

Abstract
The accelerated course of hepatic fibrosis that occurs in some patients after liver transplantation is an important clinical problem. Activation of hepatic stellate cell (HSCs) is the dominant event in hepatic fibrosis. Previous studies have shown that treatment with mammalian target of rapamycin (mTOR) inhibitors was more effective in reducing the progression of fibrosis than treatment with calcineurin inhibitors, suggesting that mTOR could be a crucial target for inhibition of fibrosis. In addition, N-acetylcysteine (NAC) has been shown to effectively suppress HSC activation-dependent expression of alpha-smooth muscle actin in HSCs, suggesting that NAC could be a candidate for the clinical treatment of hepatic fibrosis. Here, we have evaluated the effects of immunosuppressive drugs and NAC in a mice model of hepatic fibrosis and on HSC activation in vitro. We demonstrated that an mTOR inhibitor significantly inhibited fibrogenic genes in cultured HSCs until day 14. In addition, co-administration of NAC with everolimus further reduced the expression of fibrogenic genes and improved the characteristic of HSCs via blockage of HSC activation and up-regulation of fibrolytic gene. Moreover, in vivo studies showed that everolimus inhibited collagen deposition and inflammation in a mouse model of fibrogenesis, as determined by histological analysis, and everolimus treatment, in combination with NAC, significantly decreased extracellular matrix deposition and improved liver histology. These findings indicated that everolimus, combined with NAC, synergistically inhibited hepatic fibrosis and thus may become a valuable option in immunosuppressant therapy.
Keywords: Hepatic stellate cells, immunosuppressive drugs, mTOR inhibitor, N-acetylcysteine, animal model, fibrosis
Introduction
Fatty liver does not show any symptoms or cause serious problems immediately; hence, it is easily overlooked. However, if left untreated for a long time, it progresses to chronic liver disease, accompanied with cirrhosis. In addition, mortality rates in patients with liver cancer and fulminant hepatic failure are high. Only liver transplantation has been shown to be effective in such cases. However, even after liver transplantation, the rate of incidence of liver fibrosis may be high, caused by metabolic disorders of the liver, physical reaction, and use of immunosuppressive drugs. Despite improvements in surgical and post-transplant care, and better immunosuppression protocols, the recurrence of fibrosis remains a problem. Thus, hepatic fibrosis, caused due to metabolic liver disease, is emerging as a new indicator for liver transplantation. Hepatic fibrosis is a consequence of recovery from repeated liver damage [1]. In chronic liver injury, hepatocyte regeneration fails, and hepatocytes are replaced by an extracellular matrix (ECM), such as collagen. Thus, hepatic fibrosis represents qualitative and quantitative changes in the ECM of the liver [2]. ECM accumulation is also affected by the increased production and decreased degradation of the extracellular matrix [3]. The reduction in ECM removal mainly results from the overexpression of the tissue inhibitor of metalloproteinase (TIMP), which suppresses the matrix metalloproteinase (MMP) involved in the removal of the ECM. Liver fibrosis is closely related to the activation of the hepatic stellate cells (HSCs). HSCs are located in the space of Disse are the main secretory cells in the ECM of the damaged liver. HSCs store vitamin A in the normal state, and get activated upon injury, change their morphology, and appear like myofibroblasts, which are positive for alpha-smooth muscle actin (α-SMA) [1]. Activated HSCs proliferate, secrete vasoconstrictors, and synthesize collagen. They also secrete various inflammatory cytokines, MMPs, and TIMPs [4]. Most liver transplant recipients are required to take immunosuppressive drugs. Calcineurin inhibitors (CNIs), cyclosporine and tacrolimus, are the most common immunosuppressive drugs used after liver transplantation. CNI is a blocker of signal-1, which induces T-cell immunity by inhibiting the function of calcineurin. Previously, it has been shown to have an antifibrotic effect in an in vitro experiment using CNIs [5,6]. However, CNI-based immunosuppressive drugs accelerate metabolic liver disease when used for a long period, owing to side effects like diabetes and hyperlipidemia [7,8]. Thus, in recent years, mTOR inhibitor-based drugs have been used to replace CNI-based drugs for immunotherapy [9,10]. Recent studies have shown that sirolimus, an mTOR-based drug, inhibited the progression of fibrosis in rate, in vivo and in vitro [11]. Piguet et al., reported that the mTOR inhibitor, everolimus, suppressed the expression of collagen (type I) and α-SMA in the hepatic stellate cell line, LX-2 [12]. In addition, studies on the effect of antioxidants on HSCs and Kupffer cells, have reported that these drugs inhibited HSC division and α-SMA expression [13,14]. After liver transplantation, the rate of hepatic fibrosis must be reduced through prophylaxis and treatment, because chronic liver damage can rapidly progress into cirrhosis and graft failure. The selection and combination of immunosuppressive drugs for reducing the rate of hepatic fibrosis are a way of preventing hepatic fibrosis resulting from metabolic liver disease. Thus, a treatment strategy considering the selection and combination of immunosuppressive drugs is necessary. Therefore, in this study, we investigated the effect of various immunosuppressive drugs, used after liver transplantation, on HSCs and established effective drug combinations. Moreover, we applied effective immunosuppressive drugs and concomitant therapies to hepatic fibrosis animal models to verify the effect of hepatic fibrosis inhibition.
Material and methods
Hepatic stellate cell isolation and drug treatments
Hepatic stellate cells were purified from male Sprague-Dawley rats using the two-step liver perfusion method [15]. Mature hepatocytes were removed from the collagenase digested liver cell suspension by centrifugation at 50×g for 1 min, and the supernatant was centrifuged at 50×g for 5 min. This procedure was repeated until no pellets were seen. The supernatant was centrifuged at 200×g for 10 min, and the pellet containing HSCs was suspended in 10 mL of DMEM (Invitrogen, Carlsbad, USA), supplemented with 10% FBS, 15 mM HEPES, and antibiotics. After a final centrifugation at 200×g for 10 min, the cells in the pellet were resuspended in DMEM, containing 10% FBS, 50 U/mL penicillin, and 50 ng/mL streptomycin; seeded in tissue culture flasks and incubated at 37°C under saturating humidity in 5% CO2. The hepatic stellate cells were washed with PBS 20 h after seeding, to remove debris and blood cells. First, we evaluated various clinically used immunosuppressive drugs, such as 10 ng/mL rapamycin (RAPA) (R&D system, MN, USA), 10 ng/mL everolimus (EVE) (Sigma Aldrich, MO, USA), 1 µM mycophenolate mofetil (MMF) (R&D system, MN, USA), 20 ng/mL FK506 (R&D system, MN, USA), 100 ng/mL cyclosporine A (CsA) (R&D system, MN, USA), and 1 mM N-acetylcysteine (NAC) (Sigma Aldrich, MO, USA), for selecting effective drugs to inhibit the activation of HSCs. Second, HSCs were treated with 10 ng/mL FK506, 10 ng/mL EVE, 1 mM NAC, or a combination of EVE and NAC for 14 d to evaluate the effects of drug combinations. The medium, containing drugs, was changed every day. Cell cultures were observed daily by phase contrast microscopy to monitor morphological changes.
Cell growth assay
HSCs were seeded into 96-well plates (4000 cells/well) in triplicate and treated with 10 ng/mL FK506, 10 ng/mL EVE, 1 mM NAC, or a combination of EVE and NAC for the indicated times in the complete culture medium. Cell growth was measured using a Cell Counting Kit-8 (CCK-8) from Dojindo Molecular Technologies, Inc. (Rockville, USA), as per the manufacturer’s instructions. Briefly, the cells were incubated with 10 µL of CCK-8 solution mixed with 90 µL of DMEM, added to each well. After 2 h, the absorbance of each well was measured at 450 nm using a microplate reader.
Lipid droplet staining
Lipid droplets within the cells were visualized using the BODIPY staining Kit according to the manufacturer’s instructions (Invitrogen, Carlsbad, USA). Briefly, stellate cells were fixed with 4% paraformaldehyde for 20 min at 25-28°C. The cells were incubated for 20 min with 1% BODIPY (in imaging solution) and 4’,6-diamidino-2-phenylindol at 37°C. The staining solution was replaced with fresh imaging solution and the cells were imaged using a fluorescence microscope. For all experiments, measurements were made at the same intensity, background level, contrast, and zoom.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was prepared from the drug treated HSCs using TRIzol® (Invitrogen, Carlsbad, USA), according to the manufacturer’s instructions. The cDNA was prepared using PrimeScriptTM 1st strand cDNA synthesis Kit (TAKARA, Shiga, Japan), according to the manufacturer’s instructions. A total of 1 µL of the cDNA reaction mixture was subjected to PCR amplification using gene specific primers (Table S1) and AccuPower® PCR Premix (Bioneer, Korea). The PCR products were analyzed on a 2% agarose gel. Image of the PCR products was acquired with an Olympus High-Performance CCD camera (Olympus Corporation, Japan), and quantification of the bands was performed with ImageJ. The mRNA levels of each gene were normalized to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Western blotting
Cells were harvested and cellular lysates were prepared by incubating in radioimmune precipitation (RIPA) buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% TritonTM X-100, 0.5% sodium deoxycholate, and 0.05% SDS) containing 10 µL/mL protease inhibitor cocktail (Thermo Scientific, IL, USA) for 30 min on ice. Protein concentrations were measured by the Bradford method (Bio-Rad, Hercules, USA). Protein samples were heated at 95°C for 5 min, and 20 µg was applied to a 10% SDS-polyacrylamide gel (7.5% SDS-polyacrylamide gels were used for collagen analysis). After electrophoresis the proteins were electrophoretically transferred onto nitrocellulose membranes. Membranes were blocked for 1 h with 5% skim milk in TBST. Thereafter, the membranes were incubated overnight at 4°C with primary antibodies, shown in Table S2. The membranes were then incubated with the secondary antibody at 25-28°C for 2 h. After incubation with the secondary antibodies, the membranes were briefly washed twice and then thrice, for 10 min each, with TBST. Immunodetected proteins were visualized using the enhanced chemiluminescent ECL assay kit, according to the manufacturer’s protocol.
Animal care and drug treatment
This study was approved by the Korea University Institutional Animal Care and Use Committee and all animals were cared for in accordance with the National guidelines for ethical animal research. Six weeks old, male CD-1 mice (20-25 g, Orient Bio, Inc., a branch of Charles River Laboratories, Seongnam, Korea) were group-housed in individual cages in a specific pathogen free (SPF) facility. The experimental animals were housed in an air-conditioned room at 21-24°C with 12 h of light, and acclimatized for one week, before the experiment. Hepatic fibrosis was induced by the co-administration of thioacetamide (TAA, 100 mg/kg body weight) intraperitoneal (I.P.) injection twice weekly and a high-fat diet during the 8 weeks. The dose of TAA was selected based on previous reports [16]. The TAA was used in the range of 100 mg/kg to 200 mg/kg, in mice or rats. A total of 50 mice were randomly divided into five groups and treated as follows: group 1 received normal saline (N.S.) injection (n = 10); group 2 received NAC I.P. injection at a dose of 50 mmol/kg (n = 10); group 3 received FK506 I.P. injection at a dose of 1 mg/kg (n = 10); group 4 received EVE I.P. injection at a dose of 3 mg/kg (n = 10); group 5 received a combination of EVE and NAC at a dose of 3 mg/kg and 50 mmol, respectively. Immunosuppressive drugs were dissolved in dimethyl sulfoxide (DMSO), which were then diluted in normal saline prior to use. All injection were administrated once daily for 8 weeks. After 8 weeks, the animals were euthanized via CO2 inhalation, after 8 h of fasting. After sacrifice, blood was withdrawn from the abdominal vein for analysis. Liver samples were also collected for histological examination.
Biochemical analysis
To assess liver injury, the activities of aspartate transaminase (AST) and alanine transaminase (ALT) and total bilirubin (TBIL) and triglyceride (TG) in serum were assayed, using automatic chemistry analyzer (Fuji Dry Chem, Japan).
Histopathology and Fibrosis scoring
After 8 weeks, the mice were sacrificed, and liver tissues were fixed with 10% buffered formalin solution. Thereafter, the liver tissues were embedded in paraffin and sectioned at 5 µm, using an automated microtome. Deparaffinized tissue sections were stained with hematoxylin & eosin and Masson’s trichrome stain. Liver fibrosis assessment was performed after staining with Masson’s trichome stain. Histological grading of fibrosis was based on the METAVIR system. Evaluation of fibrosis was based on the following parameters: portal/perisinusoidal fibrosis (stage 1), characterized by mild fibrous expansion of portal tracts; perisinusoidal and periportal fibrosis (stage 2), characterized by fine strands of connective tissue with only rare portal-portal septa; septal or bridging fibrosis (stage 3), characterized by connective tissue bridges that link portal tracts with other portal tracts and central veins; and cirrhosis (stage 4), characterized by bridging fibrosis.
Statistical analysis
Statistical analyses were performed using SPSS statistics 23.0 (IBM Corporation, Armonk, NY, USA) for Windows. All experiments were repeated four times, unless otherwise specified. All data have been presented as mean ± SEM. Data were analyzed by one-way ANOVA using Bonferroni’s correction for multiple comparisons where required. For in vivo experiments, statistical analysis of data was performed using the Kruskal-Wallis and Mann-Whitney U tests. Data were considered statistically significant when P < 0.05.
Results
Combination treatment with everolimus and N-acetylcysteine inhibits activation and proliferation of hepatic stellate cells
The cellular morphology results from combination treatment were not significantly different from the results of single-drug treatment (Figure S1A). HSC morphology appeared unchanged until day 3 in culture; the cell grew larger and appeared like a myofibroblast with longer incubation time (Figure 1A). The presence of lipid droplets inside HSCs was confirmed by day 14 in the EVE+NAC treated group, as compared to the single drug-treated group (Figure 1B). It was difficult to compare the degree of cellular activation in the single-drug and combination-drug treated groups using morphology alone; therefore, we performed RT-PCR, which revealed a significant decrease in the expression levels of collagen and α-SMA for the experimental group treated with both EVE and NAC at the same time, compared to the group treated with EVE alone (Figure 2). HSC activation affected HSCs proliferation. Analysis of cellular proliferation for each experimental group revealed that proliferation was significantly more disrupted in groups receiving mTOR-inhibitors than in other groups, while the NAC-treated groups showed decreased proliferation at day 10 and 14 (Figure S2).
Figure 1.
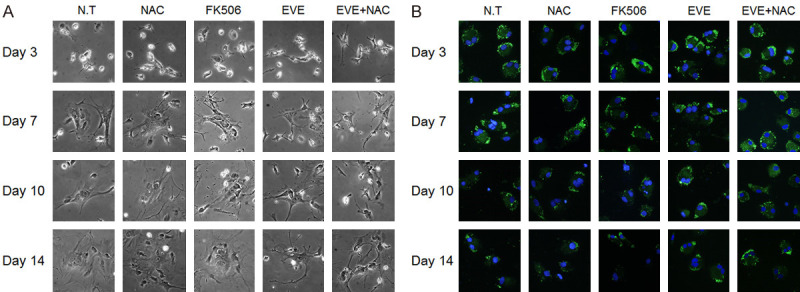
Effect of combination drugs on morphological features of HSCs. Combined everolimus and N-acetylcysteine treatment preserved morphological features of HSCs. A. Morphology of drug treated HSCs upon culturing, as visualized by phase contrast microscopy. HSCs are spontaneously activated as a result of growth in untreated and treated groups. B. Lipid droplets in the cytoplasm of HSCs, incubated with BODIPY. Green fluorescence indicates lipid droplets and blue indicates the nucleus. A considerable number of lipid droplets remain in the combination treatment group, until day 14. Morphological observation conducted at 200 times magnification.
Figure 2.
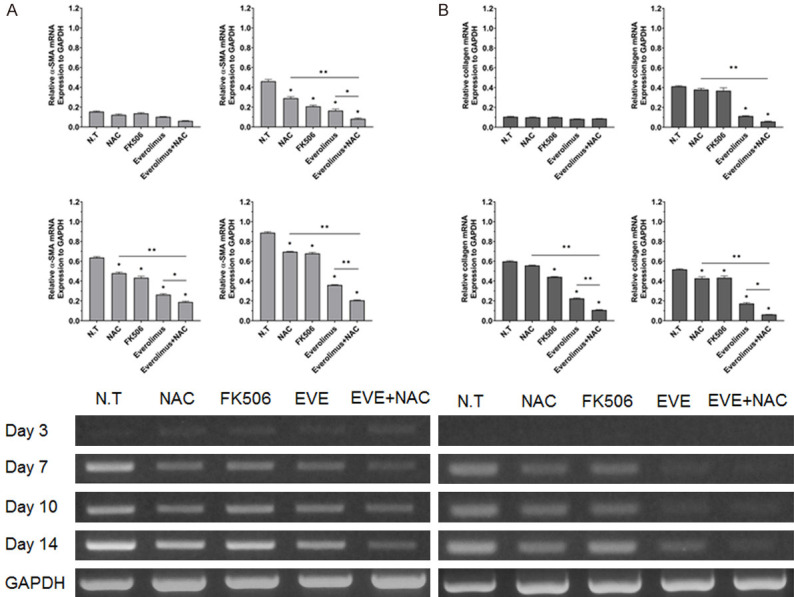
Effect of combination drugs on fibrosis related genes. Expression of α-SMA (A) and collagen (B) mRNA in drug treated HSCs for the indicated period, using RT-PCR. Combination treatment on HSCs markedly inhibits the hepatic fibrosis related genes. Representative agarose gel showing mRNA expression of fibrogenic markers in HSCs. Quantification of expression of α-SMA and collagen, relative to glyceraldehyde 3-phosphate dehydrogenase, from day 3 to day 14. The ratio between each sample and glyceraldehyde 3-phosphate dehydrogenase was calculated to normalize for initial variation in sample concentration and as a control for reaction efficiency. Values are shown as mean ± SEM. *P < 0.05, **P < 0.001.
The effect of combination treatment, with everolimus and N-acetylcysteine, on the expression of MMPs and TIMPs
The effect of combined therapy on the expression of MMPs and TIMP1s was confirmed by RT-PCR, 14 d post-treatment. In groups that were not treated with everolimus or NAC, increased expression of MMP2 and TIMP1 and increased HSC activation was seen. In the treatment group, however, expression of MMP2 and TIMP1 decreased. In the EVE+NAC treated group, MMP2 and TIMP1 expression were significantly more inhibited than in the single drug-treated group (Figure 3A, 3D). MMP13 expression decreased in the untreated group and increased in the treated groups, more significantly in the combined treatment group than in the single drug-treated group (Figure 3C). However, there was no difference in MMP9 mRNA level among the groups.
Figure 3.
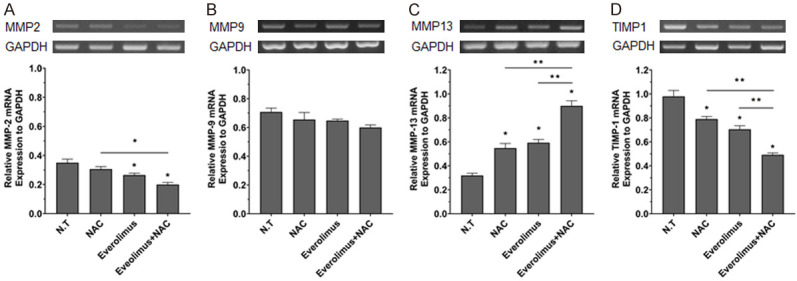
Treatment with everolimus and N-acetylcysteine regulates the expression of ECM protein degradation and rearrangement associated genes in HSCs. HSCs were treated with drugs for 14 d and the mRNA expression of MMP-2 (A), MMP-9 (B), MMP-13 (C), and TIMP-1 (D) were analyzed by PCR and the mRNA level of each gene were normalized to that of glyceraldehyde 3-phosphate dehydrogenase. Values are shown as mean ± SEM. *P < 0.05, **P < 0.001.
Combined action of everolimus and N-acetylcysteine affects the activity of signaling molecules and synthesis of collagen
To identify the mechanism underlying the inhibitory effect of drug combinations on HSC activation, we measured the expression of Akt/p70S6K in TGF-β-induced HSCs using western blot (Figure 4A, 4B). TGF-β significantly upregulated the phosphorylation of Akt and p70S6K. However, in groups receiving EVE treatment alone, phosphorylation of Akt and p70S6K was significantly inhibited. In the NAC-only treated group, Akt phosphorylation was significantly inhibited, while p70S6K was downregulated. However, in the EVE+NAC treatment group, Akt and p70S6K were both significantly downregulated. We measured collagen levels, using western blot, in HSCs treated for 7 d and 14 d (Figure 4C). Collagen synthesis appeared to increase at day 7 in the N.T. and NAC- and FK506-treated groups, but this increase was not significant. At day 14, however, collagen synthesis increased in the N.T. and NAC- and FK506-treated groups. In addition, collagen synthesis was inhibited in the EVE-only and EVE+NAC groups.
Figure 4.
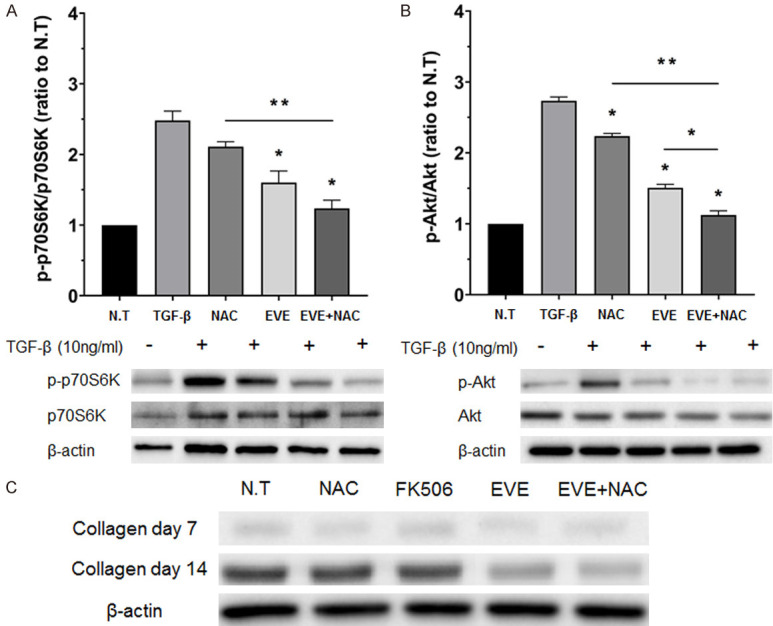
Effect of combination drugs on signaling molecules and collagen synthesis. A, B. Effects of combination treatment on TGF-β induced Akt/p70S6kinase phosphorylation in HSCs shown by western blotting. Akt/p70S6kinase phosphorylation after 1 h incubation with culture medium alone (N.T.), drugs, and 10 ng/mL TGF-β. The densities of protein bands were normalized against β-actin and expressed as a ratio. Values are expressed as mean ± SEM. *P < 0.05, **P < 0.001. C. Effects of combination treatment on collagen protein level as quantitated by western blot analysis. HSCs were treated with EVE and NAC alone, and with EVE and NAC. HSCs were obtained 14 d after treatment.
Combination of everolimus and N-acetylcysteine synergistically inhibits fibrosis in a thioacetamide and high-fat diet induced liver fibrosis mouse model
The liver function test revealed no significant difference in aminotransferase (AST) across all groups (Figure 5Aa). However, alanine aminotransferase (ALT) significantly increased with treatments involving EVE (Figure 5Ab). The mean ratio of AST/ALT was found to be 1.48, 1.30, 1.56, 0.85, and 0.74, in the N.S., NAC, FK506, EVE, and EVE+NAC groups, respectively (Figure 5B). From the AST/ALT ratio, we could conclude that hepatic fibrosis had been induced after 8 weeks and the ratio decreased in all treatment groups, except for FK506, compared to the N.S. group. Moreover, the AST/ALT ratio significantly decreased, relative to N.S. group, in the EVE+NAC treated group than in the single drug-treated group, suggesting that liver fibrosis was effectively inhibited in the combined treatment groups. Total bilirubin tended to decrease across all treatment groups, and decreased significantly in the EVE+NAC treated group (Figure 5Ac). Furthermore, triglycerides tended to decrease in all treatment groups, excluding the FK506 group, and decreased significantly in the EVE+NAC treated group, compared to the single-drug treated group (Figure 5Ad). To investigate the histopathology of livers in TAA- and high fat diet-induced models of liver fibrosis, we conducted hematoxylin and eosin staining (Figure 6A; First and second panel), which revealed the presence of fibrosis and inflammatory response around the central and portal veins in the N.S. and NAC- and FK506-treated groups. However, in the groups receiving EVE treatments, the inflammatory response was minor, relative to the N.S. group. To evaluate the degree of collagen formation in the tissue, we conducted Masson’s trichrome staining, which revealed high levels of collagen deposited around the central and portal veins in the N.S. and NAC- and FK506-treated groups (Figure 6A; Third and fourth panel). In the EVE-treated group, collagen formation, around the central and portal veins, decreased relative to the control group. Furthermore, in the EVE+NAC treated group, a small amount of collagen, comparable to normal liver tissue, was formed. We scored and compared the extent of liver fibrosis in tissues based on the results of Masson’s trichrome staining and found that the EVE+NAC treatment group had a significantly lower score than the EVE-only group (Figure 6B). Through these results we demonstrated that combination therapy with EVE and NAC, has an inhibitory effect on liver fibrosis and protective function for the liver in TAA- and high fat diet-induced models of liver fibrosis, compared to single-drug therapy.
Figure 5.
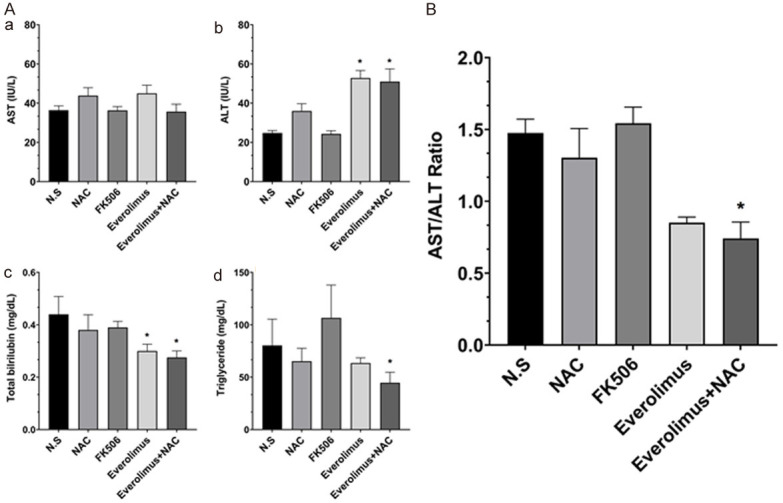
A. Level of serum aminotransferase (AST), aminotransferase (ALT), total bilirubin, and triglyceride in TAA and high fat diet induced fibrosis models and drug treatment groups. B. AST/ALT ratio decreased in the combination treatment group. All values are shown as mean ± SEM. *P < 0.05, **P < 0.001.
Figure 6.
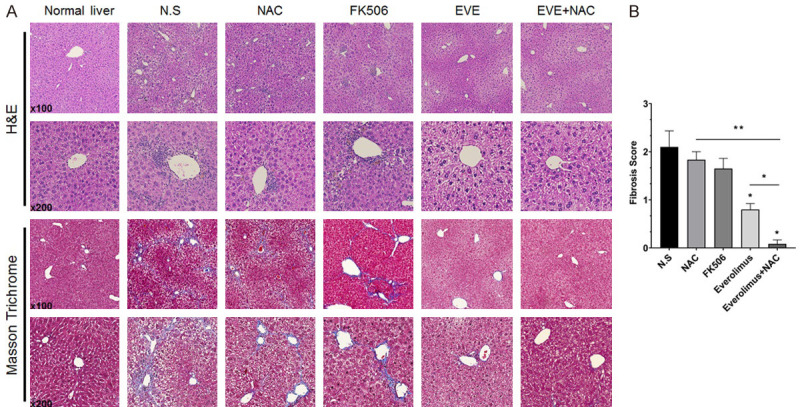
Effect of combination drugs on liver fibrogenesis and hepatic inflammation. A. Microscopy findings in rat liver sections. Progression of liver fibrosis is accompanied by inflammation and excessive extracellular matrix deposition. Everolimus, an mTOR inhibitor, prevented liver fibrosis induced by TAA and a high fat diet; liver morphology demonstrated by H&E staining. ECM production detected by Masson trichrome staining of mouse livers after 8 weeks of treatment, with co-administration of TAA and high-fat diet. Drug treatment, especially everolimus and its combination with NAC, suppressed collagen deposition in a fibrosis model. B. Fibrosis score evaluated by Masson trichrome-stained sections of mouse livers after 8 weeks. Data are represented as the mean ± SEM. *P < 0.05, **P < 0.001.
Discussion
Recurrence of liver fibrosis after liver transplantation is a serious problem. Liver fibrosis may be a sign of post-transplantation recurrence or chronic liver disease. Development of liver fibrosis after transplantation negatively affects the success of transplantation and the survival of the patient, and it is of high clinical importance, considering the potential need of re-transplantation [17,18]. Liver fibrosis is a liver condition, characterized by the excessive formation of extracellular matrix proteins [19]. Inflammation is induced by multiple factors and results in the accumulation of various inflammatory cells, which release various cytokines that help activate and proliferate the HSCs [20,21]. The activated HSCs synthesize collagen, which accumulates in the ECM and causes liver fibrosis. In healthy livers, hepatocytes, sinusoidal endothelial cells, and HSCs contribute to the production of ECM; however, following liver damage, activated HSCs become the major producers of ECM. Activated HSCs show an increased response to various growth factors, are converted into myofibroblast-like cells, and synthesize large amounts of collagen. Type I collagen further activates the HSCs. Among all cytokines, the transforming growth factor-β and platelet-derived growth factor act as the most important factors for HSC activation [22-24]. It has been reported that in liver cirrhosis patients there is an approximately 10-fold increase in the total amount of collagen in tissues than in the baseline [1,2]. Therefore, one of the most important goals for the treatment of liver fibrosis is to inhibit HSC activation and collagen formation in tissues.
Previous studies have investigated the inhibitory effects of immunosuppressants and antioxidants on cells and fibrosis-related mediators critically implicated in the fibrosis process. Mycophenolic acid has been reported to show anti-inflammatory and anti-fibrotic effects in liver endothelial and Kupffer cells [25], and the calcineurin inhibitors, cyclosporine and tacrolimus mediate anti-fibrotic effects by inhibiting HSC growth and collagen synthesis [6]. Studies in animal models of CCl4-induced liver fibrosis have reported that the mTOR inhibitors, rapamycin and everolimus, decrease the levels of TGF-β1 mRNA and pro-α2 collagen [11]. In addition, pharmacokinetic studies have demonstrated the potential of antioxidants, such as vitamin E or silymarin, for fibrosis therapy [21,26]. However, studies on the effects of various drugs on fibrosis have mostly been conducted in vivo, and to date there is either limited in vivo data on the candidate anti-fibrotic drugs or the effects have not yet been clearly identified. Moreover, no treatment method exists using drug combinations that can minimize the side effects of immunosuppressants and maximize the inhibitory effect on fibrosis. In this study, we selected an immunosuppressant with the greatest inhibitory effect on HSC activation and combined it with another drug that showed no interaction, to establish an effective combination therapy for the inhibition of HSC activation.
In the quiescent state, HSCs contain lipid droplets in the cytoplasm (Figure S3), but in the activated state they are capable of collagen synthesis. The cells become fibroblast-like in morphology and express α-SMA in the cytoplasm. α-SMA is an indicator of HSC activation and as liver fibrosis progresses, stellate cells become enlarged and strongly positive for α-SMA [27]. In addition, levels of type I collagen increases as the basement membrane is damaged by cytokines, such as TGF-β1 or PDGF, that are released from injured stellate cells [28]. Therefore, we conducted lipid droplet staining and RT-PCR to verify whether the isolated cells were quiescent HSCs. Thereafter, we treated the cells with immunosuppressants, used clinically, for 10 days to select the most effective immunosuppressant for inhibiting HSC activation and found that the mTOR inhibiting drugs were most effective in lowering the mRNA levels of α-SMA and collagen. We selected the most effective drug for inhibiting HSC activation through preliminary testing of everolimus and combined it with NAC for 14 days, and observed that the lipid droplets remained and HSC activation was significantly inhibited in groups singly-treated with everolimus or NAC, compared to the control group. These results suggested that the everolimus and NAC combination treatment had an excellent inhibitory effect on HSC activation compared to single-drug treatment. Fibrosis is a process of scar healing. Normally, if the causal factor of liver damage is temporary, the HSC that had been activated for healing the scarred tissue undergoes apoptosis and the ECM is digested by MMPs to return the liver to its normal state [29]. In the human body, excess ECM production is immediately followed by extracellular protease-mediated proteolysis. MMPs are important proteases and many different types are produced in the human liver [30]. The HSCs activated during fibrosis, not only function to digest ECM, but also secrete TIMP. In other words, activated HSCs are the primary producers of MMPs and contribute to matrix digestion in fibrotic livers [31]; at the same time they worsen fibrosis by activating TIMP1 and TIMP2, which inhibit MMPs and cause ECM accumulation [32]. Recently, MMPs and TIMPs have been actively researched in the context of HSC activation. In this study, we also explored the effects of EVE and NAC combination treatment on mRNA expression of the genetic factors of HSC activation, including MMP2, MMP13, MMP9, and TIMP1. In the EVE+NAC combination treatment group, expression of MMP2 and TIMP1 mRNA were more significantly decreased than in the single drug treated or control groups, while expression of MMP13 mRNA was significantly increased. MMP2 is responsible for the autocrine proliferation and migration of HSCs and its expression increases in HSCs stimulated by TGF-β and ROS, as seen in chronic liver disease. It has been reported that chronic overexpression of MMP2 in proliferative and migrative HSCs can accelerate fibrosis [31,33,34]. Thus, EVE and NAC treatment effectively inhibited the activation and the subsequent proliferation of HSCs, by inhibiting MMP2 expression, compared to single-drug treatment. MMP13 is expressed in the initial phase (day 0-3) of HSC primary culture, and it is downregulated as incubation time progresses [35]. A recent study has reported on the massive induction of MMP13 in the recovery stage of liver fibrosis. This suggests that MMP13 is critically implicated in the prevention of fibrosis [36]. In this study, MMP13 expression increased in HSCs treated with everolimus and NAC, while TIMP1 expression decreased. Therefore, it can be concluded that everolimus and NAC combination treatment acts in two ways to inhibit HSC activation and collagen synthesis: first, by decreasing the HSC activation marker (decreased production of collagen and α-SMA by the activated HSC), and second by increasing collagenase activity to promote fibrillar matrix digestion. In addition, regulation of proliferation and quiescence related intracellular signaling pathway attenuated HSC growth resulting in inhibition of activation of HSCs.
Animal models of liver cirrhosis are important for understanding the mechanism of liver cirrhosis and its treatment. Animal models of liver cirrhosis can be classified on the basis of the method used for induction of fibrosis, including hepatotoxicants, immunological damage [37], bile duct ligation [38], and alcohol [39]. Commonly used hepatotoxicants include CCl4 [40], DMN [41], and TAA [42]. Of these, TAA-induced models of liver damage share common morphological and biochemical features with human models, and thus identical lesions can be induced [43]. In particular, the TAA model exhibits regenerative nodules, fibrosis, and other histological characteristics, similar to human liver cirrhosis, as compared to the CCl4 models [44]. However, the TAA models requires a long time to induce liver fibrosis [45]. Therefore, we used animal models of liver fibrosis induced by TAA and a high fat diet, and conducted an in vivo study of established drug combination therapies in groups treated with normal saline, FK506, NAC, EVE, and EVE+NAC, to investigate the effects of immunosuppressants on fibrosis inhibition. We monitored the change in weight for 8 weeks and observed temporary weight loss with every peritoneal injection of TAA across all experimental groups, although a net increase in weight occurred. The liver function test showed no significant change in serum AST and ALT in the treatment group. Since aminotransferase level decreases with improvement of liver steatosis or inflammation, even when fibrosis progresses, it is not very helpful for diagnosing fibrosis [46]. Moreover, aminotransferase levels can be normal at any stage of fatty liver, including cirrhosis [47]. This can explain why AST and ALT levels were relatively unchanged, compared to the control group. A recent study proposed the increase in AST/ALT ratio as the clinical risk factor of liver fibrosis in nonalcoholic steatohepatitis (NASH) [48].
In this study, we confirmed a significant decrease in the AST/ALT ratio in the EVE+NAC treatment group, which suggested inhibition of liver fibrosis in this group compared to other treatment groups. TBIL and TG also significantly decreased in the EVE+NAC treatment group, suggesting that combination treatment of immunosuppressant and antioxidant has a protective effect on the liver. Furthermore, liver histology showed that TAA and high fat diet induced collagen formation, while treatments involving EVE tended to inhibit collagen formation. Specifically, in the EVE+NAC treatment group a significant inhibition of collagen formation was verified by the fibrosis score. Based on the serological and histological findings, we confirmed high AST/ALT ratio, TBIL, and TG levels in fibrotic liver tissue and more collagen accumulation, which causes liver fibrosis. However, with combined drug therapy, the fibrosis-related factors improved. Together, these results suggest that EVE+NAC therapy inhibits activation and proliferation of HSCs, which play an integral part in liver fibrosis, and the inhibitory effect was confirmed in animal models of TAA and high fat diet-induced liver fibrosis.
Conclusion
This study reported the effect of combined treatment, using everolimus (EVE) with N-acetylcysteine (NAC). Combination treatment significantly down-regulated fibrogenic genes, via blockage of HSCs activation, and markedly attenuated extracellular matrix deposition in mice with thioacetamide and high-fat diet induced liver fibrosis. Combination treatment with EVE with NAC may become a valuable strategy for fibrosis inhibition under immunosuppression.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (NRF-2017 R1A2B2005754).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arthur MJ. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G245–G249. doi: 10.1152/ajpgi.2000.279.2.G245. [DOI] [PubMed] [Google Scholar]
- 4.Knittel T, Mehde M, Grundmann A, Saile B, Scharf JG, Ramadori G. Expression of matrix metalloproteinases and their inhibitors during hepatic tissue repair in the rat. Histochem Cell Biol. 2000;113:443–453. doi: 10.1007/s004180000150. [DOI] [PubMed] [Google Scholar]
- 5.Manojlovic Z, Blackmon J, Stefanovic B. Tacrolimus (FK506) prevents early stages of ethanol induced hepatic fibrosis by targeting LARP6 dependent mechanism of collagen synthesis. PLoS One. 2013;8:e65897. doi: 10.1371/journal.pone.0065897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakamuta M, Kohjima M, Fukushima M, Morizono S, Kotoh K, Kobayashi N, Enjoji M. Cyclosporine suppresses cell growth and collagen production in hepatic stellate cells. Transplant Proc. 2005;37:4598–4602. doi: 10.1016/j.transproceed.2005.10.104. [DOI] [PubMed] [Google Scholar]
- 7.Kasiske BL, Snyder JJ, Gilbertson D, Matas AJ. Diabetes mellitus after kidney transplantation in the United States. Am J Transplant. 2003;3:178–185. doi: 10.1034/j.1600-6143.2003.00010.x. [DOI] [PubMed] [Google Scholar]
- 8.Menegazzo LA, Ursich MJ, Fukui RT, Rocha DM, Silva ME, Ianhez LE, Sabbaga E, Wajchenberg BL. Mechanism of the diabetogenic action of cyclosporin A. Horm Metab Res. 1998;30:663–667. doi: 10.1055/s-2007-978954. [DOI] [PubMed] [Google Scholar]
- 9.De Simone P, Nevens F, De Carlis L, Metselaar HJ, Beckebaum S, Saliba F, Jonas S, Sudan D, Fung J, Fischer L, Duvoux C, Chavin KD, Koneru B, Huang MA, Chapman WC, Foltys D, Witte S, Jiang H, Hexham JM, Junge G H2304 Study Group. Everolimus with reduced tacrolimus improves renal function in de novo liver transplant recipients: a randomized controlled trial. Am J Transplant. 2012;12:3008–3020. doi: 10.1111/j.1600-6143.2012.04212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forgacs B, Merhav HJ, Lappin J, Mieles L. Successful conversion to rapamycin for calcineurin inhibitor-related neurotoxicity following liver transplantation. Transplant Proc. 2005;37:1912–1914. doi: 10.1016/j.transproceed.2005.02.101. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Wu J, Frizell E, Liu SL, Bashey R, Rubin R, Norton P, Zern MA. Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology. 1999;117:1198–1204. doi: 10.1016/s0016-5085(99)70406-3. [DOI] [PubMed] [Google Scholar]
- 12.Piguet AC, Majumder S, Maheshwari U, Manjunathan R, Saran U, Chatterjee S, Dufour JF. Everolimus is a potent inhibitor of activated hepatic stellate cell functions in vitro and in vivo, while demonstrating anti-angiogenic activities. Clin Sci (Lond) 2014;126:775–784. doi: 10.1042/CS20130081. [DOI] [PubMed] [Google Scholar]
- 13.Hasegawa T, Yoneda M, Nakamura K, Makino I, Terano A. Plasma transforming growth factor-beta1 level and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: a pilot study. Aliment Pharmacol Ther. 2001;15:1667–1672. doi: 10.1046/j.1365-2036.2001.01083.x. [DOI] [PubMed] [Google Scholar]
- 14.Kawada N, Seki S, Inoue M, Kuroki T. Effect of antioxidants, resveratrol, quercetin, and N-acetylcysteine, on the functions of cultured rat hepatic stellate cells and Kupffer cells. Hepatology. 1998;27:1265–1274. doi: 10.1002/hep.510270512. [DOI] [PubMed] [Google Scholar]
- 15.Riccalton-Banks L, Bhandari R, Fry J, Shakesheff KM. A simple method for the simultaneous isolation of stellate cells and hepatocytes from rat liver tissue. Mol Cell Biochem. 2003;248:97–102. doi: 10.1023/a:1024184826728. [DOI] [PubMed] [Google Scholar]
- 16.Wallace MC, Hamesch K, Lunova M, Kim Y, Weiskirchen R, Strnad P, Friedman SL. Standard operating procedures in experimental liver research: thioacetamide model in mice and rats. Lab Anim. 2015;49:21–29. doi: 10.1177/0023677215573040. [DOI] [PubMed] [Google Scholar]
- 17.Crespo G, Lens S, Gambato M, Carrion JA, Marino Z, Londono MC, Miquel R, Bosch J, Navasa M, Forns X. Liver stiffness 1 year after transplantation predicts clinical outcomes in patients with recurrent hepatitis C. Am J Transplant. 2014;14:375–383. doi: 10.1111/ajt.12594. [DOI] [PubMed] [Google Scholar]
- 18.Firpi RJ, Clark V, Soldevila-Pico C, Morelli G, Cabrera R, Levy C, Machicao VI, Chaoru C, Nelson DR. The natural history of hepatitis C cirrhosis after liver transplantation. Liver Transpl. 2009;15:1063–1071. doi: 10.1002/lt.21784. [DOI] [PubMed] [Google Scholar]
- 19.Gressner OA, Weiskirchen R, Gressner AM. Biomarkers of liver fibrosis: clinical translation of molecular pathogenesis or based on liver-dependent malfunction tests. Clin Chim Acta. 2007;381:107–113. doi: 10.1016/j.cca.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 20.Gressner AM. Hepatic fibrogenesis: the puzzle of interacting cells, fibrogenic cytokines, regulatory loops, and extracellular matrix molecules. Z Gastroenterol. 1992;30(Suppl 1):5–16. [PubMed] [Google Scholar]
- 21.Li D, Friedman SL. Liver fibrogenesis and the role of hepatic stellate cells: new insights and prospects for therapy. J Gastroenterol Hepatol. 1999;14:618–633. doi: 10.1046/j.1440-1746.1999.01928.x. [DOI] [PubMed] [Google Scholar]
- 22.Breitkopf K, Haas S, Wiercinska E, Singer MV, Dooley S. Anti-TGF-beta strategies for the treatment of chronic liver disease. Alcohol Clin Exp Res. 2005;29:121S–131S. doi: 10.1097/01.alc.0000189284.98684.22. [DOI] [PubMed] [Google Scholar]
- 23.Rockey DC. Antifibrotic therapy in chronic liver disease. Clin Gastroenterol Hepatol. 2005;3:95–107. doi: 10.1016/s1542-3565(04)00445-8. [DOI] [PubMed] [Google Scholar]
- 24.Tangkijvanich P, Yee HF Jr. Cirrhosis--can we reverse hepatic fibrosis? Eur J Surg Suppl. 2002:100–112. [PubMed] [Google Scholar]
- 25.Greupink R, Bakker HI, Reker-Smit C, van Loenen-Weemaes AM, Kok RJ, Meijer DK, Beljaars L, Poelstra K. Studies on the targeted delivery of the antifibrogenic compound mycophenolic acid to the hepatic stellate cell. J Hepatol. 2005;43:884–892. doi: 10.1016/j.jhep.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Song G, Minuk GY. Effects of hepatic stimulator substance, herbal medicine, selenium/vitamin E, and ciprofloxacin on cirrhosis in the rat. Gastroenterology. 1996;110:1150–1155. doi: 10.1053/gast.1996.v110.pm8613004. [DOI] [PubMed] [Google Scholar]
- 27.Lee KS, Lee SJ, Park HJ, Chung JP, Han KH, Chon CY, Lee SI, Moon YM. Oxidative stress effect on the activation of hepatic stellate cells. Yonsei Med J. 2001;42:1–8. doi: 10.3349/ymj.2001.42.1.1. [DOI] [PubMed] [Google Scholar]
- 28.Baroni GS, D’Ambrosio L, Curto P, Casini A, Mancini R, Jezequel AM, Benedetti A. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology. 1996;23:1189–1199. doi: 10.1002/hep.510230538. [DOI] [PubMed] [Google Scholar]
- 29.Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, Hovell C, Arthur MJ. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–549. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman SL, Bansal MB. Reversal of hepatic fibrosis -- fact or fantasy? Hepatology. 2006;43:S82–88. doi: 10.1002/hep.20974. [DOI] [PubMed] [Google Scholar]
- 31.Benyon RC, Hovell CJ, Da Gaca M, Jones EH, Iredale JP, Arthur MJ. Progelatinase A is produced and activated by rat hepatic stellate cells and promotes their proliferation. Hepatology. 1999;30:977–986. doi: 10.1002/hep.510300431. [DOI] [PubMed] [Google Scholar]
- 32.Knittel T, Mehde M, Kobold D, Saile B, Dinter C, Ramadori G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: regulation by TNF-alpha and TGF-beta1. J Hepatol. 1999;30:48–60. doi: 10.1016/s0168-8278(99)80007-5. [DOI] [PubMed] [Google Scholar]
- 33.Ikeda K, Wakahara T, Wang YQ, Kadoya H, Kawada N, Kaneda K. In vitro migratory potential of rat quiescent hepatic stellate cells and its augmentation by cell activation. Hepatology. 1999;29:1760–1767. doi: 10.1002/hep.510290640. [DOI] [PubMed] [Google Scholar]
- 34.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 35.Iredale JP, Benyon RC, Arthur MJ, Ferris WF, Alcolado R, Winwood PJ, Clark N, Murphy G. Tissue inhibitor of metalloproteinase-1 messenger RNA expression is enhanced relative to interstitial collagenase messenger RNA in experimental liver injury and fibrosis. Hepatology. 1996;24:176–184. doi: 10.1002/hep.510240129. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe T, Niioka M, Hozawa S, Kameyama K, Hayashi T, Arai M, Ishikawa A, Maruyama K, Okazaki I. Gene expression of interstitial collagenase in both progressive and recovery phase of rat liver fibrosis induced by carbon tetrachloride. J Hepatol. 2000;33:224–235. doi: 10.1016/s0168-8278(00)80363-3. [DOI] [PubMed] [Google Scholar]
- 37.Kimura K, Ando K, Ohnishi H, Ishikawa T, Kakumu S, Takemura M, Muto Y, Moriwaki H. Immunopathogenesis of hepatic fibrosis in chronic liver injury induced by repeatedly administered concanavalin A. Int Immunol. 1999;11:1491–1500. doi: 10.1093/intimm/11.9.1491. [DOI] [PubMed] [Google Scholar]
- 38.Lee SS, Girod C, Braillon A, Hadengue A, Lebrec D. Hemodynamic characterization of chronic bile duct-ligated rats: effect of pentobarbital sodium. Am J Physiol. 1986;251:G176–180. doi: 10.1152/ajpgi.1986.251.2.G176. [DOI] [PubMed] [Google Scholar]
- 39.Giavarotti L, D’Almeida V, Giavarotti KA, Azzalis LA, Rodrigues L, Cravero AA, Videla LA, Koch OR, Junqueira VB. Liver necrosis induced by acute intraperitoneal ethanol administration in aged rats. Free Radic Res. 2002;36:269–275. doi: 10.1080/10715760290019282. [DOI] [PubMed] [Google Scholar]
- 40.Jeong DH, Jang JJ, Lee SJ, Lee JH, Lim IK, Lee MJ, Lee YS. Expression patterns of cell cycle-related proteins in a rat cirrhotic model induced by CCl4 or thioacetamide. J Gastroenterol. 2001;36:24–32. doi: 10.1007/s005350170150. [DOI] [PubMed] [Google Scholar]
- 41.Jenkins SA, Grandison A, Baxter JN, Day DW, Taylor I, Shields R. A dimethylnitrosamine-induced model of cirrhosis and portal hypertension in the rat. J Hepatol. 1985;1:489–499. doi: 10.1016/s0168-8278(85)80747-9. [DOI] [PubMed] [Google Scholar]
- 42.Li X, Benjamin IS, Alexander B. Reproducible production of thioacetamide-induced macronodular cirrhosis in the rat with no mortality. J Hepatol. 2002;36:488–493. doi: 10.1016/s0168-8278(02)00011-9. [DOI] [PubMed] [Google Scholar]
- 43.Muller A, Machnik F, Zimmermann T, Schubert H. Thioacetamide-induced cirrhosis-like liver lesions in rats--usefulness and reliability of this animal model. Exp Pathol. 1988;34:229–236. doi: 10.1016/s0232-1513(88)80155-5. [DOI] [PubMed] [Google Scholar]
- 44.Perez Tamayo R. Is cirrhosis of the liver experimentally produced by CCl4 and adequate model of human cirrhosis? Hepatology. 1983;3:112–120. doi: 10.1002/hep.1840030118. [DOI] [PubMed] [Google Scholar]
- 45.Seong J, Han KH, Park YN, Nam SH, Kim SH, Keum WS, Kim KS. Lethal hepatic injury by combined treatment of radiation plus chemotherapy in rats with thioacetamide-induced liver cirrhosis. Int J Radiat Oncol Biol Phys. 2003;57:282–288. doi: 10.1016/s0360-3016(03)00540-6. [DOI] [PubMed] [Google Scholar]
- 46.Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42:132–138. doi: 10.1016/j.jhep.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Mofrad P, Contos MJ, Haque M, Sargeant C, Fisher RA, Luketic VA, Sterling RK, Shiffman ML, Stravitz RT, Sanyal AJ. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology. 2003;37:1286–1292. doi: 10.1053/jhep.2003.50229. [DOI] [PubMed] [Google Scholar]
- 48.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–1362. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.