

Abstract
Aim:
Factor VII deficiency is one of the hereditary coagulation disorders that has autosomal reccessive inheritance and is observed relatively frequently (1/500 000). It is clinically heterogeneous, and may be asymptomatic or lead to life-threatening bleeding. Thus, there is no correlation between FVII activity and clinical findings. Plasma-derived and recombinant FVII concentrates are currently used for treatment. In countries where access to these products is lacking, fresh frozen plasma and prothrombin complex concentrates are also used, though they contain low amounts of factor FVII. In this study, we present the clinical properties, treatments, and surgical interventions used in patients followed up in our clinic with a diagnosis of factor FVII deficiency.
Material and Methods:
Patients who were diagnosed as FVII deficiency in Division of Pediatric Hematology and Oncology between July 1997 and July 2018, were included in the study. The patients’ demographic characteristics, symptoms at presentation, PT, aPTT, and FVII values, types of bleeding, and treatments and surgical interventions used, were recorded. The bleedings observed in the patients were classified by severity as asymptomatic, minor, and major.
Results:
A total of 18 patients (7 girls and 11 boys) with a mean age of 9.64±9.63 years were included in the study. The mean follow-up time was found as 78.06±54.4 months. When the bleedings were classified clinically, no bleeding was observed in eight patients (44.4%). The factor FVII level was found as <10% in three of these eight asymptomatic patients and above 20% in the others. Minor bleeding was observed in nine patients (50%) and major bleeding was observed in one patient. When the patients were classified as asymptomatic and symptomatic, there was no significant difference between the two groups in terms of FVII level (p=0.57). A total of 21 surgical interventions were performed in 14 (78%) of 18 patients who were being followed up.
Conclusion:
FVII deficiency has a very wide spectrum both clinically and in terms of approach to surgical interventions. Therefore, patients with factor FVII deficiency should be followed up and treated by comprehensive care centers with close collaboration of multiple disciplines.
Keywords: Clinic, factor VII, surgery
Abstract
Amaç:
Faktör VII eksikliği otozomal çekinik kalıtılan ve nispeten sık görülen (1/500 000) kalıtsal pıhtılaşma bozukluklarındandır. Klinik olarak heterojendir, asemptomatik seyredebildiği gibi yaşamsal kanamalara da yol açabilmektedir. Nitekim FVII aktivitesi ile klinik bulguların arasında korelasyon bulunmamaktadır. Tedavisinde, plazma kaynaklı ve rekombinan FVII konsantreleri kullanılırsa da, bunlara erişimin olmadığı ülkelerde düşük miktarda FVII içermelerine rağmen taze donmuş plazma ve protrombin kompleks konsantreleri de kullanılmaktadır. Bu çalışmada, kliniğimizde izlenen FVII eksikliği tanısı almış hastaların klinik özellikleri, tedavileri ve uygulanan cerrahi girişimler sunulmaktadır.
Gereç ve Yöntemler:
İstanbul Üniversitesi Onkoloji Enstitüsü, Çocuk Hematolojisi ve Onkolojisi Bilim Dalı’nda Temmuz 1997- Temmuz 2018 tarihleri arasında FVII eksikliği tanısı alan hastalar çalışmaya alındı. Hastaların demografik özellikleri, başvuru yakınmaları, PT, aPTT ve FVII değerleri, kanama tipi, tedavileri ve uygulanan cerrahi girişimler kaydedildi. Hastalarda görülen kanamalar ağırlıklarına göre asemptomatik, minör ve majör olarak sınıflandırıldı.
Bulgular:
Ortalama tanı yaşları 9,64±9,63 olan 7’si kız 11’i erkek toplam 18 hasta çalışmaya alındı. Hastaların ortalama izlem süresi 78,06±54,4 ay idi. Kanamalar klinik olarak sınıflandırıldığında; sekiz hastada (%44.4) herhangi bir kanama gözlenmezken, asemptomatik seyreden sekiz hastanın üçünde FVII düzeyi <10, diğerlerinde %20’nin üzerinde idi. Dokuz (%50) hastada minör kanama, ve bir hastada major kanama kaydedildi. Hastalar asemptomatik ve semptomatik olarak sınıflandırıldığında iki grup arasında FVII düzeyi açısından anlamlı farklılık yoktu (p=0,57). İzlenen 18 hastanın 14’üne (%78) toplam 21 cerrahi girişim uygulandı.
Çıkarımlar:
FVII eksikliği gerek klinik olarak gerekse cerrahi girişimlere yaklaşım olarak çok geniş bir yelpazede yer almaktadır. Bu nedenle, FVII eksikliği tanısı almış hastaların, uzmanlaşmış bir kanama bozukluğu merkezi ve birden çok bilim dalı ile yakın işbirliği içinde izlem ve tedavi edilmesi gerekmektedir.
Introduction
Factor VII (FVII) deficiency is one of the rare hereditary coagulation disorders (1/500 000) that has autosomal recessive inheritance (1, 2). It is clinically heterogeneous, it may be asymptomatic or may lead to life-threatening bleeding (3). Thus, there is no strong correlation between FVII activity and clinical findings (1, 4). Some patients with very low FVII activity have frequent bleeding episodes, and no bleeding episodes are observed in others. The most common symptoms include epistaxis and abnormal uterine bleeding (e.g. menorrhagia). Musculo-skeletal bleedings and life-threatening bleedings are relatively rare (1, 3, 4). Bleedings is generally not observed in patients with a factor level of 10% and above, and spontaneous bleedings is not expected when this level is 20% and above (4, 5). The diagnosis is made with a prolonged prothrombin time (PT), normal activated partial thromboplastin time (aPTT), and low FVII level (6). Plasma-derived and recombinant FVII (rFVIIa) concentrates are currently used for treatment. In countries where access to these products is lacking, fresh frozen plasma (FFP) and prothrombin complex concentrates (PCC) are also being used, though they contain low amounts of FVII. (3, 7). The short half-lives of FVII concentrates that are used in treatment render standard prophylactic applications difficult and therefore, prophylactic treatment is controversial. Accordingly, prophylaxis should only be considered in patients with frequently recurring bleedings who have a severe course (8). There are a limited number of studies on surgical interventions and their outcomes in patients with FVII deficiency. In this study, we present the clinical properties, treatments, and surgical interventions used in patients with factor FVII deficiency followed up in our clinics, because FVII is a rare coagulation disorder, and there is no clear consensus in terms of treatment, in contrast to hemophilia.
Material and Methods
Patients who were diagnosed as having FVII deficiency in Istanbul University Oncology Institute, Division of Pediatric Hematology and Oncology between July 1997 and July 2018, were included in the study. The patients’ demographic characteristics; symptoms at presentation; PT, aPTT, and FVII values; types of bleedings; and treatments and surgical interventions used, were recorded retrospectively from the patients’ records. The bleedings observed in the patients were classified by severity as asymptomatic, minor, and major according to the FVII Deficiency Seven Treatment Evaluation Registry (STER) (Table 1) (9).
Table 1.
Classification of the bleedings (9)
| Degree of bleeding | Definition |
|---|---|
| Asymptomatic | Absence of recorded bleeding |
| Minor bleeding | Skin, mucosal, soft-tissue bleedings (ecchymosis, gingival bleeding and epistaxis, menorrhagia, hematuria-excluding GIS bleeding) |
| Major bleeding | symptomatic bleeding in a critical area or organ (musculoskeletal system bleedings, GIS and CNS bleedings) |
Surgical interventions were classified as major and minor surgical interventions. Orthopedic, cardiovascular, neurologic system interventions, and open abdominal surgery are major surgical interventions, whereas endoscopic procedures, biopsy procedures, and dental procedures are minor surgical interventions. Factor treatment doses administered during surgical interventions, dose numbers, treatment period, and postoperative complications (bleeding, infection, mortality with rates higher than expected) were recorded. The study was approved by Istanbul University Istanbul Medical Faculty Clinical Researches Ethics Committee (07.02.2020/294). The study was conducted in accordance with the Decleration of Helsinki.
Statistical Analysis
The Windows SPSS 22.0 program was used for statistical analysis. Descriptive statistics were used for listing of the clinical properties belonging to the entire population. The Mann-Whitney U test was used for the assessment of inter-group differences for continuous variables and the Spearman correlation test was used to investigate correlations between continuous variables. In all analyses, p values <0.05 were considered statistically significant.
Results
Eighteen patients (7 girls and 11 boys) were included in the study. The mean age of the patients was 9.64±9.63 years. The demographic characteristics of the study population are shown in Table 2.
Table 2.
Demographic properties of the patients included in the study (n=18)
| n | % | |
|---|---|---|
| Age (years) (Mean±SD) | 9.64±9.6 | |
| Sex | ||
| Female | 7 | 39 |
| Male | 11 | 61 |
| Bleeding phenotype | ||
| Asymptomatic | 8 | 44 |
| Minor | 9 | 50 |
| Major | 1 | 6 |
| FVII level | ||
| <10 | 6 | 33 |
| 10–20 | 0 | 0 |
| >20 | 12 | 67 |
SD: Standard deviation
The patients’ mean follow-up time was 78.06±54.4 months. The ages at the time of diagnosis, symptoms at presentation, and laboratory findings are shown in Table 3.
Table 3.
The patients’ general characteristics
| No | Sex | Age at the time of diagnosis (years) | Complaint at presentation | PT (s) | aPTT (s) | FVII: C (%) |
|---|---|---|---|---|---|---|
| 1 | M | 1.5 | Epistaxis+Hemarthrosis | 68 | 23.3 | 0.2 |
| 2 | M | 29 | No bleeding | 26 | 20.4 | 6 |
| 3 | F | 38 | Menorrhagia | 27.8 | 27 | 6.3 |
| 4 | M | 1.5 | No bleeding | 17.5 | 28 | 7 |
| 5 | F | 1 | Epistaxis | 39 | 27 | 8 |
| 6 | M | 4 | No bleeding | 24 | 30 | 8.7 |
| 7 | F | 10 | Menoraji | 30.2 | 32 | 24 |
| 8 | M | 5 | Ecchymosis | 16 | 33 | 25 |
| 9 | M | 6 | No bleeding | 14 | 35 | 27 |
| 10 | M | 5.5 | No bleeding | 16 | 31 | 30 |
| 11 | M | 4.5 | No bleeding | 18.4 | 26 | 30 |
| 12 | M | 9 | Epistaxis | 16.5 | 30 | 31 |
| 13 | F | 12 | No bleeding | 13.8 | 36.4 | 33.9 |
| 14 | F | 15 | No bleeding | 15.2 | 27 | 36.5 |
| 15 | F | 6 | Epistaxis | 15 | 30 | 38 |
| 16 | M | 8 | Epistaxis | 18 | 27 | 39 |
| 17 | F | 12.5 | Menorrhagia | 27 | 23 | 40 |
| 18 | M | 5 | Epistaxis | 15.9 | 32 | 47 |
aPTT: Activated partial thromboplastin time; PT: Prothrombin time; FVII: C: Factor VII activity; M: Male; F: Female
When the patients’ symptoms at presentation were evaluated, it was observed that there was no bleeding at presentation (asymptomatic) in eight (44.4%) patients, and the diagnosis was made when examination was performed because of prolonged PT time found during preoperative investigations. The FVII level was found to be below 10% (severe) in three of these eight patients and above 20% in the others (mild).
Ten patients presented because of bleeding, and were diagnosed. Minor bleedings were observed in nine (50%) patients and major bleedings were observed in one patient. The FVII level was found as 0.2% in the patient who had major bleeding, and serious joint bleeding was observed this patient. PT was found to be prolonged and aPTT was found to be normal in all patients. The mean PT value at the time of diagnosis was found as 23.23 (range, 13.8–68).
When the patients were classified as asymptomatic (eight patients) and symptomatic (10 patients=nine minor bleeding, one major bleeding), the median FVII level was found as 28.5% (25th percentile=7.4%, 75th percentile=33%) in the asymptomatic group and 28% (25th percentile=7.6%, 75th percentile=39.2%) in the symptomatic group; there was no significant difference between the two groups (p=0.57).
The median FVII level was found as 33.9% (25th percentile=8, 75th percentile=38) in the female patients and 27% (25th percentile=7, 75th percentile=31) in the male patients; there was no significant difference between the sexes (p=0.497). There was no significant difference between factor level and age at the time of diagnosis (r=0.229, p=0.362).
When the patients’ bleedings observed during follow-up were evaluated, it was found that the first severe bleedings developed in diffuse wound sites in the back (Fig. 1) and in the left elbow joint at the age of 25 years in patient #1 who had a FVII activity of 0.2%, and the bleedings were controlled using aPCC. A single dose of rFVIIa (35 μg/kg/dose) was used for bleeding in recurrent gingival bleeding in this patient, and a very good response was obtained.
Figure 1.
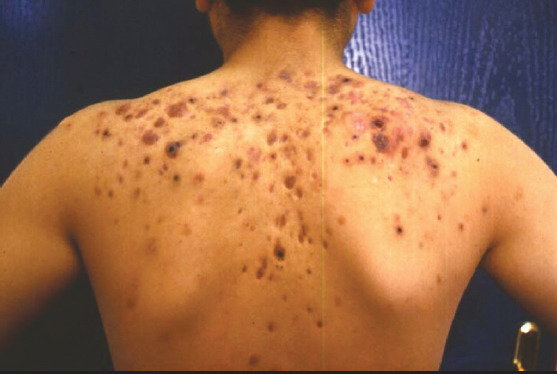
Bleeding at diffuse wound sites on the back of patient #1
In patient #15, who had FVII activity of 38%, 12 attacks of epistaxis, one attack of thoracic hematoma, and two attacks of severe menstrual bleeding were observed throughout a 14-year follow-up period, and these attacks were treated with FFP.
Patient #17 used a single dose of rFVIIa (30 μg/kg) because of severe menstrual bleeding on one occaision, and bleeding was controlled.
In the other patients who were followed up, no severe bleedings requiring factor replacement were observed. Local and/or oral tranexamic acid was used when some mild problems developed.
A total of 21 surgical interventions (seven major, 14 minor) were performed in 14 (78%) of 18 patients who were followed up. The surgical interventions are shown in Table 4. No bleeds or complications were observed with rates higher than expected in any of the patients.
Table 4.
Surgical interventions performed
| No | FVII: C (%) | Age at the time of surgical procedure (years) | Surgical procedure | Treatment | Number of treatment daysa | Number of total dosesa | Total dose (μg/kg)a |
|---|---|---|---|---|---|---|---|
| 1 | 0.2 | 15 | Circumcision | aPCC | 7a | 17a | 96 IU/kg |
| 20 | Hydradenitis excision | rFVII | 66a | 141a | 3948a | ||
| +Flap turning | |||||||
| 24 | Laparoscopic | rFVIIa | 7 | 27 | 540 | ||
| cholecystectomy | |||||||
| 34 | Dental scaling | rFVIIa | 2 | 2 | 70 | ||
| 2 | 6 | 30 | Tooth extraction | rFVIIa | 1 | 1 | 30 |
| 4 | 7 | 4 | Hypospadias | rFVIIa | 2 | 15 | 375 |
| 6 | Tooth extraction | rFVIIa | 1 | 1 | 30 | ||
| 6 | 8.7 | 5 | Circumcision+Right | rFVIIa | 14 | 42 | 1470 |
| inguinal hernia | |||||||
| 7 | 24 | 10 | Tooth extraction | rFVIIa | 1 | 1 | 30 |
| 8 | 25 | 8 | Circumcision | rFVIIa | 14 | 42 | 1470 |
| 8 | Tooth extraction | rFVIIa | 1 | 1 | 30 | ||
| 9 | 27 | 9 | Tooth extraction | rFVIIa | 1 | 1 | 30 |
| 13 | Bilateral tube replacement | rFVIIa | 3 | 11 | 440 | ||
| in the ears+Adeneidectomy | |||||||
| 11 | 30 | 5 | Bilateral inguinal hernia | TA | – | – | – |
| 12 | 31 | 8 | Circumcision | rFVIIa | 3 | 12 | 360 |
| 8 | Tooth extraction | rFVIIa | 1 | 1 | 30 | ||
| 13 | 33.9 | 13 | Tonsillectomy | rFVIIa | 4 | 10 | 400 |
| +Adenoidectomy | |||||||
| 14 | 36.5 | 15 | Concha operation | rFVIIa | 2 | 6 | 180 |
| 15 | 38 | 11 | Tooth extraction | TA | – | – | – |
| 17 | 40 | 15 | Tooth extraction | rFVIIa | 1 | 1 | 30 |
| 18 | 47 | 5 | Circumcision | TA+Ankaferd | – | – | – |
a: The details of patient-based treatment are given in the text; FVII: C: Factor VII activity; TA: Tranexamic acid
In all procedures, tranexamic acid was given additionally at a dosage of 40 mg/kg/day in three or four doses.
aPCC was administered to patient #1 at a dose of 7 IU/kg four times on the day of the circumcision operation, three times on the following three days (20 IU/kg/day), twice on the fifth day (15 IU/kg/day), and only once on the sixth and seventh days (7 IU/kg/day). No bleeding or additional complications were observed. During hydradenitis excision and flap turning operations performed in the same patient (Fig. 2), rFVIIa was used for a total of 66 days with diminishing doses (28 μg/kg/dose; every 6 hours in the first 10 days, every 8 hours for 17 days, every 12 hours for 11 days, and a single prophylactic dose for 28 days). In laproscopic cholecystectomy, rFVIIa was administered in six doses on the day of surgery, in four doses on the 2nd–4th days, and in three doses on the 5th–7th days at a dose of 20 μg/kg/dose. For dental scaling, rFVIIa was used as a single dose (35 μg/kg) on the day before the procedure and on the day after the procedure.
Figure 2.
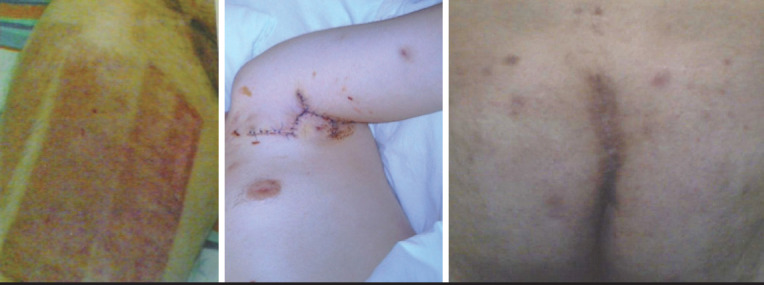
Hidradenitis excision and rotation flap operation
In patients #6 and #8 who underwent circumcision, rFVIIa was used in six doses on the first two days and then, in four doses for 4 days, in three doses for 2 days, in two doses for 2 days, and in a single dose for 4 days at a dose of 35 μg/kg until wound healing was completed. No bleeding or additional complications were observed. rFVIIa was used for three days with diminishing doses only in patient #12 who underwent circumcision, because the level of FVII was >30%.
In the patient #4 who had FVII activity of 7% and underwent hypospadias surgery, rFVIIa was administered in three doses daily (25 μg/kg/dose) and no complications were observed.
In patient #1, bilateral inguinal hernia surgery was completed without complications with tranexamic acid alone, because the FVII level was 30% and there was no previous history of bleeding.
In all patients who underwent tooth extraction, a single dose of rFVIIa (30 μg/kg/dose) was used 1 hour before the procedure.
rFVIIa was used in major and minor surgical interventions in all patients with a factor level of ≤30% (aPCC was used additionally in the patient #1).
Discussion
Factor VII (FVII) is one of the elements included in the coagulation system that is also known as proconvertin, circulates freely in plasma, and belongs to the family of serin proteases. It is synthesized in the liver in a vitamin K-dependent manner. Its half-life is 4–6 hours. Active FVII in the circulation provides activation of coagulation by binding to the tissue factor (TF) that emerges following injuries. Tissue factor and FVII in association, implement fibrin formation by activating FIX and FX (6, 10).
Factor VII deficiency is the most common rare factor deficiency and was described in 1951 for the first time (11). Bleeding symptoms are considerably variable in terms of both location and severity, and may have a heterogenous spectrum ranging from asymptomatic conditions to serious/life-threatening bleeds. There is a very low correlation betwen clinical status and FVII level (12, 13). Hence, we found no difference between asymptomatic and symptomatic patients in terms of FVII levels in our study (p=0.57).
According to the results of the European Network of Rare Bleeding Disorders (ENRBD) study conducted by Peyvandi et al. (13), 224 patients with FVII deficiency were recorded, and it was concluded that the FVII level should be >26% for patients to have an asymptomatic course. However, it was shown that traumatic bleeds occured at with FVII levels of >10% and spontaneous bleeds occured rarely at FVII levels of >20% (4). In another study, 212 patients who had FVII levels above 26% were evaluated, and it was reported that 65.1% were asymptomatic, 31.6% had minor bleeding, and only 3.3% had major bleeding (9). In our study, five (50%) of 10 patients who had FVII activity >26% were asymptomatic, and five (50%) had minor bleedings. On the other hand, only three (37.5%) of eight patients who had FVII activity <26% were asymptomatic and these patients were referred to us because of prolonged PT detected during preoperative investigations. Serious spontaneous bleeding was recorded only in severe FVII deficiency with FVII activity of 0.2%.
Prophylaxis in FVII deficiency is not used routinely because the half-life of FVII is short, in contrast to patients with hemophilia A and B. Prophylaxis is used in selected patients who have had gastrointestinal system or central nervous system bleeding (14). The recommended dose is 90 μg/kg two or three times weekly (7). Clinical experience related to prophylaxis is mostly limited to case reports (8, 15). In our study, there were no patients without a history of severe and uncontrollable bleeding who received prophylaxis.
In patients with FVII deficiency, ideal replacement treatment should be administered with rFVIIa (NovoSeven RT®) in the event of bleeding and surgical interventions. However, plasma-derived factor or aPCC can be given for treatment in countries where rFVIIa is not available. In countries where access to factor is lacking, FFP is another option (16–18). FFP had also been used in our country, because access to rFVIIa is possible since 2001. FFP was used in one patient in our patient group who had epistaxis that could not be controlled with tranexamic acid, and thoracic hematoma. However, joint bleeding, circumcision surgery, and wound site bleeds were treated with aPCC in our patient who had severe FVII deficiency in the pre-rFVIIa period.
Treatments for bleedings and surgical interventions are being pursued according to expert opinions obtained from the systems in which case reports and multinational-multicenter patient data are recorded, and a rFVIIa dosage of 30–60 μg/kg/day is recommended for most bleedings (19). The recommended rFVIIa dosage for severe bleedings, trauma or surgical interventions is 15–30 μg/kg every 4–6 hours for the first 24 hours and every 8–12 hours thereafter, with a target FVII level of >20% (7). In our series, a single dose of rFVIIa (30 μg/kg) was used in a patient with severe FVII deficiency who had gingival bleeding and in a patient who had menorrhagia, and the bleeding response was recorded as very good.
In patients included in the recording system established for patients with FVII deficiency (STER) and who underwent surgical procedures, use of rFVIIa was evaluated in 41 elective surgical procedures (24 major, 17 minor) in 34 patients and it was shown that the lowest effective dose of rFVIIa for hemostasis was 13 μg/kg on the day of surgery, and at least three doses were needed (20). In another international multicenter study, 110 surgical interventions (61 major, 49 minor) performed on 95 patients were examined, and it was shown that neither FVII level nor surgical procedure influenced rFVIIa replacement treatment, and only the patient’s phenotype of bleeding was effective in replacement treatment (21). In the same study, factor replacement was shown to be effective and safe, and was recommended before minor surgery, also in asymptomatic patients (21). In our study, a single dose of rFVIIa was administered before dental treatment in four patients who were asymptomatic, and no complications were recorded.
In conclusion, FVII deficiency has a very wide spectrum both clinically and in terms of the approach to surgical interventions. Therefore, patients with FVII deficiency should be followed up and treated by comprehensive care centers in close collaboration with a multidisciplinary team.
Footnotes
Ethics Committee Approval: The study was approved by İstanbul University İstanbul Medical Faculty Clinical Research Ethics Committee (07.02.2020/294).
Informed Consent: In accordance with the Declaration of Helsinki.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - B.Z., B.K.; Design - B.Z., B.K.; Supervision - B.Z.; Materials - B.Z., B.K.; Data Collection and/or Processing - B.K.; Analysis and/or Interpretation - B.Z., B.K.; Literature Review - B.K.; Writing - B.Z., B.K.; Critical Review - B.Z.
Conflict of Interest: The authors have no conflicts of interest to declare.
Financial Disclosure: The authors declared that this study has received no financial support.
Etik Kurul Onayı: İstanbul Üniversitesi İstanbul Tıp Fakültesi Klinik Araştırmalar Etik Kurulu tarafından onaylanmıştır (07.02.2020/294). Helsinki Bildirgesi’ne uygun yapılmıştır.
Hasta Onamı: Helsinki Bildirgesi’ne uygun yapılmıştır.
Hakem Değerlendirmesi: Dış bağımsız.
Yazar Katkıları: Fikir - B.Z., B.K.; Tasarım - B.Z., B.K.; Denetleme - B.Z.; Malzemeler - B.Z., B.K.; Veri Toplanması ve/veya İşlemesi - B.K.; Analiz ve/veya Yorum - B.Z., B.K.; Literatür Taraması - B.K.; Yazıyı Yazan - B.Z., B.K.; Eleştirel İnceleme - B.Z.
Çıkar Çatışması: Yazarlar çıkar çatışması bildirmemişlerdir.
Mali Destek: Yazarlar bu çalışma için mali destek almadıklarını beyan etmişlerdir.
References
- 1.Mariani G, Herrmann FH, Dolce A, et al. Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. Thromb Haemost. 2005;93:481–7. doi: 10.1160/TH04-10-0650. [DOI] [PubMed] [Google Scholar]
- 2.Perry DJ. Factor VII deficiency. Br J Haematol. 2002;118:689–700. doi: 10.1046/j.1365-2141.2002.03545.x. [DOI] [PubMed] [Google Scholar]
- 3.Palla R, Peyvandi F, Shapiro AD. Rare bleeding disorders:diagnosis and treatment. Blood. 2015;125:2052–61. doi: 10.1182/blood-2014-08-532820. [DOI] [PubMed] [Google Scholar]
- 4.Peyvandi F, Palla R, Menegatti M, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders:results from the European Network of Rare Bleeding Disorders. J Thromb Haemost. 2012;10:615–21. doi: 10.1111/j.1538-7836.2012.04653.x. [DOI] [PubMed] [Google Scholar]
- 5.Herrmann FH, Wulff K, Auerswald G, et al. Factor VII deficiency:clinical manifestation of 717 subjects from Europe and Latin America with mutations in the factor 7 gene. Haemophilia. 2009;15:267–80. doi: 10.1111/j.1365-2516.2008.01910.x. [DOI] [PubMed] [Google Scholar]
- 6.Sevenet PO, Kaczor DA, Depasse F. Factor VII deficiency:from basics to clinical laboratory diagnosis and patient management. Clin Appl Thromb Hemost. 2017;23:703–10. doi: 10.1177/1076029616670257. [DOI] [PubMed] [Google Scholar]
- 7.Mariani G, Konkle BA, Ingerslev J. Congenital factor VII deficiency:therapy with recombinant activated factor VII –a critical appraisal. Haemophilia. 2006;12:19–27. doi: 10.1111/j.1365-2516.2006.01180.x. [DOI] [PubMed] [Google Scholar]
- 8.Napolitano M, Giansily-Blaizot M, Dolce A, et al. Prophylaxis in congenital factor VII deficiency:indications, efficacy and safety. Results from the Seven Treatment Evaluation Registry (STER) Haematologica. 2013;98:538–44. doi: 10.3324/haematol.2012.074039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Minno MN, Dolce A, Mariani G STER Study Group. Bleeding symptoms at disease presentation and prediction of ensuing bleeding in inherited FVII deficiency. Thromb Haemost. 2013;109:1051–9. doi: 10.1160/TH12-10-0740. [DOI] [PubMed] [Google Scholar]
- 10.Mariani G, Bernardi F. Factor VII Deficiency. Semin Thromb Hemost. 2009;35:400–6. doi: 10.1055/s-0029-1225762. [DOI] [PubMed] [Google Scholar]
- 11.Alexander B, Goldstein R, Landwehr G, Cook CD. Congenital SPCA deficiency:a hitherto unrecognized coagulation defect with hemorrhage rectified by serum and serum fractions. J Clin Invest. 1951;30:596–608. doi: 10.1172/JCI102477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Napolitano M, Siragusa S, Mariani G. Factor VII deficiency:clinical phenotype, genotype and therapy. J Clin Med. 2017;6:38. doi: 10.3390/jcm6040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peyvandi F, Mannucci PM, Asti D, Abdoullahi M, Di Rocco N, Sharifian R. Clinical manifestations in 28 Italian and Iranian patients with severe factor VII deficiency. Haemophilia. 1997;3:242–6. doi: 10.1046/j.1365-2516.1997.00137.x. [DOI] [PubMed] [Google Scholar]
- 14.Siboni SM, Biguzzi E, Mistretta C, Garagiola I, Peyvandi F. Long-term prophylaxis in severe factor VII deficiency. Haemophilia. 2015;21:812–9. doi: 10.1111/hae.12702. [DOI] [PubMed] [Google Scholar]
- 15.Salcioglu Z, Akcay A, Sen HS, et al. Factor VII deficiency:a single-center experience. Clin Appl Thromb Hemost. 2012;18:588–93. doi: 10.1177/1076029611435091. [DOI] [PubMed] [Google Scholar]
- 16.Lapecorella M, Mariani G International Registry on Congenital Factor VII Deficiency. Factor VII deficiency:defining the clinical picture and optimizing therapeutic options. Haemophilia. 2008;14:1170–5. doi: 10.1111/j.1365-2516.2008.01844.x. [DOI] [PubMed] [Google Scholar]
- 17.De Moerloose P, Schved JF, Nugent D. Rare coagulation disorders:fibrinogen, factor VII and factor XIII. Haemophilia. 2016;22:61–5. doi: 10.1111/hae.12965. [DOI] [PubMed] [Google Scholar]
- 18.Robinson KS. An overview of inherited factor VII deficiency. Transfus Apher Sci. 2019;58:569–71. doi: 10.1016/j.transci.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Mariani G, Napolitano M, Dolce A, et al. Replacement therapy for bleeding episodes in factor VII deficiency. A prospective evaluation. Thromb Haemost. 2013;109:238–47. doi: 10.1160/TH12-07-0476. [DOI] [PubMed] [Google Scholar]
- 20.Mariani G, Dolce A, Batorova A, et al. STER and the International Factor VII Deficiency. Study Groups. Recombinant, activated factor VII for surgery in factor VII deficiency:a prospective evaluation - the surgical STER. Br J Haematol. 2011;152:340–6. doi: 10.1111/j.1365-2141.2010.08287.x. [DOI] [PubMed] [Google Scholar]
- 21.Di Minno MN, Napolitano M, Dolce A, Mariani G STER Study Group. Role of clinical and laboratory parameters for treatment choice in patients with inherited FVII deficiency undergoing surgical procedures:evidence from the STER registry. Br J Haematol. 2018;180:563–70. doi: 10.1111/bjh.15055. [DOI] [PubMed] [Google Scholar]