

Abstract
Accurate diagnosis of sepsis remains challenging, new markers or combinations of markers are urgently needed. In the present study, we screened differentially expressed genes (DEGs) between sepsis and non-sepsis blood samples across three previously published gene expression data sets. Common upregulated and downregulated DEGs were ranked according to their average functional similarity. The ten genes (OLFM4, ORM1, CEP55, S100A12, S100P, LRG1, CEACAM8, MS4A4A, PLSCR1, and IL1R2) with the largest average functional similarity among the common upregulated genes and another ten genes (THEMIS, IL2RB, CD2, IL7R, CD3E, KLRB1, PVRIG, CCRR3, TGFBR3, and PLEKHA1) with the largest average functional similarity among the common downregulated genes were separately identified as the upregulated crucial gene set and the downregulated crucial gene set. Gene set variation analysis (GSVA) was used to obtain the GSVA index of each sample against the two crucial gene sets. Both the two crucial GSVA indexes may be robust markers for sepsis with high area under ROC curve. The diagnostic utility of the upregulated GSVA index was validated in another independent data set. Functional analyses revealed several sepsis-related pathways. In conclusion, we proposed two sepsis-related gene sets across multiple data sets and created two GSVA indexes with promising diagnostic value.
Keywords: Sepsis, systemic inflammatory response syndrome, biomarker, gene set variation analysis
Introduction
Sepsis is a heterogeneous disease involving a dysregulated systemic response to infections caused by bacteria, fungi or viruses, which may lead to organ dysfunction [1]. No specific treatment currently exists for sepsis [2]. Sepsis is a common illness and one of the ten leading causes of death worldwide [3]. Even though there has been some progress in the treatment of sepsis in the last years. The mortality rate of sepsis is still terrible, the risk of death from sepsis is as high as 30%, and risk of death rises to 50% in the case of severe sepsis and around 80% in the case of septic shock [4]. The diagnosis of sepsis can be complicated due to its clinical similarities with systemic inflammatory response syndrome (SIRS), which can also lead to multiple organ failure and death [5]. SIRS can be caused by infection, trauma, burns, pancreatitis, or a variety of other injuries [6]. A previous study proposed that screening ward patients using SIRS criteria for identifying those with sepsis would be impractical [7], thus, one of the challenges facing sepsis research is to determine which patients are truly infected.
Antibiotics are currently the standard treatment for sepsis [8]. However, delays in the use of antibiotics can result in increased mortality [9]. Since sepsis and non-bacterial SIRS are hard to distinguish in the early stages, their clinical symptoms were very similar. Antibiotics are often abused, which may further contribute to the spread of drug resistance. Blood purification techniques, immunomodulatory drugs and treatments targeting other systems including the heart, endothelial cells or coagulation cascades are also used for sepsis management [10], but they lack specificity. The biomarkers such as c-reactive protein (CRP) [11] and procalcitonin (PCT) [12] could be used to distinguish bacterial sepsis from other inflammatory conditions, but they had some limitation [13]. There was another biomarker, lactate which could use to diagnosis sepsis, however, it lacks specificity [14]. Therefore, in order to establish a targeted treatment for sepsis, it is necessary to first establish a diagnostic model able to distinguish sepsis and non-sepsis conditions.
In this study, we identified differentially expressed genes (DEGs) between sepsis and non-sepsis blood samples across multiple data sets and created two gene sets separately comprised of ten genes as diagnostic models of sepsis. The receiver operating characteristic (ROC) curve analysis was used to explore the potential diagnostic value of these two gene set variation analysis indexes for sepsis, and further validated this in an independent data set. Moreover, we explored the potential role of these genes in sepsis pathways using the Kyoto Encyclopedia of Genes and Genomes (KEGG), and their potential involvement in dysregulated systemic responses.
Materials and methods
Data collection and processing
Gene Expression Omnibus (GEO) datasets were downloaded from https://www.ncbi.nlm.nih.gov/geo/ and used to compare gene expression in sepsis and non-sepsis. GSE57065 [15] included whole blood gene expression profiles of 28 sepsis samples and 25 non-sepsis healthy samples, based on the GPL570 platform. Whole blood gene expression profiles of GSE69528, which included 53 sepsis samples and 85 non-sepsis (uninfected type 2 diabetes mellitus, uninfected healthy and septicemic melioidosis), were based on the GPL10558 platform. Whole blood gene expression profiles of GSE95233 [9], based on GPL16791, included 51 sepsis samples and 22 non-sepsis (healthy) samples. The normalize Between Arrays function in the limma package in R [16] was used to normalize the gene expression expression profiles. If a gene corresponded to multiple probes, the average expression value of these probes was considered to reflect the expression of the gene. The workflow of the present study was shown in Figure 1.
Figure 1.

Flowchart of the present study.
Principal component analysis (PCA) and differentially expressed gene (DEG) analysis
To evaluate whether differences in gene expression patterns in whole blood can distinguish between sepsis and non-sepsis, the prcomp [17] function was used to perform PCA [18], and the ggbiplot package in R [19] was applied to visualize the results. Compared with non-sepsis, DEG were screened using limma package, and |log fold change (FC)| > 1.5 and P adjusted by the false discovery rate (FDR) < 0.01 were considered significant. We applied Venn diagram analysis [20] to find upregulated and downregulated genes common to the three data sets.
Identification of crucial genes, Gene Set Variation Analysis (GSVA) and ROC curve analysis
Crucial genes were screened by semantic similarity in the upregulated and downregulated gene sets of all three GEO data sets. According to the semantic similarities of Gene Ontology (GO) terms used for gene annotation, we ranked the gene as used to inside the interactome by the average functional similarities between the gene and its interaction partners. Genes with the highest average functional similarity were considered crucial genes [21]. GSVA was used to assess the relative enrichment of gene sets across samples using a non-parametric approach [22]. The function of gsva was used with gsva method to score each sample for the crucial gene sets, and then each sample received a value of the upregulated GSVA index and a value of the downregulated GSVA index. In addition, ROC curve analysis was performed using pROC package [23] to evaluate the diagnostic value of the GSVA indexes for sepsis in GSE57065, GSE69528 and GSE95233 datasets.
Validation of diagnostic utility of the upregulated GSVA index
GSE123729, which comprised 15 sepsis samples and 27 non-sepsis samples (11 presurgical and 16 SIRS) based on GPL21970, was used as the validation set. Similarly, each sample was assigned a value for the upregulated GSVA index using the GSVA method. The ROC curve analysis was also applied to evaluate the diagnostic value of the upregulated GSVA index for sepsis in GSE123729. PCA was used to explore the utility of the classification between sepsis and non-sepsis using the crucial upregulated genes.
Functional enrichment analysis
Standard pathway analysis may have limited ability to reveal regulatory mechanisms of key genes hidden in long pathways or sub-pathways [24]. Therefore, in order to identify potential risk sub-pathways in sepsis, sub-pathway enrichment analysis was performed using the Subpathway Miner [25] package in R for the upregulated and downregulated genes in all three data sets. Gene set enrichment analysis (GSEA) [26] was performed using the normalized gene expression profile to explore KEGG pathways related to sepsis. GSEA software was used for this analysis (http://software.broadinstitute.org/gsea/index.jsp), and c2.cp.kegg.v6.2. symbols.gmt, which come from the Molecular Signatures Database (MSigDB) was used as the reference gene set [26,27]. A nominal value of P < 0.05 was considered statistically significant. The ggplot2 package [28] in R was used to visualize the results of the GSEA.
Results
DEGs in sepsis
The results of the PCA indicated that sepsis and non-sepsis whole blood samples had significantly different gene expression patterns (Figure 2A). Compared with non-sepsis samples, a total of 298 DEGs were found in GSE57065, 174 of which were upregulated and 124 downregulated. A total of 343 DEGs were observed in GSE69528, 198 upregulated and 145 downregulated. A total of 428 DEGs were found in GSE95233, 257 regulated and 171 downregulated (Figure 2B).
Figure 2.

Differentially expressed genes in sepsis compared to non-sepsis. A. Principal component analysis of sepsis vs. non-sepsis. B. Manhattan plot of differentially expressed genes in sepsis and non-sepsis. The top ten upregulated and downregulated genes with the highest significance (ranked by P value) are highlighted. Gray represents genes that are not significantly differentially expressed. C. Venn diagram showing upregulated genes and downregulated genes. D. Expression heatmap of upregulated and downregulated genes common to the three data sets.
A total of 88 DEGs were upregulated in all three data sets, while a total of 32 downregulated DEGs were common to the three data sets (Figure 2C). The heatmap showing upregulated and downregulated gene sets shows their potential to distinguish between sepsis and non-sepsis (Figure 2D).
Crucial GSVA index may have diagnostic for sepsis
We ranked genes by their average functional similarity relationships with other genes within the interactome. In the three GEO data sets, the ten genes with the largest average functional similarity among the common upregulated genes of sepsis were OLFM4, ORM1, CEP55, S100A12, S100P, LRG1, CEACAM8, MS4A4A, PLSCR1 and IL1R2 (Figure 3A), while THEMIS, IL2RB, CD2, IL7R, CD3E, KLRB1, PVRIG, CCRR3, TGFBR3 and PLEKHA1 showed the largest functional similarity among the common downregulated genes (Figure 3B). The heatmap showed that these DEG expression patterns could distinguish sepsis from non-sepsis (Figure 3C). In the ROC curve analysis, the GSVA index of upregulated genes had an AUC = 0.9849 in GSE57065, an AUC = 0.8276 in GSE69528, and an AUC = 0.7669 in GSE95233 (Figure 3D, 3E). The GSVA index of downregulated genes had an AUC = 0.9712 in GSE57065, AUC = 0.8469 in GSE69528, and AUC = 0.6087 in GSE95233.
Figure 3.

The two crucial gene set variation analysis (GSVA) indexes in sepsis. A. Crucial upregulated genes. B. Crucial downregulated genes. C. Heatmap of crucial genes and GSVA index in all three data sets. D. Receiver operating characteristic (ROC) curve analysis of upregulated GSVA index. E. ROC curve analysis of downregulated GSVA index.
Validation of upregulated GSVA index in an independent data set
The ROC analysis suggested that the upregulated GSVA index may be a robust marker for sepsis, with AUC = 0.929 (Figure 4A) in the independent data set. Compared with non-sepsis samples, the upregulated GSVA index in sepsis samples was significantly higher (Figure 4B). Subsequently, we found that sepsis and non-sepsis could be well differentiated in GSE123729 based on the expression patterns of the crucial upregulated genes (Figure 4C).
Figure 4.
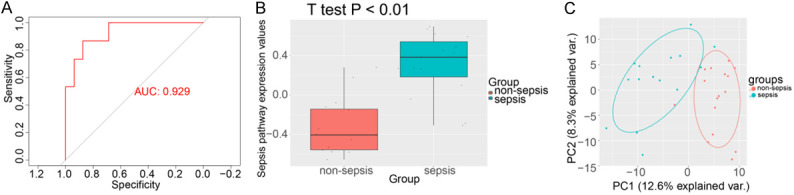
Validation of the upregulated gene set variation analysis (GSVA) index using the GSE123729 expression data set. A. ROC curve analysis of the upregulated gene set. B. Comparison of GSVA score in sepsis vs. non-sepsis in GSE123729. C. Principal component analysis of the crucial upregulated gene set in GSE123729.
Dysfunctional pathways in sepsis
The sub-pathway enrichment analysis indicated that the common upregulated genes were involved in various sepsis-related pathways, such as coagulation cascades, P53 signaling pathways and MAPK signaling pathways. The common downregulated genes were involved in Jak-STAT, PI3K-Akt and T cell receptor signaling pathways (Figure 5A). The GSEA results also confirmed that complement and coagulation cascades and P53 signaling pathway were enriched in sepsis samples in all three data sets. In contrast, allograft rejection, autoimmune thyroid disease and T-cell receptor signaling pathway were enriched in the non-sepsis samples in the three data sets (Figure 5B).
Figure 5.
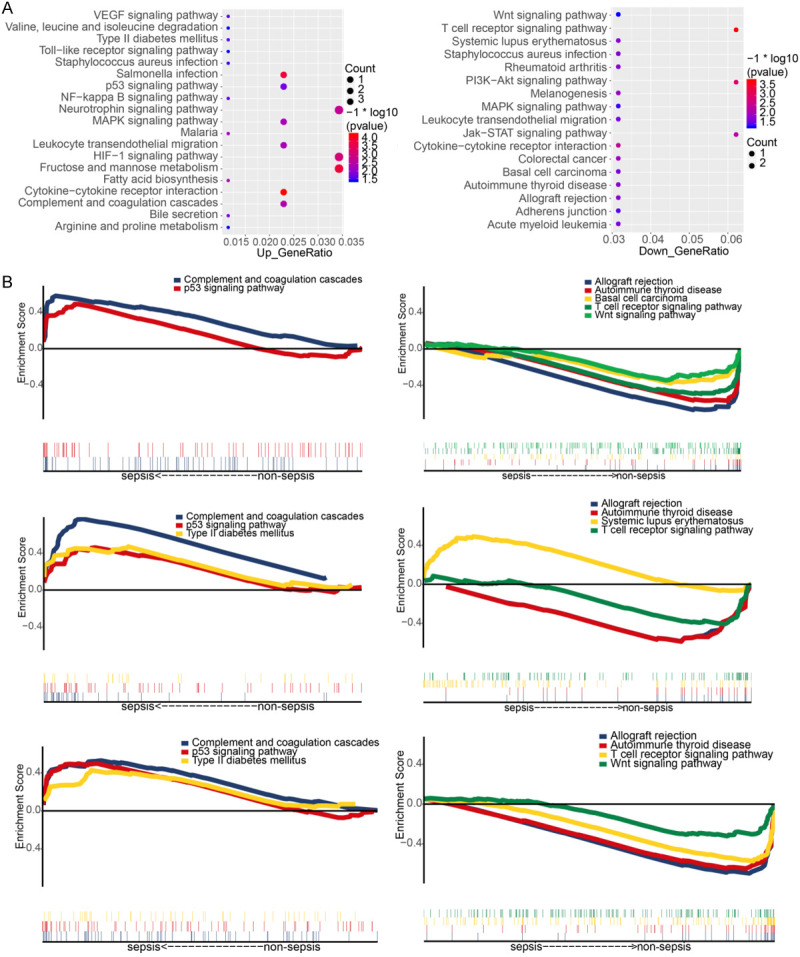
Functional enrichment analysis. A. Sub-pathway analysis for common upregulated/downregulated genes in sepsis. B. Pathways enriched in sepsis or non-sepsis in the three gene expression data sets.
Discussion
In original study of GSE57065, the correlation between gene expression pattern and clinical severity of sepsis was evaluated by severity score [29]. In original study based on GSE95233, several candidate biomarkers, including CX3CR1 and Lilrb 2, were identified as prognostic biomarkers of sepsis [9]. The aim of original study based on GSE69528 was to find biomarkers to distinguish bacterial-induced sepsis and sepsis caused by other pathogens [30]. Compared with these three original studies, we pay more attention to the variation of the crucial gene sets rather than the aberration of a single molecule. In the present study, we screened common upregulated or downregulated genes across three gene expression data sets of patients with sepsis in comparison with non-sepsis individuals. We identified a crucial gene set according to the average functional similarity among genes upregulated or downregulated across the various data sets. The crucial upregulated gene set comprised OLFM4, ORM1, CEP55, S100A12, S100P, LRG1, CEACAM8, MS4A3A, PLSCR1 and IL1R2. A previous studies have shown that OLFM4 can negatively regulate the defense response against bacterial infections [31]. ORM1 was found to be significantly upregulated in sepsis [32,33], similarly to S100P [34] and LRG1 [35], and they may have a role as sepsis biomarkers. S100A12 and CEACAM8 may be involved in natural immunity against sepsis [36,37]. PLSCR1 has been shown to take part in innate protective mechanisms against a bacterial pore-forming toxin [38]. IL1R2 is a potential biomarker for diagnosis of sepsis [39]. In our present study, CEP55 and MS4A4A were upregulated in sepses and identified as crucial genes, this indicates CEP55 and MS4A4A may be closely associated with sepsis. OLFM4 could promote leukocyte mediated migration, neutrophil activation and degranulation process [40]. CEP55 can cause the proliferation of cytotoxic T lymphocytes in vivo [41]. S100A12 plays an important role in promoting the formation of osteoclasts, which have a certain effect on systemic inflammation [42]. High expression of PLSCR1 could inhibit phagocytosis of macrophages [43]. Tumor infiltrating regulatory T cells (Treg) can inhibit tumor antigen-specific T cells, and IL1R2 was found to be expressed on the cell surface of Treg [44]. Most of the genes are found to be related to immune cells, Therefore, the variation of the crucial gene sets may reflect the response to sepsis in the host immune system.
The crucial downregulated gene set included THEMIS, IL2RB, CD2, IL7R, CD3E, KLRB1, PVRIG, CCRR3, TGFBR3 and PLEKHA1. Previous work found that IL2RB and CD3E were negatively correlated with sepsis organ failure and mortality [45], which is consistent with our study. In other study [46], IL7R expression was also found to be downregulated in sepsis. TGFBR3 was suggested to help identify a “bacteremia-prone” phenotype in sickle cell anemia [47]. There were few studies reporting a relationship of THEMIS, CD2, KLRB1, PVRIG, CCRR3 or PLEKHA1 with sepsis. Our result suggests that the downregulation of these genes may be response of the host to sepsis. THEMIS was the key in the development of T-cell [48]. IL12RB was associated with impaired T cell [49]. CD2 is a glycoprotein on the surface of T lymphocytes, and its abnormality may be due to the activation of lymphocytes [50]. IL7R could develop into plasma like dendritic cells [51]. CD3 affinity is closely related to the distribution of T cell rich tissue [52]. PVRIG was a checkpoint receptor that can induce T cell enhancement [53]. TGFBR3 was found to be preferentially expressed in CD4+ T cells [54]. Thus, the aberrant expression of genes may be caused by the blood cell composition change. Nevertheless, compared with the cell population level, our present study may access these subtle changes due to the variation of crucial gene sets were determined. In clinical practice, procalcitonin (PCT) assays are widely used for the diagnosis of sepsis, [55] although a previous meta-analysis reported that PCT has a sensitivity of only 76% and a specificity of 70% for bacteremia [56]. Therefore, more specific markers are urgently needed for effective diagnosis of sepsis. In the present study, we created two GSVA indexes and found that they may be robust biomarkers for sepsis, as suggested by their high AUC. The upregulated GSVA was also validated with an independent data set. The two GSVA indexes may be worthy of further exploration.
The occurrence of certain diseases is not always caused by the abnormality of the whole pathway involved in the biological process, but it can be caused by the dysfunction of a sub-pathway [57]. Several upregulated genes in sepsis were enriched in the sub-pathway analysis, including complement and coagulation cascades, which is consistent with other research [58]. Complement and coagulation cascades can facilitate the containment and destruction of pathogens to protect against bacterial spreading within the body. Inhibition of p53 expression can significantly inhibit cardiomyocyte apoptosis induced by sepsis [59], and p53 signaling was upregulated in our sepsis data, which means that it may promote the development of the disease. Kidney transplant recipients developing sepsis showed inferior patient survival and allograft function, and therefore the identification of differences in alloreactivity may be useful to identify transplant recipients at increased risk [60]. Chronic critical illness from sepsis has been associated with an enhanced T-cell receptor response [61]. These pathways may play a role associated with sepsis according to our analyses.
As our best knowledge, our study is the first study to explore the diagnostic value of gene set variation index in sepsis. However, our work has several limitations. Firstly, we were unable to validate the downregulated gene set using another independent data sets. One obvious reason is the heterogeneity of the three non-septic samples which included healthy volunteers, patients with type 2 diabetes mellitus. Secondly, the upregulated and downregulated GSVA indexes have different AUC values in different data sets due to the different numbers of sample in the data sets. However, all the values of AUC were above 0.7 for the upregulated GSVA index in the all data sets. In addition, we did not experimentally validate the potential of the two gene sets for the diagnosis of sepsis. Therefore, it is not clear whether these genes are causal or merely markers for sepsis. Nevertheless, our study may provide a preliminary basis for the exploration of new biomarkers for the diagnosis of sepsis.
In conclusion, we identified two crucial gene sets across multiple data sets based different platforms in sepsis patients, and we created two GSVA indexes with promising diagnostic value for sepsis.
Acknowledgements
The authors would like to thank Life-Ontology Biological Technology (Nanning, China) for assisting with bioinformatics analysis. This study was supported by the National Natural Science Foundation of China (Grant numbers: 81660132 and 81960343); the Key Research and Development Project of Guangxi (Grant number: Guike AB17195002); Guangxi Natural Science Foundation (Grant numbers: 2017GXNSFBA198043 and 2017GXNSFAA198249); the Scientific Research Funding from Population and Family Planning Commission of Guangxi Zhuang Autonomous Region (Grant number: S2017009); the Reserve Cadre Training Program Science Foundation of the Second Affiliated Hospital of Guangxi Medical University (Grant number: HBRC201805) and the High-level Medical Expert Training Program of Guangxi “139” Plan Funding (Grant number: G201903027).
Disclosure of conflict of interest
None.
References
- 1.Nunnally ME. Sepsis for the anaesthetist. Br J Anaesth. 2016;117:iii44–iii51. doi: 10.1093/bja/aew333. [DOI] [PubMed] [Google Scholar]
- 2.Rello J, Valenzuela-Sanchez F, Ruiz-Rodriguez M, Moyano S. Sepsis: a review of advances in management. Adv Ther. 2017;34:2393–2411. doi: 10.1007/s12325-017-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hawiger J, Veach RA, Zienkiewicz J. New paradigms in sepsis: from prevention to protection of failing microcirculation. J Thromb Haemost. 2015;13:1743–1756. doi: 10.1111/jth.13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sganga G. [Surgical sepsis] . Urologia. 2015;82:75–83. doi: 10.5301/uro.5000113. [DOI] [PubMed] [Google Scholar]
- 5.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–874. [PubMed] [Google Scholar]
- 6.Balk RA. Systemic inflammatory response syndrome (SIRS): where did it come from and is it still relevant today? Virulence. 2014;5:20–26. doi: 10.4161/viru.27135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Churpek MM, Zadravecz FJ, Winslow C, Howell MD, Edelson DP. Incidence and prognostic value of the systemic inflammatory response syndrome and organ dysfunctions in ward patients. Am J Respir Crit Care Med. 2015;192:958–964. doi: 10.1164/rccm.201502-0275OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sherwin R, Winters ME, Vilke GM, Wardi G. Does early and appropriate antibiotic administration improve mortality in emergency department patients with severe sepsis or septic shock? J Emerg Med. 2017;53:588–595. doi: 10.1016/j.jemermed.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Venet F, Schilling J, Cazalis MA, Demaret J, Poujol F, Girardot T, Rouget C, Pachot A, Lepape A, Friggeri A, Rimmele T, Monneret G, Textoris J. Modulation of LILRB2 protein and mRNA expressions in septic shock patients and after ex vivo lipopolysaccharide stimulation. Hum Immunol. 2017;78:441–450. doi: 10.1016/j.humimm.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 10.Heming N, Lamothe L, Ambrosi X, Annane D. Emerging drugs for the treatment of sepsis. Expert Opin Emerg Drugs. 2016;21:27–37. doi: 10.1517/14728214.2016.1132700. [DOI] [PubMed] [Google Scholar]
- 11.Povoa P. C-reactive protein: a valuable marker of sepsis. Intensive Care Med. 2002;28:235–243. doi: 10.1007/s00134-002-1209-6. [DOI] [PubMed] [Google Scholar]
- 12.Clec’h C, Ferriere F, Karoubi P, Fosse JP, Cupa M, Hoang P, Cohen Y. Diagnostic and prognostic value of procalcitonin in patients with septic shock. Crit Care Med. 2004;32:1166–1169. doi: 10.1097/01.ccm.0000126263.00551.06. [DOI] [PubMed] [Google Scholar]
- 13.O’Grady NP, Barie PS, Bartlett JG, Bleck T, Carroll K, Kalil AC, Linden P, Maki DG, Nierman D, Pasculle W, Masur H American College of Critical Care Medicine; Infectious Diseases Society of America. Guidelines for evaluation of new fever in critically ill adult patients: 2008 update from the American college of critical care medicine and the infectious diseases society of America. Crit Care Med. 2008;36:1330–1349. doi: 10.1097/CCM.0b013e318169eda9. [DOI] [PubMed] [Google Scholar]
- 14.Fan SL, Miller NS, Lee J, Remick DG. Diagnosing sepsis - The role of laboratory medicine. Clin Chim Acta. 2016;460:203–210. doi: 10.1016/j.cca.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tabone O, Mommert M, Jourdan C, Cerrato E, Legrand M, Lepape A, Allaouchiche B, Rimmele T, Pachot A, Monneret G, Venet F, Mallet F, Textoris J. Endogenous retroviruses transcriptional modulation after severe infection, trauma and burn. Front Immunol. 2018;9:3091. doi: 10.3389/fimmu.2018.03091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lakota K, Thallinger GG, Sodin-Semrl S, Rozman B, Ambrozic A, Tomsic M, Praprotnik S, Cucnik S, Mrak-Poljsak K, Ceribelli A, Cavazzana I, Franceschini F, Vencovsky J, Czirjak L, Varju C, Steiner G, Aringer M, Stamenkovic B, Distler O, Matucci-Cerinic M, Kveder T. International cohort study of 73 anti-Ku-positive patients: association of p70/p80 anti-Ku antibodies with joint/bone features and differentiation of disease populations by using principal-components analysis. Arthritis Res Ther. 2012;14:R2. doi: 10.1186/ar3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.David CC, Jacobs DJ. Principal component analysis: a method for determining the essential dynamics of proteins. Methods Mol Biol. 2014;1084:193–226. doi: 10.1007/978-1-62703-658-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Oliveira LA, da Silva CP, Nuvunga JJ, da Silva AQ, Balestre M. Bayesian GGE biplot models applied to maize multi-environments trials. Genet Mol Res. 2016;15 doi: 10.4238/gmr.15028612. [DOI] [PubMed] [Google Scholar]
- 20.Shade A, Handelsman J. Beyond the Venn diagram: the hunt for a core microbiome. Environ Microbiol. 2012;14:4–12. doi: 10.1111/j.1462-2920.2011.02585.x. [DOI] [PubMed] [Google Scholar]
- 21.Han Y, Yu G, Sarioglu H, Caballero-Martinez A, Schlott F, Ueffing M, Haase H, Peschel C, Krackhardt AM. Proteomic investigation of the interactome of FMNL1 in hematopoietic cells unveils a role in calcium-dependent membrane plasticity. J Proteomics. 2013;78:72–82. doi: 10.1016/j.jprot.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 22.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koumakis L, Kanterakis A, Kartsaki E, Chatzimina M, Zervakis M, Tsiknakis M, Vassou D, Kafetzopoulos D, Marias K, Moustakis V, Potamias G. MinePath: mining for phenotype differential sub-paths in molecular pathways. PLoS Comput Biol. 2016;12:e1005187. doi: 10.1371/journal.pcbi.1005187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C, Li X, Miao Y, Wang Q, Jiang W, Xu C, Li J, Han J, Zhang F, Gong B, Xu L. SubpathwayMiner: a software package for flexible identification of pathways. Nucleic Acids Res. 2009;37:e131. doi: 10.1093/nar/gkp667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito K, Murphy D. Application of ggplot2 to Pharmacometric Graphics. CPT Pharmacometrics Syst Pharmacol. 2013;2:e79. doi: 10.1038/psp.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cazalis MA, Lepape A, Venet F, Frager F, Mougin B, Vallin H, Paye M, Pachot A, Monneret G. Early and dynamic changes in gene expression in septic shock patients: a genome-wide approach. Intensive Care Med Exp. 2014;2:20. doi: 10.1186/s40635-014-0020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pankla R, Buddhisa S, Berry M, Blankenship DM, Bancroft GJ, Banchereau J, Lertmemongkolchai G, Chaussabel D. Genomic transcriptional profiling identifies a candidate blood biomarker signature for the diagnosis of septicemic melioidosis. Genome Biol. 2009;10:R127. doi: 10.1186/gb-2009-10-11-r127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu W, Yan M, Liu Y, McLeish KR, Coleman WG Jr, Rodgers GP. Olfactomedin 4 inhibits cathepsin C-mediated protease activities, thereby modulating neutrophil killing of Staphylococcus aureus and Escherichia coli in mice. J Immunol. 2012;189:2460–2467. doi: 10.4049/jimmunol.1103179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutov AM, Veselova IR, Malova IM. [Effect of sodium hydrocarbonate on vestibular nystagmus] . Kosm Biol Aviakosm Med. 1978;12:78–80. [PubMed] [Google Scholar]
- 33.Raju MS, V J, Kamaraju RS, Sritharan V, Rajkumar K, Natarajan S, Kumar AD, Burgula S. Continuous evaluation of changes in the serum proteome from early to late stages of sepsis caused by Klebsiella pneumoniae. Mol Med Rep. 2016;13:4835–4844. doi: 10.3892/mmr.2016.5112. [DOI] [PubMed] [Google Scholar]
- 34.Yang YX, Li L. Identification of potential biomarkers of sepsis using bioinformatics analysis. Exp Ther Med. 2017;13:1689–1696. doi: 10.3892/etm.2017.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hashida T, Nakada TA, Satoh M, Tomita K, Kawaguchi R, Nomura F, Oda S. Proteome analysis of hemofilter adsorbates to identify novel substances of sepsis: a pilot study. J Artif Organs. 2017;20:132–137. doi: 10.1007/s10047-016-0936-3. [DOI] [PubMed] [Google Scholar]
- 36.Foell D, Wittkowski H, Kessel C, Luken A, Weinhage T, Varga G, Vogl T, Wirth T, Viemann D, Bjork P, van Zoelen MA, Gohar F, Srikrishna G, Kraft M, Roth J. Proinflammatory S100A12 can activate human monocytes via Toll-like receptor 4. Am J Respir Crit Care Med. 2013;187:1324–1334. doi: 10.1164/rccm.201209-1602OC. [DOI] [PubMed] [Google Scholar]
- 37.Ribon M, Mussard J, Semerano L, Singer BB, Decker P. Extracellular chromatin triggers release of soluble CEACAM8 upon activation of neutrophils. Front Immunol. 2019;10:1346. doi: 10.3389/fimmu.2019.01346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lizak M, Yarovinsky TO. Phospholipid scramblase 1 mediates type i interferon-induced protection against staphylococcal alpha-toxin. Cell Host Microbe. 2012;11:70–80. doi: 10.1016/j.chom.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lang Y, Jiang Y, Gao M, Wang W, Wang N, Wang K, Zhang H, Chen G, Liu K, Liu M, Yang M, Xiao X. Interleukin-1 receptor 2: a new biomarker for sepsis diagnosis and gram-negative/gram-positive bacterial differentiation. Shock. 2017;47:119–124. doi: 10.1097/SHK.0000000000000714. [DOI] [PubMed] [Google Scholar]
- 40.Banerjee A, Shukla S, Pandey AD, Goswami S, Bandyopadhyay B, Ramachandran V, Das S, Malhotra A, Agarwal A, Adhikari S, Rahman M, Chatterjee S, Bhattacharya N, Basu N, Pandey P, Sood V, Vrati S. RNA-Seq analysis of peripheral blood mononuclear cells reveals unique transcriptional signatures associated with disease progression in dengue patients. Transl Res. 2017;186:62–78. e9. doi: 10.1016/j.trsl.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 41.Inoda S, Hirohashi Y, Torigoe T, Nakatsugawa M, Kiriyama K, Nakazawa E, Harada K, Takasu H, Tamura Y, Kamiguchi K, Asanuma H, Tsuruma T, Terui T, Ishitani K, Ohmura T, Wang Q, Greene MI, Hasegawa T, Hirata K, Sato N. Cep55/c10orf3, a tumor antigen derived from a centrosome residing protein in breast carcinoma. J Immunother. 2009;32:474–485. doi: 10.1097/CJI.0b013e3181a1d109. [DOI] [PubMed] [Google Scholar]
- 42.Nishida M, Saegusa J, Tanaka S, Morinobu A. S100A12 facilitates osteoclast differentiation from human monocytes. PLoS One. 2018;13:e0204140. doi: 10.1371/journal.pone.0204140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herate C, Ramdani G, Grant NJ, Marion S, Gasman S, Niedergang F, Benichou S, Bouchet J. Phospholipid scramblase 1 modulates FcR-mediated phagocytosis in differentiated macrophages. PLoS One. 2016;11:e0145617. doi: 10.1371/journal.pone.0145617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, Moro M, Crosti M, Mazzara S, Vaira V, Bosari S, Palleschi A, Santambrogio L, Bovo G, Zucchini N, Totis M, Gianotti L, Cesana G, Perego RA, Maroni N, Pisani Ceretti A, Opocher E, De Francesco R, Geginat J, Stunnenberg HG, Abrignani S, Pagani M. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016;45:1135–1147. doi: 10.1016/j.immuni.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almansa R, Heredia-Rodriguez M, Gomez-Sanchez E, Andaluz-Ojeda D, Iglesias V, Rico L, Ortega A, Gomez-Pesquera E, Liu P, Aragon M, Eiros JM, Jimenez-Sousa MA, Resino S, Gomez-Herreras I, Bermejo-Martin JF, Tamayo E. Transcriptomic correlates of organ failure extent in sepsis. J Infect. 2015;70:445–456. doi: 10.1016/j.jinf.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 46.Mohnle P, Hirschberger S, Hinske LC, Briegel J, Hubner M, Weis S, Dimopoulos G, Bauer M, Giamarellos-Bourboulis EJ, Kreth S. MicroRNAs 143 and 150 in whole blood enable detection of T-cell immunoparalysis in sepsis. Mol Med. 2018;24:54. doi: 10.1186/s10020-018-0056-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adewoye AH, Nolan VG, Ma Q, Baldwin C, Wyszynski DF, Farrell JJ, Farrer LA, Steinberg MH. Association of polymorphisms of IGF1R and genes in the transforming growth factor-beta/bone morphogenetic protein pathway with bacteremia in sickle cell anemia. Clin Infect Dis. 2006;43:593–598. doi: 10.1086/506356. [DOI] [PubMed] [Google Scholar]
- 48.Brockmeyer C, Paster W, Pepper D, Tan CP, Trudgian DC, McGowan S, Fu G, Gascoigne NR, Acuto O, Salek M. T cell receptor (TCR)-induced tyrosine phosphorylation dynamics identifies THEMIS as a new TCR signalosome component. J Biol Chem. 2011;286:7535–7547. doi: 10.1074/jbc.M110.201236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roifman CM. Human IL-2 receptor alpha chain deficiency. Pediatr Res. 2000;48:6–11. doi: 10.1203/00006450-200007000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Selvaraj P, Plunkett ML, Dustin M, Sanders ME, Shaw S, Springer TA. The T lymphocyte glycoprotein CD2 binds the cell surface ligand LFA-3. Nature. 1987;326:400–403. doi: 10.1038/326400a0. [DOI] [PubMed] [Google Scholar]
- 51.Rodrigues PF, Alberti-Servera L, Eremin A, Grajales-Reyes GE, Ivanek R, Tussiwand R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol. 2018;19:711–722. doi: 10.1038/s41590-018-0136-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mandikian D, Takahashi N, Lo AA, Li J, Eastham-Anderson J, Slaga D, Ho J, Hristopoulos M, Clark R, Totpal K, Lin K, Joseph SB, Dennis MS, Prabhu S, Junttila TT, Boswell CA. Relative target affinities of T-cell-dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol Cancer Ther. 2018;17:776–785. doi: 10.1158/1535-7163.MCT-17-0657. [DOI] [PubMed] [Google Scholar]
- 53.Murter B, Pan X, Ophir E, Alteber Z, Azulay M, Sen R, Levy O, Dassa L, Vaknin I, Fridman-Kfir T, Salomon R, Ravet A, Tam A, Levin D, Vaknin Y, Tatirovsky E, Machlenkin A, Pardoll D, Ganguly S. Mouse PVRIG Has CD8(+) T cell-specific coinhibitory functions and dampens antitumor immunity. Cancer Immunol Res. 2019;7:244–256. doi: 10.1158/2326-6066.CIR-18-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kagamu H, Kitano S, Yamaguchi O, Yoshimura K, Horimoto K, Kitazawa M, Fukui K, Shiono A, Mouri A, Nishihara F, Miura Y, Hashimoto K, Murayama Y, Kaira K, Kobayashi K. CD4(+) T-cell immunity in the peripheral blood correlates with response to anti-PD-1 therapy. Cancer Immunol Res. 2020;8:334–344. doi: 10.1158/2326-6066.CIR-19-0574. [DOI] [PubMed] [Google Scholar]
- 55.Yealy DM, Fine MJ. Measurement of serum procalcitonin: a step closer to tailored care for respiratory infections? JAMA. 2009;302:1115–1116. doi: 10.1001/jama.2009.1318. [DOI] [PubMed] [Google Scholar]
- 56.Jones AE, Fiechtl JF, Brown MD, Ballew JJ, Kline JA. Procalcitonin test in the diagnosis of bacteremia: a meta-analysis. Ann Emerg Med. 2007;50:34–41. doi: 10.1016/j.annemergmed.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 57.Liu J, Jing L, Tu X. Weighted gene co-expression network analysis identifies specific modules and hub genes related to coronary artery disease. BMC Cardiovasc Disord. 2016;16:54. doi: 10.1186/s12872-016-0217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lupu F, Keshari RS, Lambris JD, Coggeshall KM. Crosstalk between the coagulation and complement systems in sepsis. Thromb Res. 2014;133(Suppl 1):S28–31. doi: 10.1016/j.thromres.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma H, Wang X, Ha T, Gao M, Liu L, Wang R, Yu K, Kalbfleisch JH, Kao RL, Williams DL, Li C. MicroRNA-125b prevents cardiac dysfunction in polymicrobial sepsis by targeting TRAF6-mediated nuclear factor kappaB activation and p53-mediated apoptotic signaling. J Infect Dis. 2016;214:1773–1783. doi: 10.1093/infdis/jiw449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schachtner T, Stein M, Reinke P. Sepsis after renal transplantation: clinical, immunological, and microbiological risk factors. Transpl Infect Dis. 2017;19 doi: 10.1111/tid.12695. [DOI] [PubMed] [Google Scholar]
- 61.Borken F, Markwart R, Requardt RP, Schubert K, Spacek M, Verner M, Ruckriem S, Scherag A, Oehmichen F, Brunkhorst FM, Rubio I. Chronic critical illness from sepsis is associated with an enhanced TCR response. J Immunol. 2017;198:4781–4791. doi: 10.4049/jimmunol.1700142. [DOI] [PubMed] [Google Scholar]