

Abstract
Ischemic stroke has been considered to be one of the major causes of disability worldwide which related to multiple pathological processes including apoptosis. Metastasis associated lung adenocarcinoma transcript 1 (MALAT1), is one of the long non-coding RNA (lncRNA) know as a regulator for cell apoptosis. However, further and deeper cellular and molecular mechanism in stroke model remains unclear. The results showed MALAT1 was down-regulated in OGD-induced apoptosis and related with miR-205-3p expression. Knockdown of MALAT1 promote OGD-induced apoptosis, decreased the cell viability, inhibit the caspase-3 activation. Moreover, MALAT1 acts as a competing endogenous RNA (ceRNA) for miR-205-3p and further regulate PTEN expression protect OGD-induced apoptosis. Altogether, these results suggest that MALAT1 may suppress the apoptosis in ischemic stroke and function as a ceRNA for miR-205-3p to modulate PTEN expression. These findings may provide a novel therapeutic target for treating ischemic stroke.
Keywords: MALAT1, LncRNA, endothelial cells, ischemic stroke, apoptosis, competing endogenous RNA, miR-205-3p, PTEN
Introduction
Ischemic stroke has been demonstrated to be a devastating event caused by the blockage of a blood vessel(s), which triggers a series of interplayed events and signaling pathways [1,2]. These cascading events may cause dysfunction of neurovascular network and damage of neuronal cells [3]. Although remarkable progress has been achieved in mechanism of cell death in stroke model, there are still lots of puzzles remain unsolved. Meanwhile, the lack of effective treatment for neuronal dysfunction needs to be enrich. Undoubtedly, a better understanding of the pathological process of this disease will contribute to discover new targets for treatment.
Cerebral vascular endothelial cells are considered to be a key component of cerebral vascular structure which undoubtedly play an important role in pathological process of ischemic stroke [4]. One of the major functions of endothelial cells is to maintain the integrity of blood brain barrier (BBB) in physiological conditions [5,6]. However, in pathological condition, stroke-induced apoptosis of endothelial cell may cause increase permeability of cerebrovascular and BBB disruption result in primary and secondary ischemic brain damage [6-8]. Thus, vascular endothelial cells become a novel therapeutic target for treating ischemic stroke.
There are emerging evidences showed that lncRNAs may play a critical role in mediating gene expression through several mechanisms in pathology of ischemic stroke [9-12]. It has been reported that increasing number of LncRNA involved in multiple biological processes in stroke-induced pathological cascade such as cell apoptosis [13], proliferation [14], autophagy [15], differentiation [16], as well as angiogenesis [17]. LncRNA are also able to compete with microRNAs (miRNAs) which name as miRNA sponges by binding to miRNAs, result in down-regulation of target mRNAs [18,19]. So far in the stroke field, a lncRNA named Metastasis associated lung adenocarcinoma transcript 1 (MALAT1) gained much attention. As a highly conserved lncRNA in mammals, MALAT1 has been found massively expressed in numerous tissues including brain tissues. Previous study showed that MALAT1 regulates cerebrovascular function in pathologic process of ischemic stroke by regulating apoptotic factors such as MCP-1, IL-6, and E-selectin [20]. However, there is lack of the evidence of LncRNA and miRNA interaction mechanism.
In our study, we try to address the role of MALAT1 in ischemic stroke by using OGD-induced endothelial apoptosis. The functions of MALAT1 and its down-stream factors, especially the interaction with miRNA were further determined by functional experiments. Our results suggest that MALAT1 may participate in stroke-induced apoptosis, thus could be a novel therapeutic target for treatment of ischemic stroke in the coming years.
Materials and methods
bEnd.3 and HE293 cell culture
The brain microvascular endothelial bEnd.3 cells were purchased from ATCC (USA), bEnd.3 cells were cultured in endothelial basal medium with supplemented antibiotics (1% penicillin-streptomycin) and fetal bovine serum (10%), and 1 ng/mL human basic fibroblast growth factor (Sigma-Aldrich, USA). All cells were maintained in proper atmosphere (95% air and 5% CO2) at 37°C. Cells from passages 3 were used for the further in vitro experiments.
The human embryonic kidney 293T cell line were also obtained from ATCC and cultured in normal DMEM with antibiotics and 10% fetal bovine serum as previously described. The temperature of incubator was set at 37°C and contained 5% CO2. The DMEM was changed every 2 days.
OGD treatment
Oxygen-glucose deprivation (OGD) is a sensitive model of ischemic stroke in vitro. Briefly, the culture medium was changed into glucose-free DMEM in bEnd.3 cells, and then transferred to anaerobic chamber. A mixture gas of 5% CO2 and 95% N2 for 3 hours. Next, after exposed to glucose-free medium and mixture gas, the cells were returned into glucose DMEM for different recovery times. Control groups were treated with normal medium in normal conditions.
Cell transfection and luciferase assay
Cell transfections were conducted based on the manufacturers’ instructions. Lipofectamine 2000 was used as transfection reagent. First, Malat1 cDNA was cloned and amplified into pcDNA3.1 (Invitrogen) and produce pcDNA-Malat1 vector. Malat1 siRNA, miR-205-3p mimic, anti-miR-205-3p or other control factors were designed by Cyagen bioscience co. (Suzhou, China). All vectors were transfected into bEnd.3 cells with Lipofectamine 2000 for 48 hours. After 48 h of transfection, total RNA and protein were extracted from the cells. Then, the samples were restored at -20°C for the future qRT-PCR and western blot detections.
For luciferase assay, the potential binding sites of miR-205-3p on Malat1 mRNA was predicted by using Targetscan and miRanda. Malat1-3’UTR fragment which contains the binding site of miR-205-3p named as pGL3-Malat1-WT, mutated miR-205-3p binding sequences was named as pGL3-Malat1-Mut. Next, 1 × 10^5 HEK 293T cells were used for transfection based on the manufacturer’s instruction.
Endothelial cell apoptosis assay and viability assay
The TUNEL assay was conducted to analyze the apoptosis of bEnd.3 cells. Before formal experiment, the coverslips were planted into the 24-well plates, thus, the bEnd.3 cells were grown on them. Then, the coverslips with cells were incubated with TDT reaction mixture for 2 h at 37°C, after that, the coverslips were transferred into Alexa Fluorr 594 dye for another 30 min. After washing with phosphate buffer saline (PBS), the slides were incubated with blocking buffer. Next, all slides were incubated in TUNEL (Germany, Merck) at 37°C followed manufacturer’s instruction. TUNEL-positive ratio D were calculated. For caspase-3 measurement, caspase-3 assay kit (Sigma, USA) was used following the manufacturer’s instruction.
MTT assay was used to measure cell viability in vitro. Briefly, bEnd.3 cells were seeded into 96-well plates. 20 μl/well of MTT (5 mg/ml) reagent (Sigma, USA) was added to the culture medium. The 96-well plates were incubated for 4 hours before measurement. The absorbance value (OD) of each well was measured at 490 nm using microplate reader (Thermo, USA). We calculated the percentage of cell viability. MTT assays were repeated at least 3 times.
Real-time polymerase chain reaction
Total RNA was extracted from bEnd.3 cells by using RNA isolation kit (Invitrogen, USA). By using RT reagent Kit, RNA was transcribed into cDNA. The primer sequences used in this study were designed by Sigma-Aldrich company. Then, RT-PCR was performed to detect target gene expression on n ABI Prism 7500 fast PCR system. U6 or GAPDH were considered as control. ΔΔCt method was used to analyze the data, relative mRNA expression levels (fold changes) between groups were calculated.
RNA immunoprecipitation (RIP)
Briefly, the different groups of cells were collected and lysed into RIP buffer. Later, cell lysates were incubated with RIP buffer containing magnetic beads conjugated with human anti-Ago2 antibody or negative control IgG (anti-IgG). After immune-precipitated RNA was extracted, the RT-PCR was underwent to measure the purified RNA including expression of MALAT1 and miR-205-3p.
Western blot analysis
Total protein samples from b. End3 cells were extracted. First, the BCA protein assay kit was used to quantify the standard procedure. Then, all samples from different groups were used for western blotting. The primary antibodies used include PTEN (1:500, Abcam, USA), GAPDH (1:2000, Sigma, USA). First, the membrane was incubated with primary antibodies overnight, after washing the membranes, HRP-conjugated secondary antibodies were added and incubated for 45 min. All data were analyzed and quantified with Image-J software.
Statistical analysis
All data were required from repeated experiments. Data were presented as mean ± standard deviation (SD) and analyzed with SPSS 21.0 software. All data were checked if they were normally distributed. The experimental results were analyzed using one-way ANOVA or the Two-tailed student’s t-test. The correlation between Malat1 and miR-205-3p were conducted by Pearson’s correlation. Figures were performed by using Graph Pad Prism 5.1 software. P value ≤ 0.05 was considered statistically significant.
Results
MALAT1 is down-regulated in OGD model and related with miR-205-3p expression
First of all, we would like to confirm the expression of MALAT1 in bEnd.3 OGD model by using RT-PCR. In Figure 1A, we found that the expression of MALAT1 was significantly decreased in OGD condition compared with control group. Moreover, the decreased expression of MALAT1 was shown at time-dependent manner. Secondly, we assessed the expression of miR-205-3p in OGD model as well. The results demonstrated that the expression of miR-205-3p was up-regulated in OGD which showed opposite result as MALAT1 (Figure 1B). Therefore, in order to address the correlation between Malat1 and miR-205-3p, we conducted Pearson’s correlation as shown in Figure 1C, these data suggested that the expression of MALAT1 and miR-205-3p have negative correlation. Since 8 h of OGD shows the peak point of change, we use 8 h OGD for further experiments.
Figure 1.
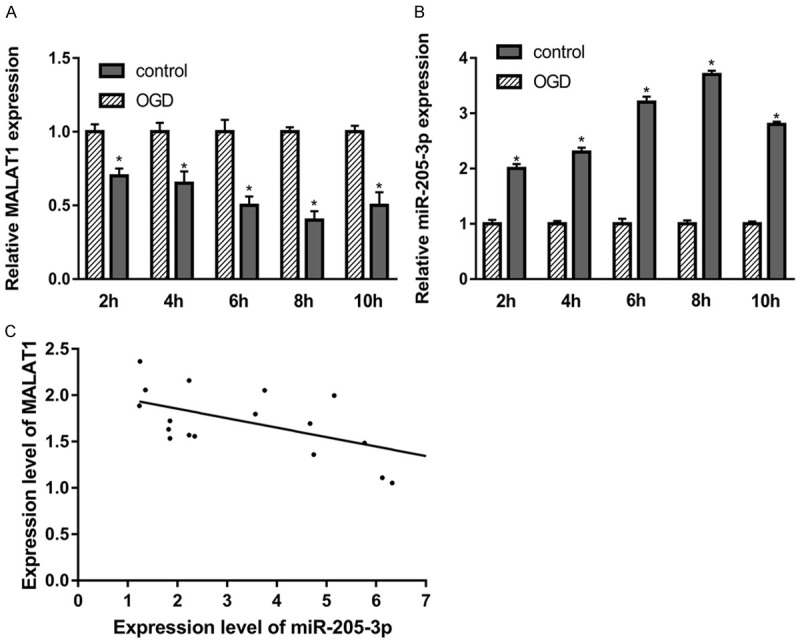
MALAT1 down-regulation is related with miR-205-3p down-regulation in OGD model. Real-time PCR assay for the expression of MALAT1 (A) and miR-205-3p (B) at each time point in OGD at 2 h, 4 h, 6 h, 8 h, and 10 h of recovery. The correlation between Malat1 and miR-205-3p was shown in (C). Data were represented as mean ± SD. *P<0.05, **P<0.01, and ***P<0.001.
Knockdown of MALAT1 promote OGD-induced apoptosis in bEnd.3 cells
In order to determine the specific role of MALAT1 in OGD model, we established si-MALAT1 which could suppress the expression of MALAT1 in bEnd.3 cells. As shown in Figure 2A, by using RT-PCR, the expression of MALAT1 were detected. The result showed that MALAT1 expression was suppressed by si-MALAT1 in both OGD and normal conditions. Moreover, we used MTT assay and TUNEL assay to detect the cell viability and apoptosis. Our data suggested that knockdown of MALAT1 suppress cell viability and increased OGD-induced endothelial cell apoptosis (Figure 2B, 2C). Furthermore, as a major factor in cell apoptosis, the expression of caspase-3 was found increased in MALAT1 knockdown cells (Figure 2D). To sum up, these functional experiments indicated that knockdown of MALAT1 promote OGD-induced apoptosis in bEnd.3 cells.
Figure 2.
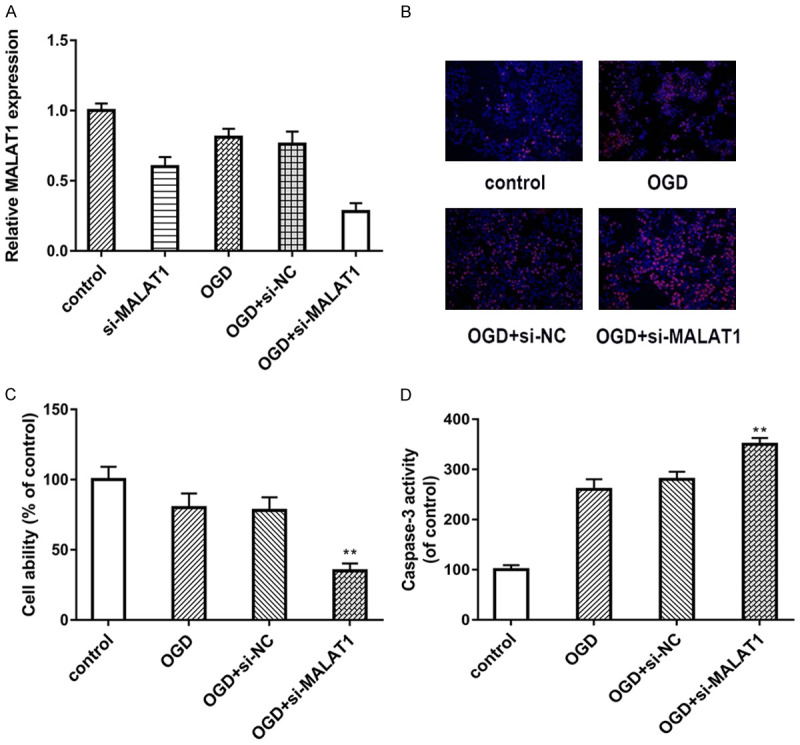
Knockdown of MALAT1 promoted OGD-induced apoptosis. A. Real-time PCR showed the expression of MALAT1 in bEnd.3 cells with different treatment. B. Cell viability was detected by MTT assay in different groups. C. TUNEL was used to assay the apoptosis of bEnd.3 cell. D. Caspase-3 activity was analyzed in bEnd.3 cell in different groups. Data were represented as mean ± SD. *P<0.05, **P<0.01, and ***P<0.001.
MALAT1 functions as a ceRNA for miR-205-3p and negatively regulates its expression
We further try to address the function and potential mechanism of MALAT1 and its potential target miRNAs. By using Starbase (http://starbase.sysu.edu.cn), which known as a database for bioinformatics analysis, we found MALAT1 contains one conserved target site of miR-205-3p, as shown in Figure 3A. Next, we used dual-luciferase assay to confirm the predicted binding site. The data demonstrated that miR-205-3p mimic significantly suppressed luciferase activity of MALAT1-WT compared to the NC-mimic. However, there was no change in MALAT1-MUT group (Figure 3B). To further investigating the relationship between MALAT1 and miR-205-3p, we tested the expression of miR-205-3p in si-MALAT1 model. As shown in Figure 3C, knockdown of MALAT1 remarkably increased the expression of miR-205-3p in OGD model. Finally, we explored whether MALAT1 and miR-205-3p were associated through miRNA ribonucleoprotein complexes. The results showed MALAT1 and miR-205-3p were preferentially enriched in the Ago2-containing miRNPs compared with the control IgG immunoprecipitates (Figure 3D). Taken together, our data suggested that MALAT1 functions as a ceRNA for miR-205-3p.
Figure 3.
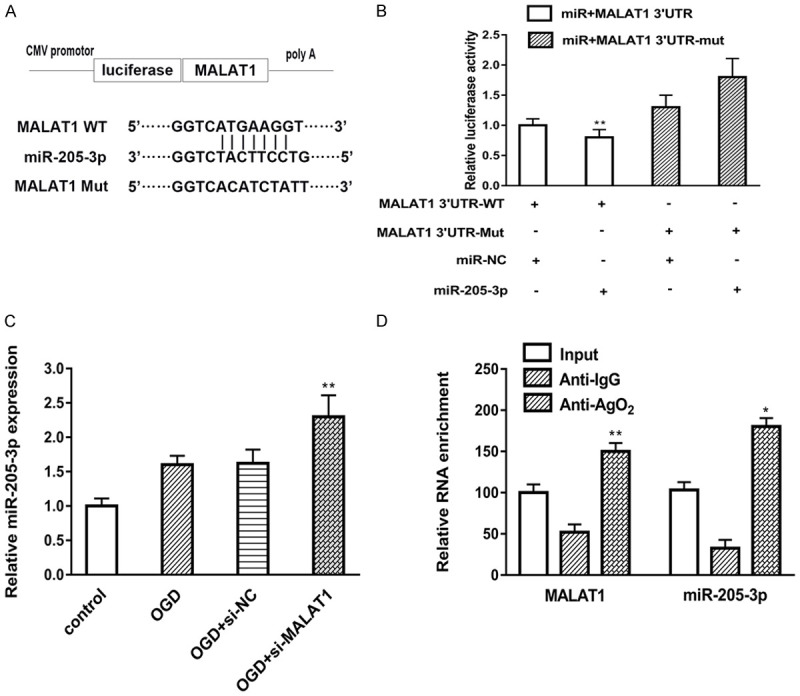
MiR-205-3p confirmed to be a direct target of MALAT1. A. The putative miR-205-3p binding sequence of the wild type and mutation sequence of MALAT1. B. Luciferase activity was detected in different groups. Wide type (MALAT1-WT) and mutant MALAT1 (MALAT1-MUT) were co-transfected with miR-205-3p mimic or control mimic (miR-NC) into bEnd.3 cells. C. The expression of miR-205-3p was detected in different groups by using RT-PCR. D. Cellular lysates from End.3 cells were used for RIP with an Ago2 antibody and IgG antibody. The expression of MALAT1 and miR-205-3p were detected by qRT-PCR. Data were represented as mean ± SD. *P<0.05, **P<0.01, and ***P<0.001.
Inhibition of MALAT1 enhance OGD-induced apoptosis by targeting miR-205-3p
In order to determine whether MALAT1 functions via modulating expression of miR-205-3p, we used miR-205-3p mimic, anti-miR-205-3p, miR-NC, anti-miR-NC and co-transfected these factors with si-MALAT1. Next, we test cell viability and cell apoptosis via MTT assay and TUNEL respectively. As shown in Figure 4A-C, anti-miR-205-3p group showed higher cell survival rate and lower apoptotic rate compared with the si-MALAT1 only group. However, miR-205-3p mimic counteract the effect of si-MALAT1 on both cell survival and apoptosis. Figure 4D showed the caspase-3 activity, which had the similar results. To sum up, these data revealed that inhibition of MALAT1 enhance OGD-induced endothelia apoptosis via up-regulating miR-205-3p expression.
Figure 4.
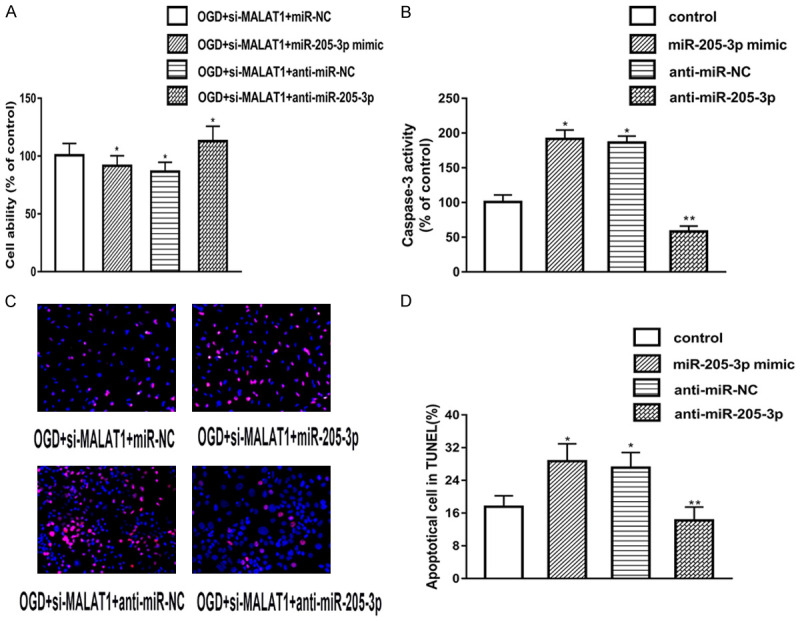
MiR-205-3p regulates OGD-induced apoptosis by targeting PTEN. A. MTT assay was used to detect the cell viability in different groups. B, C. TUNEL was used to detect cell apoptosis in different groups. D. Caspase-3 activity was measured in different groups. Data were represented as mean ± SD. *P<0.05, **P<0.01, and ***P<0.001.
MALAT1 enhances PTEN expression by binding miR-205-3p
According to previous results, PTEN may act as an important role in ischemic stroke, PTEN also confirmed to be active in cerebral ischemic conditions. Up to date, PTEN already confirmed to be a target of miR-205 in different studies [21-24]. Therefore, the correlation between MALAT1 and PTEN is worth to explore. As shown in Figure 5A, MALAT1 and PTEN share the same site of miR-205-3p. Next, we measured the expression of mRNA PTEN in different groups basically showed in Figure 5B. The results demonstrated that PTEN mRNA expression can be suppressed by si-MALAT1 compared with control group. The protein level of PTEN tested by Western blot showed the similar tendency (Figure 5C, 5D). Finally, we would like to investigate the direct relationship with MALAT1 and PTEN in cell death. We used pcDNA3.1-MALAT1 to up-regulate the expression of MALAT1, which confirmed in Figure 5E. Meanwhile, PTEN-siRNA was also used to decrease the expression of PTEN. Cell viability was measured via MTT assay. Data showed that MALAT1 upregulation increase cell viability after OGD, this effect was abolished by PTEN knockdown, which presented in Figure 5F, 5G. Collectively, all these results suggested that MALAT1 enhances PTEN expression by binding miR-205-3p and involved in cell viability.
Figure 5.

MALAT1 enhances PTEN expression by binding miR-205-3p. A. The putative binding sites of PTEN and MALAT1 with miR-205-3p. B. Real-time PCR was used to determine PTEN mRNA expression in different groups. C-E. The protein levels of PTEN in different groups were measured by Western blot. D. MALAT1 expression was detected after transfection with pcDNA3.1-MALAT1 plasmid. E. mRNA expression of PTEN was detected after transfection with si-PTEN. F. Cell viability was detected by MTT in different groups. G. Data were represented as mean ± SD. *P<0.05, **P<0.01, and ***P<0.001.
Discussion
Endothelial cell apoptosis induced by ischemic stroke plays a key role in BBB dysfunction, which causes secondary damage and leading to a poor prognosis in ischemic stroke patients [25]. For the underline mechanisms of apoptosis, the regulation by lncRNA or miRNA are often attributed to targeting key factors or the vital pathways which related to apoptosis. Meanwhile, the research on the interactions between lncRNA and miRNA has become more and more prevalent on stroke field. For instance, lncRNA SNHG14 confirmed to be play a role promoting microglia activation in cerebral infarction via regulating miR-145-5p/PLA2G4A [26]. In another study, the author identified that LncRNA GAS5 acts as a ceRNA for miR-137 to modulate the Notch1 signaling pathway [27]. However, the relationship between lncRNA MALAT1 and miR-205-3p in endothelial cells remain unclear.
MALAT1 is one of the first identified lncRNAs associated with human disease [28], which originally clarified in non-small cell lung cancer by Tano K et al [29]. Recently, emerging evidence indicates that MALAT1 plays a vital role in ischemic stroke by targeting many pathways and factors. In Zhang et al’s study [30], they used microvascular endothelial cells, and further induce OGD to mimic stroke. After 16 h of OGD exposure, they measured the expression levels of 10,677 lncRNAs. Among them, the most highly upregulated lncRNAs include MALAT1, which suggested that MALAT1 significantly changed under pathological condition of ischemic stroke. Furthermore, MALAT1 also confirmed to be involved in multiple bioprocess in cerebral ischemic stroke, as far as we know, MALAT1 mainly regulate neuronal autophagy and endothelial apoptosis. However, the function of MALAT1 is quite controversial, for instance, a study demonstrated that down-regulation of MALAT1 can attenuate neuronal cell apoptosis by targeting miR-30a expression and suppressing Beclin1-dependent autophagy [31], which means, the overexpression of MALAT1 may deteriorate neuronal cell death. Conversely, in another study, MALAT1 play a protecting role in ischemic stroke. The results showed that MALAT1 served as a transcriptional target of KLF4, activation of MALAT1 protect cerebral microvascular endothelial cells from ischemic insult [32]. Li et al’s study [33] showed the similar results, their results identified that MALAT1 act as an endogenous sponge to inhibit miR-26b expression, promoting BMEC autophagy and microvascular endothelial cell survival. In addition, MALAT1 was identified binding Bim and E-selectin both in vitro and in vivo, and regulating MCP-1, IL-6, and E-selectin, which suggested that MALAT1 may have protective effect on ischemic stroke [20]. Furthermore, MALAT1 also confirmed targeting blood-tumor barrier and regulate its permeability [34]. To sum up all these studies of MALAT1 in ischemic stroke, MALAT1 may have a protective effect on endothelial cells, while in neurons, MALAT1 may induce cell death.
In our study, we first identified that in OGD model, which can mimic the ischemic stroke in vitro, the expression of MALAT1 was decreased, conversely, the expression of miR-205-3p was increased compared with control. Furthermore, there was a negatively correlation between MALAT1 and miR-205-3p (Figure 1C). To further investigate the relationship between those two factors, we used bioinformatics analysis and further identified miR-205-3p was a direct target of MALAT1 by using Luciferase assay. MALAT1 served as a ceRNA and was able to down-regulate miR-205-3p’s expression. Moreover, we used a series of gain- and loss-function experiment in order to identify the role of MALAT1 in in vitro stroke model. We found that knockdown of MALAT1 suppressed the cell viability and promote cell apoptosis in OGD condition. The effect of si-MALAT1 can be changed by miR-205-3p mimic and anti-miR-205-3p. Finally, we explored the PTEN, which can be regulated by miR-205-3p, we found that MALAT1 enhances PTEN expression by binding miR-205-3p. Taken together, we story showed that MALAT1 regulates apoptosis in ischemic stroke by sponging miR-205-3p and modulating PTEN expression. MALAT1 can be a protect factor in OGD-induced endothelial injury.
In summary, this study provides the evidence of MALAT1 as a regulator of endothelial cell apoptosis during ischemic stroke. MALAT1 may have anti-apoptotic effect in ischemic stroke and function as a ceRNA for miR-205-3p to modulate PTEN expression. Our study demonstrated a novel lncRNA-miRNA-mRNA regulatory network, may contribute to a better understanding the mechanism of ischemic stroke.
Acknowledgements
This study was supported by The Affiliated Huaian No. 1 People’s Hospital of Nanjing Medical University.
Disclosure of conflict of interest
None.
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2017 update: a report from the american heart association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Ay H, Arsava EM, Andsberg G, Benner T, Brown RD Jr, Chapman SN, Cole JW, Delavaran H, Dichgans M, Engstrom G, Giralt-Steinhauer E, Grewal RP, Gwinn K, Jern C, Jimenez-Conde J, Jood K, Katsnelson M, Kissela B, Kittner SJ, Kleindorfer DO, Labovitz DL, Lanfranconi S, Lee JM, Lehm M, Lemmens R, Levi C, Li L, Lindgren A, Markus HS, McArdle PF, Melander O, Norrving B, Peddareddygari LR, Pedersen A, Pera J, Rannikmae K, Rexrode KM, Rhodes D, Rich SS, Roquer J, Rosand J, Rothwell PM, Rundek T, Sacco RL, Schmidt R, Schurks M, Seiler S, Sharma P, Slowik A, Sudlow C, Thijs V, Woodfield R, Worrall BB, Meschia JF. Pathogenic ischemic stroke phenotypes in the NINDS-stroke genetics network. Stroke. 2014;45:3589–3596. doi: 10.1161/STROKEAHA.114.007362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guell K, Bix GJ. Brain endothelial cell specific integrins and ischemic stroke. Expert Rev Neurother. 2014;14:1287–1292. doi: 10.1586/14737175.2014.964210. [DOI] [PubMed] [Google Scholar]
- 5.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 6.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Ishikawa M, Zhang JH, Nanda A, Granger DN. Inflammatory responses to ischemia and reperfusion in the cerebral microcirculation. Front Biosci. 2004;9:1339–1347. doi: 10.2741/1330. [DOI] [PubMed] [Google Scholar]
- 8.Ochoa-Callejero L, Pozo-Rodrigalvarez A, Martinez-Murillo R, Martinez A. Lack of adrenomedullin in mouse endothelial cells results in defective angiogenesis, enhanced vascular permeability, less metastasis, and more brain damage. Sci Rep. 2016;6:33495. doi: 10.1038/srep33495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dharap A, Pokrzywa C, Vemuganti R. Increased binding of stroke-induced long non-coding RNAs to the transcriptional corepressors Sin3A and coREST. ASN Neuro. 2013;5:283–289. doi: 10.1042/AN20130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saugstad JA. Non-coding RNAs in stroke and neuroprotection. Front Neurol. 2015;6:50. doi: 10.3389/fneur.2015.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dykstra-Aiello C, Jickling GC, Ander BP, Shroff N, Zhan X, Liu D, Hull H, Orantia M, Stamova BS, Sharp FR. Altered expression of long noncoding RNAs in blood after ischemic stroke and proximity to putative stroke risk loci. Stroke. 2016;47:2896–2903. doi: 10.1161/STROKEAHA.116.013869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bao MH, Szeto V, Yang BB, Zhu SZ, Sun HS, Feng ZP. Long non-coding RNAs in ischemic stroke. Cell Death Dis. 2018;9:281. doi: 10.1038/s41419-018-0282-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y, Huang D, Yang F, Tian M, Wang Y, Shen D, Wang Q, Chen Q, Zhang L. Long noncoding RNA highly upregulated in liver cancer regulates the tumor necrosis factor-alpha-induced apoptosis in human vascular endothelial cells. DNA Cell Biol. 2016;35:296–300. doi: 10.1089/dna.2015.3203. [DOI] [PubMed] [Google Scholar]
- 14.Liao B, Chen R, Lin F, Mai A, Chen J, Li H, Xu Z, Dong S. Long noncoding RNA HOTTIP promotes endothelial cell proliferation and migration via activation of the Wnt/beta-catenin pathway. J Cell Biochem. 2018;119:2797–2805. doi: 10.1002/jcb.26448. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Cao B, Han D, Sun M, Feng J. Long non-coding RNA H19 induces cerebral ischemia reperfusion injury via activation of autophagy. Aging Dis. 2017;8:71–84. doi: 10.14336/AD.2016.0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang B, Wang D, Ji TF, Shi L, Yu JL. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-kappaB signaling pathway in a rat model. Oncotarget. 2017;8:17347–17359. doi: 10.18632/oncotarget.14468. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Zhan R, Xu K, Pan J, Xu Q, Xu S, Shen J. Long noncoding RNA MEG3 mediated angiogenesis after cerebral infarction through regulating p53/NOX4 axis. Biochem Biophys Res Commun. 2017;490:700–706. doi: 10.1016/j.bbrc.2017.06.104. [DOI] [PubMed] [Google Scholar]
- 18.Soreq H. Novel roles of non-coding brain RNAs in health and disease. Front Mol Neurosci. 2014;7:55. doi: 10.3389/fnmol.2014.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaur H, Sarmah D, Saraf J, Vats K, Kalia K, Borah A, Yavagal DR, Dave KR, Ghosh Z, Bhattacharya P. Noncoding RNAs in ischemic stroke: time to translate. Ann N Y Acad Sci. 2018;1421:19–36. doi: 10.1111/nyas.13612. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Tang X, Liu K, Hamblin MH, Yin KJ. Long noncoding RNA Malat1 regulates cerebrovascular pathologies in ischemic stroke. J Neurosci. 2017;37:1797–1806. doi: 10.1523/JNEUROSCI.3389-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xin W, Liu X, Ding J, Zhao J, Zhou Y, Wu Q, Hua K. Long non-coding RNA derived miR-205-5p modulates human endometrial cancer by targeting PTEN. Am J Transl Res. 2015;7:2433–2441. [PMC free article] [PubMed] [Google Scholar]
- 22.Lei L, Huang Y, Gong W. miR-205 promotes the growth, metastasis and chemoresistance of NSCLC cells by targeting PTEN. Oncol Rep. 2013;30:2897–2902. doi: 10.3892/or.2013.2755. [DOI] [PubMed] [Google Scholar]
- 23.Qu C, Liang Z, Huang J, Zhao R, Su C, Wang S, Wang X, Zhang R, Lee MH, Yang H. MiR-205 determines the radioresistance of human nasopharyngeal carcinoma by directly targeting PTEN. Cell Cycle. 2012;11:785–796. doi: 10.4161/cc.11.4.19228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bai J, Zhu X, Ma J, Wang W. miR-205 regulates A549 cells proliferation by targeting PTEN. Int J Clin Exp Pathol. 2015;8:1175–1183. [PMC free article] [PubMed] [Google Scholar]
- 25.Shimotake J, Derugin N, Wendland M, Vexler ZS, Ferriero DM. Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke. 2010;41:343–349. doi: 10.1161/STROKEAHA.109.564229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi X, Shao M, Sun H, Shen Y, Meng D, Huo W. Long non-coding RNA SNHG14 promotes microglia activation by regulating miR-145-5p/PLA2G4A in cerebral infarction. Neuroscience. 2017;348:98–106. doi: 10.1016/j.neuroscience.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Chen F, Zhang L, Wang E, Zhang C, Li X. LncRNA GAS5 regulates ischemic stroke as a competing endogenous RNA for miR-137 to regulate the Notch1 signaling pathway. Biochem Biophys Res Commun. 2018;496:184–190. doi: 10.1016/j.bbrc.2018.01.022. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Hamblin MH, Yin KJ. The long noncoding RNA Malat1: its physiological and pathophysiological functions. RNA Biol. 2017;14:1705–1714. doi: 10.1080/15476286.2017.1358347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tano K, Mizuno R, Okada T, Rakwal R, Shibato J, Masuo Y, Ijiri K, Akimitsu N. MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett. 2010;584:4575–4580. doi: 10.1016/j.febslet.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Yuan L, Zhang X, Hamblin MH, Zhu T, Meng F, Li Y, Chen YE, Yin KJ. Altered long non-coding RNA transcriptomic profiles in brain microvascular endothelium after cerebral ischemia. Exp Neurol. 2016;277:162–170. doi: 10.1016/j.expneurol.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo D, Ma J, Yan L, Li T, Li Z, Han X, Shui S. Down-regulation of lncrna MALAT1 attenuates neuronal cell death through suppressing beclin1-dependent autophagy by regulating mir-30a in cerebral ischemic stroke. Cell Physiol Biochem. 2017;43:182–194. doi: 10.1159/000480337. [DOI] [PubMed] [Google Scholar]
- 32.Yang H, Xi X, Zhao B, Su Z, Wang Z. KLF4 protects brain microvascular endothelial cells from ischemic stroke induced apoptosis by transcriptionally activating MALAT1. Biochem Biophys Res Commun. 2018;495:2376–2382. doi: 10.1016/j.bbrc.2017.11.205. [DOI] [PubMed] [Google Scholar]
- 33.Li Z, Li J, Tang N. Long noncoding RNA Malat1 is a potent autophagy inducer protecting brain microvascular endothelial cells against oxygen-glucose deprivation/reoxygenation-induced injury by sponging miR-26b and upregulating ULK2 expression. Neuroscience. 2017;354:1–10. doi: 10.1016/j.neuroscience.2017.04.017. [DOI] [PubMed] [Google Scholar]
- 34.Ma J, Wang P, Yao Y, Liu Y, Li Z, Liu X, Li Z, Zhao X, Xi Z, Teng H, Liu J, Xue Y. Knockdown of long non-coding RNA MALAT1 increases the blood-tumor barrier permeability by up-regulating miR-140. Biochim Biophys Acta. 2016;1859:324–338. doi: 10.1016/j.bbagrm.2015.11.008. [DOI] [PubMed] [Google Scholar]