

Abstract
An impaired epithelial barrier is often observed in allergic rhinitis (AR), which facilitates the infiltration of allergens. The aim of this study was to investigate the role of autophagy in the impaired epithelial barrier in AR and the related signalling pathways. A human nasal epithelial cell line was treated with dust mite allergen (Derp1). Autophagy was evaluated by GFP-LC3 adenovirus transfection and measurement of autophagy-related proteins. Epithelial barrier function was evaluated by measuring tight junction protein expression, transepithelial electrical resistance, and fluorescein isothiocyanate-dextran (FD4) permeability. Next, miR-125b inhibitor, miR-125b mimics, shRNA targeting FoxP3, pcDNA3.1 expressing FoxP3, and inhibitor of C-X-C motif chemokine receptor type 4 (CXCR4) were used to investigate the roles of miR-125b, FoxP3, and CXCR4 in epithelial cell autophagy and epithelial barrier function. An in vivo AR model was generated by exposing rat nasal mucosa to an allergen. Derp1 exposure enhanced autophagy and impaired the epithelial barrier in epithelial cells. Upregulation of miR-125b expression led to enhanced autophagy and impaired epithelial barrier through inhibition of FoxP3. Derp1 exposure increased miR-125b expression by increasing the expression and activation of CXCR4, which downregulated FoxP3 expression and led to enhanced autophagy and an impaired epithelial barrier. In vivo analysis confirmed the role of the CXCR4/miR-125b/FoxP3 axis in the impaired epithelial barrier in AR. This study demonstrates that the CXCR4/miR-125b/FoxP3 axis may participate in the pathogenesis of AR by regulating autophagy in epithelial cells and dysfunction of the epithelial barrier.
Keywords: Allergic rhinitis, CXCR4, miR-125b, FoxP3, autophagy, epithelial barrier
Introduction
Allergic rhinitis (AR), also known as hay fever, is one of the most prevalent chronic diseases worldwide and affects one in four people worldwide [1,2]. Although it is not a lethal disease, AR leads to symptoms of nasal itching, sneezing, watery discharge, and many complications, such as asthma, nasosinusitis, and nasal polyps, which severely affect quality of life [1,2]. The pathogenesis of AR has not yet been fully clarified, but allergen exposure may play major roles in this disease [2].
Previous studies have shown that allergens and autophagy are related in nasal epithelial cells [3-6]. Autophagy is an essential cellular mechanism that plays important “housekeeping” roles by removing long-lived proteins and damaged organelles in the cytoplasm [7]. Autophagy is involved in many cellular processes [7]. In epithelial cells, autophagy was also shown to regulate the expression of tight junction-related proteins [8]. Tight junctions between epithelial cells are closely associated with the proper function of the epithelial barrier, the first barrier against allergen infiltration [9]. These findings support the hypothesis that allergen-induced dysfunction of the epithelial barrier via autophagy could be a potential pathogenesis mechanism of AR. However, the underlying mechanism of how allergen exposure regulates the autophagy of epithelial cells in AR remains unclear.
MicroRNAs (miRNAs) are a group of short noncoding regulatory RNAs that target mRNAs for cleavage, resulting in translational repression [10]. MiRNAs are involved in many biological processes, including inflammation, cell cycle regulation, apoptosis, differentiation, migration, and autophagy [11]. Several miRNAs have been found to regulate the autophagy cascade in various ways. For example, miR-20a and miR-106b were reported to inhibit leucine deprivation-induced autophagy through suppression of ULK1 expression [12]. MiR-30a and miR-216a were found to downregulate the expression of Beclin-1, resulting in decreased autophagy activity [13,14], and miR-216a was also found to regulate autophagy by suppressing the expression of ATG4C [15]. A recent study reported that miR-125b could induce autophagy by suppressing FoxP3 expression [16]. In addition, miR-125b was reported to be differentially expressed in AR patients, asthma patients, and healthy subjects [17]. These findings suggest a potential role of miR-125b in the regulation of allergen-induced autophagy in epithelial cells and dysfunction of the epithelial barrier.
Inhibition of C-X-C motif chemokine receptor type 4 (CXCR4) expression resulted in significant downregulation of inflammation and allergen-induced responses in bronchial asthma [18-20]. CXCR4 is a chemokine receptor and has been shown to be involved in many pathological conditions, including allergic airway diseases [21]. Overexpression or activation of CXCR4 induces the activation of many signalling pathways, including ERK1/2, p38, AKT, and mTOR, among others [22]. A recent study indicated that CXCR4 could regulate miR-125b expression in colorectal cancer [23], but it is not clear whether CXCR4 also regulates the function of the epithelial barrier through miR-125b and autophagy.
Here, we hypothesized that allergens could activate the CXCR4/miR-125b/FoxP3 axis, leading to enhanced autophagy of epithelial cells and dysfunction of the epithelial barrier in AR. The aim of our studies was to shed some light on the mechanism of AR, which could serve as a foundation for future discovery of new therapeutic methods for AR.
Materials and methods
Cell culture and in vitro AR model
The human nasal epithelial cell line (HNEpC) RPMI 2650 was purchased from American Type Culture Collection (ATCC, USA) and cultured in minimum essential medium (MEM, Gibco, USA) with 10% foetal calf serum (FCS, Gibco, USA) as described [24]. Cells were seeded in 6-well plates with MEM and 10% FCS and cultured until 60-70% confluence. The in vitro AR model was generated by 24-hour treatment with 5 μg/mL dust mite allergen, Derp1 (Greer Laboratories, USA). 3-Methyladenine (3-MA, Sigma-Aldrich, USA) or AMD3100 (Sigma-Aldrich, USA) was administered to the culture medium at a final concentration of 5 mmol/L (3-MA) or 20 μM (AMD3100).
Cell transfection and construction of GFP-LC3 cells
MiR-125b mimics and miR-125b inhibitor were purchased from GenePharma (Shanghai, China). The shRNA-FoxP3 and pcDNA3.1-FoxP3 vectors were obtained from GeneChem (Shanghai, China). Cells were transfected using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s protocols. RPMI 2650 cells expressing GFP-LC3 were constructed by transfection with GFP-LC3 adenovirus (Hanbio, China) for 24 hours.
Extraction of total RNA and quantitative PCR
TRIzol RNA isolation reagent (Invitrogen, CA, USA) was used to extract total RNA, and 1 μg RNA from each sample was reverse-transcribed using SuperScript IV VILO Master Mix (Thermo Scientific, USA). Quantitative PCR (qPCR) was then performed using SYBR Premix Dimmer Eraser Kit (Takara, China) on an Applied Biosystems 7500 System (ABI, USA). The results were calculated using the 2-ΔΔCt method. U6 snRNA was used as an internal reference for miR-125b, while β-actin was used as an internal reference for the mRNAs measured. Specifically, the primers were listed in Table 1.
Table 1.
Primers for quantitative real-time PCR
| Gene | Primers | Sequences (5’-3’) |
|---|---|---|
| CXCR4-h | Forward | CTCCTCTTTGTCATCACGCTTCC |
| Reverse | GGATGAGGACACTGCTGTAGAG | |
| miR-125b-h | Forward | CCTGAGACCCTAACTTG |
| Reverse | GAACATGTCTGCGTATCTC | |
| FoxP3-h | Forward | GGCACAATGTCTCCTCCAGAGA |
| Reverse | CAGATGAAGCCTTGGTCAGTGC | |
| Occludin-h | Forward | ATGGCAAAGTGAATGACAAGCGG |
| Reverse | CTGTAACGAGGCTGCCTGAAGT | |
| Claudin-1-h | Forward | GTCTTTGACTCCTTGCTGAATCTG |
| Reverse | CACCTCATCGTCTTCCAAGCAC | |
| ZO-1-h | Forward | GTCCAGAATCTCGGAAAAGTGCC |
| Reverse | CTTTCAGCGCACCATACCAACC | |
| JAM-A-h | Forward | GTGAAGTTGTCCTGTGCCTACTC |
| Reverse | ACCAGTTGGCAAGAAGGTCACC | |
| β-actin-h | Forward | CTCCATCATGAAGTGTGACGTT |
| Reverse | ATCTCCTTCTGCATCCTGTCAG | |
| U6 snRNA-h | Forward | CGCAAGGATGACACGCAAATTC |
| Reverse | GTGCAGGGTCCGAGGT | |
| CXCR4-r | Forward | GGATGGTGGTGTTCCAGTTCTCCCCACGTAATACGGTAGC |
| Reverse | TCCCCACGTAATACGGTAGC | |
| miR-125b-r | Forward | ACACTCCAGCTGGGTCCCTGAGACCCTAACTT |
| Reverse | TGTCGTGGAGTCGGCAATTC | |
| FoxP3-r | Forward | CCCAAATTCCTGCCTGTGCTTA |
| Reverse | GCTGAAGACGTGTGCATCCTAT | |
| Occludin-r | Forward | CAGACCACTATGAAACCGACTA |
| Reverse | TCTCCAGCAACCAGCATC | |
| Claudin-1-r | Forward | GGGACAACATCGTGACTGCT |
| Reverse | CCACTAATGTCGCCAGACCTG | |
| ZO-1-r | Forward | CACGATGCTCAGAGACGAAGG |
| Reverse | TTCTACATATGGAAGTTGGGGATC | |
| JAM-A-r | Forward | ATCCCACAACAGGAGAGCTGCTCGGCTATAGGCAAACCAG |
| Reverse | CTCGGCTATAGGCAAACCAG | |
| β-actin-r | Forward | GGGAAATCGTGCGTGACATT |
| Reverse | CAGGAAGGAAGGCTGGAAGA | |
| U6 snRNA-r | Forward | GCTTCGGCAGCACATATACTAAAATGGCAGCACATATACTAAAAT |
| Reverse | CGCTTCAGAATTTGCGTGTCAT |
h, Human; r, Rat; CXCR4, C-X-C motif chemokine receptor type 4; ZO-1, zonula occludens-1; JAM-A, junctional adhesion molecule-A.
Protein extraction and western blot
Cells were first homogenized in HLB buffer with protease inhibitors. Cell lysates were prepared in SDS lysis buffer and boiled. After protein concentrations were determined using BCA Protein Assay Reagent (Thermo Scientific, USA), equal amounts of protein were loaded and separated in SDS-polyacrylamide gels by electrophoresis. After transferring the protein to nitrocellulose membranes and blocking in 5% skimmed milk, membranes were incubated overnight with a primary antibody against Beclin-1 (ab207612, Abcam), light chain 3 (LC3, ab128025, Abcam), Occludin (ab216327, Abcam), Claudin-1 (ab15098, Abcam), ZO-1 (zonula occludens-1, ab96587, Abcam), JAM-A (junctional adhesion molecule-A, ab180821, Abcam), FoxP3 (ab191416, Abcam), phospho-CXCR4 (p-CXCR4, ab74012, Abcam), CXCR4 (ab181020, Abcam), or β-actin (ab8227, Abcam). Membranes were then incubated with goat anti-rabbit IgG (ab6721, Abcam, USA) secondary antibody for 2 hours. Relative protein levels were determined by band intensities quantitated with the Image J software.
Prediction of miRNA targets
Prediction of the target genes of miR-125b was performed using the TargetScan (www.targetscan.org) and PicTar (www.pictar.org) databases.
Dual-luciferase reporter assay
To estimate whether miR-125b could bind to the FoxP3 3’-UTR, cells were cotransfected with a FoxP3-wild type or FoxP3-mutant reporter plasmid and control miRNA or miR-125b mimics using Lipofectamine 2000 (Invitrogen, USA). Luciferase activities were then measured using the Promega Dual-Luciferase® Reporter Assay System (Promega, USA) according to the manufacturer’s instructions.
Measurement of transepithelial resistance (TER)
Cells were first seeded on polyester transwell inserts (Corning, USA) at 105 cells per well. After the cells reached complete confluence, TER was then measured using an EVOM/EndOhm system (WPI Inc, USA).
Fluorescein isothiocyanate-dextran (FD4) permeability assay
FD4 was purchased from Sigma-Aldrich (USA). FD4 was added to the apical side of the cells cultured in transwell inserts at 2 mg/mL. FD4 intensity in the fluid on the basolateral side was measured after 4 hours using a FLUOstar Omega fluorescence plate reader (BMG Labtech, Germany).
Establishment of the in vivo AR model
For initial exposure, the nasal mucosa of rats was exposed to 1 μg/mL dust mite allergen, Derp1, on day 0. On days 7 to 11, a high dose of Derp1 (10 μg/mL) was administered to generate the AR model. Normal saline was used in the control group. AMD3100 was injected subcutaneously daily at 5 mg/kg, while PBS was used for the control group. All procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Hainan General Hospital (Hainan, China).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, USA). Data are shown as the mean ± standard deviation (SD). Student’s t test was conducted for comparisons between two groups. One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was carried out to compare three or more groups. All experiments were repeated at least 3 times. Values of P < 0.05 were considered statistically significant.
Results
Dust mite allergen promoted autophagy in nasal epithelial cells and damaged the function of the epithelial barrier
After 24 hours of exposure to 5 μg/mL dust mite allergen, Derp1, the number of autophagosomes in human nasal epithelial cells (RPMI 2650) was increased compared to that in the control group, while treatment with 3-MA, an autophagy inhibitor, resulted in a lower number of autophagosomes in Derp1-treated RPMI 2650 cells (Figure 1A). The expression levels of the autophagy-related proteins LC3 II/I and Beclin-1 were also increased after Derp1 exposure and were inhibited by 3-MA (Figure 1B). These results indicated that Derp1 exposure could promote autophagy in nasal epithelial cells.
Figure 1.

Dust mite allergen (Derp1) promoted autophagy of nasal epithelial cells and damaged the function of the epithelial barrier. A. GFP-LC3 cells treated with PBS (Control), Derp1 (Derp1), or both Derp1 and 3-MA (Derp1+3-MA), LC3 expression was tested by fluorescence microscope. B. Western blot analysis of LC3 II/I and Beclin-1 in cells treated with PBS (Control), Derp1 (Derp1), or both Derp1 and 3-MA (Derp1+3-MA). C. qPCR measurements of Occludin, Claudin-1, ZO-1, and JAM-A in cells treated with PBS (Control), Derp1 (Derp1), or both Derp1 and 3-MA (Derp1+3-MA). D. Western blot analysis of Occludin, Claudin-1, ZO-1, and JAM-A in cells treated with PBS (Control), Derp1 (Derp1), or both Derp1 and 3-MA (Derp1+3-MA). E. TER measurement on cells treated with PBS (Control), Derp1 (Derp1), or both Derp1 and 3-MA (Derp1+3-MA). F. FD4 permeability assay in cells treated with PBS (Control), Derp1 (Derp1), or both Derp1 and 3-MA (Derp1+3-MA). Data were shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01.
Measurement of the expression levels of epithelial barrier-related proteins showed that Occludin, Claudin-1, ZO-1 and JAM-A expression was significantly inhibited in the Derp1 group at both the mRNA and protein levels, which was reversed by 3-MA (Figure 1C and 1D). TER is commonly used to measure the integrity of the epithelial barrier, and a high TER usually indicates high integrity of the epithelial barrier [25]. In our results, TER was also significantly lower in the group treated with Derp1 than the control group, indicating damage to the integrity of the epithelial barrier, while 3-MA treatment partially recovered the function of the epithelial barrier (Figure 1E). In addition, the FD4 permeability assay showed that Derp1 treatment significantly increased the permeability of the epithelial barrier, which was also partially recovered by 3-MA treatment (Figure 1F). These results supported the hypothesis that allergen exposure could cause dysfunction of the nasal epithelial barrier by promoting autophagy.
MiR-125b regulated autophagy and the function of the nasal epithelial barrier
A previous study showed that miR-125b promoted autophagy in cancer cells [23]. In our study, we also investigated the regulatory effect of miR-125b on autophagy in nasal epithelial cells. As shown in Figure 2A, Derp1 exposure resulted in increased miR-125b expression in RPMI 2650 cells compared to the control group, indicating that allergen exposure could upregulate the expression of miR-125b in nasal epithelial cells. Using miR-125b mimics or inhibitor, we successfully overexpressed or knocked down miR-125b expression, respectively (Figure 2A). The expression levels of the autophagy-related proteins LC3 II/I and Beclin-1 were significantly higher in the miR-125b mimics group than in the control group, while the miR-125b inhibitor resulted in significantly decreased expression of LC3 II/I and Beclin-1 compared to the control (Figure 2B). Additional 3-MA treatment significantly decreased the expression of LC3 II/I and Beclin-1 compared to the miR-125b mimics group (Figure 2B). These results indicated that miR-125b could promote autophagy in nasal epithelial cells. On the other hand, the expression levels of epithelial barrier-related proteins, including Occludin, Claudin-1, ZO-1, and JAM-A, were significantly lower in the miR-125b mimics group and higher in the miR-125b inhibitor group than in the control group (Figure 2C and 2D). Further treatment with 3-MA in the miR-125b mimics group led to recovery of the expression of epithelial barrier proteins (Figure 2C and 2D). These results supported the notion that miR-125b could inhibit the expression of essential proteins in the epithelial barrier by inducing autophagy.
Figure 2.

MiR-125b regulated autophagy and the function of the epithelial barrier. A. qPCR measurement of miR-125b in cells treated with PBS (Control), Derp1, or cells transfected with miR-125b mimics or inhibitor. B. Western blot analysis of LC3 and Beclin-1 in cells treated with PBS (Control), cells transfected with miR-125b mimics or inhibitor, or cells transfected with miR-125b mimics and treated with 3-MA (miR-125b mimics+3-MA). C. qPCR measurement of Occludin, Claudin-1, ZO-1, and JAM-A in cells treated with PBS (Control), cells transfected with miR-125b mimics or inhibitor, or cells transfected with miR-125b mimics and treated with 3-MA (miR-125b mimics+3-MA). D. Western blot analysis of Occludin, Claudin-1, ZO-1, and JAM-A in cells treated with PBS (Control), cells transfected with miR-125b mimics or inhibitor, or cells transfected with miR-125b mimics and treated with 3-MA (miR-125b mimics+3-MA). Data were shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.01.
MiR-125b regulated autophagy in epithelial cells via FoxP3
Using online bioinformatics tools (TargetScan and PicTar), we investigated the binding site of miR-125b on FoxP3, and the results showed that miR-125b was able to bind to the 3’UTR of FoxP3 (Figure 3A). Dual-luciferase reporter assay also showed that cells transfected with both FoxP3-wild type plasmid and miR-125b mimics showed significantly decreased luciferase activity compared to control cells and cells transfected with FoxP3-mutant plasmid and miR-125b mimics, indicating the potential binding of miR-125b and FoxP3 (Figure 3B). The expression levels of FoxP3 were also significantly lower when cells were treated with miR-125b mimics and significantly higher when cells were treated with miR-125b inhibitor at both the mRNA and protein levels (Figure 3C and 3D), indicating that miR-125b could inhibit the expression of FoxP3 in nasal epithelial cells. Western blot results showed that inhibition of FoxP3 expression reversed the downregulation of autophagy by the miR-125b inhibitor, while FoxP3 overexpression inhibited the upregulation of LC3 II/I and Beclin-1 expression by the miR-125b mimics (Figure 3E). These results indicated that the enhancement of autophagy by miR-125b in epithelial cells was possibly through inhibition of FoxP3 expression.
Figure 3.

MiR-125b regulated autophagy of epithelial cells via FoxP3. A. Binding site of miR-125b on the 3’UTR of FoxP3. B. Dual luciferase reporter assay on FoxP3-wild type cells (WT) and FoxP3-mutant cells (MUT) transfected with miR-125b mimics, non-specific miRNA (mimics NC), or not transfected (Control). C. qPCR measurement of FoxP3 in cells transfected with miR-125b mimics or inhibitor. D. Western blot analysis of FoxP3 in cells transfected with miR-125b mimics or inhibitor and untransfected control cells. E. Western blot analysis of LC-3 II/I and Beclin-1 in cells transfected with miR-125b inhibitor, both miR-125b inhibitor and sh-FoxP3 (miR-125b inhibitor+sh-FoxP3), or cells transfected with both miR-125b mimics and pcDNA3.1-FoxP3 (miR-125b mimics+pcDNA3.1-FoxP3). Data were shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01.
Allergen enhanced miR-125b expression and autophagy by upregulating CXCR4
Both CXCR4 mRNA and protein expression levels were found to be significantly increased in nasal epithelial cells treated with Derp1, while treatment with both Derp1 and a CXCR4 inhibitor (AMD3100) resulted in significantly lower CXCR4 expression levels than Derp1 treatment only (Figure 4A and 4B). In addition, the level of p-CXCR4 was also increased in the Derp1 group, and additional treatment with AMD3100 significantly decreased the level of p-CXCR4 (Figure 4B). Nasal epithelial cells treated with Derp1 also showed an increased number of autophagosomes compared to the control group, which could be reversed by AMD3100 (Figure 4C). All these results indicated that an allergen could increase the expression and activation of CXCR4, which may participate in the upregulation of autophagy upon allergen exposure. Furthermore, nasal epithelial cells treated with Derp1 showed significantly increased miR-125b expression level compared to the control, which could be reversed by AMD3100 (Figure 4D). These results suggested an important role of CXCR4 in the regulation of miR-125b expression by allergen exposure.
Figure 4.
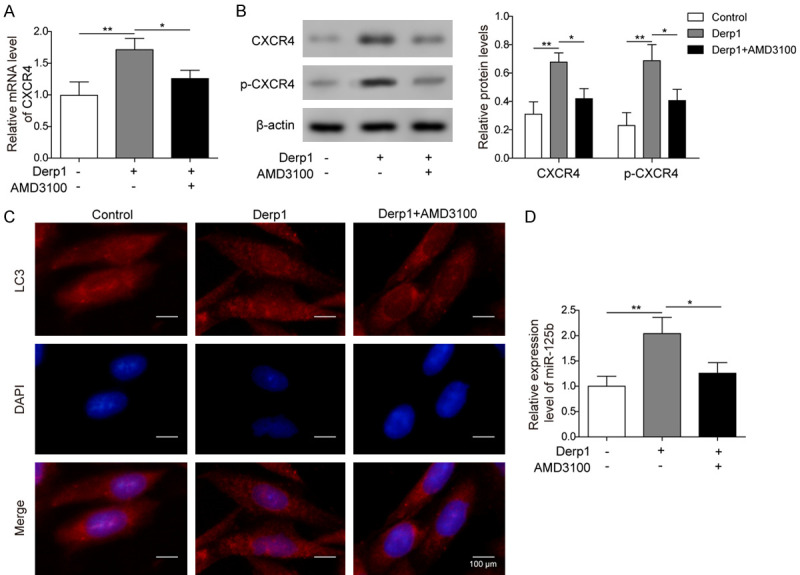
Allergen exposure enhanced miR-125b expression and autophagy by upregulating CXCR4. A. qPCR measurement of CXCR4 in cells treated with PBS, Derp1, or both Derp1 and AMD3100. B. Western blot analysis of CXCR4 and p-CXCR4 in cells treated with PBS, Derp1, or both Derp1 and AMD3100. C. GFP-LC3 cells treated with PBS, Derp1, or both Derp1 and AMD3100, LC3 expression was tested by fluorescence microscope. D. qPCR measurement of miR-125b in cells treated with PBS, Derp1, or both Derp1 and AMD3100. Data were shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01.
Allergens influence the autophagy of epithelial cells and the function of the epithelial barrier by regulating the CXCR4/miR-125b/FoxP3 axis
To further clarify the roles of CXCR4, miR-125b and FoxP3 in the regulation of autophagy in epithelial cells and their function in the epithelial barrier, we used AMD3100 to treat epithelial cells transfected with miR-125b mimics or sh-FoxP3. Western blot results showed that after treatment with AMD3100, the expression levels of autophagy-related proteins (LC3 II/I and Beclin-1) were significantly decreased compared to those in the control, and transfection with miR-125b mimics or sh-FoxP3 significantly increased the levels of LC3 II/I and Beclin-1 (Figure 5A). These results suggested that CXCR4 may regulate autophagy through miR-125b and FoxP3.
Figure 5.

Allergen affected autophagy in epithelial cells and the function of the epithelial barrier by regulating the CXCR4/miR-125b/FoxP3 axis. A. Western blot analysis of LC3 II/I and Beclin-1 in cells treated with PBS or AMD3100 and in cells transfected with miR-125b mimics or sh-FoxP3 and treated with AMD3100. B. qPCR measurement of Occludin, Claudin-1, ZO-1, and JAM-A in cells treated with PBS or AMD3100 and in cells transfected with miR-125b mimics or sh-FoxP3 and treated with AMD3100. C. Western blot analysis of Occludin, Claudin-1, ZO-1, and JAM-A in cells treated with PBS or AMD3100 and in cells transfected with miR-125b mimics or sh-FoxP3 and treated with AMD3100. D. Measurement of TER in cells treated with PBS or AMD3100 and in cells transfected with miR-125b mimics or sh-FoxP3 and treated with AMD3100. E. FD4 permeability assay in cells treated with PBS or AMD3100 and in cells transfected with miR-125b mimics or sh-FoxP3 and treated with AMD3100. Data were shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01.
The expression levels of epithelial barrier-related proteins were also investigated, and the results showed that after treatment with AMD3100, the expression levels of those proteins were higher than those in the control group, and these effects were reversed by transfection with miR-125b mimics or sh-FoxP3 (Figure 5B and 5C), suggesting that CXCR4 could also regulate the epithelial barrier through miR-125b and FoxP3. Measurement of the TER and the FD4 permeability assay showed significantly increased TER and decreased permeability in nasal epithelial cells treated with AMD3100, and transfection with miR-125b mimics or sh-FoxP3 reversed the effects of AMD3100 treatment on TER and permeability of the epithelial barrier (Figure 5D and 5E), which further supported the roles of CXCR4, miR-125b and FoxP3 in the regulation of nasal epithelial barrier function. Taken together, these results in Figures 4 and 5 indicated that allergens could regulate the autophagy of epithelial cells and the function of the nasal epithelial barrier through regulation of CXCR4 expression and subsequent changes in miR-125b and FoxP3 levels.
Validation of the regulatory effects of the CXCR4/miR-125b/FoxP3 axis on autophagy and the epithelial barrier in an in vivo AR model
Derp1 was used to generate an in vivo AR model. Normal saline was used instead in the control group. In the other two groups, either AMD3100 or 3-MA was used together with Derp1 (Derp1+AMD3100 and Derp1+3-MA) to treat the animals. The results showed that the expression levels of autophagy-related proteins (LC3 II/I and Beclin-1) were significantly increased and those of epithelial barrier-related proteins (Occludin, Claudin-1, ZO-1, and JAM-A) were significantly decreased in nasal epithelial cells from animals treated with Derp1 only compared to the control group, and treatment with AMD3100 or 3-MA reversed the effects of Derp1 on autophagy-related proteins and epithelial barrier-related proteins (Figure 6A-C). These results suggested that inhibition of CXCR4 or autophagy could help reduce the effect of allergens on epithelial cell autophagy and the epithelial barrier in an animal model of AR.
Figure 6.

Validation of the regulatory effect of the CXCR4/miR-125b/FoxP3 axis on autophagy and the epithelial barrier in an in vivo AR model. A. Western blot analysis of LC3 II/I and Beclin-1 in animals treated with normal saline, Derp1, both Derp1 and AMD3100, or both Derp1 and 3-MA. B. qPCR measurement of Occludin, Claudin-1, ZO-1, and JAM-A in animals treated with normal saline, Derp1, both Derp1 and AMD3100, or both Derp1 and 3-MA. C. Western blot analysis of Occludin, Claudin-1, ZO-1, and JAM-A in animals treated with normal saline, Derp1, both Derp1 and AMD3100, or both Derp1 and 3-MA. D. FD4 permeability assay in animals treated with normal saline, Derp1, both Derp1 and AMD3100, or both Derp1 and 3-MA. Data were shown as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01.
On day 12, FD4 was administered to the nasal mucosa of all four groups of animals, and serum levels of FD4 fluorescence intensity were measured. The results showed that serum from animals treated with Derp1 had the highest fluorescence intensity compared to the control group, while the addition of AMD3100 or 3-MA resulted in less serum fluorescence intensity compared to the Derp1 only group (Figure 6D). These results indicated that allergens could impair the function of the epithelial barrier and autophagy of epithelial cells through the CXCR4/miR-125b/FoxP3 axis. In addition, inhibition of autophagy or CXCR4 could enhance the epithelial barrier in AR.
Discussion
Allergen exposure plays important roles in the pathogenesis of AR. The nasal epithelial barrier serves as the first physiological barrier against the infiltration of allergens. However, dysfunction of this barrier, e.g., impaired tight junction structure, has been observed in AR patients, which may contribute to the pathogenesis of the disease [9]. It is therefore important to understand the underlying mechanism of the disruption of the epithelial barrier in AR, which could possibly provide new therapeutic targets in the future.
Autophagy of epithelial cells was found to regulate the expression of tight junction-related proteins and therefore the function of the epithelial barrier [8]. Epithelial barrier dysfunction has been observed in AR caused by diesel exhaust [26], and alternaria [27], and interferon-inducible proteins were found to play essential roles in patients with AR [28]. Our results showed that allergen exposure could enhance autophagy in nasal epithelial cells (Figure 1A and 1B). In addition, allergen exposure could also inhibit the expression of epithelial barrier-related proteins, leading to decreased electronic resistance and increased permeability of the nasal epithelial barrier (Figure 1C-F). Subsequent in vivo studies also showed enhanced autophagy and damaged epithelial barrier functions in the AR animal model (Figure 6). These results were consistent with previous findings that house dust mite-induced AR patients had impaired nasal epithelial barrier function and decreased expression of tight junction proteins (Occludin and ZO-1) [29]. Our findings further suggested a possible role of autophagy in allergen-induced epithelial barrier dysfunction and the pathogenesis of AR.
MiRNAs are important regulatory molecules that are involved in many biological processes and diseases, including autophagy and AR [17,30]. Several miRNAs were found to be differentially expressed in patients with AR and asthma, including miR-125b, miR-16, miR-299-5p, miR-126, miR-206, and miR-133b [17]. Among those miRNAs, miR-16, miR-125b and miR-381-3p are involved in the dysregulation of the intestinal epithelial barrier [31,32], and miR-143 and miR-145 are involved in the disruption of the cervical epithelial barrier [33]. Our results showed that miR-125b also induced autophagy of nasal epithelial cells and decreased the expression of epithelial barrier proteins, which was similar to observations in the Derp1-treated in vitro AR model (Figure 2). Further investigation showed that those effects of miR-125b on autophagy and the epithelial barrier were possibly through suppression of FoxP3 expression (Figure 3), which was similar to the findings in thyroid cancer [16]. These findings suggested that the abnormal expression levels of miR-125b in AR could contribute to its impaired epithelial barrier functions by inducing autophagy in epithelial cells.
A previous study found that CXCR4 expression was significantly higher in the nasal tissue of symptomatic AR patients than in the normal population [34]. A specific inhibitor of CXCR4, AMD3100, was reported to attenuate allergic lung inflammation and airway hyperreactivity [20], and the underlying mechanism included downregulation of MMP-9 and ERK1/2 [19] or inhibition of the T cell-related immune response [18]. Another study showed that the CXCL12/CXCR4 axis could enhance autophagy via miR-125b in colorectal cancer [23]. Similarly, our results showed that the upregulation of miR-125b by dust mite allergen occurred through upregulation and activation of CXCR4 (Figures 4 and 5), which led to the downregulation of FoxP3 expression, increase in autophagy in epithelial cells and dysfunction of the epithelial barrier.
Overall, our results suggest a role for the CXCR4/miR-125b/FoxP3 axis in the pathogenesis of AR, which regulates autophagy of epithelial cells, as well as the function of the epithelial barrier. Future investigations may involve validation of this hypothesis in clinical subjects, and factors in this pathway could serve as potential therapeutic targets for AR in the future.
Acknowledgements
This work was supported by Regional Science Fund Project of National Natural Science Foundation of China [grant number 81760054, 81760186, 81760187]; Key Research and Development Program of Hainan Province [grant number ZDYF2017115]; Hainan Natural Science Foundation Project [grant number 818MS8130]; and Scientific Research Project of Hainan Health and Family Planning Industry [grant number 18A200022].
Disclosure of conflict of interest
None.
Abbreviations
- AR
allergic rhinitis
- miRNA
microRNA
- CXCR4
C-X-C motif chemokine receptor type 4
- HNEpC
human nasal epithelial cell
- ATCC
American Type Culture Collection
- MEM
minimum essential medium
- FCS
foetal calf serum
- 3-MA
3-Methyladenine
- qPCR
quantitative PCR
- ZO-1
zonula occludens-1
- JAM-A
junctional adhesion molecule-A
- p-CXCR4
phosphor-CXCR4
- FD4
fluorescein isothiocyanate-dextran
- SD
standard deviation
- ANOVA
analysis of variance
References
- 1.Greiner AN, Hellings PW, Rotiroti G, Scadding GK. Allergic rhinitis. Lancet. 2011;378:2112–2122. doi: 10.1016/S0140-6736(11)60130-X. [DOI] [PubMed] [Google Scholar]
- 2.Eifan AO, Durham SR. Pathogenesis of rhinitis. Clin Exp Allergy. 2016;46:1139–1151. doi: 10.1111/cea.12780. [DOI] [PubMed] [Google Scholar]
- 3.Wang BF, Cao PP, Wang ZC, Li ZY, Wang ZZ, Ma J, Liao B, Deng YK, Long XB, Xu K, Wang H, Wang H, Zeng M, Lu X, Liu Z. Interferon-gamma-induced insufficient autophagy contributes to p62-dependent apoptosis of epithelial cells in chronic rhinosinusitis with nasal polyps. Allergy. 2017;72:1384–1397. doi: 10.1111/all.13153. [DOI] [PubMed] [Google Scholar]
- 4.Tsai YG, Wen YS, Wang JY, Yang KD, Sun HL, Liou JH, Lin CY. Complement regulatory protein CD46 induces autophagy against oxidative stress-mediated apoptosis in normal and asthmatic airway epithelium. Sci Rep. 2018;8:12973. doi: 10.1038/s41598-018-31317-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Assani K, Shrestha CL, Rinehardt H, Zhang S, Robledo-Avila F, Wellmerling J, Partida-Sanchez S, Cormet-Boyaka E, Reynolds SD, Schlesinger LS, Kopp BT. AR-13 reduces antibiotic-resistant bacterial burden in cystic fibrosis phagocytes and improves cystic fibrosis transmembrane conductance regulator function. J Cyst Fibros. 2019;18:622–629. doi: 10.1016/j.jcf.2018.10.010. [DOI] [PubMed] [Google Scholar]
- 6.Trybus E, Krol G, Obarzanowski T, Trybus W, Kopacz-Bednarska A, Obarzanowski M, Krol T. In vivo and in vitro studies on multidirectional mechanism of anti-allergic activity of budesonide. J Physiol Pharmacol. 2017;68:907–919. [PubMed] [Google Scholar]
- 7.Ravanan P, Srikumar IF, Talwar P. Autophagy: the spotlight for cellular stress responses. Life Sci. 2017;188:53–67. doi: 10.1016/j.lfs.2017.08.029. [DOI] [PubMed] [Google Scholar]
- 8.Nighot P, Ma T. Role of autophagy in the regulation of epithelial cell junctions. Tissue Barriers. 2016;4:e1171284. doi: 10.1080/21688370.2016.1171284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukuoka A, Yoshimoto T. Barrier dysfunction in the nasal allergy. Allergol Int. 2018;67:18–23. doi: 10.1016/j.alit.2017.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Su Z, Yang Z, Xu Y, Chen Y, Yu Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget. 2015;6:8474–8490. doi: 10.18632/oncotarget.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314. doi: 10.1146/annurev-pathol-012513-104715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu H, Wang F, Hu S, Yin C, Li X, Zhao S, Wang J, Yan X. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell Signal. 2012;24:2179–2186. doi: 10.1016/j.cellsig.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, Liu CG, Yang JM. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2009;5:816–823. doi: 10.4161/auto.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menghini R, Casagrande V, Marino A, Marchetti V, Cardellini M, Stoehr R, Rizza S, Martelli E, Greco S, Mauriello A, Ippoliti A, Martelli F, Lauro R, Federici M. MiR-216a: a link between endothelial dysfunction and autophagy. Cell Death Dis. 2014;5:e1029. doi: 10.1038/cddis.2013.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korkmaz G, le Sage C, Tekirdag KA, Agami R, Gozuacik D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy. 2012;8:165–176. doi: 10.4161/auto.8.2.18351. [DOI] [PubMed] [Google Scholar]
- 16.Wang S, Wu J, Ren J, Vlantis AC, Li MY, Liu SYW, Ng EKW, Chan ABW, Luo DC, Liu Z, Guo W, Xue L, Ng SK, van Hasselt CA, Tong MCF, Chen GG. MicroRNA-125b interacts with Foxp3 to induce autophagy in thyroid cancer. Mol Ther. 2018;26:2295–2303. doi: 10.1016/j.ymthe.2018.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panganiban RP, Wang Y, Howrylak J, Chinchilli VM, Craig TJ, August A, Ishmael FT. Circulating microRNAs as biomarkers in patients with allergic rhinitis and asthma. J Allergy Clin Immunol. 2016;137:1423–1432. doi: 10.1016/j.jaci.2016.01.029. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Xu X, Teng J, Cheng S, Bunjhoo H, Cao Y, Liu J, Xie J, Wang C, Xu Y, Xiong W. CXCR4 inhibitor attenuates ovalbumin-induced airway inflammation and hyperresponsiveness by inhibiting Th17 and Tc17 cell immune response. Exp Ther Med. 2016;11:1865–1870. doi: 10.3892/etm.2016.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H, Xu X, Teng J, Cheng S, Bunjhoo H, Cao Y, Liu J, Xie J, Wang C, Xu Y, Xiong W. CXCR4 inhibitor attenuates allergen-induced lung inflammation by down-regulating MMP-9 and ERK1/2. Int J Clin Exp Pathol. 2015;8:6700–6707. [PMC free article] [PubMed] [Google Scholar]
- 20.Lukacs NW, Berlin A, Schols D, Skerlj RT, Bridger GJ. AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol. 2002;160:1353–1360. doi: 10.1016/S0002-9440(10)62562-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pozzobon T, Goldoni G, Viola A, Molon B. CXCR4 signaling in health and disease. Immunol Lett. 2016;177:6–15. doi: 10.1016/j.imlet.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Scala S. Molecular pathways: targeting the CXCR4-CXCL12 axis-untapped potential in the tumor microenvironment. Clin Cancer Res. 2015;21:4278–4285. doi: 10.1158/1078-0432.CCR-14-0914. [DOI] [PubMed] [Google Scholar]
- 23.Yu X, Shi W, Zhang Y, Wang X, Sun S, Song Z, Liu M, Zeng Q, Cui S, Qu X. CXCL12/CXCR4 axis induced miR-125b promotes invasion and confers 5-fluorouracil resistance through enhancing autophagy in colorectal cancer. Sci Rep. 2017;7:42226. doi: 10.1038/srep42226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albano GD, Bonanno A, Cavalieri L, Ingrassia E, Di Sano C, Siena L, Riccobono L, Gagliardo R, Profita M. Effect of high, medium, and low molecular weight hyaluronan on inflammation and oxidative stress in an in vitro model of human nasal epithelial cells. Mediators Inflamm. 2016;2016:8727289. doi: 10.1155/2016/8727289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henry OYF, Villenave R, Cronce MJ, Leineweber WD, Benz MA, Ingber DE. Organs-on-chips with integrated electrodes for trans-epithelial electrical resistance (TEER) measurements of human epithelial barrier function. Lab Chip. 2017;17:2264–2271. doi: 10.1039/c7lc00155j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukuoka A, Matsushita K, Morikawa T, Takano H, Yoshimoto T. Diesel exhaust particles exacerbate allergic rhinitis in mice by disrupting the nasal epithelial barrier. Clin Exp Allergy. 2016;46:142–152. doi: 10.1111/cea.12597. [DOI] [PubMed] [Google Scholar]
- 27.Shin SH, Ye MK, Lee DW, Che MH. Alternaria-induced barrier dysfunction of nasal epithelial cells: role of serine protease and reactive oxygen species. Int Forum Allergy Rhinol. 2019;9:514–521. doi: 10.1002/alr.22266. [DOI] [PubMed] [Google Scholar]
- 28.Ndika J, Airaksinen L, Suojalehto H, Karisola P, Fyhrquist N, Puustinen A, Alenius H. Epithelial proteome profiling suggests the essential role of interferon-inducible proteins in patients with allergic rhinitis. J Allergy Clin Immunol. 2017;140:1288–1298. doi: 10.1016/j.jaci.2017.05.040. [DOI] [PubMed] [Google Scholar]
- 29.Steelant B, Farre R, Wawrzyniak P, Belmans J, Dekimpe E, Vanheel H, Van Gerven L, Kortekaas Krohn I, Bullens DMA, Ceuppens JL, Akdis CA, Boeckxstaens G, Seys SF, Hellings PW. Impaired barrier function in patients with house dust mite-induced allergic rhinitis is accompanied by decreased occludin and zonula occludens-1 expression. J Allergy Clin Immunol. 2016;137:1043–1053. e5. doi: 10.1016/j.jaci.2015.10.050. [DOI] [PubMed] [Google Scholar]
- 30.Aredia F, Scovassi AI. A new function for miRNAs as regulators of autophagy. Future Med Chem. 2017;9:25–36. doi: 10.4155/fmc-2016-0173. [DOI] [PubMed] [Google Scholar]
- 31.Martinez C, Rodino-Janeiro BK, Lobo B, Stanifer ML, Klaus B, Granzow M, Gonzalez-Castro AM, Salvo-Romero E, Alonso-Cotoner C, Pigrau M, Roeth R, Rappold G, Huber W, Gonzalez-Silos R, Lorenzo J, de Torres I, Azpiroz F, Boulant S, Vicario M, Niesler B, Santos J. miR-16 and miR-125b are involved in barrier function dysregulation through the modulation of claudin-2 and cingulin expression in the jejunum in IBS with diarrhoea. Gut. 2017;66:1537–1538. doi: 10.1136/gutjnl-2016-311477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu L, Yao J, Li Z, Zu G, Feng D, Li Y, Qasim W, Zhang S, Li T, Zeng H, Tian X. miR-381-3p knockdown improves intestinal epithelial proliferation and barrier function after intestinal ischemia/reperfusion injury by targeting nurr1. Cell Death Dis. 2018;9:411. doi: 10.1038/s41419-018-0450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anton L, DeVine A, Sierra LJ, Brown AG, Elovitz MA. miR-143 and miR-145 disrupt the cervical epithelial barrier through dysregulation of cell adhesion, apoptosis and proliferation. Sci Rep. 2017;7:3020. doi: 10.1038/s41598-017-03217-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eddleston J, Christiansen SC, Zuraw BL. Functional expression of the C-X-C chemokine receptor CXCR4 by human bronchial epithelial cells: regulation by proinflammatory mediators. J Immunol. 2002;169:6445–6451. doi: 10.4049/jimmunol.169.11.6445. [DOI] [PubMed] [Google Scholar]