

Abstract
According to the WHO new renal tumor classification (2016), the clinical and pathologic characteristics, immunophenotype and molecular genetic characteristics of 2 cases of succinate dehydrogenase (SDH)-deficient renal cell carcinoma were retrospectively analyzed, and the relevant literature was reviewed. In 2 cases, there was 1 male and 1 female, the average age was 52.5 years old. The renal tumor average length was 4.2 cm. Tumor cut surface was solid, grayish yellow and soft. The tumor boundary was clear, and the cells were arranged in solid, nested, or small tubular growth. The cytoplasm was vacuolated or contained eosinophilic or light-stained flocculent substance, with a regular nucleus and no obvious nucleoli, showing low-grade nuclei. No atypical mitotic figures or necrosis were found. SDH-deficient renal cell carcinoma has a characteristic morphologic manifestation, and lack of SDHB expression in the immunophenotype. During the clinical diagnosis and treatment, the patient’s condition and family genetic history should be asked for in detail, and genetic detection should be performed to confirm the diagnosis if necessary.
Keywords: Renal tumor, succinate dehydrogenase, immunohistochemistry, genetic detection
Introduction
Succinate dehydrogenase-deficient renal cell carcinoma is a new classification in WHO renal tumors for 2016 [1], that occurs in young people and slightly more in men than in women. The tumor is highly genetically related. Patients often have germline mutations of SDH-related genes, with SDHB mutations the most common, followed by SDHC, SDHA and SDHD more rarely, causing mitochondrial complex II function defects resulting in tumorigenesis. About 30% of patients present with multifocal or bilateral renal tumors. SDH-deficient renal cell carcinoma is a rare renal tumor, with few reports. This article retrospectively analyzes 2 cases of SDHB-deficient renal cell carcinoma and reviews relevant literature. The purpose is to improve clinicians’ and pathologists’ understanding of the clinical and pathological characteristics of such tumors.
Materials and methods
Case selection and histologic review
Two cases of SDHB-deficient renal cell carcinomas were collected from Department of Pathology of the First Affiliated Hospital of Bengbu Medical College from January 2017 to September 2019. Clinical and pathologic data available from the patients’ medical records were reviewed. All available histologic and immunohistologic sections were independently reviewed by two experienced pathologists according to the new 2016 WHO classification of renal tumors.
Pursuant to Research Ethics Committee approval at the First Affiliated Hospital of Bengbu Medical College, follow-up information was obtained by review of medical records or direct communication with patients or their family by telephone.
Clinical information
First case: The patient, a 58-year-old female, was admitted to the hospital in April 2019 for “left renal mass lesion on physical examination about one week”. The patient had no frequent urination, urgency, dysuria, gross hematuria, no waist and abdominal pain or chills and fever. Physical examination showed no abnormalities. Abdominal CT in the other hospital showed that the left renal inferior pole was occupied, renal carcinomas was possible; and there were mixed density nodules in front of the left kidney. The patient had a previous physical examination and had a right thigh mass resection in a hospital in January 2016. Postoperative pathology in the other hospital showed that the mass was cystic and solid, about 5.5 cm × 5.0 cm × 3.5 cm in size, and the contents of the cystic area had been lost. Pathologic diagnosis was poorly differentiated synovial sarcoma (Rhabdomyoid differentiation) of right thigh. Immunohistochemical results: CKP and Vimentin were all positive, LCA, CD3, CD20, and CD79α were all negative, and Ki-67 was about 10%. Two of the five siblings had a history of renal tumors and had renal tumor resection (pathologically suggestive of renal carcinoma), but they had no other tumors. Their parents had no physical examination and the specifics were unknown.
Second case: The patient, a 47-year-old male, was admitted to the hospital in July 2017 for “left waist pain for more than a month”. The patient had no frequent urination, urgency, dysuria, gross hematuria, no chills and fever. Physical examination showed no abnormalities. The enhanced CT of the abdomen in our hospital suggested that the left inferior pole had significant uneven enhancement, and renal carcinomas was possible.
Immunohistochemistry and genetic testing
Specimens were fixed in 10% neutral formalin, embedded in conventional paraffin, 4 μm thick serial sections, stained with H&E, and observed under a light microscope. For immunohistochemical staining, envision two-step method was used, and TBS buffer was used instead of the primary antibody as a blank control. Primary antibodies PAX8, CK (AE1/AE3), EMA, P504s, CD117, CD10, CK8, Vimentin, CK7, CA9, Claudin-7, SDHB, HMB45, TFE-3, ALK-1, and MyoD1 all were purchased from Fuzhou Maixin Biological Technology Development Co., Ltd. The secondary antibody is a mouse and rabbit universal antibody, and DAB was the chromogen. With the first patient’s informed consent, the patient’s wax block was sent to Anhui Boao Medical Genetic Testing Co., Ltd. for complete exon gene sequencing, and then further verified by one-generation sequencing.
Results
Gross
First case: The left nephrectomy specimen was 7.0 cm × 5.0 cm × 4.0 cm in size, and a gray-yellow nodule was observed on the section, the size was 2.5 cm × 2.0 cm × 2.0 cm, and soft texture with a capsule in some areas.
Second case: The left nephrectomy specimen was 8.5 cm × 6.3 cm × 4.7 cm in size, and a brown-yellow nodule was observed on the cut surface; the size was 5.8 cm × 4.3 cm × 3.2 cm, with soft texture with a capsule in some areas.
Microscopic examination
First case: The tumor boundary was clear. The cells were arranged in solid, nest-like, small tubular growths. Cytoplasm was vacuolated, and the cytoplasm contained eosinophilic or light-stained flocculent substances. The nucleus was regular with light staining and inconspicuous nucleoli. Atypical mitotic figuresand necrosis were not seen (Figure 1A). The surgical margin was negative. WHO/ISUP classification: G2, pT1a.
Figure 1.
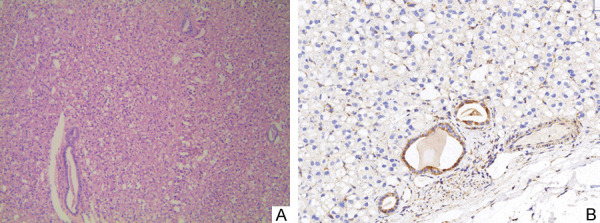
A. Tumor formations are solid, nested, or small tube-shaped. There is abundant cytoplasm, mildly eosinophilic, vacuole-like or flocculent/feathery. Normal renal tubules are engulfed in the stroma (H&E stain × 400). B. Immunohistochemical expression of SDHB in tumor tissues was negative, while engulfed normal renal tubules were positive (magnification, × 400).
Second case: The tumor boundary was clear. The cells were arranged irregularly. Cytoplasm was lightly stained with eosinophilic particles. The nuclei was regular, the staining was fine, and the nucleoli were not obvious, consistent with low-grade (Figure 2A). The surgical margin was negative.
Figure 2.
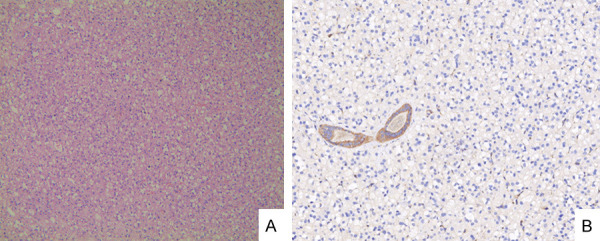
A. Tumor cells are irregularly arranged, and the cytoplasm is vacuolated or mildly eosinophilic (H&E stain × 400). B. Immunohistochemical expression of SDHB in tumor tissues was negative, while engulfed normal renal tubules were positive (magnification, × 400).
Immunohistochemical features
First case: PAX8, CK (AE1/AE3), EMA, P504s CD117, CD10, CK8, and MyoD1 were positive; Vimentin, CK7, CA9, Claudin-7, SDHB (Figure 1B), HMB45, TFE-3, and ALK-1 were all negative.
Second case: PAX8, CK, P504s, CD117, and CD10 were positive. Vimentin, CK7, SDHB (Figure 2B), HMB45, and TFE-3 were all negative.
Genetic testing
For the first patient who had a clear family history of renal tumors, genetic testing of this family lineage was recommended. The whole exon was sequenced and the results indicated that a heterozygous mutation was found in the exon region of the SDHB gene (c.725G > Ap.R242H): c.725G > A (guanine > adenine), which led to amino acid changes p.R242H (arginine > histidine). This mutation site has been reported as a pathogenic mutation [2]. Further application of one-generation sequencing verification, gave results that were consistent (Figure 3).
Figure 3.
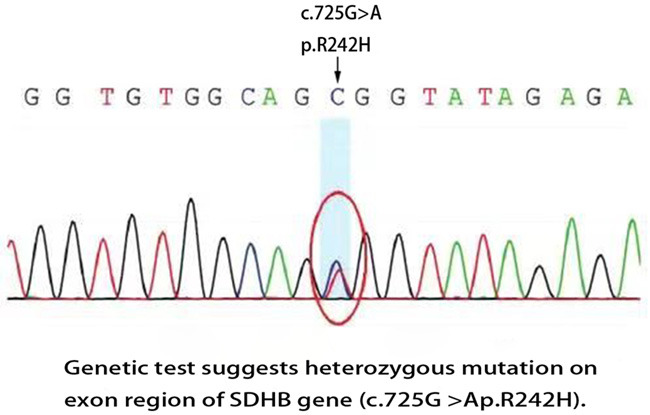
Genetic test suggests heterozygous mutation on exon region of SDHB gene (c.725G > Ap.R242H).
Pathologic diagnosis: Succinate dehydrogenase (SDH)-deficient renal cell carcinoma.
Discussion
Succinate dehydrogenase (SDH) is a mitochondrial enzyme complex composed of four protein subunits: SDHA, SDHB, SDHC and SDHD, and participates in the tricarboxylic acid cycle and mitochondrial electron transport chain transport. It catalyzes the oxidation of succinate to fumaric acid and is also involved in electron transport in the respiratory chain [3-5]. Among them, the genes encoding SDHA [6] (5p15.33), SDHB (1p36.13), SDHC (1q23.3) and SDHD [7] (11q23) are located on different chromosomes, and the deletion of any one subunit will cause these complex to be unstable, causing mitochondrial complex II function deficiency and inducing tumorigenesis. SDHA gene mutation is related to the occurrence of gastrointestinal stromal tumors (GIST); SDHB gene mutation is closely related to abdominal or head and neck paraganglioma and renal carcinoma; SDHC gene mutation is related to head and neck paraganglioma and renal carcinoma; SDHD mutations are associated with tumor formation in head and neck or abdominal paraganglioma, adrenal pheochromocytoma, and gastrointestinal stromal tumor. Studies reported that mutations or abnormal expression of SDH-related genes are closely related to the occurrence of various diseases, that is SDH-deficient tumors, such as pituitary adenoma, pulmonary chondroma, neuroblastoma, renal eosinophilic adenoma, papillary thyroid carcinoma, seminoma, and pancreatic neuroendocrine tumor.
Vanharanta et al. [8] first presented SDH-deficient renal cell carcinoma in 2004. This type of renal cell carcinoma has a rare incidence, often occurs in young adults, and has obvious heredity. Tumors develop unilaterally in most patients. About 30% of patients present with multifocal tumors or bilateral nephrogenesis [9]. Most patients present with germline mutations in SDH-related genes; SDHB mutation is the most common, followed by SDHC, SDHA and SDHD mutations rarely. The clinical features of SDH-deficient renal cell carcinoma are mainly lumbar pain or accidental discovery. There may be a personal or familial clinical history of paraganglioma, or the presence of SDH-deficient GIST [10]. About 1/3 of them may metastasize, only as low-level forms are rare.
Histopathologic manifestations showed clear tumor boundaries, solid red or brown appearance, and varying degrees of polycystic changes, usually without necrosis. Lesions were confined to kidney without invasion of the sinus, vein, or perirenal adipose tissue. Microscopic tumor boundaries were distinct. Renal tubular entrapment was common. Tumor cells are arranged in solid, nested or small tubes, scattered in small sacs, and contain eosinophils, regular nucleation, fine staining, inconspicuous nucleoli, and the characteristic histologic manifestation of cytoplasmic vacuoles plus eosinophilic or light-stained flocculent substances. Under ultrastructure, these inclusions are megamitochondria. In most cases (75%), the nucleus is of low nuclear grade and lacks coagulative necrosis; a few are high nuclear grade and may even be accompanied by sarcoma-like changes. In addition, mast cell infiltration can be seen in the tumor stroma. The immune phenotype of this type of renal carcinoma is characterized by a loss of SDHB expression. When rare renal cell carcinoma with SDHA gene mutation occurs, both SDHA and SDHB immunohistochemical markers are negative. Immunohistochemical interpretation of SDHB expression needs to be compared with the surrounding normal renal tissue, and some cytoplasmic renal cell carcinomas have weaker SDHB staining rather than true negatives, so that SDHB-deficient renal cell carcinoma cannot be diagnosed. However, the diagnostic value of other immunohistochemical markers is limited. For example, only 30% of cases are positive for CK; PAX8 and Ksp-cad are mostly positive, CK7 is negative in most cases, and neuroendocrine markers are negative [11].
SDH-deficient renal cell carcinoma needs to be distinguished from the following types of renal carcinoma: clear cell renal cell carcinoma, chromophobe renal cell carcinoma, oncocytoma/renal eosinophilic adenoma, eosinophil/chromophobe renal cell carcinoma mixed, acquired cystic disease-associated renal cell carcinoma, hereditary leiomyomatosis and renal cell carcinoma, and PTEN hamartoma syndrome. If necessary, DNA can be extracted from tumor tissue or patient blood for genetic testing to further confirm the diagnosis [12].
There is no evidence-based medicine guideline for the treatment of SDH-deficient renal cell carcinoma. Surgical resection is still the preferred treatment. If the tumor is at an early stage or the volume is small, nephron-sparing surgery can be selected, including partial nephrectomy or tumor ablation. For advanced renal cell carcinoma, a comprehensive examination is recommended first to determine the current growth of the tumor. Considering the patient’s condition, surgical resection or molecular targeted drug therapy can be considered [13]. In addition, these patients need to be monitored for cancer in other susceptible organs and followed up regularly. In clinical diagnosis and treatment, for patients with renal tumors, especially young patients should be asked about their condition and family history of tumors in detail. Genetic testing is necessary to confirm the diagnosis, which is helpful to guide clinical treatment decisions and promote eugenics [14]. The first case has the special feature that the patient reported that she had a history of right thigh synovial sarcoma. It is possible that further study of SDH-deficient renal cell carcinoma will disclose new associations.
Acknowledgements
This study was supported by the National Innovation and Entrepreneurship Program for College Students (201810367025) and Natural Science Foundation of Bengbu Medical College (BYKY1822ZD).
Disclosure of conflict of interest
None.
References
- 1.Gill AJ, et al. Succinate dehydrogenase-deficient renal cell carcinoma. In: Moch H, Humphrey PA, Ulbright TM, Reuter V, editors. WHO classification of Tumours of the urinary system and male genital organs. 4th edition. Lyon, France: International Agency for Research on Cancer; 2016. [Google Scholar]
- 2.Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peçzkowska M, Szmigielski C, Eng C Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 3.Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathol. 2012;44:285–292. doi: 10.1097/PAT.0b013e3283539932. [DOI] [PubMed] [Google Scholar]
- 4.Rao Q, Xia QY, Cheng L, Zhou XJ. Molecular genetics and immunohistochemistry characterization of uncommon and recently described renal cell carcinomas. Chin J Cancer Res. 2016;28:29–49. doi: 10.3978/j.issn.1000-9604.2016.01.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paik JY, Toon CW, Benn DE, High H, Hasovitz C, Pavlakis N, Clifton-Bligh RJ, Gill AJ. Renal carcinoma associated with succinate dehydrogenase B mutation:a new and unique subtype of renal carcinoma. J. Clin. Oncol. 2014;32:e10–e13. doi: 10.1200/JCO.2012.47.2647. [DOI] [PubMed] [Google Scholar]
- 6.Yakirevich E, Ali SM, Mega A, McMahon C, Brodsky AS, Ross JF, Allen J, Elvin JA, Safran H, Resnick MB. A novel SDHA deficient renal cell carcinoma revealed by comprehensive genomic profiling. Am J Surg Pathol. 2015;39:858–63. doi: 10.1097/PAS.0000000000000403. [DOI] [PubMed] [Google Scholar]
- 7.Andrews KA, Ascher DB, Pires DEV, Barnes DR, Vialard L, Casey RT, Bradshaw N, Adlard J, Aylwin S, Brennan P, Brewer C, Cole T, Cook JA, Davidson R, Donaldson A, Fryer A, Greenhalgh L, Hodgson SV, Irving R, Lalloo F, McConachie M, McConnell VPM, Morrison PJ, Murday V, Park SM, Simpson HL, Snape K, Stewart S, Tomkins SE, Wallis Y, Izatt L, Goudie D, Lindsay RS, Perry CG, Woodward ER, Antoniou AC, Maher ER. Tumour risks and genotype phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J Med Genet. 2018;55:384–394. doi: 10.1136/jmedgenet-2017-105127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peçzkowska M, Morrison CD, Lehtonen R, Januszewicz A, Järvinen H, Juhola M, Mecklin JP, Pukkala E, Herva R, Kiuru M, Nupponen NN, Aaltonen LA, Neumann HP, Eng C. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. 2004;74:153–159. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72:106–16. doi: 10.1111/his.13277. [DOI] [PubMed] [Google Scholar]
- 10.Gill AJ, Chou A, Vilain R, Clarkson A, Lui M, Jin R, Tobias V, Samra J, Goldstein D, Smith C, Sioson L, Parker N, Smith RC, Sywak M, Sidhu SB, Wyatt JM, Robinson BG, Eckstein RP, Benn DE, Clifton-Bligh RJ. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into two distinct types. Am J Surg Pathol. 2010;34:636–44. doi: 10.1097/PAS.0b013e3181d6150d. [DOI] [PubMed] [Google Scholar]
- 11.Agaimy A. Succinate dehydrogenase (SDH)-deficient renal cell carcinoma. Pathologe. 2016;37:144–152. doi: 10.1007/s00292-016-0158-8. [DOI] [PubMed] [Google Scholar]
- 12.Kim E, Wright MJ, Sioson L, Novos T, Gill AJ, Benn DE, White C, Dwight T, Clifton-Bligh RJ. Utility of the succinate: fumarate ratio for assessing SDH dysfunction in different tumor types. Mol Genet Metab Rep. 2017;10:45–49. doi: 10.1016/j.ymgmr.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuroda N, Yorita K, Nagasaki M, Harada Y, Ohe C, Jeruc J, Raspollini MR, Michal M, Hes O, Amin MB. Review of succinate dehydrogenase-deficient renal cell carcinoma with focus on clinical and pathobiological aspects. Pol J Pathol. 2016;67:3–7. doi: 10.5114/pjp.2016.59227. [DOI] [PubMed] [Google Scholar]
- 14.Saxena N, Maio N, Crooks DR, Ricketts CJ, Yang Y, Wei MH, Fan TW, Lane AN, Sourbier C, Singh A, Killian JK, Meltzer PS, Vocke CD, Rouault TA, Linehan WM. SDHB-deficient cancers: the role of mutations that impair iron sulfur cluster delivery. J Natl Cancer Inst. 2016;108:djv287. doi: 10.1093/jnci/djv287. [DOI] [PMC free article] [PubMed] [Google Scholar]