

Abstract
Background and Aim: Myeloid-derived suppressor cells are a heterogeneous cell population that expand during several pathogenic conditions. However, their role in non-alcoholic steatohepatitis remains unclear. This study aimed to examine the systemic effects of myeloid-derived suppressor cells, to determine the role of Gr-1highLy6G+MDSCs and their correlation with the CXCL12/CXCR4 axis in non-alcoholic steatohepatitis. Methods: We established a non-alcoholic steatohepatitis model and detected inflammatory factors IL-6, PGE2, and INF-γ, using an enzyme-linked immunosorbent assay. Proportions of lymphocyte subsets in peripheral blood, CD11b+Gr-1+myeloid-derived suppressor cells and its subsets in the blood, spleen, liver, and bone marrow were identified using flow cytometry. Adoptive transfer and depletion experiments for MDSCs were performed. Immunohistochemistry, migration assays, and in vivo experiments were used to analyze the role of CXCL12/CXCR4 in non-alcoholic steatohepatitis. Results: The proportion of CD11b+Gr-1+MDSCs changed in the bone marrow, spleen, blood, and liver in the non-alcoholic steatohepatitis model. CD4+ and CD8+ T lymphocytes were significantly reduced in non-alcoholic steatohepatitis. Compared with control mice, a significant decrease in ALT and AST levels was observed in Gr-1highLy6G+MDSCs-treated model mice. The migration ability of AMD3100-treated MDSCs was significantly reduced, but was restored as CXCL12 levels increased. CXCL12 and CXCR4 protein levels increased significantly in the non-alcoholic steatohepatitis livers. Conclusions: Exogenous Gr-1highLy6G+MDSCs improved liver function during non-alcoholic steatohepatitis. The CXCR4/CXCL12 axis could be the key pathway mediating the attraction of myeloid-derived suppressor cells into the non-alcoholic steatohepatitis environment in mice.
Keywords: Non-alcoholic steatohepatitis, myeloid-derived suppressor cells, CXCR4/CXCL12 axis
Introduction
The global prevalence of non-alcoholic fatty liver disease (NAFLD) is 25.24% [1]. The term NAFLD includes a spectrum of hepatic disorders, ranging from simple or bland fatty liver (NAFL, non-alcoholic fatty liver) to non-alcoholic steatohepatitis (NASH) [2]. Therapeutic options for NAFLD and NASH are limited mainly to dietary and lifestyle interventions. Myeloid-derived suppressor cells (MDSCs), a heterogeneous population of myeloid cells that are a subset of innate immune cells, can alter adaptive immunity and produce immune suppression [3]. MDSCs are a prominent leukocyte subpopulation involved not only in tumor-associated immune suppression, but also in regulation of the immune system [4,5]. MDSCs emerge in various bodily compartments, such as the blood, bone marrow (BM) or spleen, especially in cancer, infections or inflammation. Their accumulation in the liver protects against liver injury and dampens T cell-mediated hepatitis [6-8]. To the best of our knowledge, the therapeutic value of MDSCs in the context of NASH has not been studied. We hypothesized that MDSCs could prevent the development of NASH. The molecular mechanisms underlying MDSCs recruitment into the tumor microenvironment and inflammatory sites remain unclear. Chemokine receptors mediate the chemotaxis of cells towards a gradient of chemokines. The migration of leukocytes from the blood into the liver during homeostasis and liver injury is mainly controlled by chemokines. CXCR4 (chemokine C-X-C motif receptor 4) selectively binds the CXC chemokine ligand-12 (CXCL12 or SDF-1), which is important in tumorigenesis, proliferation, metastasis and angiogenesis in cancers [9,10]. CXCR4 expression is low or absent in many healthy tissues, but is highly expressed in various tumors, and is considered the most widely expressed chemokine receptor in cancer [11]. CXCL12 is highly induced in inflamed tissues, where it attracts activated CXCR4+ T cells and monocytes, thereby enhancing local inflammatory responses. CXCL12, or its receptor CXCR4, has been associated with chronic inflammatory disease pathogenesis, such as inflammatory bowel diseases [12]. It would be interesting to determine the biological functions of CXCR4 in MDSCs. CXCR4+ cancer cells can be recruited to CXCL12-rich mesenchymal stroma niches. This recruitment mimics the homing of normal stem cells to the BM [13,14]. However, the role of CXCL12/CXCR4 axis in the migration of MDSCs in NASH remains unclear. In this study, we examined MDSCs distribution and function to elucidate their roles in NASH, analyzed the link between the CXCL12/CXCR4 axis and the migration of MDSCs. Our findings will help to explain the association of MDSCs and CXCL12/CXCR4 with NASH.
Materials and methods
Mice and animal experiments
Male C57BL/6 mice (6-8-weeks-old) were obtained from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). All animal care and handling protocols were approved by the Animal Ethics Committee of Shanxi Medical University. All experiments were carried out in Shanxi Medical University Translational Medicine Center and the department of Otorhinolaryngology Head and Neck Tumor key Laboratory of Shanxi Province. After receiving normal food (NF) for at least 1 week, the mice were divided into three groups: mice fed with CDAA (choline-deficient L-amino acid-defined diet), control CSAA (choline-sufficient amino acid-defined diet) diet, and NF. The CDAA and CSAA diets contain higher calories, fat and carbohydrates than NF [15]. C57BL mice were fed the CDAA diet for 12 weeks to induce NASH. Mice were sacrificed by cervical dislocation.
Biochemical analyses and ELISA
Blood samples were collected from each mouse by removing their eyeball. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglycerides (TG), urea nitrogen (BUN), and blood glucose, total cholesterol (T-CHO), and total bilirubin (TBIL) were measured using an automatic analyzer. The levels of interleukin (IL)-6, prostaglandin E2 (PGE2), and interferon (INF)-γ in serum were measured using commercial mouse ELISA kits (GeneSci. Co. Ltd, Beijing, China).
Immunohistochemical and western blot analyses
Immunohistochemical (IHC) analyses of CXCR4 and CXCL12 (Abcam, USA) were performed using paraffin-embedded sections. To evaluate IHC staining, the scan scope digital scanning system (Aperio company, USA), Nuclear v9, and Cytoplasmic v2 software were used. For western blotting, primary antibodies against CXCR4 (1:1000), CXCL12 (1:1000) and glyceraldehyde-3-phosphate (GAPDH; 1:5000) were incubated with membranes containing separated proteins at 4°C overnight. After incubation with the corresponding peroxidase-conjugated secondary antibodies (1:5000), protein bands were detected using chemiluminescence. Data were analyzed using the Image J software.
Flow cytometry analysis
After single cell suspensions were prepared, red blood cells (RBCs) were lysed using 1 ×RBC Lysis Buffer (eBioscience, San Diego, CA, USA). Cells were stained with antibodies against the following mouse proteins: CD11b (Clone M1/70), Gr-1 (RB6-8C5), Ly6G (1A8), Ly6C (HK1.4) CD3 (145-2C11), CD4 (RM4-5), CD8 (53-6.7), and CD184 (CXCR4) (2B11), and with Rat IgG2bκ isotype control-fluorescein isothiocyanate (FITC) and Rat IgG2bκ isotype control-PE (eBioscience). Flow cytometry was performed on a BD FACS Calibur instrument (BD Biosciences, San Diego, CA, USA). Data were analyzed using Cellquest software.
Magnetic bead purification
Positive selection of MDSCs was performed using MicroBeads and MACS MS columns (MiltenyiBiotec) according to the manufacturer’s protocol. The purity of the selected cells was verified by flow cytometry. The purity of the isolated population was 95.12% for Gr-1+CD11b+ cells and 78% for CD11b+Gr-1highLy6G+MDSCs.
In vivo experiments
(1) Adoptive transfer of MDSCs: CD11b+Gr-1highLy6G+MDSCs were purified from the BM of normal donor mice using magnetic beads. Recipient mice were injected via the tail vein with 1×106 cells in 500 μL of saline. The same amount of PBS was used as a control. After 72 h, the recipient mice were sacrificed.
(2) MDSCs depletion: Mice were injected intraperitoneally with a single dose of 120 mg/kg of gemcitabine (Selleck) diluted in saline. Control mice received saline alone [16]. After 4 days, the mice were sacrificed.
(3) AMD3100 (Sigma), a specific CXCR4 antagonist, was injected intraperitoneally into NASH model mice for 3 consecutive days. AMD3100 was dissolved in normal saline at 100 μg/mL. The dose of AMD3100 was 1 mg/kg/day, which is sufficient to block CXCR4 without causing stem cell mobilization [17]. The same amount of normal saline was used in the control group.
In vitro MDSCs migration assay
In vitro cell invasion assays were performed using 24-well multiplates with 8-μm pore and 5-μm pore transwell filter chamber inserts (Corning, USA). Isolated Gr-1highLy6G+ cells (1×105/well) were added to the upper compartment of each transwell. An aliquot of 150 μL of medium containing 10, 100, or 200 ng/mL murine recombinant chemokine ligand 2 (CXCL12) (Peprotech, New Jersey, USA) was added to the feeder tray. The kit was incubated at 37°C in a 5% CO2 cell culture incubator. After 48 h, cells that had migrated were stained using crystal violet, counted, and photographed under a light microscope. All experiments were repeated at least three times. After dissolving the crystals using 33% acetic acid on ice, the OD values were recorded using a spectrophotometer.
Statistical analysis
Values are presented as the mean ± SD. Comparisons between groups were made using two-way analysis of variance (ANOVA) or unpaired Student’s t tests. P < 0.05 was considered as statistically significant. Pearson correlation was used to explore the relationship between the level of ALT, AST, and related parameters.
Results
Consuming the CDAA diet for 12 weeks induced NASH in mice
Histological analysis showed that the mice fed the CDAA diet had higher levels of steatosis (90% compared with 30-60% in the control CSAA livers). Moderate inflammation occurred in the livers of mice fed the CDAA diet. Histological scoring of NASH was determined as described previously [18]. Mice fed the control CSAA diet scored 1-3 points, whereas CDAA diet group exhibited higher NAS (5 points) (Figure 1A). Mice fed the CDAA diet showed significant hepatic steatosis based on Oil red O staining (Figure 1B) and hepatic fibrosis was shown by Sirius Red staining (Figure 1C). Body weights increased similarly in the NF, CDAA, and CSAA diet groups (Figure 1D). The serum levels of ALT and AST were significantly higher in the CDAA diet group compared with the control group (P < 0.05) (Figure 1E). There was no significant difference in BUN, TG, T-CHO, and TBIL among the groups (P > 0.05) (Table 1). Next, we measured inflammatory cytokines in the blood to examine systemic cytokine release. Importantly, their basal expression was very low; however, NASH induced significant elevation of IL-6, INF-γ, and PGE2 compared with that in NF mice (P < 0.05, Figure 1F). Elevated levels of IL-6, INF-γ, and PGE2 are reliable indicators of low-grade chronic inflammation in NASH in vivo.
Figure 1.
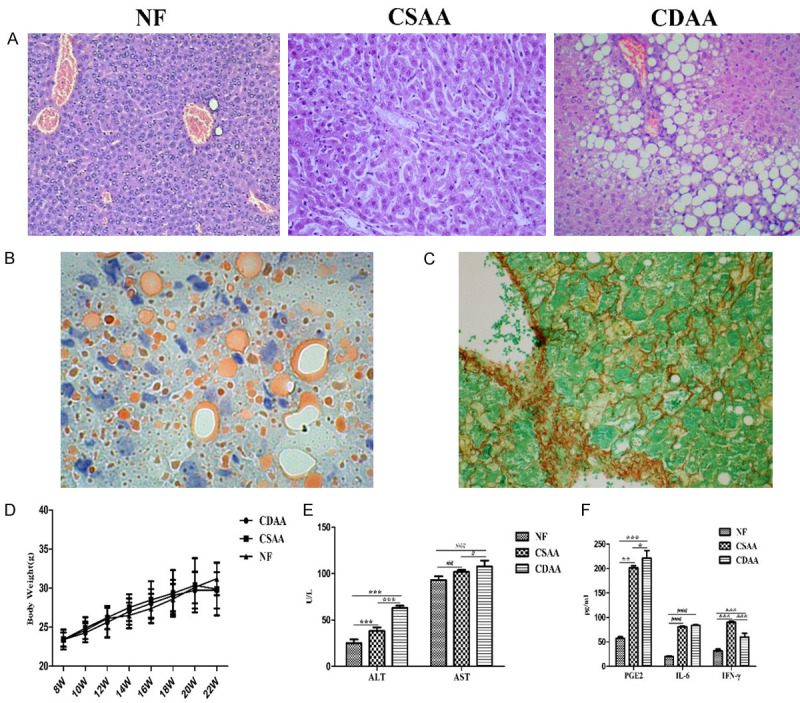
Representative liver histopathology, body weight change and serological test results in C57BL/6J mice fed NF, CSAA or CDAA diet for 12 weeks. A. NASH liver sections were stained with hematoxylin and eosin (HE, 100×). B. Representative image of oil red o staining of liver tissues was shown (400×). C. Hepatic fibrosis was evaluated based on Sirius Red (400×). D. Changes in body weight of mice fed the NF, CSAA or CDAA diet. E. Serum biochemical markers in mice fed for 12 weeks. ALT, AST levels increased in NASH model. F. Cytokine secretion profiles of NASH models. Serum samples from NE, CSAA and CDAA mice were analyzed for IL-6, INF-γ and PGE2. Data are expressed as means ± SD, by one-way ANOVA. ALT, PGE2 * P < 0.05, ** P < 0.01, *** P < 0.001: AST, IL-6 # P < 0.05, ## P < 0.01, ### P < 0.001; INF-γ & P < 0.05, && P < 0.01, &&& P < 0.001.
Table 1.
Serum biochemical markers in NASH mice
| NF (n = 6) | CSAA diet (n = 6) | CDAA diet (n = 6) | F | P-value | |
|---|---|---|---|---|---|
| ALT (U/L)* | 25.33 ± 3.78 | 38.17 ± 4.07 | 63.17 ± 2.32 | 184.065 | < 0.001 |
| AST (U/L)# | 93 ± 4.00 | 101.83 ± 2.14 | 107.50 ± 6.53 | 15.192 | < 0.001 |
| BUN (mmol/L) | 5.51 ± 0.94 | 6.14 ± 0.47 | 5.65 ± 1.35 | 0.649 | > 0.05 |
| Glucose (mmol/L) | 10.17 ± 0.49 | 10.86 ± 0.45 | 16.12 ± 1.16 | 106.906 | < 0.001 |
| TG (mmol/L) | 1.41 ± 0.37 | 1.05 ± 0.38 | 1.02 ± 0.15 | 2.787 | > 0.05 |
| T-CHO (mmol/L) | 2.55 ± 0.40 | 2.91 ± 0.82 | 3.25 ± 0.44 | 2.138 | > 0.05 |
| TBIL (vmol/L) | 5.98 ± 1.29 | 4.66 ± 1.40 | 5.17 ± 1.18 | 1.605 | > 0.05 |
Values are means ± SD. P-values reflect ANOVA.
P< 0.001 CDAA diet VS. NF.
P< 0.001 CDAA diet VS. NF.
Changes in T cell subsets in NASH
Combining the lymphocyte counts with the flow cytometric analysis indicated that the proportions of CD4+ and CD8+ T lymphocytes in the blood of CSAA and CDAA mice were significantly reduced, respectively, compared with that in NF mice. The proportions of peripheral blood lymphocyte subsets are shown in Figure 2.
Figure 2.

Numbers of T cell subsets in the blood. (A-D) Numbers of peripheral blood T lymphocyte subsets in the isotypi control (A) NF (B), CSAA (C) and CDAA (D) groups. These data are from a single experiment which is representative of at least three independent experiments. (E) The changes in peripheral blood T cell subgroup. Numbers of peripheral blood *CD4+ and *CD8+ T lymphocyte subsets decreased significantly in the CSAA and CDAA micc. CD4+ (* P < 0.05, ** P < 0.01, *** P < 0.001 vs. the NF group); CD8+ (# P < 0.05, ## P < 0.01, ### P < 0.001 vs. NF group).
CD11b+Gr-1+MDSCs changed in peripheral tissues of NASH mice
The numbers of liver CD11b+Gr-1+MDSCs changed compared with those in normal mice. The number of MDSCs increased significantly in the BM compared with that in the NF group (P < 0.05). The numbers of MDSCs in liver were significantly lower in the CDAA or CSAA group than in the NF group (P < 0.001). The number of MDSCs decreased in the blood of the NASH group. However, no significant differences were observed in the number of MDSCs in the spleen and blood between these groups (Figure 3).
Figure 3.

Representative images of flow cytometric staining of Gr-1+CD11b+MDSCs in the peripheral blood, bone marrow (BM), spleen, and liver from CDAA, CSAA and NF food mice. A. The dot plots showing CD11b+Gr-1+MDSCs. These data are from a single experiment, which is representative of at least three independent experiments. B. Mouse IgG2bκ antibodies were used as isotype controls. C. Numbers of Gr-1+CD11b+MDSCs in peripheral tissues (n = 5). *BM (* P < 0.05, ** P < 0.01, *** P < 0.001 vs. the NF group); #liver (# P < 0.05, ## P < 0.01, ### P < 0.001 vs. the NF group).
MDSCs polarization links their phenotypic and functional changes to disease development [19], for example, the granulocytic CD11b+Gr-1highLy6G+ population and the monocytic CD11b+Gr-1dimLy6G- population. Representative results of three independent experiments are shown in Figure 4. We found that Gr-1highLy6G+MDSCs were significantly expanded in the BM upon NASH (P < 0.001, Figure 4A, 4D). The percentage of Gr-1dimLy6G-MDSCs was decreased in NASH mice compared with that in normal mice. A similar expansion of Gr-1highLy6G+MDSCs was observed in the spleen of a NASH mouse model (P < 0.001, Figure 4B, 4E). However, there was no obvious change inGr-1highLy6G+MDSCs in the blood (P > 0.05 Figure 4C, 4F).
Figure 4.

Change in MDSCs subsets in NASH. (A) The dot plots showing MDS Cs subsets in the BM. (B) Dot plots showing MDSCs subsets in the spleen. (C) Dot plots showing MDSCs subsets in blood. The number of CD11b+Gr-1highLy6G+MDSCs increased significantly in the BM (D) and spleen (E) of NASH compared with that in normal mice. *BM (* P < 0.05, ** P < 0.01, *** P < 0.001 vs. the NF group); #spleen (# P < 0.05, ## P < 0.01, ### P < 0.001 vs. the NF group ). (F) CD11b+Gr-1highLy6G+MDSCs in blood showed no statistically significant difference compared with those in the NF group.
Correlation analysis
Although the numbers of CD4+ T cells did not correlate with ALT and AST levels, the numbers of CD8+ T cells correlated negatively with ALT (r = -0.811; P = 0.004), and AST (r = -0.840; P = 0.002) in mice (Figure 5A, 5B). The number of MDSCs in the BM correlated negatively with numbers of CD8+ T cells (r = -0.701; P = 0.024, Figure 5C). The results indicated that immunosuppression of T cell immunity during NASH involves MDSCs expansion.
Figure 5.

The number of CD8+ T cells correlates with ALT and AST levels and MDSCs of the bone marrow (BM) in NASH. A, B. CD8+ T cells correlated negatively with ALT and AST levels (r = -0811, P < 0.01: r = -0840, P < 0.01). C. CD8+ T cells correlated negatively with MDSCs numbers in the BM (r = 0.701, P < 00.05). Correlation was assessed by Pearson’s correlation coefficient.
Gr-1highLy6G+MDSCs treatment ameliorated NASH in mice
The purity of MACS-sorted cells was assessed using flow cytometry; The purified Gr-1+CD11b+MDSCs were over 95% pure, and the Gr-1highLy6G+MDSCs achieved 78% purity (Figure 6A). ALT and AST levels were determined in the serum of adoptively transferred and control mice at day 3 after cell transfer. Compared with those in control mice, AST and ALT levels were significantly decreased (P * = 0.01 and P # = 0.048 respectively) in the Gr-1highLy6G+MDSCs-treated mice (Figure 6C). After the Gr-1highLy6G+MDSCs intervention, the serum PGE2 level increased (P * = 0.021), the INF-γ level decreased (P # = 0.004), while there was no significant change in the IL-6 level, compared with that in the control mice (Figure 6D). Histological examination showed that hepatic inflammatory degree was reduced (Figure 6E). These results demonstrated that exogenous Gr-1highLy6G+MDSCs could improve liver function during NASH.
Figure 6.
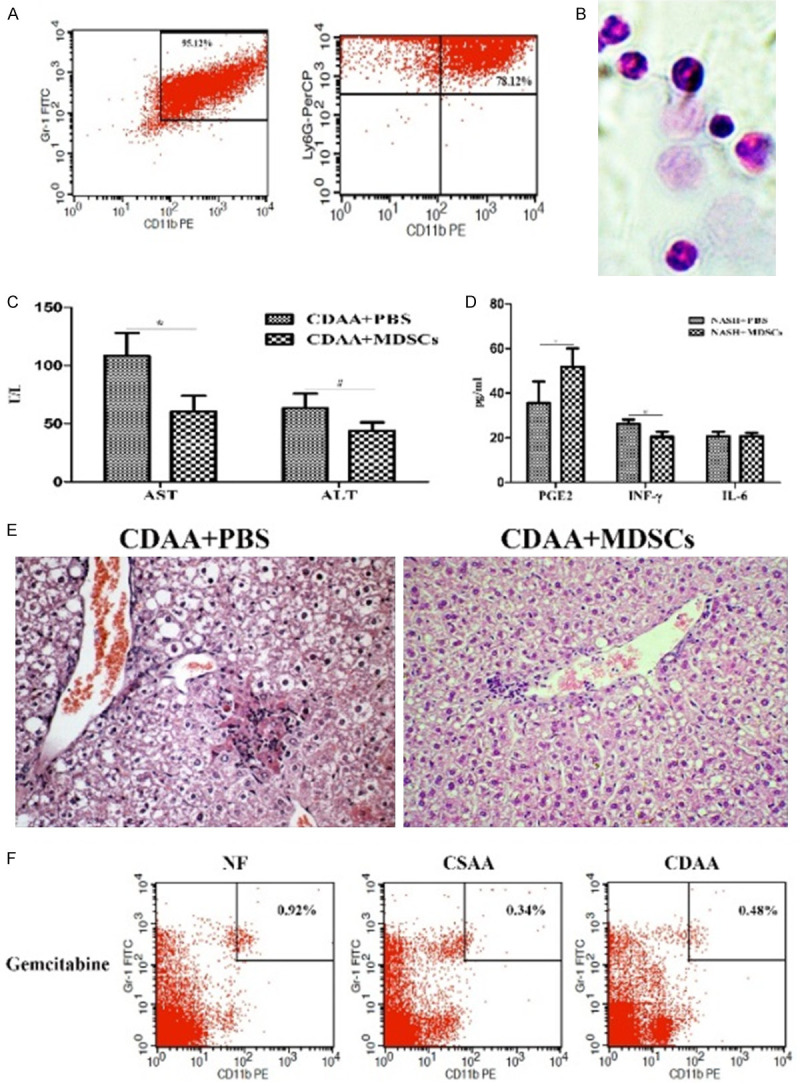
Adoptively transferred CD11b+Gr-1highLy6G+MDSCs alleviate the liver lesions in NASH. A. Flow cytometric analysis of CD11b+Gr-1highLy6G+MDSCs purified using the MDSC-specific magnetic beads, which were subsequently used in the adoptive transfer experiments. Note that over 95.12% total cells were Gr-1+CD11b+MDSCs, among which about 78% cells were CD11b+Gr-1highLy6G+MDSCs. B. Morphology of purified CD11b+Gr-1highLy6G+MDSCs by HE staining (400×). C. Analysis of the levels of ALT and AST after adoptive transfer. *AST (* P < 0.05, ** P < 0.01, *** P < 0.001 vs. the normal food (NF) group); #ALT (# P < 0.05, ## P < 0.01, ### P < 0.001 vs. the NF group). D. Changes in the levels of PGE2, INF-γ and IL-6 in the serum after CD11b+Gr-1highLy6G+MDSCs intervention. *PGE2 (* P < 0.05, ** P < 0.01, *** P < 0.001 vs. PBS group); #INF-γ (# P < 0.05, ## P < 0.01, ### P < 0.001 vs. the PBS group). E. Histological examination showed a slight reduction in the degree of NASH lesions (HE, 100×). F. Depletiontif MDSCs in vivo by gemcitabine markedly reduced the MDSCs population in blood of NASH mice. Data are representative of five mice in each cohort from three independent expriments. Data are expressed as the mean ± SD.
An alternative approach to demonstrate the function of MDSCs in the NASH in vivo was MDSCs depletion, which was achieved using by gemcitabine [16]. The numbers of CD4+ T cells and CD8+ T cells were unchanged after gemcitabine treatment in all models (data not shown). Depletion of MDSCs did not affect the level of transaminase (P > 0.05, data not show).
CD11b+Gr-1highLy-6G+MDSCs might be recruited to the NASH liver via the CXCR4/CXCL12 pathway
Multicolor fluorescence-activated cell sorting (FACS) analysis showed that the BM, liver, spleen, and blood from NASH mice contained significantly higher percentages of CXCR4+MDSCs compared with those from normal subjects (P < 0.001, Figure 7A, 7B). IHC staining of livers from NASH mice showed extensive positivity for CXCL12 and CXCR4. In normal liver, CXCR4 was largely restricted to sinus endothelial cells and stellate cells. In NASH liver, the CXCR4 protein level was increased, and was observed in infiltrating single cells (Figure 7C-E). Western blotting results also showed the same trend (Figure 7F).
Figure 7.

CXCR4 expression of MDSCs and the expression of CXCR4 and CXCL12 in liver tissue. (A) MFI (mean fluorescence intensity) of CXCR4 on MDSCs in blood, BM, spleen and liver. (B) Diagram of the MFI analysis. (*BM, * P < 0.05, ** P < 0.01, *** P < 0.001 vs. the NF group; #spleen, # P < 0.05, ## P < 0.01, ### P < 0.001 vs. the NE group; &liver, & P < 0.05, && P < 0.01, &&& P < 0.001 vs. the NF group). (C) IHC staining of CXCL12 in liver tissues of the NF, CSAA and CDAA group. (D) IHC staining of CXCR4 in liver tissues of the NF, CSAA, and CDAA groups. Magnification for (C, D) 200×. (E) Image analysis results showing the expression of CXCR4 and CXCL12 in liver tissue. (F) The protein levels of CXCL12 and CXCR4 in the livers were investigated by western blotting (n = 5). GAPDH was used as a loading control. All data are expressed as the mean ± SD and are representative of three independent experiments.
To further investigate the role of the CXCL12/CXCR4 axis in MDSCs migration, an MDSCs chemotaxis assay was performed. MDSCs migrated in response to CXCL12 in a dose-dependent manner. The number of migrated cells decreased when the cells were pre-incubated with AMD3100 (Figure 8A-D). This can be explained by the fact that intraperitoneal injection of AMD3100 (1 mg/kg/day) for 3 consecutive days adjusted MDSCs distribution in NASH by promoting BM MDSCs release, which increased the number of MDSCs in peripheral blood (Figure 8E-H).
Figure 8.

The CXCL12/CXCR4 pathway mediates the migration of MDSCs to the NASH liver. A. Migration assays revealed that CD11b+Gr-1highLy6G+MDSCs migrated in response to CXCL12 in vitro. B. The abrogation of CXCR4 inhibits the invasion and migration ability of MDSCs. C. The number of migrating MDSCs increased as the CXCL12 concentration increased. D. The diagram of the count analysis of the AMD3100 (+) group compared with the AMD3100 (-) group, ***P < 0.001. E. The result of 3 consecutive days of abdominal injection of AMD3100. Flow cytometry results showing that the numbers of bone marrow MDSCs from mice treated with AMD3100 were significantly lower than those of the control. F. A histogram showing that the number of MDSCs from the BM in the medication group was significantly lower than that in the control group. G. After administering AMD3100, the numbers of peripheral blood MDSCs were obviously higher compared with those of the control group (n = 5). H. Histogram showing that the numbers of peripheral blood MDSCs in the medication group were significantly higher than those in the control group.
Discussion
NAFLD causes a selective loss of intrahepatic CD4+ T cell and promotes hepatic arcinogenesis [20]. Notably, we found a remarkable decrease in the proportions of CD4+CD3+ and CD8+CD3+ T cells in mice with NASH (Figure 2). The number of CD8+ T cells was negatively correlated with the levels of ALT and AST. The hallmark of MDSCs is their immune suppressive function. The percentages of BM MDSCs in NASH and CD8+ T cells were negatively correlated. Thus, circulating T-lymphocyte subsets showed significant and consistent changes in their numbers during NASH.
MDSCs have been linked to a wide range of inflammation-associated pathological processes. Elevated serum concentrations of the proinflammatory cytokines IFN-γ and TNF-α detected in chronically inflamed mice correlated with extensive recruitment and expansion of MDSCs [21]. Proinflammatory cytokines such as IL-6 and PGE2 also mediate MDSCs accumulation at tumor sites, displaying characteristics of a chronic inflammatory environment [22-24]. Our study showed during NASH, the expressions of IFN-γ, PGE2 and IL-6 were upregulated (Figure 1F). The accumulation of MDSCs in response to chronic inflammation, coupled with their immunomodulatory activities in cancer, led us to hypothesize that the low-grade inflammation present during NASH might be associated with changes in MDSCs that function to maintain immune homeostasis.
The liver is a primary site of MDSCs in vivo, and modulating MDSCs functionality might represent a promising therapeutic target for liver diseases. BM-derived MDSCs can ameliorate hepatofibrogenesis [25]. MDSCs have been studied in the context of acute liver inflammation and are usually associated with protective functions in this setting [26,27]. Depletion of liver MDSCs enhanced fibrosis markers [28], suggesting a protective role for MDSCs in liver fibrosis. Mice bearing liver tumors showed increased numbers of MDSCs in the liver, spleen, and BM [29-31]. Thus, MDSCs play an important role in inflammation, fibrosis, and liver cancer, but have different roles at different stages.
However, the therapeutic value of MDSCs in the context of NASH has not been previously studied. We identified that Gr-1+CD11b+MDSCs are enriched and accumulated in peripheral tissues, including both BM and spleen, during NASH (Figure 3). The percentages of BM MDSCs in NASH mice were significantly higher than those in the CSAA and NF groups. Moreover, among MDSCs subsets, CD11b+Gr1highLy6G+MDSCs were the predominant subset. Taken together, our data showed that MDSCs are enriched in the BM and spleen during NASH. However, mobilization of MDSCs into the blood circulation and liver lesion did not increase, suggesting that NASH involves an immune imbalance.
The observation that adoptively transferred MDSCs preferentially home to the liver [24] favors this approach, allowing directed delivery of MDSCs to the site of inflammation. Exogenous addition of purified BM Gr-1high Ly6G+MDSCs induced decreased levels of ALT and AST and contributed to ameliorating NASH. Recently, an anti-Gr-1 antibody failed to deplete MDSCs in the liver [32]. We observed that MDSCs depletion using gemcitabine alone did not affect ALT and AST in NASH mice. How can we explain the discrepancy between the gain-of-function experiment and the loss-of-function experiment? One possible explanation is that there are fewer MDSCs in the blood of NASH environment. Application of gemcitabine cleared the endogenous MDSCs, which could not play a role in the short term, while the added exogenous MDSCs could directly suppress the immune function. Indeed, cytokines might also cause hepatocyte death and liver injury directly without harnessing immune cells as effector cells. We cannot exclude the possibility that other immune cells are involved when Ly6G+MDSCs were injected.
The transfer experiments in conjunction with the depletion experiments established a direct link between CD11b+Gr-1highLy6G+MDSCs and NASH, emphasizing the significance of supplementing these cells in restoring liver function, and preventing excessive liver damage. Therefore, enhancing CD11b+Gr-1highLy6G+MDSCs numbers in vivo might be used to treat mice with NASH.
The mechanisms that regulate the essential step of migration and retention of MDSCs in the liver during NASH are unknown. The CCL2/CCR2 chemokine axis plays a pivotal role in the migration of MDSCs in cancer, and impairment of CCL2/CCR2 signaling inhibits tumor growth [33]. CXCL12 is locally increased within the BM following total body irradiation or chemotherapy, suggesting that it also serves as a stress signal for circulating cells, recruiting them to the damaged tissue. Given the fact that CXCR4 receptors are expressed on MDSCs and CXCL12 is expressed in liver tissue, it was not surprising that the expression of CXCR4 in MDSCs significantly increased as NASH progressed (Figure 7). CXCL12 was significantly upregulated in liver of NASH mice, which might contribute to stimulating the increase in the numbers of CD11b+Gr1highLy6G+MDSCs in the BM. This would result in increased influx of MDSCs into the NASH liver. This was further confirmed by the migration of Ly6G+MDSCs into the CXCL12 conditions was significantly blocked by AMD3100 in transwell study (Figure 8A-D). CXCL12 is upregulated in NASH liver tissues, and a local increase in the expression of this chemokine, together with specific local or circulating stress signals, enhances the motility and response sensitivity of CXCR4+MDSCs to CXCL12 signaling. Therefore, we hypothesized that CXCL12/CXCR4 interactions are implicated in NASH in vivo and thus, we sought to identify the mechanisms that mediate the in vivo migration of MDSCs into the liver during NASH. Indeed, inhibition of CXCR4 exerted anti-inflammatory effects in experimental models of joint inflammation, colitis, and allergic lung inflammation. AMD3100 (also termed plerixafor) inhibits CXCL12-mediated migration in vitro by blocking CXCL12 binding to its major receptor CXCR4. AMD3100 rapidly mobilizes immature progenitor cells from the BM into the blood in mice. We observed a significant increase in Gr-1+CD11b+MDSCs in the blood and a significant decrease in Gr-1+CD11b+MDSCs in the BM of these mice after AMD3100 treatment (Figure 8E-H), indicating that AMD3100 can mobilize MDSCs in vivo. However, systemic administration of AMD3100 had no obvious effect on NASH. These results are consistent with previous studies on murine hepatic injury. It is difficult to explain these results. Other cells show CXCR4 expression, such as CD4+ T cells. The effect of AMD3100 was restricted to the recruitment of lymphocytes into the liver. One explanation is that AMD3100 inhibits CXCR4 non-selectively. Thus, it is likely that these cells also interact with AMD3100.
In conclusion, to the best of our knowledge, this study is the most comprehensive analysis of the MDSCs profile of NASH mice, and provide evidence that CD11b+Gr-1highLy6G+MDSCs protect liver function. Our findings suggested that clinically, granulocytic MDSCs supplementation might be effective in NASH. NASH conditions induce CXCR4 ligand expression in liver tissue. High levels of CXCL12 attract and retain CXCR4-expressing MDSCs from the BM into the liver, suggesting that the CXCR4/CXCL12 pathway is involved in MDSCs chemotaxis in NASH.
The present study aids our understanding of the biology of MDSCs during NASH. MDSCs represent promising therapeutic targets in the treatment of NASH; however, there also exists some defects such as the model we use. Still, further extensive research is needed before these approaches can be used clinically.
Acknowledgements
This work was supported by the Health and Family Planning Commission Foundation of Shanxi Province, China (No. 2015029). Science and Technology Department Foundation of Shanxi Province, China (No. 201701D221276). Scientific and technological achievements transformation guidance special of Shanxi Province, China (No. 201604D132042).
Disclosure of conflict of interest
None.
References
- 1.Rinella M, Charlton M. The globalization of nonalcoholic fatty liver disease: prevalence and impact on world health. Hepatology. 2016;64:19–22. doi: 10.1002/hep.28524. [DOI] [PubMed] [Google Scholar]
- 2.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 3.Stewart TJ, Smyth MJ. Improving cancer immunotherapy by targeting tumor-induced immune suppression. Cancer Metastasis Rev. 2011;30:125–140. doi: 10.1007/s10555-011-9280-5. [DOI] [PubMed] [Google Scholar]
- 4.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 5.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrad E, Resch TK, Gogesch P, Kalinke U, Bechmann I, Bogdan C, Waibler Z. Protection against RNA-induced liver damage by myeloid cells requires type I interferon and IL-1 receptor antagonist in mice. Hepatology. 2014;59:1555–1563. doi: 10.1002/hep.26915. [DOI] [PubMed] [Google Scholar]
- 7.Sarra M, Cupi ML, Bernardini R, Ronchetti G, Monteleone I, Ranalli M, Franzè E, Rizzo A, Colantoni A, Caprioli F, Maggioni M, Gambacurta A, Mattei M, Macdonald TT, Pallone F, Monteleone G. IL-25 prevents and cures fulminant hepatitis in mice through a myeloid-derived suppressor cell-dependent mechanism. Hepatology. 2013;58:1436–1450. doi: 10.1002/hep.26446. [DOI] [PubMed] [Google Scholar]
- 8.Zuo D, Yu X, Guo C, Wang H, Qian J, Yi H, Lu X, Lv ZP, Subjeck JR, Zhou H, Sanyal AJ, Chen Z, Wang XY. Scavenger receptor A restrains T-cell activation and protects against concanavalin A-induced hepatic injury. Hepatology. 2013;57:228–238. doi: 10.1002/hep.25983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 10.Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG, Walenkamp AM. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219–230. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 11.Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol. 2013;14:e218–228. doi: 10.1016/S1470-2045(12)70582-X. [DOI] [PubMed] [Google Scholar]
- 12.Werner L, Guzner-Gur H, Dotan I. Involvement of CXCR4/CXCR7/CXCL12 Interactions in Inflammatory bowel disease. Theranostics. 2013;3:40–46. doi: 10.7150/thno.5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J. Clin. Oncol. 2011;29:591–599. doi: 10.1200/JCO.2010.31.0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, Osterreicher CH, Schnabl B, Seki E, Brenner DA. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 2009;137:1467–1477. e1465. doi: 10.1053/j.gastro.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 17.Theiss HD, Vallaster M, Rischpler C, Krieg L, Zaruba MM, Brunner S, Vanchev Y, Fischer R, Gröbner M, Huber B, Wollenweber T, Assmann G, Mueller-Hoecker J, Hacker M, Franz WM. Dual stem cell therapy after myocardial infarction acts specifically by enhanced homing via the SDF-1/CXCR4 axis. Stem Cell Res. 2011;7:244–255. doi: 10.1016/j.scr.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 19.Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40:2969–2975. doi: 10.1002/eji.201040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, Stroncek DF, Terabe M, Kapoor V, ElGindi M, Han M, Thornton AM, Zhang H, Egger M, Luo J, Felsher DW, McVicar DW, Weber A, Heikenwalder M, Greten TF. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253–257. doi: 10.1038/nature16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaknin I, Blinder L, Wang L, Gazit R, Shapira E, Genina O, Pines M, Pikarsky E, Baniyash M. A common pathway mediated through Toll-like receptors leads to T- and natural killer-cell immunosuppression. Blood. 2008;111:1437–1447. doi: 10.1182/blood-2007-07-100404. [DOI] [PubMed] [Google Scholar]
- 22.Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–10026. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–4513. doi: 10.1158/0008-5472.CAN-06-4174. [DOI] [PubMed] [Google Scholar]
- 24.Ilkovitch D, Lopez DM. The liver is a site for tumor-induced myeloid-derived suppressor cell accumulation and immunosuppression. Cancer Res. 2009;69:5514–5521. doi: 10.1158/0008-5472.CAN-08-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suh YG, Kim JK, Byun JS, Yi HS, Lee YS, Eun HS, Kim SY, Han KH, Lee KS, Duester G, Friedman SL, Jeong WI. CD11b(+) Gr1(+) bone marrow cells ameliorate liver fibrosis by producing interleukin-10 in mice. Hepatology. 2012;56:1902–1912. doi: 10.1002/hep.25817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu K, Zhang N, Guo N, Yang J, Wang J, Yang C, Yang C, Zhu L, Xu C, Deng Q, Zhu R, Wang H, Chen X, Shi Y, Li Y, Leng Q. SSC(high)CD11b(high)Ly-6C(high)Ly-6G(low) myeloid cells curtail CD4 T cell response by inducible nitric oxide synthase in murine hepatitis. Int J Biochem Cell Biol. 2014;54:89–97. doi: 10.1016/j.biocel.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Liu G, Bi Y, Wang R, Yang H, Zhang Y, Wang X, Liu H, Lu Y, Zhang Z, Chen W, Chu Y, Yang R. Targeting S1P1 receptor protects against murine immunological hepatic injury through myeloid-derived suppressor cells. J Immunol. 2014;192:3068–3079. doi: 10.4049/jimmunol.1301193. [DOI] [PubMed] [Google Scholar]
- 28.Höchst B, Mikulec J, Baccega T, Metzger C, Welz M, Peusquens J, Tacke F, Knolle P, Kurts C, Diehl L, Ludwig-Portugall I. Differential induction of Ly6G and Ly6C positive myeloid derived suppressor cells in chronic kidney and liver inflammation and fibrosis. PLoS One. 2015;10:e0119662. doi: 10.1371/journal.pone.0119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapanadze T, Gamrekelashvili J, Ma C, Chan C, Zhao F, Hewitt S, Zender L, Kapoor V, Felsher DW, Manns MP, Korangy F, Greten TF. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol. 2013;59:1007–1013. doi: 10.1016/j.jhep.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider C, Teufel A, Yevsa T, Staib F, Hohmeyer A, Walenda G, Zimmermann HW, Vucur M, Huss S, Gassler N, Wasmuth HE, Lira SA, Zender L, Luedde T, Trautwein C, Tacke F. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut. 2012;61:1733–1743. doi: 10.1136/gutjnl-2011-301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gauttier V, Judor JP, Le Guen V, Cany J, Ferry N, Conchon S. Agonistic anti-CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int J Cancer. 2014;135:2857–2867. doi: 10.1002/ijc.28943. [DOI] [PubMed] [Google Scholar]
- 32.Ma C, Kapanadze T, Gamrekelashvili J, Manns MP, Korangy F, Greten TF. Anti-Gr-1 antibody depletion fails to eliminate hepatic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biology. 2012;92:1199–1206. doi: 10.1189/jlb.0212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boelte KC, Gordy LE, Joyce S, Thompson MA, Yang L, Lin PC. Rgs2 mediates pro-angiogenic function of myeloid derived suppressor cells in the tumor microenvironment via upregulation of MCP-1. PLoS One. 2011;6:e18534. doi: 10.1371/journal.pone.0018534. [DOI] [PMC free article] [PubMed] [Google Scholar]