

Abstract
Aim:
To determine the frequency of sinopulmonary infections, detect changes in the respiratory system, and measure functional capacity of the lungs in our patients with humoral immunodeficiency.
Material and Methods:
Fifty-six patients with humoral immunodeficiency were enrolled in this study. The clinical, laboratory, and radiologic data, and pulmonary function tests of the subjects were evaluated from their file records, retrospectively.
Results:
The distribution of our patients was as follows: 25 patients had common variable immune deficiency, three patients had X-linked agammaglobulinemia, five patients had hyper immunoglobulin M syndrome, 19 patients had deficiency of immunoglobulin G subset, and four patients had selective immunoglobulin A deficiency. The most common symptom of the patients was chronic cough (n=47, 83.9%). The most common pathologies on high-resolution computed tomography of the chest were atelectasis and bronchiectasis (27.7%). The most common pathology in pulmonary function tests was the presence of moderate obstructive patterns along with restrictive patterns (n=6,12.5%). The FEV 1, FVC, and FEF 25–75 values were significantly lower in patients with common variable immunodeficiency compared with the patients who had IgG subset deficiencies (p=0.001, p=0.01, p=0.01). Among the patients who were treated with intravenous immunoglobulin, the age at the diagnosis of immunodeficiency was higher in patients with bronchiectasis (14.2±8.4 years) compared with those without bronchiectasis (10.1±11.4 years) (p=0.04).
Conclusion:
Clinical findings are not sufficient to monitor the structural and functional changes in the respiratory system, and patients should be evaluated using high-resolution computed tomography of the chest and pulmonary function tests.
Keywords: Humoral immunodeficiency, pulmonary findings, radiologic evaluation
Abstract
Amaç:
Humoral immün yetmezlik tanılı olgularımızda, sinopulmoner enfeksiyonların sıklığı ve bunların sonucunda solunum sisteminde meydana gelen değişikliklerin saptanması ve fonksiyonel akciğer kapasitelerinin ölçülmesi amaçlanmıştır.
Gereç ve Yöntemler:
Çalışmaya humoral immün yetmezlik tanılı 56 olgu alındı. Olguların dosya kayıtlarından klinik, laboratuvar, radyolojik görüntüleme bulguları ve solunum fonksiyon testi verileri geriye dönük olarak incelendi.
Bulgular:
Olguların tanılara göre dağılımı; yaygın değişken immün yetmezlik tanılı 25 olgu, X’e bağlı agamaglobulinemi tanılı üç olgu, Hiper immunoglobulin M sendromu tanılı beş olgu, İgG alt grup eksikliği tanılı 19 olgu ve selektif immunoglobulin A eksikliği tanılı dört olgu şeklindedir. Olguların 37’si (%66,1) erkek 19’u (%33,9) kadın, yaş ortalaması 14,1±10,6 yıldı. Olguların en sık yakınması kronik öksürük idi (n=47, %83,9). Akciğerlerin yüksek çözünürlüklü bilgisayarlı tomografilerinde en sık görülen patoloji atelektazi ve bronşiektaziydi (%27,7). Solunum fonksiyon testlerinde en sık görülen anormallik orta obstrüktif ve orta restriktif patern birlikteliğiydi (n=6,%12,5). Solunum fonksiyon testi bulguları ile radyolojik bulguların her zaman tutarlı olmadığı görüldü. İntravenöz immünglobulin alan olgular arasında yaygın değişken immün yetmezlik tanılı olgularda, immünglobulin G alt grup eksikliği olan olgulara göre solunum fonksiyon testlerinde; FEV 1, FVC ve FEF 25–75 değerlerinin anlamlı düşük olduğu saptandı (p=0,001, p=0,01, p=0,01). İntravenöz immünglobulin tedavisi alan olgularda; bronşiektazisi olanların immün yetmezlik tanı yaşı (14,2±8,4 yıl), olmayanlara (10,1±11,4 yıl) göre anlamlı olarak büyüktü (p=0,04).
Çıkarımlar:
Klinik bulgular solunum sistemindeki yapısal ve fonksiyonel değişiklikleri izlemek için yeterli değildir ve olgular gereğinde akciğer yüksek çözünürlüklü bilgisayarlı tomografi ve solunum fonksiyon testleri ile değerlendirilmelidir.
Introduction
Primary immunodeficiencies (PID) are characterized by infectious diseases, allergy, autoimmune diseases, and disorders in the function of the immune system with tendency to malignancies (1, 2). Although the prevalence and distribution vary by region, humoral immune system diseases constitute 55.2% of PIDs in Europe (3). In Turkey, the prevalence of humoral immune deficiency was found as 73.5% in a prevalence study based on the records of the European Society for Immunodeficiencies (ESID) (4). In a study conducted by Yorulmaz et al. (5), the frequency of humoral immunodeficiency was reported as 92.8% in 1054 patients with PID.
Humoral immunodeficiencies are characterized by insufficient antibody production and the main subgroups are as follows: common variable immune deficiency (CVID), X-linked agammaglobulinemia (XLA), immunoglobulin (Ig) class-switching defect/hyper IgM syndrome, selective IgA, IgG, IgM deficiencies, IgG subgroup deficiencies, specific antibody production defects, and other hypogammaglobulinemias (6).
In primary immunodeficiencies, different complications related to the respiratory system including recurrent pneumonia, bronchitis, chronic atelectasis, empyema, interstitial pulmonary fibrosis, pulmonary arterial hypertension, cor pulmonale, hilar lymphadenopathy, bronchiectasis, lymphoid interstitial pneumonia, pulmonary abscess, lymphoreticular malignancies, and bronchiolitis obliterans may occur (7–9). In individuals who are affected, Haemophilus influenzae, Streptococcus pneumoniae, and Staphylococcus aureus are frequently responsible for pyogenic infections (10–12).
In this study, it was aimed to evaluate the frequency of sinopulmonary infections and pathologic changes occurring in the respiratory tract as a result of these infections in patients with humoral immunodeficiency.
Material and Methods
The files of 56 patients who were followed up in Uludağ University, Faculty of Medicine, Pediatric Immunology Clinic with a diagnosis of humoral immunodeficiency were examined, retrospectively. The study was conducted in accordance with the principles of the Declaration of Helsinki. Approval was obtained from Uludağ University, Faculty of Medicine, Medical Researches Ethics Committee (June 2009, Decision number: 2009-11/63).
The data related to age, demographic properties, clinical characteristics, intravenous immunoglobulin (IVIG) treatment periods, infections, number of hospitalizations due to respiratory tract infection, sputum cultures, treatments administered, and comorbid allergic, autoimmune, and malignant diseases were recorded.
The Ig levels at the time of presentation and final Ig levels were evaluated. Cough lasting for at least 8 weeks was considered chronic cough. The upper limit was considered 100 IU/L for total IgE and 0.35 IU/L for specific IgE.
Evaluation of imaging findings
Lung radiography and high-resolution computed tomography (HRCT) findings were reevaluated by the same radiologist. Lung radiographs that were not obtained with an interval of shorter than two months, were not evaluated because of the necessity of assessing lung radiography and HRCTs in association. Diffuse bronchiectasis was defined as bronchiectasis formations in three or more lobes. The patients’ pulmonary nodules were classified as parenchymal, peribronchial, and diffuse nodules.
Pulmonary function test
Pulmonary function tests (PFT) were performed using a spirometer (Jaeger MasterScope, Germany) and the data were interpreted according to the American Thoracic Society guidelines (13). Vital capacity (VC), forced vital capacity (FVC), forced expiratory volume in one second (FEV1), forced expiratory flow during the middle portion of a forced expiration (FEF 25–75%), and the ratio of FEV1 to FVC as percentage, were calculated.
Statistical Analysis
The SPSS 23 program was used for statistical analysis of the data. Descriptive statistics and/or frequency distribution were calculated by variable types. The Kolmogorov-Smirnov and Shapiro-Wilk tests were performed. Wilcoxon’s signed-rank test was used for the comparison of the median values of two dependent groups, and the Mann-Whitney U test was used for the comparison of the median values of two independent groups. Fisher’s exact test was used for the analysis of categorical variables. A p value of <0.05 was considered statistically significant.
Results
The subjects’ distribution by diagnosis was as follows: 25 subjects with CVID, three subjects with XLA, five subjects with hyper IgM syndrome, and 23 subjects with IgG subgroup deficiency, partial or selective immunoglobulin A deficiency.
Thirty-seven (66.1%) of the subjects were male and 19 (33.9%) were female. The mean age was found as 14.2±10.6 years, and the mean age at the time of diagnosis was 9.7±9.8 years. The patients’ distribution by diagnosis, age, age at the time of diagnosis, number of hospitalizations, and IVIG treatment periods are shown in Table 1.
Table 1.
Mean age, age at the time of diagnosis, and number of hospitalizations by diagnosis groups
| Number of patients | Age of the patients (years) Mean±SD (min.–max.) | Age at the time of diagnosis (years) Mean±SD (min.–max.) | Number of hospitalizations (years) Mean±SD (min.–max.) | IVIG treatment period (years) Mean±SD (min.–max.) | |
|---|---|---|---|---|---|
| CVID | 25 | 18.8±10.6 (6–43) | 12.0±11.0 (1–34) | 3.8±3.0 (0–13) | 6.7±4.6 (0.5–20) |
| Hyper IgM syndrome | 5 | 25.2±15.6 (14–51) | 21.9±13.8 (10–45) | 2.8±2.3 (0–6) | 3.3±2.4 (0.5–6) |
| XLA | 3 | 13.0±8.0 (5–21) | 5.3±4.0 (3–10) | 1.0±1.7 (0–3) | 7.6±4.9 (2–11) |
| Selective IgA deficiency | 4 | 6.5±1.7 (4–8) | 4.7±1.5 (2.5–6) | 0.8±0.9 (0–2) | 0.0±0.0 (0–0) |
| IgG subgroup deficiencies | |||||
| IgG3 deficiency | 10 | 7.3±2.7 | 5.2±1.0 | 1.1±1.7 | 0.0±0.0 |
| IgG2 deficiency | 4 | (4–13) | (4–10) | (0–6) | (0–0) |
| IgG3 and IgG1 deficiency | 2 | ||||
| IgG3 +partial IgA deficiency | 2 | ||||
| IgG1 deficiency | 1 | ||||
| Total (n=56) | 56 | 14.2±10.6 (2–51) | 9.7±9.8 (1–45) | 2.4±2.7 (0–13) | 6.2±4.5 (0.5–20) |
Hyper IgM: Hyperimmunoglobulin M syndrome; Ig: Immunoglobulin; IVIG: Intravenous immunoglobulin; SD: Standard deviation; CVID: Common variable immunodeficiency; XLA: X-linked agammaglobulinemia
Forty-seven (83.9%) of the subjects had chronic cough (productive and non-productive), 33 (58.9%) produced sputum, and 10 (17.9%) had dyspnea. Growth in culture was found in only four subjects who were diagnosed as having pneumonia (Haemophilus influenzae in two subjects, Acinetobacter baumannii in one subject, Candida albicans in one subject).
Sixteen (28.5%) subjects had asthma, 30.3% (n=17) had allergic rhinitis, and 16% (n=9) had both asthma, and allergic rhinitis. The total IgE level was elevated in 11 subjects (19.6%) and specific IgE levels were elevated in seven subjects (mite in two subjects, egg white in two subjects, grass in two subjects, and fungus in one subject). Two of the subjects with positive specific IgE had asthma, one had allergic rhinitis, one had allergic conjunctivitis, and three had association of asthma, allergic rhinitis, and conjunctivitis. Twelve subjects (21.4%) had a family history of atopy, and 32 (57.1%) subjects had a family history of smoking.
The subjects’ annual frequencies of sinopulmonary infection before and after IVIG treatment were compared. When the total of 33 subjects who were given intravenous immunoglobulin were examined, the annual frequency of pulmonary infection was found as 4.5±3.0 before IVIG treatment and 0.8±0.8 after IVIG treatment (p=0.001). The annual frequency of upper respiratory tract infection was found as 11.3±6.4 before IVIG treatment and 2.2±1.8 after IVIG treatment (p=0.001). The infection frequencies of the subjects with humoral immunodeficiency by subtypes before and after IVIG are shown in Table 2.
Table 2.
Number of annual sinopulmonary infections before and after treatment by diagnosis groups in the subjects who received intravenous immunoglobulin
| Number of lung infections before IVIG/year Mean (min.–max.) | Number of lung infections after IVIG/year Mean (min.–max.) | p | Number of URTI before IVIG/year Mean (min.–max.) | Number of URTI after IVIG/year Mean (min.–max.) | p | |
|---|---|---|---|---|---|---|
| CVID (n=25) | 5 (1–14) | 1 (0–2) | <0.001 | 10 (2–30) | 2 (0–10) | <0.001 |
| Hyper IgM (n=5) | 4 (1–8) | 0 (0–3) | NA | 10 (7–15) | 3 (0–4) | NA |
| XLA (n=3) | 1 (0–2) | 0 (0–1) | NA | 4 (2–10) | 0 (0–3) | NA |
| Total (n=33) | 3 (0–14) | 0 (0–3) | <0.001 | 10 (2–30) | 5 (0–10) | <0.001 |
Ig: Immunoglobulin; IVIG: Intravenous immunoglobulin; Hyper IgM: Hyperimmunoglobulin M syndrome; CVID: Common variable immunodeficiency; XLA: X-linked agammaglobulinemia; Test: Wilcoxon signed-rank test
Forty-one percent of the subjects (n=23) were using antibiotic prophylaxis (18 subjects trimethoprim-sulfamethoxazole, two subjects ciprofloxacin, three subjects clarithromycin). Long-acting β2 agonist with inhaled steroid was given to patients with bronchoconstriction, and chest physiotherapy was applied to patients with bronchiectasis. In addition, rituximab and steroid were given to one patient who had nodular pulmonary findings, and abatacept, steroid, and sirolimus were given to another patient, and steroid and sirolimus were given to a third patient.
Chest radiography
Among 27 subjects who had an antero-posterior lung radiography obtained in the last two months, 17 (62.9%) had normal findings, five had consolidation, three had bronchial thickening, one had volume loss in one lung, and one had bronchiectasis.
High-resolution computed tomography (HRCT)
Lung HRCTs of 54 (96.4%) subjects could be obtained and 55.5% of these subjects (n=30) had normal lung HRCT. The most common pathologies were found to be atelectasis and bronchiectasis (27.7%, n=15). The distribution of lung HRCT findings by order of frequency is shown in Figure 1. The distribution of these findings by diagnoses is presented in Table 3. When the regions in which bronchiectasis was found were evaluated by order of frequency, the most commonly involved region was found to be the right lower lobe (18.5%), followed by the left lower lobe (16.7%) and the right middle lobe (11.1%); the least commonly involved region was found to be the left upper lobe (5.6%).
Figure 1.
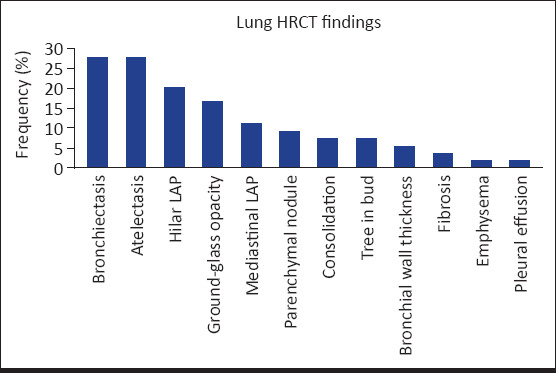
Distribution of the findings on high resolution computed tomography of the lung by order of frequency
Table 3.
Assessment of high-resolution computed tomography of the lung and pulmonary function test findings in the subjects with humoral immunodeficiency by diagnosis groups
| CVID | Hyper IgM | XLA | Selective IgA deficiency | IgG subgroup deficiency | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | n | % | n | % | |
| Lung HRCT | ||||||||||||
| Ground-glass opacity | 7 | 30.4 | 2 | 40 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 16.6 |
| Atelectasis | 11 | 47.8 | 2 | 40 | 1 | 33.3 | 0 | 0 | 1 | 5 | 15 | 27.7 |
| Bronchiectasis | 11 | 47.8 | 3 | 60 | 1 | 33.3 | 0 | 0 | 0 | 0 | 15 | 27.7 |
| Consolidation | 2 | 8.6 | 2 | 40 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 7.4 |
| Bronchial wall thickness | 3 | 13 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 5.5 |
| Parenchymal nodule | 3 | 13 | 2 | 40 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 9.2 |
| Pleural effusion | 1 | 4.3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1.8 |
| Tree in bud | 4 | 17.3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 7.4 |
| Fibrosis | 1 | 4.3 | 1 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 3.7 |
| Emphysema | 0 | 0 | 0 | 0 | 1 | 33.3 | 0 | 0 | 0 | 0 | 1 | 1.8 |
| Hilar LAP | 6 | 26 | 2 | 40 | 0 | 0 | 1 | 25 | 2 | 10 | 11 | 20.3 |
| Mediastinal LAP | 5 | 21.7 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 5 | 6 | 11.1 |
| Total | 23 | 100 | 5 | 100 | 3 | 100 | 4 | 100 | 19 | 100 | 54 | 100 |
| Pulmonary function tests | ||||||||||||
| Normal | 7 | 31.8 | 2 | 40 | 1 | 33.3 | 3 | 100 | 15 | 100 | 28 | 58.3 |
| Mild obstructive | 2 | 9.0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 4.1 |
| Moderate obstructive | 2 | 9.0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 6.2 |
| Marked obstructive | 2 | 9.0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 6.2 |
| Mild restrictive | 3 | 13.6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 6.2 |
| Moderate restrictive | 2 | 5.3 | 1 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 4.1 |
| Mild obstructive+restrictive | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2.0 |
| Moderate obstructive +restrictive | 4 | 18 | 2 | 40 | 1 | 33.3 | 0 | 0 | 0 | 0 | 6 | 12.5 |
| Total (Number of subjects who underwent pulmonary function test) | 22 | 100 | 5 | 100 | 3 | 100 | 3 | 100 | 15 | 100 | 48 | 100 |
Hyper IgM: Hyperimmunoglobulin M syndrome; HRCT: High-resolution computed tomography; CVID: Common variable immunodeficiency; XLA: X-linked agammaglobulinemia; LAP: Lymphadenopathy
All subjects with bronchiectasis had a history of recurrent pulmonary infection before the diagnosis. The frequencies of bronchiectasis by diagnoses were as follows: 47.8% in CVID (n=11), 60% in hyper IgM syndrome (n=3), and 33.3% in XLA (n=1). The appearances of bronchiectasis, atelectasis, ground-glass opacity, and parenchymal nodules observed on the patients’ lung HRCTs are shown in Figure 2.
Figure 2.
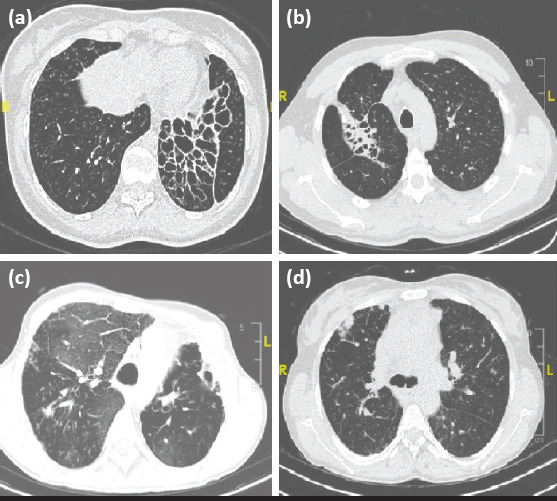
Appearances of the findings on high-resolution computed tomography of the lung. (a) Cystic bronchiectasis. (b) Bronchiectasis and atelectasis. (c) Ground-glass opacity. (d) Parenchymal nodule
Among the subjects who received intravenous immunoglobulin treatment, the IgG level was found as 800 mg/dL and above in 57.5% (n=19), and below 800 mg/dL in 42.5% (n=14) after treatment. Bronchiectasis was found to be less frequent in subjects who had IgG levels of 800 mg/dL and above (n=3, 15.7%) compared with those who had a IgG levels below 800 mg/dL (n=4, 33.3%), but this difference was not statistically significant (p=0.42).
Five of the subjects with diffuse bronchiectasis (n=7, 46.6%) were being followed up with a diagnosis of CVID, and two were being followed up with a diagnosis of hyper IgM syndrome. Three (17.6%) of the subjects who had diffuse bronchiectasis were diagnosed as having asthma. All these three subjects had moderate obstructive pattern findings in PFTs (mixed in two subjects).
Among 33 subjects who received intravenous immunoglobulin treatment, the age at the time of diagnosis was significantly older in the subjects who had bronchiectasis (14.2±8.4 years) compared with those without bronchiectasis (10.1±11.4 years) (p=0.04).
The file records revealed that follow-up lung HRCT was obtained in 11 subjects. The shortest time between shootings was found to be four years, and the longest time between shootings was 9 years and 6 months. It was found that previous bronchiectasis persisted in five of 11 patients, and bronchiectasis was newly found in three patients. Bronchiectasis was not observed on new lung HRCTs in three patients who did not have bronchiectasis previously. It was observed that atelectasis persisted in three of six subjects who had atelectasis and regressed in three. Development of new atelectasis areas was observed in two patients. Ground-glass opacity persisted in two of four patients with ground-glass opacity and regressed in two. New ground-glass areas were observed in four patients. ‘Tree-in-bud’ appearance regressed in two patients who had this finding and new ‘Tree-in-bud’ appearance developed in one patient. Among two patients who had nodular findings in the lung, these findings persisted in one and new nodular findings were found in five patients who did not have nodular appearance previously.
Pulmonary function test
PFTs were found to be within the normal limits in 58.3% of the subjects (n=28). SFTs were found to be normal in all patients who had a diagnosis of selective IgA deficiency and IgG subgroup deficiency. The most common pathology observed in pulmonary function tests was association of moderate obstructive and moderate restrictive pattern (n=6, 12.5%). Assessments of 48 subjects who underwent pulmonary function test are shown in Table 3.
PFTs revealed a significant reduction in the FEV1, FVC, and FEF 25–75 values in subjects who had common variable immunodeficiency compared with the subjects who had IgG subgroup deficiency (p=0.001, p=0.01, p=0.01) (Table 4). All measurements in PFTs were found to be lower in subjects who had X-linked agammaglobulinemia compared with subjects who had CVID and hyper IgM syndrome, but statistical analysis could not be performed because the number of subjects was low.
Table 4.
Distribution of the test findings by diagnoses in the subjects in whom pulmonary function tests could be performed
| CVID n=25 | IgG subgroup deficiencies n=19 | p | |
|---|---|---|---|
| FEV 1 median (min.–max.) | 74.5 (19–124) | 99 (74–122) | 0.001 |
| FVC median (min.–max.) | 73.5 (48–127) | 90 (63–125) | 0.01 |
| FEV 1/FVC median (min.–max.) | 84.5 (30–117) | 86 (84–111) | 0.15 |
| VC median (min.–max.) | 74.5 (42–134) | 88 (66–113) | 0.06 |
| FEF 25–75 median (min.–max.) | 79 (5–140) | 104 (83–175) | 0.01 |
FEF 25–75: Forced expiratory flow at 25–75% of forced vital capacity; FEV1: Forced expiratory volume in 1 second; FVC: Forced vital capacity; Hyper IgM: Hyperimmunoglobulin M syndrome; VC: Vital capacity; CVID: Common variable immunodeficiency; XLA: X-linked agammaglobulinemia; Test: Mann-Whitney U test
The FEV1, FEV1/FVC, and FEF 25–75 values were lower in subjects who had bronchiectasis compared with those without bronchiectasis, though the difference was not statistically significant (p>0.05). The FEF 25–75% value was found to be lower in subjects who had diffuse bronchiectasis as compared with those without diffuse bronchiectasis (p=0.01). No significant difference was found between the two groups in terms of other PFT measurements. PFT findings showed no significant difference between the subjects who did and did not have atelectasis on lung HRCT (p>0.05).
No correlation was found between PFTs and lung pathologies in five subjects who had parenchymal nodules (three CVID, two Hyper IgM). PFTs were found as normal in two of these subjects. Marked obstructive pattern was observed in one subject who had abnormal PFTs and parenchymal nodules, and a moderate obstructive pattern and mixed pattern were found in the other two patients, respectively.
Discussion
Although immunoglobulin treatment reduces the risk of development of chronic pulmonary disease by reducing the frequency of infection in humoral immune deficiencies, pulmonary problems are an important cause of morbidity in these cases (1, 14).
Chronic cough is an important marker for diseases such as chronic bronchitis, bronchiectasis, and sinusitis (15). In our study, the most common respiratory symptom in our cases was found to be chronic cough (83.9%, n=47). In different studies, the frequency of cough has been reported to range between 30% and 40% (16–18).
The defect that occurs in mucosal immunity facilitates inhaled allergen sensitivity, bronchial hyperreactivity, and thus development of asthma (19). The pathogenesis of asthma in patients with common variable immunodeficiency is still not clear; intrinsic asthma (non-atopic) is observed mostly (20). In different studies, the frequency of asthma has been reported to range between 9% and 15% in patients with CVID (15, 21–23). In our study, the percentage of general asthma was found as 24% and the percentage of atopic asthma was found as 4% in cases of CVID. Elevated total IgE was found too with a rate of 19.6% in the whole group, and with a rate of 37.5% in subjects with asthma. Elevated specific IgE was found in 31.6% of the subjects with asthma.
In IgG subgroup deficiency, respiratory tract infections occur frequently as a result of insufficient immune response against infectious agents. In two studies conducted with children, an association of IgG subgroup deficiency with asthma was reported with a rate of 7% and with a rate of 34% (24, 25). Asthma was observed with a rate of 31.5% in our subjects who had IgG subgroup deficiency. In our study, IgG3 deficiency was found in 50% of the subjects who had asthma and IgG subgroup deficiency. Thus, the most common subgroup deficiency was found as IgG3 deficiency in children with severe asthma in the study conducted by de Moraes Lui et al. (26).
Increased serum IgG levels were shown to have a healing effect in terms of lung involvement in patients with humoral immunodeficiency (27). In our study, the percentage of bronchiectasis was found lower in patients who had an IgG threshold level above 800 mg/dL, in accordance with the literature, though the difference was not statistically significant. This was thought to be associated with the low number of patients.
Bronchiectasis is irreversible dilatation of the airways that develops as a result of inflammatory destruction. The best method to make a diagnosis of bronchiectasis is lung HRCT (28, 29). In many previous studies, the frequency of bronchiectasis has been reported to range between 38.1% and 71% in patients with CVID (28, 30–33). In two studies conducted in our country, the rates of bronchiectasis were reported as 41.6% and 68%, respectively, in patients with CVID (8, 9). In our study, the frequency of bronchiectasis was found as 27.7% in all subjects, whereas this rate was 47.8% in subjects with CVID, similar to the literature. Bronchiectasis was found with a rate of 33.3% in patients with a diagnosis of X-linked agammaglobulinemia and with a rate of 60% in patients hyper IgM syndrome.
In patients with humoral immunodeficiency, bronchiectasis is observed most commonly in the middle and lower lobes (29, 30, 34). In different studies, different areas of involvement including the upper lobes, right middle lobe, and lower lobes, have been reported (29, 30, 35). In our study, bronchiectasis was observed more frequently in the lower lobes (57.5%) in accordance with the literature [especially in the right lower lobe (18.5%)].
In primary immunodeficiencies, respiratory findings occur at the age of 3–5 years in 20% of children. Therefore, permanent lung injury might have already developed when the diagnosis was made at advanced ages in these subjects (36). In addition, positive effects of early diagnosis of immunodeficiency and early initiation of treatment in terms of protecting lung parenchyma have been shown in various studies (30, 37, 38). In this study, the age at the time of diagnosis was found to be significantly higher in subjects with bronchiectasis compared with those without bronchiectasis.
In patients with common variable immunodeficiency, granulomas observed in the lung and other organs were thought to occur as a result of an inflammatory process (39). The incidence of granuloma in patients with CVID was reported as 5.4–10% by Mechanic et al. (40) and 50% by Park et al. (41). In our study, parenchymal nodules were found in 8.9% (n=5) of all subjects [three patients with CVID (12%) and two patients with hyper IgM syndrome (40%)]. All nodules were smaller than 5 mm and showed irregular distribution.
Lymphocytic interstitial pneumonia is a common finding in lymphoproliferative diseases and characterized by diffuse interstitial accumulation in plasma cells. Radiologic findings of the disease are variable, ranging from interstitial infiltrations to perihilar involvement, and from parenchymal nodules to patchy infiltration and ground-glass appearance (42). In patients with common variable immunodeficiency, lymphocytic interstitial pneumonia is characterized by diffuse or multifocal reactive lymphoid infiltrations, especially in the alveolar interstitium (43–45). These appearances are mostly nonspecific and cannot be differentiated from opportunistic infections. In our study, interstitial fibrosis was found in two patients with hyper IgM syndrome and in one subject with CVID.
Early interstitial ground-glass opacity showing airway involvement represents incomplete filling of the parenchymal interstitium or alveoli. Early recognition of this finding, which is generally observed in interstitial pneumonia, is important, because it shows active or treatable disease phases (46). Ground-glass appearance was found with a rate of 10% on lung HRCTs by Boloursaz et al. (47) and 42% by Obregon et al. (34) in patients with CVID. In our study, ground-glass opacity on lung HRCT was found with a rate of 16.6% (n=9) in the entire study population (30% in patients with CVID and 40% in patients with hyper IgM syndrome).
Although ‘tree in bud,’ which is a term used for dilated centrilobular bronchioles that are filled with fluid on lung HRCT, is generally a marker of infection, it may also be observed when the bronchioles are obstructed with mucus plaque in patients with pulmonary tuberculosis, bronchiectasis, and asthma (48, 49). ‘Tree-in-bud’ appearance was found in only four patients who had CVID (17.3%) in our study group, and all these patients had bronchiectasis additionally.
In patients with common variable immunodeficiency, restrictive lung disease may develop secondary to progression of primary or obstructive diseases (21). Watts et al. (50) found restrictive pattern on PFTs in 40% of subjects with CVID. Kainulainen et al. (28) reported that restrictive pattern on PFT most commonly developed secondary to pulmonary fibrosis. Restrictive pattern alone was observed in five of our subjects who had common variable immunodeficiency, and association of restrictive pattern and obstructive pattern was found in four subjects. Restrictive pattern on PFT in the absence of any pathology on lung HRCT is a clue for interstitial lung diseases (21). In our study, restrictive pattern was found in 12% of the subjects who had CVID despite normal lung CT findings, and association of restrictive and obstructive pattern was found in one subject with hyper IgM syndrome.
Although lung radiography is used in the initial assessment and follow-up of patients with immunodeficiency with respiratory problems, patients with chronic symptoms should be evaluated using CT. Hence, bronchiectasis was found on 3.7% of lung radiographs in our study, whereas this percentage was 27.7% in patients who were assessed with HRCT. Again, parenchymal nodules were found with a rate of 9.2% on HRCT, whereas no parenchymal nodules were detected in lung radiography. Although it is thought that HRCT should be obtained with certain intervals in patients with common variable immunodeficiency, there is no consensus on the issue in the literature of when to obtain the initial HRCT and what the intervals should be. In addition, the effect of radiation on the development of cancer should also be kept in mind in patients with immunodeficiency.
The limitations of our study included the study’s retrospective design, its low number of patients, and the fact that atopy was detected only through specific IgE levels.
In conclusion, assessment of lung findings in patients with humoral immunodeficiency and planning treatment accordingly, are very important in terms of prognosis. Patients with immunodeficiency should be evaluated with certain intervals in terms of parenchymal diseases. Assessment by lung HRCT and PFT may be needed even if lung radiography is normal.
Footnotes
Ethics Committee Approval: Approval was obtained from Uludağ University, Faculty of Medicine, Medical Research Ethics Committee for the study (June 2009, Decision number: 2009-11/63).
Informed Consent: Informed consent was not obtained due to the retrospective design of the study.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - S.Ş.K.G., Z.K.; Design - S.Ş.K.G., Z.K.; Supervision - S.Ş.K.G.; Funding - S.Ş.K.G., Z.K.,Y.K.; Materials - S.Ş.K.G., Z.K., Y.K.; Data Collection and/or Processing - S.Ş.K.G., Z.K., Y.K., Z.Y., N.S., Y.C.; Analysis and/or Interpretation - S.Ş.K.G., Z.K., Y.K., Ş.Ç.; Literature Review - S.Ş.K.G., Z.K., Y.K.; Writing - Z.K.; Critical Review - S.Ş.K.G., Z.K., Y.K., Ş.Ç.
Conflict of Interest: The authors have no conflicts of interest to declare.
Financial Disclosure: The authors declared that this study has received no financial support.
Etik Kurul Onayı: Çalışma için Uludağ Üniversitesi Tıp Fakültesi tıbbi araştırmalar etik kurulundan onay alındı (Haziran 2009 yılı, Karar No: 2009-11/63).
Hasta Onamı: Çalışmanın geriye dönük tasarımından dolayı hasta onamı alınmamıştır.
Hakem Değerlendirmesi: Dış bağımsız.
Yazar Katkıları: Fikir - S.Ş.K.G., Z.K.; Tasarım - S.Ş.K.G., Z.K.; Denetleme - S.Ş.K.G.; Kaynaklar - S.Ş.K.G., Z.K., Y.K.; Malzemeler - S.Ş.K.G., Z.K., Y.K.; Veri Toplanması ve/veya İşlemesi S.Ş.K.G., Z.K., Y.K., Z.Y., N.S., Y.C.; Analiz ve/veya Yorum - S.Ş.K.G., Z.K., Y.K., Ş.Ç.; Literatür Taraması - S.Ş.K.G., Z.K., Y.K.; Yazıyı Yazan - Z.K.; Eleştirel İnceleme - S.Ş.K.G., Z.K., Y.K., Ş.Ç.
Çıkar Çatışması: Yazarlar çıkar çatışması bildirmemişlerdir.
Mali Destek: Yazarlar bu çalışma için mali destek almadıklarını beyan etmişlerdir.
References
- 1.McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol. 2018;14:61. doi: 10.1186/s13223-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stiehm ER, Ochs HD, Winkelstein J. Immunodeficiency disorders;general consideration. In: Ochs HD, Stiehm ER, Winkelstein J, editors. Immunologic disorders in infant and children. 5th ed. Pennsylvania: Elsevier Saunders Company; 2004. pp. 652–84. [Google Scholar]
- 3.Gathmann B, Grimbacher B, Beauté J, et al. ESID Registry Working Party. The European internet-based patient and research database for primary immunodeficiencies:results 2006-2008. Clin Exp Immunol. 2009;157:3–11. doi: 10.1111/j.1365-2249.2009.03954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kilic SS, Ozel M, Hafizoglu D, Karaca NE, Aksu G, Kutukculer N. The prevalences [correction] and patient characteristics of primary immunodeficiency diseases in Turkey--two centers study. J Clin Immunol. 2013;33:74–83. doi: 10.1007/s10875-012-9763-3. [DOI] [PubMed] [Google Scholar]
- 5.Yorulmaz A, Artaç H, Kara R, Keleş S, Reisli İ. Primer immün yetmezlikli 1054 olgunun retrospektif değerlendirilmesi. Astım Allerji İmmünoloji. 2008;6:127–34. [Google Scholar]
- 6.Bousfiha A, Jeddane L, Picard C, et al. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2018;38:129–43. doi: 10.1007/s10875-017-0465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yazdani R, Abolhassani H, Asgardoon MH, et al. Infectious and Noninfectious Pulmonary Complications in Patients With Primary Immunodeficiency Disorders. J Investig Allergol Clin Immunol. 2017;27:2130–224. doi: 10.18176/jiaci.0166. [DOI] [PubMed] [Google Scholar]
- 8.Alkan G, Keles S, Reisli İ. Evaluation of Clinical and Immunological Characteristics of Children with Common Variable Immunodeficiency. Int J Pediatr. 2018;2018:3527480. doi: 10.1155/2018/3527480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Çalişkaner AZ, Reisli İ, Arslan Ş, Uçar R, Ataseven H, Selçuk NY. Common variable immunodeficiency in adults requires reserved protocols for long-term follow-up. Turk J Med Sci. 2016;46:430–6. doi: 10.3906/sag-1412-108. [DOI] [PubMed] [Google Scholar]
- 10.Cinetto F, Scarpa R, Rattazzi M, Agostini C. The broad spectrum of lung diseases in primary antibody deficiencies. Eur Respir Rev. 2018;27:180019. doi: 10.1183/16000617.0019-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sperlich JM, Grimbacher B, Workman S, et al. Respiratory Infections and Antibiotic Usage in Common Variable Immunodeficiency. J Allergy Clin Immunol Pract. 2018;6:159–68.e3. doi: 10.1016/j.jaip.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mooney D, Edgar D, Einarsson G, Downey D, Elborn S, Tunney M. Chronic lung disease in common variable immune deficiency (CVID):A pathophysiological role for microbial and non-B cell immune factors. Crit Rev Microbiol. 2017;43:508–19. doi: 10.1080/1040841X.2016.1268568. [DOI] [PubMed] [Google Scholar]
- 13.Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J. 2005;26:948–68. doi: 10.1183/09031936.05.00035205. [DOI] [PubMed] [Google Scholar]
- 14.Jesenak M, Banovcin P, Jesenakova B, Babusikova E. Pulmonary manifestations of primary immunodeficiency disorders in children. Front Pediatr. 2014;2:77. doi: 10.3389/fped.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rusconi F, Panisi C, Dellepiane RM, et al. Pulmonary and sinus diseases in primary humoral immunodeficiencies with chronic productive cough. Arch Dis Child. 2003;88:1101–5. doi: 10.1136/adc.88.12.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency:respiratory manifestations, pulmonary function and high-resolution CT scan findings. QJM. 2002;95:655–62. doi: 10.1093/qjmed/95.10.655. [DOI] [PubMed] [Google Scholar]
- 17.Aghamohammadi A, Allahverdi A, Abolhassani H, et al. Comparison of pulmonary diseases in common variable immunodeficiency and X-linked agammaglobulinaemia. Respirology. 2010;15:289–95. doi: 10.1111/j.1440-1843.2009.01679.x. [DOI] [PubMed] [Google Scholar]
- 18.Costa-Carvalho BT, Wandalsen GF, Pulici G, Aranda CS, Solé D. Pulmonary complications in patients with antibody deficiency. Allergol Immunopathol (Madr) 2011;39:128–32. doi: 10.1016/j.aller.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Papadopoulou A, Mermiri D, Taousani S, Triga M, Nicolaidou P, Priftis KN. Bronchial hyper-responsiveness in selective IgA deficiency. Pediatr Allergy Immunol. 2005;16:495–500. doi: 10.1111/j.1399-3038.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 20.Milota T, Bloomfield M, Parackova Z, Sediva A, Bartunkova J, Horvath R. Bronchial Asthma and Bronchial Hyperresponsiveness and Their Characteristics in Patients with Common Variable Immunodeficiency. Int Arch Allergy Immunol. 2019;178:192–200. doi: 10.1159/000494128. [DOI] [PubMed] [Google Scholar]
- 21.Busse PJ, Farzan S, Cunningham-Rundles C. Pulmonary complications of common variable immunodeficiency. Ann Allergy Asthma Immunol. 2007;98:1–43. doi: 10.1016/S1081-1206(10)60853-8. [DOI] [PubMed] [Google Scholar]
- 22.Agondi RC, Barros MT, Rizzo LV, Kalil J, Giavina-Bianchi P. Allergic asthma in patients with common variable immunodeficiency. Allergy. 2010;65:510–5. doi: 10.1111/j.1398-9995.2009.02211.x. [DOI] [PubMed] [Google Scholar]
- 23.Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON):Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract. 2016;4:38–59. doi: 10.1016/j.jaip.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loftus BG, Price JF, Lobo-Yeo A, Vergani D. IgG subclass deficiency in asthma. Arch Dis Child. 1988;63:1434–7. doi: 10.1136/adc.63.12.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JH, Ye YM, Ban GY, et al. Effects of Immunoglobulin Replacement on Asthma Exacerbation in Adult Asthmatics with IgG Subclass Deficiency. Allergy Asthma Immunol Res. 2017;9:526–33. doi: 10.4168/aair.2017.9.6.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Moraes Lui C, Oliveira LC, Diogo CL, Kirschfink M, Grumach AS. Immunoglobulin G subclass concentrations and infections in children and adolescents with severe asthma. Pediatr Allergy Immunol. 2002;13:195–202. doi: 10.1034/j.1399-3038.2002.00058.x. [DOI] [PubMed] [Google Scholar]
- 27.Ballow M. Practical aspects of immunoglobulin replacement. Ann Allergy Asthma Immunol. 2017;119:299–303. doi: 10.1016/j.anai.2017.07.020. [DOI] [PubMed] [Google Scholar]
- 28.Kainulainen L, Varpula M, Liippo K, Svedström E, Nikoskelainen J, Ruuskanen O. Pulmonary abnormalities in patients with primary hypogammaglobulinemia. J Allergy Clin Immunol. 1999;104:1031–6. doi: 10.1016/s0091-6749(99)70085-0. [DOI] [PubMed] [Google Scholar]
- 29.Curtin JJ, Webster AD, Farrant J, Katz D. Bronchiectasis in hypogammaglobulinaemia--a computed tomography assessment. Clin Radiol. 1991;44:82–4. doi: 10.1016/s0009-9260(05)80501-x. [DOI] [PubMed] [Google Scholar]
- 30.Dukes RJ, Rosenow EC, 3rd, Hermans PE. Pulmonary manifestations of hypogammaglobulinaemia. Thorax. 1978;33:603–7. doi: 10.1136/thx.33.5.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martínez García MA, de Rojas MD, Nauffal Manzur MD, et al. Respiratory disorders in common variable immunodeficiency. Respir Med. 2001;95:191–5. doi: 10.1053/rmed.2000.1020. [DOI] [PubMed] [Google Scholar]
- 32.Feydy A, Sibilia J, De Kerviler E, et al. Chest high resolution CT in adults with primary humoral immunodeficiency. Br J Radiol. 1996;69:1108–16. doi: 10.1259/0007-1285-69-828-1108. [DOI] [PubMed] [Google Scholar]
- 33.Sweinberg SK, Wodell RA, Grodofsky MP, Greene JM, Conley ME. Retrospective analysis of the incidence of pulmonary disease in hypogammaglobulinemia. J Allergy Clin Immunol. 1991;88:96–104. doi: 10.1016/0091-6749(91)90306-9. [DOI] [PubMed] [Google Scholar]
- 34.Obregon RG, Lynch DA, Kaske T, Newell JD, Jr, Kirkpatrick CH. Radiologic findings of adult primary immunodeficiency disorders Contribution of CT. Chest. 1994;106:490–5. doi: 10.1378/chest.106.2.490. [DOI] [PubMed] [Google Scholar]
- 35.Reiff DB, Wells AU, Carr DH, Cole PJ, Hansell DM. CT findings in bronchiectasis:limited value in distinguishing between idiopathic and specific types. AJR Am J Roentgenol. 1995;165:261–7. doi: 10.2214/ajr.165.2.7618537. [DOI] [PubMed] [Google Scholar]
- 36.Primary immunodeficiency diseases. Report of an IUIS Scientific Committee. International Union of Immunological Societies. Clin Exp Immunol. 1999;118:1–28. doi: 10.1046/j.1365-2249.1999.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cunningham-Rundles C. Clinical and immunologic analyses of 103 patients with common variable immunodeficiency. J Clin Immunol. 1989;9:22–33. doi: 10.1007/BF00917124. [DOI] [PubMed] [Google Scholar]
- 38.Cunningham-Rundles C, Bodian C. Common Variable imunodeficiency:clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 39.Mullighan CG, Fanning GC, Chapel HM, Welsh KI. TNF and lymphotoxin-alpha polymorphisms associated with common variable immunodeficiency:role in the pathogenesis of granulomatous disease. J Immunol. 1997;159:6236–41. [PubMed] [Google Scholar]
- 40.Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med. 1997;127:613–7. doi: 10.7326/0003-4819-127-8_part_1-199710150-00005. [DOI] [PubMed] [Google Scholar]
- 41.Park JE, Beal I, Dilworth JP, Tormey V, Haddock J. The HRCT appearances of granulomatous pulmonary disease in common variable immune deficiency. Eur J Radiol. 2005;54:359–64. doi: 10.1016/j.ejrad.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 42.Gibson M, Hansell DM. Lymphocytic disorders of the chest:pathology and imaging. Clin Radiol. 1998;53:469–80. doi: 10.1016/s0009-9260(98)80165-7. [DOI] [PubMed] [Google Scholar]
- 43.Davies CW, Juniper MC, Gray W, Gleeson FV, Chapel HM, Davies RJ. Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinaemia treated with cyclosporin A. Thorax. 2000;55:88–90. doi: 10.1136/thorax.55.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia:a survey of clinical manifestations and complications. Q J Med. 1993;86:31–42. [PubMed] [Google Scholar]
- 45.Buckley RH. Pulmonary complications of primary immunodeficiencies. Paediatr Respir Rev. 2004;5:S225–33. doi: 10.1016/s1526-0542(04)90043-7. [DOI] [PubMed] [Google Scholar]
- 46.Tuncel E. Ankara: Nobel Tıp GüneşYayınları; 2008. Klinik radyoloji; pp. 278–9. [Google Scholar]
- 47.Boloursaz MR, Khalilzadeh S, Nezhad PR, et al. Chest CT Manifestations in Children with CVID:A 10-Year Report. Tanaffos. 2012;11:56–9. [PMC free article] [PubMed] [Google Scholar]
- 48.Webb WR, Higgins CB. Philadelphia: Lippincott Williams and Wilkins; 2005. Thoracic Imaging:Pulmonary and Cardiovascular Radiology; pp. 321–2. [Google Scholar]
- 49.Miller WT, Jr, Panosian JS. Causes and imaging patterns of tree-in-bud opacities. Chest. 2013;144:1883–92. doi: 10.1378/chest.13-1270. [DOI] [PubMed] [Google Scholar]
- 50.Watts WJ, Watts MB, Dai W, Cassidy JT, Grum CM, Weg JG. Respiratory dysfunction in patients with common variable hypogammaglobulinemia. Am Rev Respir Dis. 1986;134:699–703. doi: 10.1164/arrd.1986.134.4.699. [DOI] [PubMed] [Google Scholar]