

Abstract
Background: Imatinib resistance is commonly associated with the activation of BCR-ABL signaling in chronic myeloid leukaemia (CML). The activation of Lyn can result in imatinib resistance by regulating the formation of BCR-ABL protein complexes. SIRT1 is a novel survival pathway activated by BCR-ABL expression in haematopoietic progenitor cells. This study aimed to investigate whether the signaling pathway of Lyn/BCR-ABL/SIRT1 could mediate imatinib resistance in CML. Methods: The MTT assay was used to detect cell viability. Apoptosis was measured by a flow cytometry assay. Protein expression was detected by Western blotting. Knockdown CML cells were constructed by shRNA interference. The CML mouse model was used to investigate the role of SIRT1 in CML in vivo. Results: Lyn was overexpressed in K562R cells. BCR-ABL phosphorylation and activation were promoted by Lyn. Imatinib suppressed BCR-ABL phosphorylation in both K562 and K562R cells. BCR-ABL positively regulated SIRT1 and Foxo1 but negatively regulated acetylated Foxo1 (Ac-Foxo1) and p53 expression. Pharmacological inhibition of SIRT1 or knockdown of SIRT1 increased apoptosis and reduced growth in vitro and in vivo. Foxo1 was downregulated by SIRT1 inhibition or knockdown, while Ac-Foxo1 and p53 were upregulated. In vivo experiments showed that imatinib and/or SIRT1 inhibition both prolonged the survival of the CML mouse model and that the effects of imatinib were enhanced in combination with SIRT1 inhibition. Conclusion: We proposed a novel molecular mechanism of imatinib resistance in CML in which the high expression of Lyn in imatinib-resistant cells inhibited Ac-Foxo1 and p53 expression through the BCR-ABL/SIRT1/Foxo1 signaling pathway, thus reducing apoptosis and mediating imatinib resistance.
Keywords: Lyn, BCR-ABL, SIRT1, imatinib resistance, chronic myeloid leukaemia
Introduction
Chronic myeloid leukaemia (CML) is a haematopoietic malignancy that accounts for approximately 15%-20% of adult leukaemias [1]. It originates from bone marrow (BM) haematopoietic stem cells and is characterized by unregulated growth of myeloid cells in BM and the accumulation in blood [2]. Approximately 95% of patients with CML express BCR-ABL fusion protein as a consequence of the translocation between chromosomes 9 and 22, which generates a shortened chromosome 22 called the Philadelphia (Ph) chromosome [3,4]. BCR-ABL usually leads to uncontrollable development of CML. Recently, targeted therapies have shown promising prospects. However, some patients may experience primary resistance or secondary resistance, resulting in no response to targeted therapies [5]. Therefore, it is essential to explore the mechanism of leukaemia drug resistance and to further increase therapeutic success by combining targeted strategies.
BCR-ABL is a kind of constitutively active tyrosine kinase expressed in Ph chromosome-positive leukaemia [6]. For most CML patients, the hyperactive fusion kinase BCR-ABL was confirmed to be the basis of targeted therapy by the tyrosine kinase inhibitor imatinib [7]. Imatinib as an initial therapy was found to induce durable responses in a high proportion of patients with CML and effectively control the disease progression of BCR-ABL-positive leukaemias [8]. As previously reported, a reduction in imatinib activity was associated with changes in BCR-ABL structure and expression, including mutations, amplification, insertions or deletions [9-11]. However, these changes alone could not account for all imatinib failures. Resistance of K562 cells to imatinib occurred independent of BCR-ABL mutations or amplification but was associated with overexpression of Lyn kinase, which is a member of the Src-family kinases (SFKs) and is unaffected by imatinib [12]. It has been reported that Lyn kinase is highly expressed and activated in patients with CML, and Lyn kinase activation increases the phosphorylation of BCR-ABL [12,13]. These results suggested that persistent Lyn kinase overexpression or activation may participate in imatinib resistance.
In addition, in Lyn-overexpressing cells after imatinib incubation, incomplete suppression of BCR-ABL signaling was found to be related to the formation of Lyn/Gab2/BCR-ABL complexes and the persistent tyrosine phosphorylation of both BCR-ABL (Y177) and Gab2 [13]. Wu, J. et al. described the detailed regulatory activities of Lyn in CML cells as follows. The silencing of Lyn or Gab2 could induce apoptosis in CML cells. Meanwhile, Lyn also binds tightly to c-Cbl, which is a tyrosyl-phosphoprotein. When Lyn expression was inhibited, c-Cbl was stabilized. Lyn regulates the formation of BCR-ABL protein complexes, and its overexpression or activation leads to inadequate control of BCR-ABL signaling and CML disease progression on imatinib therapy [13].
SIRT1, a nicotinamide adenine dinucleotide-dependent protein deacetylase, is overexpressed in various solid tumours and haematopoietic malignancies [2,3]. The inactivation of SIRT1 could inhibit proliferation and promote cell apoptosis in human tumour cells [14]. SIRT1 was transcriptionally activated by BCR-ABL to promote the expression of Foxo1. SIRT1 is a novel survival pathway activated by BCR-ABL expression in haematopoietic progenitor cells that promotes leukaemogenesis and oncogenic transformation [15]. However, whether SIRT1 plays a critical role in imatinib resistance in CML has not been elucidated.
The aim of this study was to investigate whether the signaling pathway of Lyn/BCR-ABL/SIRT1 mediates the imatinib resistance of CML and to explore the potential molecular mechanism. In the present study, we propose a novel molecular mechanism of imatinib resistance in CML in which the high expression of Lyn in imatinib-resistant cells inhibits Foxo1 acetylation and p53 expression through the BCR-ARL/SIRT1/Foxo1 signaling pathway, thus reducing apoptosis and resulting in imatinib resistance. To our knowledge, this is the first study focused on the signaling pathway of Lyn/BCR-ABL/SIRT1 in imatinib-resistant CML. Thus, our study provides a novel perspective to study the mechanism of imatinib resistance and a new target for the targeted therapy of CML. Meanwhile, our results suggest that it may be beneficial to combine SIRT1 inhibitors with approved ABL tyrosine kinase inhibitors (TKIs), such as imatinib, for patients with CML.
Materials and methods
Materials and reagents
Imatinib mesylate (IM) and sirtinol were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Antibodies to Lyn, BCR-ABL, SIRT1, Foxo1, acetylated-Foxo1 (Ac-Foxo1) and p53 for immunoblotting were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Rabbit monoclonal pY177-BCR-ABL and pY396-Lyn (Epitomics, Burlingame, CA) were used to detect activated BCR-ABL and activated Lyn. RPMI-1640 and foetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY).
Cell lines and cell culture
Human CML cell lines, namely, KCL-22, K562 (imatinib-sensitive), and K562R (imatinib-resistant), were obtained from the American Type Culture Collection (ATCC, USA). KCL-22, K562 and K562R cells were grown in RPMI-1640 medium supplemented with 10% FBS, 100 μg/ml streptomycin and 100 units/ml penicillin. The medium was changed twice a week, and the cells were cultured in a humidified incubator at 37°C with 5% (v/v) CO2/95% (v/v) air. The cells were sub-cultured using trypsin (2.5 g/l) when confluent (> 80% confluence).
Cell viability assays and apoptosis analysis
The MTT assay was used to detect the effects of IM on the proliferation of CML cells. The MTT assay was performed using the Cell Proliferation Reagent Kit I (MTT; Roche Applied Science) according to the manufacturer’s protocol. A total of 1×104 cells/well were seeded in 96-well plates. Four hours after incubation, according to experimental grouping, cells were treated with IM at different concentrations (0, 0.1, 1, 5, and 10 μM) and incubated at 37°C for 48 hours. After IM treatment, MTT solution (5 mg/mL) was added to each well and incubated for another 6 hours. Then, the medium was removed, and 150 μL/well DMSO was added. The optical density (OD) value was measured at 490 nm.
Cell apoptosis was analysed with the Annexin-V FITC apoptosis kit (BD Biosciences, CA, USA). Cells were stained with FITC-annexin V and propidium iodide (PI) according to the manufacturer’s protocol. The apoptosis rate was measured through flow cytometry (BD FACSAria™ Fusion).
Western blotting
Whole-cell lysates were prepared using RIPA lysis buffer. Protein lysates were separated by 15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, USA). Then, the PVDF membrane was blocked in skim milk at 25°C for 2 hours and subsequently incubated at 4°C overnight with specific primary antibodies against Lyn, p-Lyn, BCR-ABL, p-BCR-ABL, SIRT1, Foxol, Ac-Foxo1, p53 or GAPDH separately. Next, the PVDF membrane was stained with HRP-linked secondary antibodies and imaged using a scanner. GAPDH was used as an endogenous control.
RNA interference using short hairpin RNA (shRNA)
RNA interference was performed using Lipofectamine 3000 Reagent (Thermo Fisher Scientific, Waltham, USA) according to the manufacturer’s instructions. Lyn, BCR-ABL, and SIRT1 shRNA constructed by the pRNAT-U6.1/Neo vector were used to transduce KCL-22, K562, and K562R cells. Cells that had a control shRNA unrelated to target sequences were used as a negative control. The knockdown of target proteins was confirmed by immunoblotting analysis after transfection.
CML mouse model
Specific-pathogen-free male wild-type C57BL/6J mice (aged 4-10 weeks and weighing 20-25 g) from the Shanghai National Center for Laboratory Animals (Shanghai, China) were used for the induction of CML. Mice were housed in a standard laboratory under a 12-hour light/dark cycle at a temperature/humidity of 25 ± 2°C/60 ± 5% and free access to food and water. For the construction of the CML mouse model, the retroviral vector MSCR-IRES-BCR-ABL-WT-eGFP was used to generate high-titre retrovirus stocks as previously described [16]. Donor mice were pre-treated with 5-fluorouracil (5-FU; 200 mg/kg) by intravenous tail injection. Then, BM cells were harvested and transduced twice with BCR-ABL retroviral stock in the presence of interleukin (IL)-3, IL-6 and stem cell factor. Subsequently, 1×105 virally transduced cells were transplanted by lateral tail vein injection into recipient mice conditioned by two doses of 550-cGy γ-irradiation, following that imatinib (200 mg/kg/day), a SIRT1 inhibitor sirtinol (50 mg/kg/day) alone or imatinib plus sirtinol also was intravenously injected into recipient mice for 14 consecutively days. Recipient mice were evaluated daily for symptoms of CML, including weight loss, body temperature, splenomegaly and overall signs of morbidity (decreased level of activity). The recipient mice were randomly divided into four groups (N=10/group): (1) group receiving intravenous injection of saline once every day (CML group), (2) group receiving intravenous injection of imatinib once every day (IM group), (3) group receiving intravenous injection of sirtinol once every day (sirtinol group), and (4) group which receiving injection of imatinib and sirtinol once every day (IM+sirtinol group). Moreover, wild-type or SIRT1-knockdown donor BM cells were transduced with BCR-ABL retroviral vectors, and 1×105 cells were transplanted into each recipient by lateral tail vein injection. Imatinib (200 mg/kg/day) was used to treat the mice to evaluate the effect of the combination of SIRT1 knockdown with imatinib on CML mouse survival. The recipient mice were also randomly divided into four groups (N=10/group): (1) CML group, (2) IM group, (3) shSIRT1 group, and (4) IM+shSIRT1 group. All animal procedures were carried out in accordance with protocols approved by the Animal and Ethics Review Committee of The First Affiliated Hospital of Harbin Medical University (Ethics approval number: IRB-AF/SC-04/01.0).
Flow cytometry for white blood cells and BM cells
Beginning at 10 days after BM transplantation, CML mice were treated with sirtinol (50 mg/kg/day) and/or imatinib (200 mg/kg/day) or the blank vehicle (controls) for 10 days. Haematopoietic cells and leukaemic cells from BM were collected from the diseased mice and analysed by fluorescence-activated cell sorting (FACS) analysis. Then, green fluorescent protein-positive (GFP+) BM leukaemic cells were analysed using an Annexin-V FITC apoptosis kit to measure cell apoptosis.
Colony formation assay
A colony formation assay was performed to detect the survival of GFP+ BM cells. Cells were exposed to 2.5 μM sirtinol and/or 1 μM IM for 24 hours. Then, the standard two-layer soft agar culture was performed, and colonies were stained with crystal violet. Colony formation (CFC) numbers were manually counted under a microscope (Olympus CKX41, Tokyo, Japan).
Statistical analysis
Statistical analysis was performed with GraphPad Prism 6 software (GraphPad Software Inc., CA, USA). Survival analysis was performed by Kaplan-Meier analysis. The results from each experiment are expressed as the mean ± standard deviation (SD) of three separate experiments. Statistical significance was determined with ANOVA followed by a Bonferroni post hoc analysis or Student’s t-test. Differences were considered statistically significant when P < 0.05.
Results
Treatment of K562 and K562R cells with imatinib
We first investigated the effect of imatinib on the proliferation and apoptosis of K562 and K562R cells. The K562 and K562R cell lines were exposed to increasing concentrations of imatinib. We observed that imatinib strongly reduced cell viability and promoted the apoptosis of K562 cells in a dose-dependent manner, while it had no significant effect on K562R cells (Figure 1A and 1B). To confirm the role of Lyn, BCR-ABL and SIRT1 in CML, we detected the effect of imatinib on protein expression using Western blotting. As shown in Figure 1C, the expression levels of Lyn, BCR-ABL and SIRT1 were elevated in imatinib-resistant K562R cells, suggesting that Lyn, BCR-ABL and SIRT1 might be involved in the imatinib resistance of CML. After imatinib treatment, the expression of Lyn, BCR-ABL and SIRT1 decreased in a drug dose-dependent manner in K562 cells, but no significant effects were found in K562R cells (Figure 1C). These results suggest that the high expression of Lyn, BCR-ABL and SIRT1 may be involved in imatinib resistance.
Figure 1.

Differential sensitivity of the K562 and K562R cell lines to imatinib.The K562 and K562R cell lines were exposed to various concentrations (0, 0.1, 1, 5, and 10 μM) of imatinib. A. The MTT assay was performed to detect the cell viability of imatinib-treated cells; B. The flow cytometric assay was performed to assess the apoptosis rates of imatinib-treated cells; C. The protein expression of Lyn, BCR-ABL and SIRT1 was measured by Western blotting, with GAPDH acting as the endogenous control. The results are expressed as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Lyn knockdown inhibits BCR-ABL expression and phosphorylation
To determine whether Lyn regulates BCR-ABL expression and phosphorylation, sh-Lyn and its shRNA negative control (sh-NC) were transfected into the K562 and K562R cell lines. As shown in Figure 2, Lyn expression was higher in imatinib-resistant (K562R) cells than in imatinib-sensitive (K562) cells. After Lyn knockdown, the expression of Lyn, BCR-ABL and p-BCR-ABL was significantly decreased in both the K562 and K562R cell lines. The results suggested that BCR-ABL was regulated by Lyn and that Lyn could promote BCR-ABL phosphorylation and activation. After treatment with imatinib in Lyn knockdown cells, BCR-ABL phosphorylation was rapidly suppressed in both the K562 and K562R cell lines, indicating that the combination of Lyn knockdown and imatinib effectively blocked BCR-ABL tyrosine phosphorylation. These results suggested that BCR-ABL phosphorylation and activation were suppressed through Lyn-directed inhibition with imatinib.
Figure 2.

Lyn knockdown inhibits BCR-ABL expression and phosphorylation.The knockdown of Lyn was induced by short hairpin RNA (shRNA) using control shRNA as a negative control (NC). Transfected cells (K562 and K562R) were treated with 5 μM imatinib. Then, the protein expression of Lyn, phosphorylated (p)-Lyn, BCR-ABL and p-BCR-ABL was measured by Western blotting, with GAPDH acting as the endogenous control. The results are expressed as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
BCR-ABL knockdown inhibits SIRT1 expression
To investigate the regulatory mechanism of SIRT1 by BCR-ABL, BCR-ABL expression was reduced by shRNA silencing. The reduction in BCR-ABL expression decreased the levels of SIRT1 and Foxo1 proteins both in K562 and K562R cells and increased the levels of Ac-Foxo1 and p53 proteins (Figure 3). In cells with BCR-ABL knockdown, imatinib was able to suppress the expression of SIRT1 and Foxo1 and promote Ac-Foxo1 and p53 expression. The results suggested that SIRT1 and Foxo1 were positively regulated by BCR-ABL, while Ac-Foxo1 and p53 were negatively regulated by BCR-ABL.
Figure 3.

BCR-ABL positively regulated SIRT1 and Foxo1 and negatively regulated Ac-Foxo1 and p53.The knockdown of BCR-ABL was induced by short hairpin RNA (shRNA), using control shRNA as a negative control (NC). Transfected cells (K562 and K562R) were treated with 5 μM imatinib. Then, Western blotting was performed to measure the protein expression of SIRT1, Foxo1, acetylated-Foxo1 (Ac-Foxo1) and p53, with GAPDH acting as the endogenous control. The results are expressed as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
SIRT1 promotes Foxo1 and inhibits Ac-Foxo1 and p53 expression
To confirm the regulatory effects of SIRT1 on downstream proteins, we treated K562 and KCL-22 cells with an inhibitor of SIRT1 (sirtinol), imatinib, or a combination of both. After imatinib or sirtinol treatment, the expression of SIRT1 was reduced in a dose-dependent manner in K562 and KCL-22 cells, while no change was observed in K562R cells (Figure 4A). Both imatinib and sirtinol suppressed cell survival and promoted cell apoptosis (Figure 4B and 4C). The combination of sirtinol with imatinib enhanced the inhibitory effects (Figure 4B and 4C). As shown in Figure 4D, the expression of SIRT1 and Foxo1 was inhibited by both imatinib and sirtinol, while Ac-Foxo1 and p53 were upregulated. The combination of sirtinol with imatinib synergistically downregulated the expression of SIRT1 and Foxo1 and upregulated Ac-Foxo1 and p53 expression.
Figure 4.

SIRT1 promotes Foxo1 and inhibits Ac-Foxo1 and p53 expression. A. Human CML cell lines (K562, K562R and KCL-2) were exposed to various concentrations of imatinib or sirtinol. Then, SIRT1 protein expression was measured by Western blotting. The KCL-2 and K562 cell lines were exposed to imatinib (1 μM) and/or sirtinol (50 μM). B. Surviving cell numbers were analysed using the MTT assay on days 2 and 4 after treatment; C. Apoptosis was analysed by flow cytometry on day 4 after treatment; D. The protein expression levels of SIRT1, Foxo1, Ac-Foxo1 and p53 were measured by Western blotting on day 4 after treatment. The knockdown of BCR-ABL in the KCL-2 and K562 cell lines was induced by short hairpin RNA (shRNA). After 48 hours of shSIRT1 interference, cells were cultured with or without imatinib (1 μM). At day 2 and day 4 after treatment. E. The surviving cell number was analysed using the MTT assay. F. The apoptosis was evaluated by flow cytometry. G. The protein expression levels of SIRT1, Foxo1, Ac-Foxo1 and p53 were measured by Western blotting on day 4 after treatment. The results are expressed as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
To further confirm the above results of SIRT1 inhibition, we examined the effect of SIRT1 knockdown. Cell proliferation was inhibited by both imatinib and SIRT1 knockdown, and SIRT1 knockdown enhanced the imatinib inhibition of cell proliferation (Figure 4E). Both imatinib and SIRT1 knockdown promoted cell apoptosis, and the combination of SIRT1 knockdown with imatinib enhanced the inhibitory effects (Figure 4F). As shown in Figure 4G, Foxo1 expression was inhibited by both imatinib and SIRT1 knockdown, while Ac-Foxo1 and p53 were upregulated. The combination of SIRT1 knockdown with imatinib synergistically downregulated Foxo1 expression and upregulated Ac-Foxo1 and p53 expression. These results suggested that SIRT1 mediates imatinib resistance by promoting Foxo1 expression and inhibiting Foxo1 deacetylation and p53 expression.
Inhibition of SIRT1 prolongs the survival of the CML mouse model
To further provide in vivo evidence for the molecular mechanism of resistance, we constructed a CML mouse model and treated it with sirtinol or imatinib to verify the role of SIRT1 in the imatinib resistance of CML. As shown in Figure 5A and 5B, both sirtinol and imatinib significantly extended CML mouse survival and reduced WBC counts. In addition, the combination of sirtinol with imatinib further extended CML mouse survival compared to single-drug treatment. To confirm whether these results were caused by species differences, BM cells were isolated from the CML mouse model and treated with drugs in vitro. Sirtinol enhanced the imatinib promotion of cell apoptosis and further suppressed CFC formation (Figure 5C and 5D). We also examined the effect of SIRT1 knockdown on CML mouse survival to assess whether SIRT1 knockdown improves imatinib effectiveness. BCR-ABL-transduced wild-type or SIRT1 knockdown BM cells were transplanted, and the recipient mice were treated with vehicle or imatinib. Compared to wild-type mice and vehicle control mice, imatinib extended the survival of CML mice (P < 0.05), similar to SIRT1 knockdown, while the combination of SIRT1 knockdown and imatinib significantly extended CML mouse survival compared to individual treatment (P < 0.05) (Figure 5E). These results suggested that specific SIRT1 inhibition may enhance imatinib effectiveness and prolong the survival of CML mice. Together, our in vivo studies further confirmed that SIRT1 mediated imatinib resistance and that SIRT1 inhibition may be a novel targeted therapy method for reversing imatinib resistance.
Figure 5.
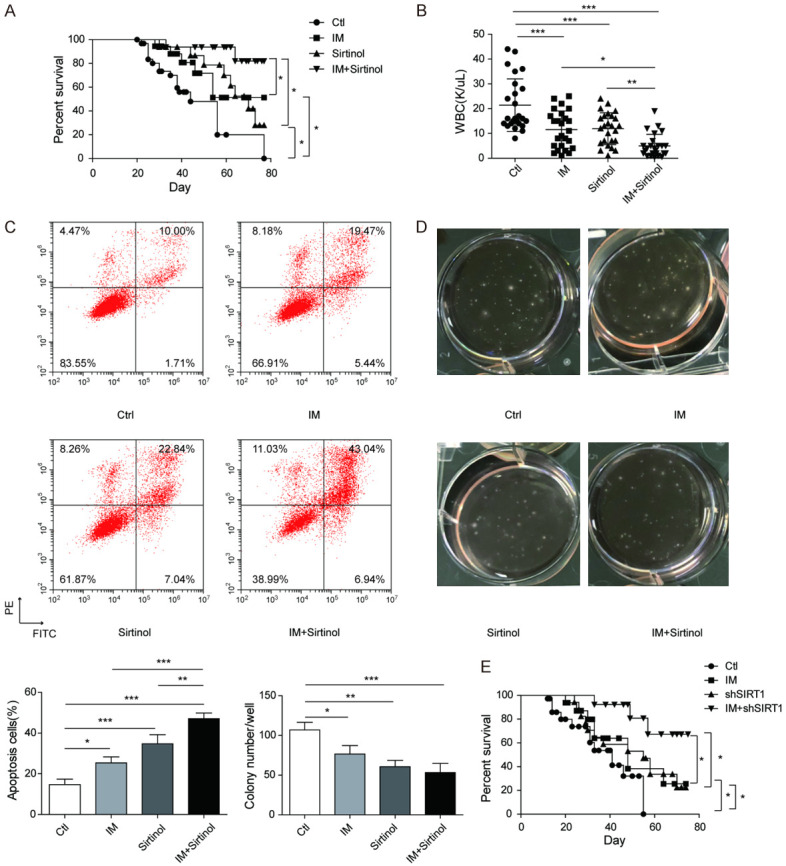
Inhibition of SIRT1 prolongs the survival of the CML mouse model. CML mice were constructed successfully and randomly divided into four groups (n=10 mice each). Sirtinol (50 mg/kg/day) and/or imatinib (200 mg/kg/day) were used to treat CML mice, using untreated mice as a blank control. A. Survival analysis was performed by Kaplan-Meier analysis; B. GFP+ white blood cell counts were analysed using flow cytometry at 3 weeks after treatment. Bone marrow (BM) cells from CML mice were cultured with sirtinol (2.5 μM) and/or imatinib (1 μM) for 3 days. C. Apoptosis was analysed in the GFP+ cell fraction. D. The colony formation ability of GFP+ BM cells was analysed using a colony formation assay. E. Wild-type and SIRT1 knockdown CML mice were used to evaluate the effect of the combination of SIRT1 knockdown with imatinib on CML mouse survival. Wild-type or SIRT1-knockdown donor BM cells were transduced with BCR-ABL retroviral vectors, and 1×105 cells were transplanted into each recipient. Imatinib (200 mg/kg/day) was used to treat the mice. Then, survival analysis was performed by Kaplan-Meier analysis. The results are expressed as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Targeted therapy with imatinib is now used as the frontline therapy for CML [17]. Nevertheless, imatinib resistance has become a major obstacle for successful treatment with chemotherapy [18]. Therefore, elucidation of the imatinib resistance mechanism in CML and reversal of imatinib resistance are critical. In the current study, we investigated the role of the Lyn/BCR-ABL/SIRT1 signaling pathway in imatinib-resistant CML cells. Our findings revealed that Lyn/BCR-ABL/SIRT1 and its downstream Foxo1/Ac-Foxo1/p53 signaling pathway were associated with imatinib resistance.
First, we detected imatinib sensitivity and protein expression in imatinib-sensitive and imatinib-resistant cell lines. The K562 cell lines were sensitive to imatinib, whereas K562R cells were resistant to imatinib with a higher cell viability and lower apoptosis than sensitive cells. Meanwhile, high expression of Lyn, BCR-ABL and SIRT1 was measured in imatinib-resistant cell lines, suggesting that Lyn, BCR-ABL and SIRT1 may be involved in imatinib resistance.
In the present study, imatinib was confirmed to reduce the expression and phosphorylation of BCR-ABL. Although this inhibitory effect also occurred in imatinib-resistant cells, resistant cells were still insensitive to imatinib and resisted apoptosis with high cell viability. On the one hand, the mechanism of resistance may be related to the overexpression of Lyn in drug-resistant cells. O’Hare T et al. previously proposed a phenomenon called transferred addiction [19]. Increased expression of Lyn and persistent Lyn activity were found in imatinib-resistant cells, thus raising the speculation that Lyn regulation is no longer primarily downstream of and dependent upon BCR-ABL signaling [19]. Imatinib blocked BCR-ABL signaling but did not suppress Lyn phosphorylation due to its unique Lyn phosphorylation sites and associated proteins [20]. Thus, the overexpression of Lyn still leads to a degree of BCR-ABL phosphorylation in imatinib-resistant cells that mediates the resistance of the cells to imatinib.
On the other hand, we speculated that imatinib resistance may be related to the BCR-ARL/SIRT1/Foxo1 pathway. In CML cells, the knockdown of BCR-ABL significantly inhibited SIRT1 expression, indicating that SIRT1 was downstream of BCR-ABL and positively regulated by BCR-ABL. SIRT1 is a multi-functional protein and is involved in gene silencing and heterochromatin formation [21]. It has been confirmed to play an important role in myeloid leukemogenesis and drug resistance [22]. Similarly, Yuan et al. previously demonstrated that SIRT1 was activated by BCR-ABL kinase in haematopoietic stem cells in STAT5 kinase-dependent or STAT5 kinase-independent manners [22]. Furthermore, the activation of SIRT1 suppressed the functions of the tumour suppressor p53 and inhibited cell apoptosis [22]. Additionally, SIRT1 has previously been shown to deacetylate p53 and thereby regulate its transcriptional activity [23]. Thus, we next explored the signaling pathway between SIRT1 and p53. Notably, we found that Foxo1, Ac-Foxo1 and p53 were also regulated by SIRT1 knockdown or sirt1 inhibition. Foxo1 was downregulated by SIRT1 knockdown or sirt1 inhibition, while p53 was upregulated with a high apoptosis rate in CML cells, suggesting that SIRT1 may positively regulate Foxo1 and thereby promote Foxo1 deacetylation and inhibit p53 expression. Yuan et al. previously also reported a similar result: Foxo1 was deacetylated in CD34+ cells upon BCR-ABL expression [22]. Thus, we concluded that BCR-ABL could enhance SIRT1, upregulate Foxo1, and finally inhibit Ac-Foxo1 and tumour suppressor p53 expression in imatinib-resistant cells. Interestingly, SIRT1 inhibition plus imatinib treatment enhanced the imatinib promotion of cell apoptosis and further suppressed CFC formation, suggesting that it may be beneficial to combine SIRT1 inhibitors with imatinib for patients with CML. The finding of the involvement of the BCR-ARL/SIRT1/Foxo1 pathway in imatinib-resistant CML cells provides a new exciting target for the potential reversal of imatinib resistance and increase in therapeutic success.
In the present study, we proposed a novel molecular mechanism of imatinib resistance in CML in which the high expression of Lyn in imatinib-resistant cells promoted Foxo1 deacetylation and then inhibited Ac-Foxo1 and p53 expression through the BCR-ARL/SIRT1/Foxo1 signaling pathway, thus reducing apoptosis and resulting in imatinib resistance. Our findings help to understand the resistance of CML cells to imatinib and provide a new target for the targeted therapy of CML. Meanwhile, our results suggest that it may be beneficial to combine SIRT1 inhibitors with imatinib for patients with CML. Therefore, a regimen containing approved ABL-TKIs and SIRT1 inhibitors may enhance the response of patients.
Acknowledgements
This work was supported by Scientific Research Subject of Heilongjiang Provincial Health and Family Planning Commission (2018120) and the First Affiliated Hospital of Harbin Medical University Scientific Research Innovation Fund (2019B02).
Disclosure of conflict of interest
None.
Abbreviations
- CML
Chronic myeloid leukemia
- Ph
Philadelphia
- SFKs
Src-family kinases
- TKIs
tyrosine kinase inhibitors
- IM
Imatinib mesylate
- FBS
fetal bovine serum
- SDS-PAGE
SDS-polyacrylamide gel electrophoresis
- PVDF
polyvinylidene fluoride
- SD
standard deviation
- shRNA
short hairpin RNA
References
- 1.Sessions J. Chronic myeloid leukemia in 2007. Am J Health Syst Pharm. 2007;64:S4–9. doi: 10.2146/ajhp070484. [DOI] [PubMed] [Google Scholar]
- 2.Ma L, Shan Y, Bai R, Xue L, Eide CA, Ou J, Zhu LJ, Hutchinson L, Cerny J, Khoury HJ, Sheng Z, Druker BJ, Li S, Green MR. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med. 2014;6:252ra121. doi: 10.1126/scitranslmed.3009073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagle M, Eiring AM, Wongchenko M, Lu S, Guan Y, Wang Y, Lackner M, Amler L, Hampton G, Deininger MW, O’Hare T, Yan Y. A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Leukemia. 2016;30:1493–1501. doi: 10.1038/leu.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halbach S, Kohler M, Uhl FM, Huber J, Zeiser R, Koschmieder S, Aumann K, Brummer T. Gab2 is essential for Bcr-Abl-mediated leukemic transformation and hydronephrosis in a chronic myeloid leukemia mouse model. Leukemia. 2016;30:1942–1945. doi: 10.1038/leu.2016.92. [DOI] [PubMed] [Google Scholar]
- 5.Hantschel O, Warsch W, Eckelhart E, Kaupe I, Grebien F, Wagner KU, Superti-Furga G, Sexl V. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol. 2012;8:285–293. doi: 10.1038/nchembio.775. [DOI] [PubMed] [Google Scholar]
- 6.Cortez D, Reuther G, Pendergast AM. The Bcr-Abl tyrosine kinase activates mitogenic signaling pathways and stimulates G1-to-S phase transition in hematopoietic cells. Oncogene. 1997;15:2333–2342. doi: 10.1038/sj.onc.1201400. [DOI] [PubMed] [Google Scholar]
- 7.Shinohara H, Kumazaki M, Minami Y, Ito Y, Sugito N, Kuranaga Y, Taniguchi K, Yamada N, Otsuki Y, Naoe T, Akao Y. Perturbation of energy metabolism by fatty-acid derivative AIC-47 and imatinib in BCR-ABL-harboring leukemic cells. Cancer Lett. 2016;371:1–11. doi: 10.1016/j.canlet.2015.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, Letvak L, Larson RA. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 9.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 10.Gupta P, Kathawala RJ, Wei L, Wang F, Wang X, Druker BJ, Fu LW, Chen ZS. PBA2, a novel inhibitor of imatinib-resistant BCR-ABL T315I mutation in chronic myeloid leukemia. Cancer Lett. 2016;383:220–229. doi: 10.1016/j.canlet.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 11.Patkar N, Ghodke K, Joshi S, Chaudhary S, Mascerhenas R, Dusseja S, Mahadik S, Gaware S, Tembhare P, Gujral S, Kabre S, Kadam-Amare P, Jain H, Dangi U, Bagal B, Khattry N, Sengar M, Arora B, Narula G, Banavali S, Menon H, Subramanian PG. Characteristics of BCR-ABL kinase domain mutations in chronic myeloid leukemia from India: not just missense mutations but insertions and deletions are also associated with TKI resistance. Leuk Lymphoma. 2016;57:2653–2660. doi: 10.3109/10428194.2016.1157868. [DOI] [PubMed] [Google Scholar]
- 12.Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, Talpaz M. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–698. doi: 10.1182/blood.V101.2.690. [DOI] [PubMed] [Google Scholar]
- 13.Wu J, Meng F, Lu H, Kong L, Bornmann W, Peng Z, Talpaz M, Donato NJ. Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib-resistant chronic myelogenous leukemia cells. Blood. 2008;111:3821–3829. doi: 10.1182/blood-2007-08-109330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 15.Yuan H, Wang Z, Li L, Zhang H, Modi H, Horne D, Bhatia R, Chen W. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood. 2012;119:1904–1914. doi: 10.1182/blood-2011-06-361691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Peng C, Hu Y, Li H, Sheng Z, Chen Y, Sullivan C, Cerny J, Hutchinson L, Higgins A, Miron P, Zhang X, Brehm MA, Li D, Green MR, Li S. The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat Genet. 2012;44:861–871. doi: 10.1038/ng.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldman JM, Melo JV. Chronic myeloid leukemia--advances in biology and new approaches to treatment. N Engl J Med. 2003;349:1451–1464. doi: 10.1056/NEJMra020777. [DOI] [PubMed] [Google Scholar]
- 18.Bixby D, Talpaz M. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. Hematology Am Soc Hematol Educ Program. 2009:461–476. doi: 10.1182/asheducation-2009.1.461. [DOI] [PubMed] [Google Scholar]
- 19.O’Hare T, Eide CA, Deininger MW. Persistent LYN signaling in imatinib-resistant, BCR-ABL-independent chronic myelogenous leukemia. J Natl Cancer Inst. 2008;100:908–909. doi: 10.1093/jnci/djn204. [DOI] [PubMed] [Google Scholar]
- 20.Wu J, Meng F, Kong LY, Peng Z, Ying Y, Bornmann WG, Darnay BG, Lamothe B, Sun H, Talpaz M, Donato NJ. Association between imatinib-resistant BCR-ABL mutation-negative leukemia and persistent activation of LYN kinase. J Natl Cancer Inst. 2008;100:926–939. doi: 10.1093/jnci/djn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaquero A. The conserved role of sirtuins in chromatin regulation. Int J Dev Biol. 2009;53:303–322. doi: 10.1387/ijdb.082675av. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, Bhatia R. Roles of SIRT1 in leukemogenesis. Curr Opin Hematol. 2013;20:308–313. doi: 10.1097/MOH.0b013e328360ab64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]