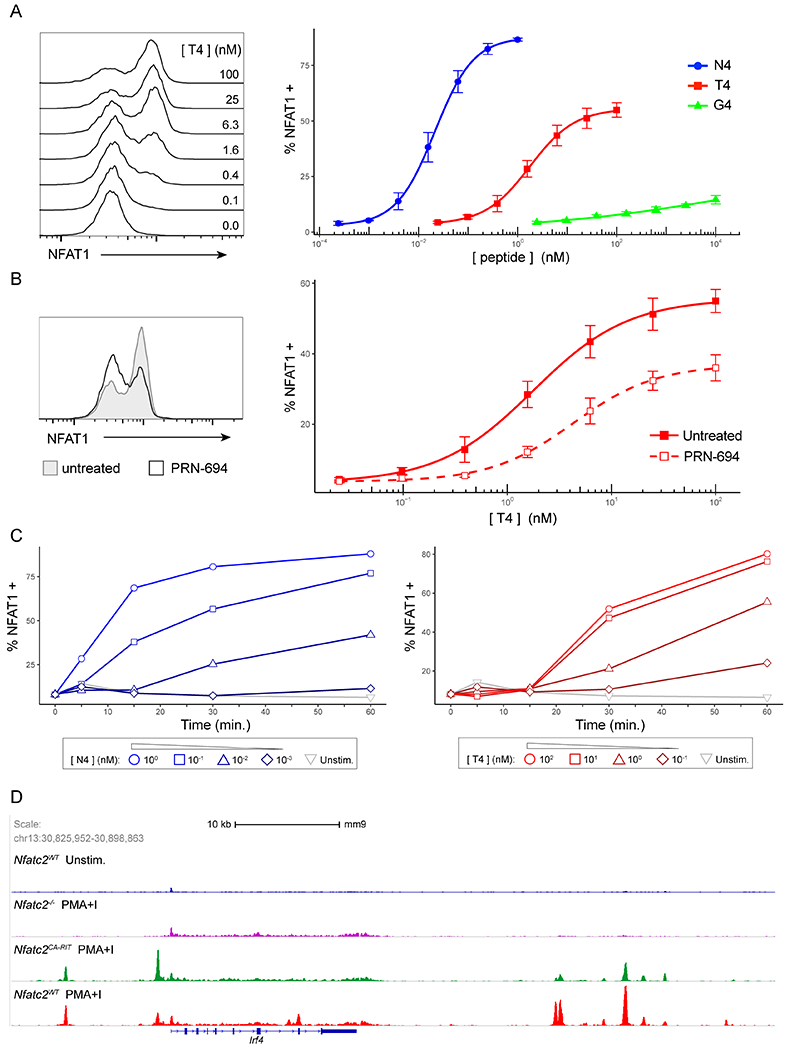
(A) Representative histograms of NFAT1 fluorescence in OT-I nuclei isolated after T cells were stimulated with B6 splenocytes pulsed with indicated doses of T4 peptide for 30m (left). Line plots of %NFAT1+ nuclei after 30m of stimulation with B6 splenocytes pulsed with indicated doses of either N4, T4 and G4 peptides (right). OT-I nuclei were identified as CellTrace Violet+ events.
(B) Representative histograms of NFAT1 fluorescence in OT-I nuclei after cells were stimulated for 30m with B6 splenocytes pulsed with 25 nM T4 peptide with or without 50nM PRN694 treatment (left). Line plots of %NFAT1+ (nuclear NFAT) values are shown for the T4 peptide dose response with and without 50nM PRN694 treatment (right). Nuclei were gated on CellTrace Violet+ events.
(C) Line plots of %NFAT1+ OT-I nuclei over a 60m timecourse after cells were stimulated B6 splenocytes pulsed with varying doses of either N4 (left) or T4 (right) peptides as indicated.
(D) NFAT1 ChIP-Seq data (GSE64409) on activated CD8 T cells from Martinez et al (21) were visualized using IGV software and a snapshot was taken of the Irf4 locus. The data represents 4 samples: (1) WT T cells, transduced with Mock construct, unstimulated (2) Nfatc2−/− T cells, transduced with Mock construct, stimulated with PMA/Ionomycin for 1h (3) Nfatc2−/− T cells, transduced with CA-RIT-NFAT (constitutively active NFAT unable to bind AP-1), stimulated with PMA/Ionomycin for 1h (4) WT T cells, transduced with Mock construct, stimulated with PMA/Ionomycin for 1h.
Data are representative of three or five experiments.
Figure 5. Reducing TCR strength, through ITK inhibition, delays Irf4 mRNA upregulation.
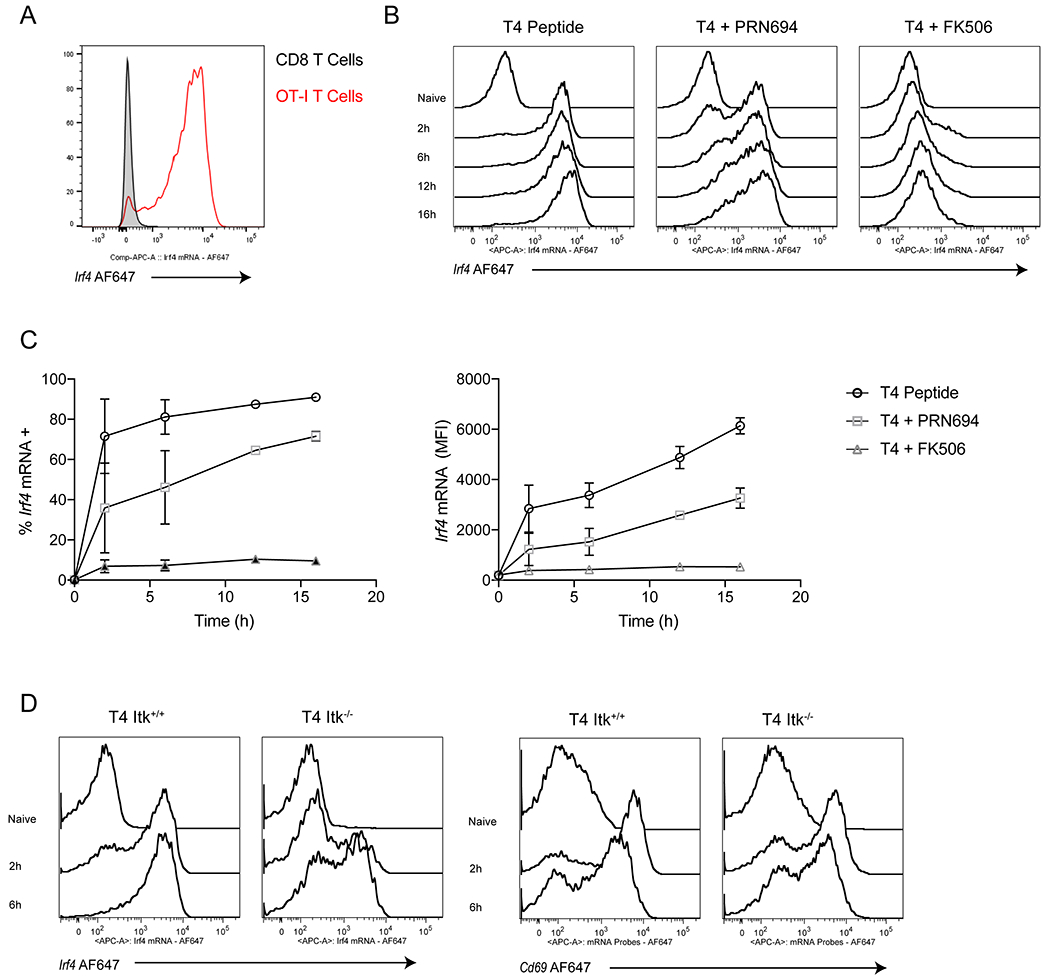
(A) OT-I T cells were labeled with CellTraceViolet and combined 5:1 with unlabeled peptide-pulsed APCs from B6 splenocytes. Representative histograms plots of Irf4 mRNA expression after 16h of 100nM T4 peptide stimulation. Cells are gates on live CD8+ TCRβ+ cells (+CellTraceViolet from OT-I mice and –CellTraceViolet from B6 mice.)
(B-C) OT-I T cells were treated with 100nM T4 peptide for timepoints from 2-16h either alone or in the presence of 100nM PRN694 or 100nM FK506. (A) Representative histogram plots of Irf4 mRNA expression. (B) Data are plotted as %Irf4 mRNA positive (left) or as Irf4 mRNA MFI (right) over time. Cells are gates on live CD8+ TCRβ+ CellTraceViolet+ cells
(C) Itk+/+ or Itk−/− cells OT-I T cells were treated with 100nM T4 peptide for 2 or 6h. Representative histograms for Irf4 and Cd69 mRNA expression are shown. Cells are gates on live CD8+ TCRβ+ CellTraceViolet+ cells
Data are representative of three experiments.
Antibodies and flow cytometry
IRF4 (eF660 and PE), CD8a (PE-eF610), Va2 (APC and APC-eF780), TCRb (APC-eF780), Eomes (PE and PE-Cy7), T-bet (PerCP-Cy5.5), CD45.1 (eF450 and APC-eF780), CD69 (FITC and PE-Cy7) were purchased from eBioscience / Thermo Fisher (San Diego, CA). CD44 (v500) and CD45.1 (BV510) were purchased from BD Biosciences (Billerica, MA). LIVE/DEAD Violet, LIVE/DEAD Aqua, goat anti-rabbit (PE) were purchased from Life Technologies (Grand Island, NY). Single cell suspensions were prepared from isolated spleens; RBCs were lysed; Fc receptors were blocked using supernatant from 2.4G2 hybridomas. Intracellular transcription factor staining was performed using eBioscience FoxP3 Transcription Factor Staining Buffer Set. Samples were analyzed on the LSR II flow cytometer (BD Bioscience) and data was analyzed on Flow Jo (Tree Star).
Nuclear flow and RNA flow
Nuclei isolation and flow cytometry staining of OT-I cells stimulated in co-cultures were performed as previously described (20). Briefly, CD8+ T cells were negatively enriched from OT-I spleens (Stemcell Technologies) and labeled with CellTrace Violet cell tracking and proliferation dye (Thermo Fisher) for 20m prior to co-culture with peptide-pulsed B6 splenocytes at a 5:1 ratio of APCs to T cells. For nuclei isolation, cells were treated and washed with sucrose and detergent buffers. The nuclei were fixed in 4% paraformaldehyde and then intranuclear staining was performed with a 0.3% Triton-X 100 detergent PBS buffer. NFAT1-AF488 antibody (clone D43B1) was purchased from Cell Signaling Technology (Danvers, MA) and NFAT2-PE antibody (clone 7A6) was purchased from BioLegend (San Diego, CA). The PrimeFlow assay kit, from Thermo Fisher, was used for RNA flow. Cd69 and Irf4 probe sets were ordered for AF647.
ChIP analysis
NFAT1 ChIP-Seq data (GSE64409) from Martinez et. al. (21) was analyzed using Integrative Genomics Viewer (Broad Institute, Cambridge, MA) (22). A snapshot was taken of the Irf4 gene locus.
Statistical analysis
Graphs represent mean ± SEM. Statistical significance indicated by * p≤0.05, ** p≤0.01, *** p≤0.001, **** p≤0.0001 (NS, p>0.05) based on one-way ANOVA followed by Dunnett’s test or unpaired Student t test. Data analysis performed using GraphPad Prism 7.0 (GraphPad, San Diego, CA).
Results
Weaker TCR signaling reduces maximum effector-associated gene expression in CD8+ T cells
In the first 24h after TCR stimulation, CD8+ T cells upregulate a variety of proteins that play key roles in T cell proliferation and cell trafficking, including CD69 and IRF4. We used the OT-I TCR transgenic line to study naive CD8+ T cell activation in response to different peptide ligands that stimulate the TCR with differing potencies. The cognate peptide for the OT-I TCR is derived from chicken Ovalbumin, with the amino acid sequence of SIINFEKL (N4) (23). By substituting amino acids at the fourth position, TCR affinity for the MHC:peptide complex can be decreased without affecting peptide binding to MHC Class I (3). For our studies, we used the SIITFEKL peptide (T4), which is approximately 100-fold less potent, and the SIIGFEKL peptide (G4), which is approximately 1000-fold less potent, than N4 (3, 24).
Initially, we performed dose response experiments in which each of the three peptides was added to unfractionated splenocytes from OT-I TCR transgenic x Rag2−/− (hereafter referred to as OT-I) mice, and cells were assessed 24h later by flow cytometry. For CD69 expression, as expected, a bimodal response was observed. At low peptide doses of T4 peptide, for instance, no CD69 upregulation was observed. As the peptide dose increased, a threshold was reached where a subset of cells transitioned from low CD69 expression to high expression (Fig. 1A, left panel). At these intermediate doses, a mixture of CD69+ and CD69− cells was observed. Plotting the percent CD69 positive fraction at each peptide dose generated a steep dose response curve, indicative of an all-or-nothing binary response (Fig. 1A, right panel).
In contrast, simultaneous examination of IRF4 expression in the same cell populations showed a markedly distinct pattern of expression. Like CD69, low peptide doses do not turn on IRF4. However, at higher peptide doses, IRF4 levels continue to increase as peptide dose increases (Fig. 1B). If CD69 expression is used as a minimal marker for activation, IRF4 levels continue to rise with higher peptide doses, even after the peak magnitude of CD69 is achieved. Simultaneously overlaying the %CD69+ and IRF4 MFI line plots on the same graph illustrates this comparison, as highlighted by the shaded area (Fig. 1C). The shallow slopes for the dose response curves for IRF4 expression, as well as the variable slopes of these curves for peptides of different potencies, indicate that IRF4 regulation is not binary in nature; rather, IRF4 expression levels are tunable by TCR signal input. Correspondingly, a significant difference in maximum IRF4 expression levels is seen when comparing the responses to the three different peptides, with maximal expression proportional to TCR affinity (Fig. 1D).
To determine whether differences in peak expression levels of IRF4 in response to stimulation with different potency peptides might result from altered kinetics of IRF4 gene expression, we examined CD69 and IRF4 levels on stimulated OT-I cells at 12, 24, 36, and 48h post-stimulation. These data clearly indicate that the affinity-based maximum expression of IRF4 was not due to a delay in the timepoint at which maximal IRF4 expression was achieved. Instead, we observed maximum levels of IRF4 at 24-36h post-stimulation, regardless of the peptide used to stimulate the T cells. Similar kinetics of CD69 induction was also observed with all 3 peptides (Fig. 1E). Overall these results show that IRF4 expression levels are a sensitive measure of TCR signal strength, and are sensitive to both the dose of antigen as well as the strength of binding of each TCR to peptide-MHC. Eomesodermin (Eomes), a transcription factor associated with memory CD8 T cells, has been shown to be downregulated by IRF4 (5). Therefore, we predicted an inverse relationship between Eomes and IRF4 expression in stimulated OT-I T cells. Consistent with this, we observed Eomes expression increasing at the low peptide doses just above the threshold needed to induce CD69 upregulation; as peptide doses increased, leading to increasing levels of IRF4, Eomes expression was reduced in a dose-dependent manner. Additionally, peak expression levels of Eomes were inversely related to peptide potency, as the highest levels of Eomes were observed after stimulating OT-I cells with the weakest potency G4 peptide (Fig. 1F).
We next examined other genes known to be regulated by TCR signaling. For these studies, we first chose Nur77 (Nr4a1) induction, a response commonly used to monitor TCR signal strength both in vitro and in vivo. (17, 25, 26) OT-I mice were crossed to the Nur77-GFP reporter line (17), and OT-I CD8+ T cells were stimulated in bulk splenocyte cultures with varying doses of each of the three Ova peptides (N4, T4, and G4). Unlike the pattern of expression observed for IRF4, Nur77-GFP peak expression levels were not dependent on the peptide potency (Fig. S1A, left and middle panels). In each case, higher doses of lower potency peptides were able to achieve the same levels of GFP expression as the strongest N4 peptide. This pattern of expression was independent of the timepoint examined, as timecourse studies showed overlapping kinetics of Nur77-GFP expression regardless of the peptide used to stimulate the T cells (Fig. S1A, right panel). Thus, while Nur77 reporter expression is closely linked to TCR activation in vitro and in vivo, we did not observe the same affinity based maximum expression relationship that was observed with IRF4 (Fig. S1A).
In contrast to Nur77-GFP, examination of CD25, the IL-2 receptor alpha chain, showed a pattern resembling IRF4, in that maximum expression levels of CD25 were proportional to peptide potency (Fig. S1B). The graded expression of CD25 was even more pronounced over time with large differences observed at 48h post-stimulation. These data confirm our findings that some gene expression responses are highly tunable by TCR signal strength, and depend on the binding interactions of individual MHC:peptide complexes with the TCR, and not just on the relative dose of antigenic peptide present on antigen-presenting-cells.
ITK activity drives graded IRF4 expression in CD8+ T cells
To address in greater detail the TCR signals responsible for graded expression of genes based on TCR signal strength, we chose to focus on the regulation of IRF4. We reasoned that binary modes of expression, such as that seen for CD69, had previously been characterized. Specifically, Das et. al. demonstrated a positive feed-forward pathway accounting for the all-or-nothing response of Cd69 to TCR stimulation. In this study, the authors showed that following TCR stimulation, the initial production of the second messenger DAG activated the Ras GTP-exchange factor Ras-GRP, leading to the generation of activated Ras molecules. These Ras-GTP molecules bound to a second Ras GTP-exchange factor SOS, acting as an allosteric modulator of SOS activity and promoting a massive enhancement in the production of activated Ras. A consequence of this feed-forward loop was that low signaling thresholds could rapidly produce maximal activation of the Ras-MAPK pathway, a key inducer of CD69 expression. The authors then linked this pathway to the bimodal expression of CD69 (27). In contrast to this well-characterized pathway producing bimodal expression of CD69, the signals accounting for graded expression of IRF4 in response to variations in TCR signal strength have not been described.
Changes in IRF4 expression were shown to be dependent on ITK activity in the context of low dose αCD3 stimulation (5). To pursue this connection further, we generated Itk−/− OT-I TCR transgenic mice. Stimulation of splenocytes from these mice with the three Ova peptide variants showed that CD69 upregulation was nearly unaffected by the absence of ITK when T cells were stimulated with varying doses of N4 or T4 peptide. Stimulation with the weakest potency peptide G4 showed an impairment of CD69 upregulation in the absence of ITK, shifting the EC50 for the G4 peptide from 11 to 37nM, but the maximal response of CD69 expression could be restored by modest increases in peptide dose (Fig. 2A). This suggests a greater ITK-dependent signaling defect with the weakest TCR stimulation.
In contrast to CD69, IRF4 upregulation was markedly reduced in stimulated Itk−/− OT-I T cells, particularly in response to T4 and G4 peptides (Fig. 2B–C). Interestingly, the overall peptide concentration-dependence of IRF4 expression was not changed; for example, for T4 peptide, the EC50 for WT OT-I cells was 1.3nM and was 1.0nM for Itk−/− cells. However, the maximum level of IRF4 expression was significantly decreased, in addition to being substantially delayed (Fig. 2D–E). To further explore the relationship between ITK and maximum IRF4 expression, we treated OT-I cells with a small molecule dual ITK/RLK Tec kinase family inhibitor PRN-694 (19). Inhibition of ITK/RLK signaling significantly reduced IRF4 expression in WT OT-I cells stimulated with either N4 or T4 stimulated peptide, with similar IC50s (20-40nM) (Fig. 2F). Further evidence that ITK/RLK activity could fine tune IRF4 expression was generated in an additional experiment, where cells were treated with multiple doses of T4 peptide, and for each peptide dose, cells were treated with a series of doses of PRN-694 in an 8×8 matrix format (Fig. 2G). Quantification of these dose-response experiments demonstrates a clear relationship between ITK/RLK activity and maximum IRF4 expression, as is evident by the stratified plateaus of the dose response curves. PRN694 also inhibited CD25 expression at a similar potency as for inhibition of IRF4 upregulation, but did not inhibit CD69 or Nur77-GFP expression (Fig. 2H). Overall, these studies demonstrated that modulation of TCR signal strength by reducing TCR affinity for MHC:peptide or by inhibiting ITK activity led to reduced IRF4 expression in each responding T cell. Furthermore, these data also showed that low affinity peptides were unable to induce the same levels of IRF4 expression as higher affinity peptides, regardless of the dose of peptide provided.
Calcium and calcineurin signaling drives graded IRF4 expression in CD8+ T cells
To determine the component(s) of TCR signaling that could account for the graded expression of IRF4 in response to variable TCR signaling, as well as the importance of ITK signaling in this response, we considered the major transcription factors activated by the TCR. Due to the demonstrated role of ITK in this phenomenon, we focused on pathways downstream of PLC-γ1, including the MAPK, NF-κB, and calcium/NFAT signaling pathways (28–30). As previous studies have demonstrated that Itk−/− T cells have a substantial defect in calcium signaling amplitude (16, 31, 32), we initially hypothesized that calcium could directly contribute to graded IRF4 expression in T cells.
To address the signaling pathways that could be contributing to increased IRF4 expression, we utilized two common pharmacological agents commonly used to stimulate T cells in vitro, phorbol 12-myristate 13-acetate (PMA) and Ionomycin. PMA is a small molecule mimetic of DAG that directly activates protein kinase C and Ras-MAPK signaling, whereas the calcium ionophore, Ionomycin, induces calcium signaling in T cells. Unlike the usual strategy of combining these two agents together to stimulate T cells, we separately treated WT OT-I cells with varying doses of PMA or Ionomycin alone. After 24h, IRF4 expression was assessed by flow cytometry (Fig. 3A). After treatment with PMA, we observed a binary response of IRF4 expression, with a modest induction of IRF4 that achieved a stable plateau. In contrast, treatment with Ionomycin recapitulated the graded induction of IRF4 expression. Only in combination do the two agents yield comparable IRF4 expression to that observed with strong peptide antigen-dependent stimulation (Fig. 3B; Fig. S2A). Furthermore, the ability of Ionomycin to promote a graded induction of gene expression was unique to IRF4, as neither CD69, Nur77-GFP, nor CD25 showed a comparable upregulation induced by calcium signaling alone (Fig. S2B–D). The calcineurin inhibitor FK-506 was also able to block IRF4 upregulation, emphasizing the dependence of this response on calcium and calcineurin signaling (Fig. 3C). These data indicated that multiple signaling pathways regulate IRF4, but that the calcium/calcineurin pathway can control the graded expression of IRF4 in response to variations in TCR signal strength.
Figure 3. Calcium and calcineurin signaling drives graded IRF4 expression in CD8+ T cells.
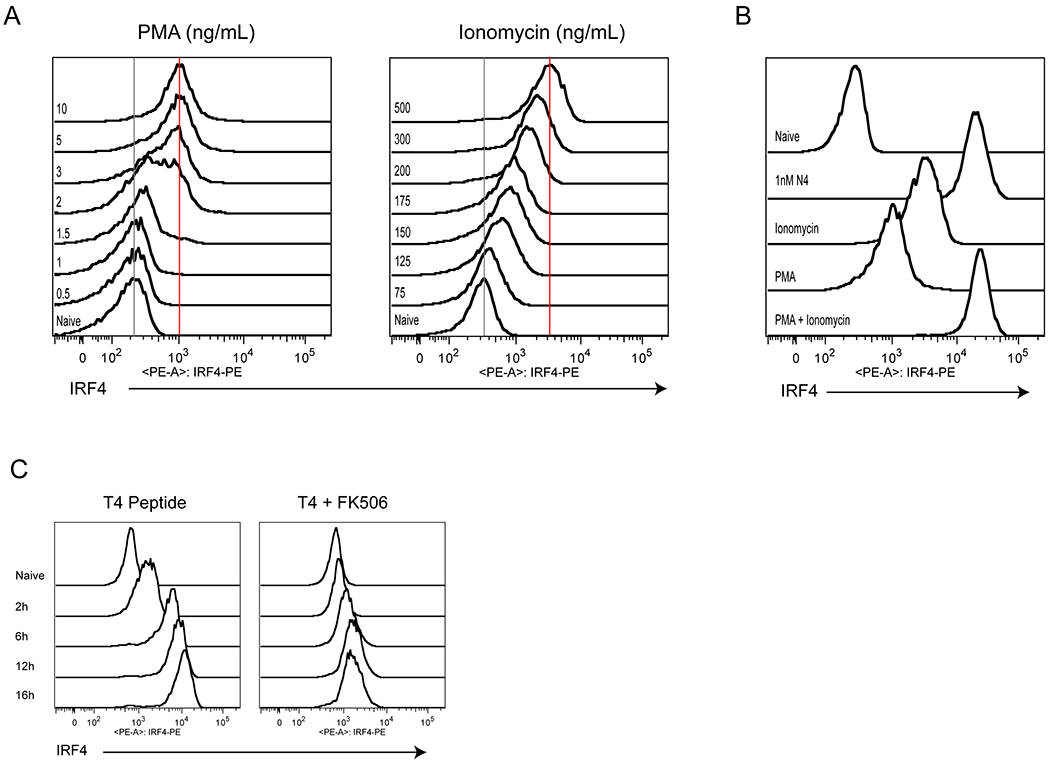
(A) OT-I T cells were treated with varying doses of PMA or Ionomycin for 24h, and cells were stained for intracellular IRF4. Representative histograms of IRF4 staining are shown.
(B) Representative histograms of IRF4 staining for OT-I cells left unstimulated (Naïve), or stimulated with 500ng/mL Ionomycin, 10ng/mL PMA, a combination of both, or 1nM N4 for 24h.
(C) Representative histograms of IRF4 staining for OT-I cells treated with 100nM T4 for timepoints from 2-16h in the absence (left) or presence (right) of 100nM FK506.
Data are representative of three experiments.
TCR signal strength and ITK activity drives digital NFAT activation in CD8+ T cells
We hypothesized that the calcium/calcineurin-mediated regulation of IRF4 expression worked through NFAT activation. NFAT is a family of transcription factors that are regulated indirectly by calcium flux through the removal of inhibitory phosphorylation sites by the calcium-activated phosphatase calcineurin (33, 34). Normally, NFAT is highly phosphorylated and sequestered in the cytoplasm. When calcineurin is active, these sites are removed, unmasking the nuclear localization domain and allowing nuclear transport where it can then bind to DNA along with other transcription factor binding partners, often AP-1 (35, 36). The two primary NFAT proteins that are critical for CD8+ T cell function are NFAT1 (encoded by NFATc2) and NFAT2 (encoded by NFATc1). NFAT2 is critical for cytotoxic function in activated CD8+ T cells, specifically through regulation of key genes like Gzmb (37). Both NFAT proteins also promote CD8+ T cell exhaustion with NFAT2 expression elevated during chronic LCMV clone 13 infection (38). However, in newly activated T cells, both proteins have similar expression with similar binding to the Irf4 gene locus (38).
To test the effects of graded TCR signaling on NFAT activation in CD8+ T cells, we utilized a nuclear flow cytometry assay to assess nuclear NFAT1 and NFAT2 levels in activated OT-I T cells (20). For these experiments, OT-I CD8+ T cells were isolated, labeled with fluorescent cell tracking dye, and then mixed with unlabeled splenocytes plus varying doses of the T4 peptide antigen. After 30 minutes, nuclei were prepared and stained with antibodies to NFAT1 and NFAT2. Histograms of NFAT1 and NFAT2 staining in nuclei of OT-I cells stimulated with varying concentrations of the T4 peptide variant showed that activation of both proteins was not graded in nature, but rather, exhibited a binary response (Fig. S3A). Both proteins had a similar response to increasing doses of T4 peptide. We also tested NFAT1 activation with varying doses of the three peptide antigens. As the dose of peptide increased, the proportion of OT-I cells with nuclear NFATc2 increased in a dose-dependent manner (Fig. 4A, right panel). While NFAT activation was binary at the single-cell level, the overall pattern of NFAT1 activation seen in the population of OT-I cells as a whole reflected both the dose of peptide as well as the binding strength of each peptide variant. When plotted as a percentage of NFAT1+ nuclei, the data for NFAT1 activation show a striking similarity to the data examining IRF4 protein expression under these same conditions. Thus, at this early 30 minute timepoint, the OT-I population shows a maximum level of NFAT1-positive nuclei that is proportional to peptide affinity with N4>T4>G4.
We then examined the effect of ITK activity on NFAT activation. As shown in Fig. 4B (left panel), ITK inhibition by PRN694 retained a bimodal response of NFAT1 activation following stimulation of OT-I cells with antigen-presenting-cells and peptide. Instead, inhibition of ITK reduced the percentage of NFAT1+ nuclei at each peptide dose tested (Fig. 4B). Inhibition was also observed in Itk-deficient OT-I T cells with reductions of both NFAT1+ and NFAT2+ nuclei (Fig. S3B). These results also mirror the effects of ITK deficiency on the expression of IRF4 protein in response to varying doses of T4 peptide.
To generate a more detailed view of the NFAT activation response, we performed a timecourse experiment, including timepoints ranging from 5 minutes to 60 minutes post-stimulation. Because both NFAT proteins had similar ITK-dependent activation, we focused on NFAT1. These data confirmed our initial observations that varying TCR signal strength had a dramatic effect on the kinetics of NFAT1 nuclear localization (Fig. 4C). Whereas higher doses of N4 peptide elicited a more rapid rise in the percentage of NFAT1+ nuclei, stimulation with lower doses of this peptide delayed the accumulation of NFAT1+ nuclei. Strikingly, the weaker peptide variant T4 was unable to induce NFAT1 nuclear localization in any cells, regardless of the peptide dose, within the first 15 minutes of stimulation (Fig 4C, right panel). This lag in NFAT nuclear localization is consistent with the delay in calcium flux onset observed in single OT-I cells stimulated with lower doses of the N4 peptide antigen (39). We reasoned that this delay in NFAT activation could dictate the graded levels of IRF4 protein seen at later timepoints. Extensive characterization of NFAT DNA binding sites in activated CD8+ T cells has been reported (21). Analysis of these published data for NFAT binding sites at the Irf4 promoter region revealed a robust peak 1.2kB upstream of exon 1 (Fig. 4D). Interestingly, this NFAT binding peak is also present in cells that expressed an NFAT protein carrying amino acid substitutions that interfere with its binding to AP-1. These data confirm that NFAT is able to bind to the Irf4 regulatory region as a homodimer in the absence of its usual binding partner, consistent with the robust control of IRF4 expression through calcium signaling alone.
To further examine the relationship between NFAT and IRF4, we took advantage of the hyper-activable NFAT mouse (18). These mice have a mutant form of NFATc2 knocked into the Rosa26 locus, named NFAT-AV. This mutant form of NFAT has reduced binding to the CK1 kinase and increased binding to the Calcineurin phosphatase, thus creating a protein more able to localize to the nucleus and bind DNA while not constitutively active in the absence of calcium signaling. We isolated CD8+ T cells from these mice and stimulated with anti-CD3 antibody. While the naïve NFAT-AV T cells had an increase in CD44hi cells, IRF4, CD69, and CD25 expression was low and comparable to WT B6 T cells prior to stimulation (Fig. S3C). In addition, stimulated WT and NFAT-AV CD8+ T cells had similar levels of IRF4, CD69, and CD44. However, there was a significant increase in CD25 expression, consistent with the known role of NFAT in driving Il2ra expression (40). WT and NFAT-AV T cells stimulated in the presence of the ITK inhibitor PRN694 showed a partial rescue of %IRF4hi cells across the entire population at 24 and 36h, further confirmation that NFAT contributes to IRF4 production, along with other transcription factors (Fig. S3D).
Graded TCR signaling alters the kinetics of Irf4 mRNA upregulation
IRF4 upregulation is detectable by 6h post-stimulation and generally peaks at 24h, with maximum levels of expression differing between T cells stimulated by ligands of different potencies. These data suggested that evaluating the kinetics of early Irf4 mRNA upregulation could be informative in understanding how cells stimulated with distinct TCR signal strengths ultimately accumulate different amounts of IRF4 protein. Using RNA flow cytometry, we measured Irf4 mRNA in activated OT-I T cells over the first 16h post-stimulation. Histograms comparing Irf4 mRNA expression in activated OT-I T cells compared to CD8+ T cells from bulk splenocytes illustrated antigen-dependent expression of the mRNA (Fig. 5A). Cells stimulated with 100nM of T4 peptide rapidly upregulated Irf4 mRNA by 2h, and levels of this mRNA were relatively constant for 16h post-stimulation (Fig. 5B–C). We also observed a sharp transition of cells going from Irf4 mRNA negative to positive in the time from 0-2h post-stimulation. Consistent with the IRF4 protein data, FK506 completely blocks upregulation of Irf4 mRNA. These findings emphasize the important role of calcium signaling and NFAT activation in Irf4 mRNA transcription, a result consistent with previous data demonstrating NFAT binding to the Irf4 gene locus in activated T cells (41, 42).
In contrast, inhibition of ITK signaling by treatment of cells with PRN694 delayed, but did not abolish, the upregulation of Irf4 mRNA. Similar to our results examining NFAT activation, we observed two distinct populations of OT-I cells at 2h post-stimulation in the presence of PRN694, one population positive for the Irf4 mRNA and one population remaining negative. Compared to the cells stimulated in the absence of the ITK inhibitor, those treated with PRN694 showed a slower transition to having Irf4 mRNA, along with a broader histogram peak, indicative of greater variability in expression within the population. The role for ITK in accelerating the rate of Irf4 mRNA upregulation was also confirmed by examination of stimulated WT versus Itk−/− OT-I T cells. Again, along with delayed kinetics of Irf4 mRNA expression in the absence of ITK, we observed a bimodal pattern of Irf4 mRNA staining (Fig. 5D). Finally, as expected based on the protein expression data, we found no major effect on Cd69 mRNA expression in the absence of ITK (Fig. 5D).
Reduced TCR signaling consistently yielded a reduction in peak expression of IRF4 protein, based on the IRF4 histogram MFI, while also showing a broader distribution within the population. To quantify this variability in expression we plotted the coefficient of variance (CV) for IRF4 MFI of OT-I cells stimulated with 100nM T4 peptide, in the absence or the presence of varying different doses of PRN694 (Fig. S4). ITK inhibition significantly increased the CV of both IRF4 protein and Irf4 mRNA, suggesting increased heterogeneity of expression across the clonal population. This was not a consequence of failing to activate a subset of cells, as >95% of cells were CD69+ under each of these conditions.
Discussion
Understanding the molecular mechanisms that control graded signaling through the TCR has been a central effort in T cell biology. A component of this effort has been studies directed at elucidating the contribution of the Tec kinase ITK to this process. Here, we utilized a combination of assays providing single cell data to dissect the signaling inputs that control the graded expression of the transcription factor IRF4, a key factor regulating the magnitude of the CD8+ effector T cell response to infection. Our studies revealed a process in which a clonal population of naïve CD8 T cells integrated different strengths of TCR stimulation into binary NFAT activation responses. Weaker TCR stimulation led to a delay in the kinetics of NFAT nuclear localization that correlated with slower upregulation of Irf4 mRNA, ultimately resulting in reduced expression of the IRF4 protein. As a consequence of this mechanism, a population of CD8+ T cells that receives weak TCR signaling is destined to undergo limited clonal expansion (9–11, 15).
Our results are in agreement with the recent studies of Richard et. al. Using state-of-the-art single cell RNA sequencing and proteomic techniques, the authors showed that the strength of TCR signaling controlled the rate at which individual cells were activated within a clonal population (43), and was responsible for greater variability in gene expression between individual cells in low affinity clonal populations. However, in analyses of cytolytic effector functions several days post-stimulation, this delay in kinetics among weakly stimulated naïve CD8+ T cells did not ultimately impair their cytolytic capacity, results that are in line with our observations that only some genes (e.g., Irf4, Il2ra) are sensitive to this delay in activation and are likely driving the magnitude of clonal expansion, while others (e.g., Cd69) are not.
Our results also expand our knowledge of how the Tec kinase ITK regulates the strength of TCR signaling. Many studies have documented that ITK is not required for T cell activation per se. Instead, T cells lacking ITK have selective defects in some T cell responses, while other responses appear unaffected by the absence of ITK. For instance, naïve Itk−/− CD4 T cells are defective in producing IL-17A or IL-9, when stimulated under TH17 or TH9 polarizing conditions, respectively (6, 44). Yet, Itk−/− mice are proficient at clearing infections of Leishmania major, and viruses such as LCMV, Influenza A, and Vaccinia, responses that require effective TH1 and cytotoxic CD8+ T cell responses, respectively (14, 45–47). Interestingly, these data also correspond nicely to the recent findings of Richard, et al, which found that expression of genes required for cytotoxic T cell function were independent of the strength of the initial T cell stimulation.
Our studies indicate that ITK functions as an accelerator in the TCR signaling pathway. In this regard, ITK is not required for TCR signaling, but acts to accelerate the process by which TCR stimulation produces a calcium response capable of inducing NFAT nuclear translocation. We speculate that ITK functions to increase the number of activated PLC-γ1 molecules produced by each stimulated TCR. As our data also show that NFAT nuclear translocation is an all-or-nothing response to TCR stimulation, we envision a sharp threshold of IP3 production by activated PLC-γ1 that is required to induce NFAT activation. When ITK signaling is engaged, the rate at which this threshold is achieved can be dramatically increased. It is likely that other key transcription factors activated by TCR stimulation have alternative modes of response to variations in TCR signal strength, possibly explaining why some genes expression responses, but not all, are sensitive to ITK signaling.
Our data indicate that NFAT is not the sole transcription factor contributing to IRF4 expression after TCR stimulation. As shown by the modest upregulation of IRF4 in response to PMA alone, as well as the combinatorial effect of PMA and Ionomycin added together, we conclude that additional DAG-dependent factors are required for optimal IRF4 upregulation in response to TCR signaling. One likely pathway contributing to the NFAT-independent component of IRF4 expression is NF-κB, as previous studies have suggested a role for both canonical NF-κB p65, as well as c-Rel in Irf4 expression (48). While these findings reveal the complex and multifactorial regulation of genes induced by TCR stimulation, they also highlight the utility of reductionist experiments, such as those described here, in identifying key inputs responsible for differing responses to variations in TCR signal strength. Our studies also demonstrate that increasing ligand dose or density on antigen-presenting-cells cannot necessarily overcome signaling deficits due to weakly stimulatory ligands. Thus, increasing the concentration of weakly potent peptide antigens is unable to generate the levels of IRF4 in naïve CD8+ T cells that can be achieved by more potent ligands. Understanding the complete program of genes that share this characteristic of Irf4 will provide critical information in the development of improved vaccines and T cells used for adoptive immunotherapy, to maximize desired effector and/or memory T cell responses.
Supplementary Material
Key Points:
Antigen potency and ITK control graded IRF4 expression.
Altered kinetics of NFAT activation provide a mechanism linking ITK and IRF4.
Acknowledgments:
We would like to thank Regina Whitehead and Sharlene Hubbard for their technical assistance. We thank Nicholas Spidale, PhD for suggesting nuclear flow cytometry. We thank Principia Biopharma Inc for generously providing PRN694. We thank the Department of Animal Medicine at the University of Massachusetts Medical School and University of Colorado Anschutz for maintaining our mouse colony.
Funding: NIH grant AI32419 (LJB), AI40127 (A.R.)
References:
- 1.Zhang N, and Bevan MJ. 2011. CD8+ T Cells: Foot Soldiers of the Immune System. Immunity 35: 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klein L, Kyewski B, Allen PM, and Hogquist KA. 2014. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol 14: 377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zehn D, Lee SY, and Bevan MJ. 2009. Complete but curtailed T-cell response to very low-affinity antigen. Nature 457: 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andreotti AH, Joseph RE, Conley JM, Iwasa J, and Berg LJ. 2018. Multidomain Control Over TEC Kinase Activation State Tunes the T Cell Response. Annu. Rev. Immunol 36: 549–578. [DOI] [PubMed] [Google Scholar]
- 5.Nayar R, Enos M, Prince A, Shin H, Hemmers S, Jiang J-K, Klein U, Thomas CJ, and Berg LJ. 2012. TCR signaling via Tec kinase ITK and interferon regulatory factor 4 (IRF4) regulates CD8+ T-cell differentiation. Proc Natl Acad Sci U.S.A 109: E2794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez-Rodriguez J, Meylan F, Handon R, Hayes ET, Anderson SM, Kirby MR, Siegel RM, and Schwartzberg PL. 2016. Itk is required for Th9 differentiation via TCR-mediated induction of IL-2 and IRF4. Nat Commun. 7: 10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang W, Solouki S, Koylass N, Zheng S-G, and August A. 2017. ITK signalling via the Ras/IRF4 pathway regulates the development and function of Tr1 cells. Nat Commun. 8: 15871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huber M, and Lohoff M. 2014. IRF4 at the crossroads of effector T-cell fate decision. Eur. J. Immunol 44: 1886–1895. [DOI] [PubMed] [Google Scholar]
- 9.Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, Brehm MA, Swain SL, Welsh RM, and Berg LJ. 2014. Graded Levels of IRF4 Regulate CD8+ T Cell Differentiation and Expansion, but Not Attrition, in Response to Acute Virus Infection. J. Immunol 192: 5881–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, Pellegrini M, Belz GT, Smyth GK, Febbraio MA, Nutt SL, and Kallies A. 2013. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol 14: 1155–1165. [DOI] [PubMed] [Google Scholar]
- 11.Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH, and Sun J. 2013. Interferon Regulatory Factor 4 Sustains CD8+ T Cell Expansion and Effector Differentiation. Immunity 39: 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, Wherry EJ, and Haining WN. 2014. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol 15: 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwata A, Durai V, Tussiwand R, Briseño CG, Wu X, Grajales-Reyes GE, Egawa T, Murphy TL, and Murphy KM. 2017. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF–IRF4 transcription factor complex. Nat Immunol 18: 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atherly LO, Brehm MA, Welsh RM, and Berg LJ. 2006. Tec Kinases Itk and Rlk Are Required for CD8+ T Cell Responses to Virus Infection Independent of Their Role in CD4+ T Cell Help. J. Immunol 176: 1571–1581. [DOI] [PubMed] [Google Scholar]
- 15.Nayar R, Schutten E, Jangalwe S, Durost PA, Kenney LL, Conley JM, Daniels K, Brehm MA, Welsh RM, and Berg LJ. 2015. IRF4 Regulates the Ratio of T-Bet to Eomesodermin in CD8+ T Cells Responding to Persistent LCMV Infection. PLoS ONE 10: e0144826–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu KQ, Bunnell SC, Gurniak CB, and Berg LJ. 1998. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J. Exp. Med 187: 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, and Hogquist KA. 2011. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med 208: 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh S, Koralov SB, Stevanovic I, Sundrud MS, Sasaki Y, Rajewsky K, Rao A, and Müller MR. 2010. Hyperactivation of nuclear factor of activated T cells 1 (NFAT1) in T cells attenuates severity of murine autoimmune encephalomyelitis. Proc Natl Acad Sci U.S.A 107: 15169–15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong Y, Dong S, Strattan E, Ren L, Butchar JP, Thornton K, Mishra A, Porcu P, Bradshaw JM, Bisconte A, Owens TD, Verner E, Brameld KA, Funk JO, Hill RJ, Johnson AJ, and Dubovsky JA. 2015. Targeting Interleukin-2-inducible T-cell Kinase (ITK) and Resting Lymphocyte Kinase (RLK) Using a Novel Covalent Inhibitor PRN694. J Biol Chem. 290: 5960–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallagher MP, Conley JM, and Berg LJ. 2018. Peptide Antigen Concentration Modulates Digital NFAT1 Activation in Primary Mouse Naive CD8 +T Cells as Measured by Flow Cytometry of Isolated Cell Nuclei. IH 2: 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, Lamperti ED, Ansel KM, Mempel TR, Lähdesmäki H, Hogan PG, and Rao A. 2015. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 42: 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotech 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, and Carbone FR. 1994. T cell receptor antagonist peptides induce positive selection. Cell 76: 17–27. [DOI] [PubMed] [Google Scholar]
- 24.Rosette C, Werlen G, Daniels MA, Holman PO, Alam SM, Travers PJ, Gascoigne NR, Palmer E, and Jameson SC. 2001. The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity 15: 59–70. [DOI] [PubMed] [Google Scholar]
- 25.Woronicz JD, Calnan B, Ngo V, and Winoto A. 1994. Requirement for the orphan steroid receptor Nur77 in apoptosis of T-cell hybridomas. Nature 367: 277–281. [DOI] [PubMed] [Google Scholar]
- 26.Baldwin TA, and Hogquist KA. 2007. Transcriptional analysis of clonal deletion in vivo. J. Immunol 179: 837–844. [DOI] [PubMed] [Google Scholar]
- 27.Das J, Ho M, Zikherman J, Govern C, Yang M, Weiss A, Chakraborty AK, and Roose JP. 2009. Digital Signaling and Hysteresis Characterize Ras Activation in Lymphoid Cells. Cell 136: 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brownlie RJ, and Zamoyska R. 2013. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol 13: 257–269. [DOI] [PubMed] [Google Scholar]
- 29.Macian F 2005. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 5: 472–484. [DOI] [PubMed] [Google Scholar]
- 30.Paul S, and Schaefer BC. 2013. A new look at T cell receptor signaling to nuclear factor. Trends Immunol. 34: 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donnadieu E, Lang V, Bismuth G, Ellmeier W, Acuto O, Michel F, and Trautmann A. 2001. Differential roles of Lck and Itk in T cell response to antigen recognition revealed by calcium imaging and electron microscopy. J. Immunol 166: 5540–5549. [DOI] [PubMed] [Google Scholar]
- 32.Miller AT, Wilcox HM, Lai Z, and Berg LJ. 2004. Signaling through Itk Promotes T Helper 2 Differentiation via Negative Regulation of T-bet. Immunity 21: 67–80. [DOI] [PubMed] [Google Scholar]
- 33.Dolmetsch RE, Xu K, and Lewis RS. 1998. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392: 933–936. [DOI] [PubMed] [Google Scholar]
- 34.Marangoni F, Murooka TT, Manzo T, Kim EY, Carrizosa E, Elpek NM, and Mempel TR. 2013. The Transcription Factor NFAT Exhibits Signal Memory during Serial T Cell Interactions with Antigen-Presenting Cells. Immunity 38: 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hogan PG, Chen L, Nardone J, and Rao A. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17: 2205–2232. [DOI] [PubMed] [Google Scholar]
- 36.Müller MR, and Rao A. 2010. NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol 10: 645–656. [DOI] [PubMed] [Google Scholar]
- 37.Klein-Hessling S, Muhammad K, Klein M, Pusch T, Rudolf R, Flöter J, Qureischi M, Beilhack A, Vaeth M, Kummerow C, Backes C, Schoppmeyer R, Hahn U, Hoth M, Bopp T, Berberich-Siebelt F, Patra A, Avots A, Müller N, Schulze A, and Serfling E. 2017. NFATc1 controls the cytotoxicity of CD8+ T cells. Nat Commun. 8: 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, Shi W, and Kallies A. 2017. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47: 1129–1141.e5. [DOI] [PubMed] [Google Scholar]
- 39.Dura B, Dougan SK, Barisa M, Hoehl MM, Lo CT, Ploegh HL, and Voldman J. 1AD. Profiling lymphocyte interactions at the single-cell level by microfluidic cell pairing. Nat Commun. 6: 1–13. [DOI] [PubMed] [Google Scholar]
- 40.Schuh K, Twardzik T, Kneitz B, Heyer J, Schimpl A, and Serfling E. 1998. The interleukin 2 receptor alpha chain/CD25 promoter is a target for nuclear factor of activated T cells. J. Exp. Med 188: 1369–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, Shi W, and Kallies A. 2017. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47: 1129–1141.e5. [DOI] [PubMed] [Google Scholar]
- 42.Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, Lamperti ED, Ansel KM, Mempel TR, Lähdesmäki H, Hogan PG, and Rao A. 2015. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 42: 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richard AC, Lun ATL, Lau WWY, Göttgens B, Marioni JC, and Griffiths GM. 2018. T cell cytolytic capacity is independent of initial stimulation strength. Nat Immunol 19: 849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, Kirby MR, Belkaid Y, and Schwartzberg PL. 2014. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J. Exp. Med 211: 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaeffer EM, Yap GS, Lewis CM, Czar MJ, McVicar DW, Cheever AW, Sher A, and Schwartzberg PL. 2001. Mutation of Tec family kinases alters T helper cell differentiation. Nat Immunol 2: 1183–1188. [DOI] [PubMed] [Google Scholar]
- 46.Atherly LO, Lucas JA, Felices M, Yin CC, Reiner SL, and Berg LJ. 2006. The Tec Family Tyrosine Kinases Itk and Rlk Regulate the Development of Conventional CD8+ T Cells. Immunity 25: 79–91. [DOI] [PubMed] [Google Scholar]
- 47.Bachmann MF, Littman DR, and Liao XC. 1997. Antiviral immune responses in Itk-deficient mice. J Virol. 71: 7253–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grumont RJ, and Gerondakis S. 2000. Rel induces interferon regulatory factor 4 (IRF-4) expression in lymphocytes: modulation of interferon-regulated gene expression by rel/nuclear factor kappaB. J. Exp. Med 191: 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.