

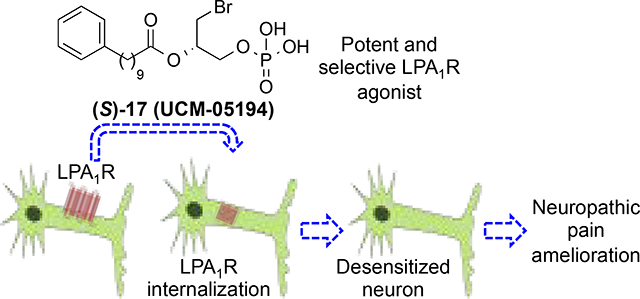
Abstract
Neuropathic pain (NP) is a complex chronic pain state with a prevalence of almost 10% in the general population. Pharmacological options for NP are limited and slightly effective, so there is a need of developing more efficacious NP attenuating drugs. Activation of the type 1 lysophosphatidic acid (LPA1) receptor is a crucial factor in the initiation of NP. Hence, it is conceivable that a functional antagonism strategy could lead to NP mitigation. Here we describe a new series of LPA1 agonists among which derivative (S)-17 (UCM-05194) stands out as the most potent and selective LPA1 receptor agonist described so far (Emax=118%, EC50=0.24 μM, KD=19.6 nM; inactive at autotaxin and LPA2–6 receptors). This compound induces characteristic LPA1-mediated cellular effects and prompts the internalization of the receptor leading to its functional inactivation in primary sensory neurons and to an efficacious attenuation of the pain perception in an in vivo model of NP.
Graphical Abstract
INTRODUCTION
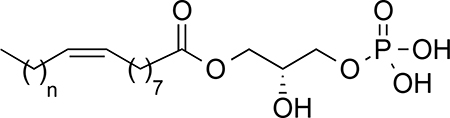
Lysophosphatidic acid (LPA, 1-acyl-sn-glycerol-3-phosphate) is a lipid mediator required for maintaining the homeostasis of numerous physiological processes but also participates in the development of various pathological conditions. Although LPA can denote a variety of lysophospholipids with saturated (16:0, 18:0) and unsaturated (16:1, 18:1, 18:2, 20:4) acyl chains, in the context of LPA as a signaling molecule and thus along this work, LPA refers to 1-oleoyl-sn-glycerol-3-phosphate. LPA acts through six different receptors (LPA1–6) that belong to the G protein-coupled receptor (GPCR) superfamily.1, 2 These six receptors can be further divided into two groups, the endothelial differentiation gene (EDG) family (LPA1–3) and the phylogenetically distant non-EDG family (LPA4–6).3, 4 Genetic deletion studies, over-expression models, and other chemical tools have provided insight into some of the many biological effects, mechanisms and medical application of LPA signaling.5–7 Because LPA signaling has been implicated in cancer,8, 9 fibrosis,10, 11 and pain,12–14 LPA receptors are regarded as promising drug targets.15, 16 Among all these effects, LPA has recently attracted attention because of its emerging role as an important factor in neuropathic pain (NP).13, 14, 17, 18 NP is a type of chronic pain caused by a lesion or disease of the somatosensory system, including peripheral fibers and central neurons. It affects around 10% of the general population and its incidence is likely to increase. Patients with NP do not usually respond to standard pain treatments such as acetaminophen, non-steroidal anti-inflammatory drugs (NSAIDs) or weak opioids. In general, available pharmacological treatments for NP are only moderately efficacious with effectiveness in less than 50% of patients, and they are also associated with adverse effects.19 Mechanistically, NP arises because peripheral sensory and central neurons show a gain of excitation and/or a loss of inhibition prompting a permanent state of hyperexcitability. In this scenario, the brain receives abnormal sensory messages that translate into continuous burning pain and abnormal sensory sensations such as allodynia (pain as a result of non-noxious stimuli) and hyperalgesia (increased response to a normally painful stimuli). Several signaling pathways have been described that contribute to NP neuropathy, including pathways activated through inflammatory molecules, growth factors, and lipid metabolites. Signaling through these pathways causes changes in the function and expression of voltage-gated sodium, calcium and potassium channels, and also leads to abnormal function of second-order nociceptive and inhibitory neurons. As this situation becomes chronic, it leads to a permanent maladaptive plasticity that none of the pharmacological therapies currently available can reverse.19 NP seriously compromises the quality of life of affected people, and clearly represents a currently unmet clinical need. Consequently, there is a pressing demand for the development of efficacious new drugs which can be used either alone or in combination with existing therapies to provide a long-term alleviation of chronic pain through novel mechanisms of action. Among the different factors involved in NP, LPA has been characterized as crucial for NP initiation.13, 18 NP is attenuated in Lpar1-deficient mice, indicating that LPA signaling through the LPA1 is responsible for pain transmission.13 However, direct antagonists of LPA1 such as the compound Ki-16425, have shown efficacy in inhibiting NP but only for limited periods of time.20 This fact suggests that initial LPA signaling through LPA1 is not only important for NP initiation, but that LPA signaling may exacerbate chronic pain through subsequent signaling mechanisms including: LPA1-dependent regulation of pain-related molecules, feed forward pain facilitation caused by the LPA3-stimulated production of LPA following the initial LPA1 activation,21 and concomitant LPA mediated activation of the transient receptor potential vanilloid 1 (TRPV1) ion channel, which also dramatically contributes to the NP onset.22–24 Therefore, we hypothesized that a longer period of LPA1 desensitization will be a more efficacious strategy for ameliorating NP. This strategy has been already successfully used in the development of fingolimod (FTY720 or Gilenya®), the first oral disease-modifying drug for treating multiple sclerosis, and a non-selective agonist of sphingosine 1-phosphate (S1P) that behaves as a functional S1P1 and S1P3–5 antagonist or receptor modulator, inducing receptor internalization and degradation after binding and activation.25–27
In this work we describe the development of a potent new LPA1 selective agonist that behaves as a functional receptor antagonist and shows efficacy in an in vivo mouse model of NP.
RESULTS AND DISCUSSION
Very few agonists of the LPA1 have been reported to date.6, 28, 29 The first LPA-based agonist was N-acylethanolamide phosphoric acid (2-[(9Z)-octadec-9-enoylamino]ethyl dihydrogen phosphate or NAEPA, 1, Figure 1), which was characterized as a dual LPA1/2 agonist.30 Several changes in its structure have led to ligands with improved activity and, in some cases, such as derivative 2, selectivity for LPA1 (Figure 1), with EC50 values of 221 nM and 1089 nM at LPA1 and LPA2, respectively.31 In an attempt to identify new structures devoid of the fatty acid moiety, Astellas Pharma reported a series of indoloquinolizines with agonist activity at the LPA1. Among them, compound 3 (Figure 1) exhibited the best EC50 value (3.6 μM),32 although selectivity was not discussed.
Figure 1.

Representative LPA1 agonists
Considering this paucity of LPA1 selective agonists and the lack of an LPA1/agonist complex crystal structure,33 we attempted to identify novel LPA1 agonists using the structure of the endogenous ligand as a starting point (Figure 2). Initially, modifications were made in two parts of the LPA molecule, the acid group and the hydrophobic moiety, keeping the LPA stereochemistry constant. With respect to the acid group, several structure-activity relationship studies suggested that changes in the LPA polar head group would be poorly tolerated.34 In particular, it has been shown that the presence of free acid groups that retain their negative charge under physiological conditions is an important requirement for activity. Hence, we decided to replace the phosphate with carboxylic and boronic acids or with a tetrazole moiety (compounds 4–11, Figure 2), all of which have been described as suitable isosteres.35–37 With respect to the hydrophobic moiety, modifications in the length of the fatty acid chain were studied (compounds 12–16, Figure 2). In addition, based on our previous results showing that fatty acid chains can be replaced by phenyl and biphenyl moieties,38, 39 we incorporated a variety of phenyl and biphenyl-containing subunits (compounds 17–26, Figure 2).
Figure 2.

Design of new LPA1 agonists 4–26
Changes in the polar head of LPA
Compounds 4–11 (Scheme 1) were obtained by coupling of oleoyl chloride with the corresponding alcohol or glycerol derivatives 27-33 (see Supporting Information for details) in the presence of 2,4,6-collidine at low temperature. For all the diols employed, this reaction was completely regioselective, and only the acylation at sn-1 position of the glycerol moiety was observed. Carboxylic acid derivatives 34-39 were then deprotected with TFA to yield the final products 4–9. In the case of the malonic acid derivatives 35 and 36, this reaction led to the formation of lactones 5 and 6 as a mixture of diastereoisomers instead of free dicarboxylic acids. Thus, in order to avoid cyclization, the hydroxy group was masked with a methyl group (compound 7) or removed (derivatives 8 and 9). Boronic acid 10 was obtained by transformation of the pinacolyl boronate ester 40 into the corresponding trifluoroborate salt, followed by hydrolysis with trimethylsilyl chloride to yield the desired boronic acid. Finally, treatment of nitrile 41 with sodium azide led to tetrazole 11.
Scheme 1. a Synthesis of Compounds 4–11.

a Reagents and conditions: (a) Glycerol or alcohol derivatives (27–33), 2,4,6-collidine, DCM, −78 °C to rt, 24 h, 36–99%; (b) TFA, DCM, rt, 16–18 h, 52−99%; (c) trimethylsilyldiazomethane, HBF4, DCM, 0 °C, 90 min, 38%; (d) i) KHF2, CH3OH:H2O, rt, 30 min; 99%, ii) TMSCl, CH3CN:H2O, rt, 1 h, 60%; (e) NaN3, NH4Cl, DMF, MW, 160 °C, 45 min, 18%.
The ability of compounds 4–11 to activate the LPA1 was assessed by measuring the extent of calcium mobilization in LPA1 stably transfected RH7777 cells, as the binding of an LPA1 agonist causes an increase in intracellular calcium levels, which can be quantified using a fluorescence-based assay. Among these compounds, only derivatives 6 and 7 were able to activate the receptor [Emax (6) = 33%; EC50 (6) = 1.7 μM and Emax (7) = 74%; EC50 (7) = 6 μM] whereas the rest of the compounds did not induce calcium mobilization at the highest concentration of compound tested (10 μM). Although these results indicate that other polar moieties can mimic the LPA phosphate group, these derivatives are less potent inducers of calcium mobilization than LPA.
Changes in the hydrophobic moiety of LPA: modification of fatty acid chain length
The hydrophobic moiety plays an important role in LPA receptor activity. To study the influence of this part of the molecule on receptor activity, comprehensive changes were made to the overall length of the fatty acid chain (compounds 12-16, Scheme 2) and aromatic ring were incorporated (compounds 17–26, Scheme 3). Modification of the fatty acid chain length required the preparation of the non-commercial carboxylic acids 42–44 by steroselective cis-hydrogenation of the corresponding alkynyl derivatives (see Supporting Information for details) and glycerol 45, obtained following a synthetic procedure previously developed in our group.40 Regioselective esterification between the corresponding fatty acid and the protected phosphorylated diol 45 yielded derivatives 46–50 which were deprotected to provide the final compounds 12–16 (Scheme 2).
Scheme 2. a Synthesis of Compounds 12–16.

a Reagents and conditions: (a) DCC, DMAP, DCM, −20 °C to rt, 16 h, 12−49%; (b) TFA, DCM, rt, 4−5 h, 90−99%.
Scheme 3. a Synthesis of Compounds 17–26.

a Reagents and conditions: (a) DCC, DMAP, DCM, −20 °C to rt, 16 h, 10−58%; (b) i) TMSBr, DCM, rt, 4 h; ii) MeOH/H2O, rt, 1h, 90%; (c) H2, 10% Pd/C, EtOH, rt, 74−99%.
The results obtained for LPA1 agonist activity (Table 1) highlight the tremendous influence exerted by the hydrophobic chain length. In this regard, compounds 12-16 show that variations of just one methylene unit can turn a compound inactive (for example derivative 15 shows an activity comparable to LPA whereas 16 is inactive) or increase its activity almost two-fold, as shown by compounds 15 (Emax = 88%; EC50 = 3.6 μM) and 14 (Emax = 202%; EC50 = 2.1 μM). These data suggest that the optimal chain is that of palmitoleic acid, present in compound 13 (Emax = 205%; EC50 = 0.45 μM), the most potent derivative within the series and more active than the endogenous ligand LPA (Emax = 100%; EC50 = 0.83 μM).
Table 1.
Agonist activities of compounds 12–16 at LPA1 receptor
 | |||
|---|---|---|---|
| Cpd | n | Emax (%)a | EC50 (μM)b |
| 12 | 3 | 127± 1 | 2.8 ± 0.1 |
| 13 | 5 | 205 ± 9 | 0.45 ± 0.01 |
| 14 | 6 | 202 ± 1 | 2.1 ± 0.3 |
| 15 | 8 | 88 ± 2 | 3.6 ± 0.2 |
| 16 | 9 | N.E.c | - |
| LPA | 7 | 100 | 0.83 ± 0.02 |
Emax = maximal efficacy of the drug/maximal efficacy of LPA, expressed as the percentage.
For Emax > 30%, EC50 values are expressed as mean ± s.e.m, from a minimum of two independent experiments, performed in triplicate.
N.E., no effect was observed at the highest concentration of compound tested (10 μM).
Changes in the hydrophobic moiety of LPA: derivatives containing aromatic rings
Previous data from our group showed that unsaturated fatty acid chains can be replaced by phenyl groups.38, 39 Our results observed with derivatives 12–16 together with studies using other lipid-binding GPCRs,41 show the importance of aliphatic chain length on activity. To determine if the distance between the aromatic rings and the phosphorylated glycerol moiety (n) influences activity, compounds 17–26 (Scheme 3) were prepared. Their synthesis required the preparation of carboxylic acids 51–56 (see Supporting Information for details) and the conveniently protected phosphorylated glycerol. Initially, looking for synthetic simplicity and high yields, the ethyl group was selected for phosphate protection (intermediate 57). However, using standard conditions to remove the ethyl groups from intermediate 59 with TMSBr yielded bromine derivative 17 instead of the expected product. The structure of 17 was determined by nuclear magnetic resonance (NMR) and high pressure liquid chromatography coupled to mass spectrometry (HPLC-MS). The benzyl group was therefore selected as the phosphate protecting group for intermediates 60-68, which, after hydrogenation, led to the target compounds 18–26 (Scheme 3).
All the synthesized compounds, including the initially unexpected derivative 17, were tested for LPA1 activity. The data obtained (Table 2) confirms that the fatty acid chain can be replaced by phenyl and biphenyl moieties and that the distance between the ester group and the aromatic subunit influences agonist activity, as small changes in the number of methylene units dramatically affects Emax and EC50 values. For example, compound 19, with n = 7, is inactive, whereas derivative 20, with n = 8, is almost as active as LPA, or the case of derivative 25, which shows the highest activity when compared to either compounds 24 or 26. In all the cases the optimal chain resulted to be the one with n=9. Finally, it is remarkable the excellent activity of compound 17 at the LPA1 receptor.
Table 2.
Agonist activities of compounds 17–26 at LPA1 receptor
 | ||||
|---|---|---|---|---|
| Cpd |  |
n | Emax (%)a | EC50 (μM)b |
| 17 | 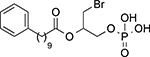 |
118 ± 14 | 0.24 ± 0.09 | |
| 18 | 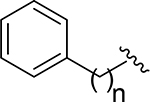 |
6 | N.E.c | - |
| 19 | 7 | N. E. | - | |
| 20 | 8 | 74 ± 4 | 2.1 ± 0.3 | |
| 21 | 9 | 112 ± 3 | 0.5 ± 0.1 | |
| 22 | 10 | 135 ± 31 | 3.2 ± 0.5 | |
| 23 | 14 | N.E. | - | |
| 24 | 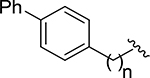 |
6 | N.E. | - |
| 25 | 9 | 127 ± 9 | 3.3 ± 0.6 | |
| 26 | 12 | 37 ± 1 | 19 ± 2 | |
| LPA | 100 | 0.83 ± 0.02 | ||
Emax = maximal efficacy of the drug/maximal efficacy of LPA, expressed as the percentage.
For Emax > 30%, EC50 values are expressed as mean ± s.e.m, from a minimum of two independent experiments, performed in triplicate.
N.E., no effect was observed at the highest concentration of compound tested (10 μM).
In summary, from the modifications carried out in the hydrophobic moiety of LPA, three compounds, derivatives 13, 17, and 21, with Emax values of 205%, 118% and 112%, respectively, and EC50 values of 0.45, 0.24, and 0.5 μM respectively, have activities greater than that of the endogenous ligand LPA (Emax = 100%; EC50 = 0.83 μM) and stand out within the series.
The possibility of optimizing the structure by combination of the best phosphate free polar heads identified so far (the fluorine containing moieties of compounds 6 and 7) with the optimal hydrophobic subunits (the aliphatic chains of palmitoleic acid, in compound 13, and of 10-phenyldecanoic acid, in derivatives 17 and 21) was assessed with the synthesis of compounds 69–72 (Figure 3, see Supporting Information for details). In the case of derivative 71, its chromatographic purification allowed the separation of the two diastereoisomers. The absolute configuration of the chiral carbon bonded to the fluorine atom was determined on the basis of NOE experiments and 1H- and 19-F-NMR spectra. Compound 72 was obtained as 1:1 mixture of diastereoisomers. Among these derivatives, only compound 69 showed some agonist activity (Emax and EC50 values of 43±6% and 1.4±0.6 μM, respectively), whereas the rest of the compounds were inactive at the highest concentration tested (10 μM).
Figure 3.

Compounds 69–72
From the obtained results, derivative 17, which contains a chiral center, was identified as the compound with the greatest activity (Emax and EC50 values of 118% of 0.24±0.09 μM, respectively). In order to establish its absolute configuration, the two enantiomers of 17 were synthesized from the corresponding (S)- or (R)-oxiran-2-ylmethanol (glycidol), which was phosphorylated using the conditions previously employed. The oxirane ring was opened and the resulting intermediates (S)- and (R)-74 coupled with 10-phenyldecanoic acid (53). Final removal of the benzyl groups yielded the target compounds (S)- and (R)-17 (Scheme 4). Comparison of the specific optical rotation values revealed that compound 17, obtained in the phosphate deproctection reaction (Scheme 3) was the S enantiomer. Therefore, after confirming that (S)-17 was indeed the active enantiomer (Table 3), we carried out some structural exploration around this compound (Scheme 5). Modifications included changing the length of its methylenic chain (derivatives 76 and 77) and replacing it with palmitoleic and oleic acid chains (compounds 78 and 79). However, none of these compounds had agonist activities that were significantly better that that obtained with (S)-17, as shown in Table 3.
Scheme 4. a Synthesis of (S)- and (R)-17.

a Reagents and conditions: (a) i) iPr2NP(OBn)2, 1H-tetrazole, DCM, rt, 2 h; ii) mCPBA, DCM, −30 °C, 90 min, 86–95%; (b) TBABr, TFA, CHCl3, rt, 10 min, 89–99%; (c) 53, DCC, DMAP, DCM, rt, 18 h, 68–73%; (d) H2, 10% Pd/C EtOH, rt, 97–99%.
Table 3.
Agonist activities of analogs of (S)-17 at LPA1 receptor
| Cpd | Structure | Emax (%)a | EC50 (μM)b |
|---|---|---|---|
| 76 | 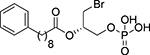 |
N.E.c | - |
| 77 | 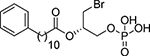 |
N.E. | - |
| 78 | N.E. | - | |
| 79 | 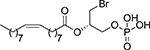 |
39 ± 3 | 3.2 ± 0.4 |
| (R)-17 | 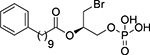 |
32 ± 3 | 26 ± 5 |
| (S)-17 | 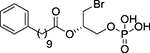 |
118 ± 14 | 0.24 ± 0.09 |
Emax = maximal efficacy of the drug/maximal efficacy of LPA, expressed as the percentage.
For Emax > 30%, EC50 values are expressed as mean ± s.e.m, from a minimum of two independent experiments, performed in triplicate.
N.E., no effect was observed at the highest concentration of compound tested (10 μM).
Scheme 5. a Synthesis of analogues of compound (S)-17.

a Reagents and conditions: (a) 52, 11-phenylundecanoic acid, or palmitoleic acid, DCC, DMAP, DCM, rt, 5 h, 55–96%; (b) oleoyl chloride, pyridine, DCM, rt, 18 h, 15%; (c) H2, 10% Pd/C, EtOH, rt, 72–80%; (d) TFA, DCM, rt, 5 h, 81–93%.
In addition, we analysed the effect of the replacement of the ester for an amide group. Hence, the synthesis of the corresponding amides (85–87) of three representative LPA1 agonists identified so far [(S)-17, and phosphate free compounds 7 and 69] was addressed as depicted in Scheme 6 and their activity at LPA1 receptor determined (Table 4). However, whereas amides 85 and 86 could be readily obtained by coupling of intermediate amine 88 with palmitoleic acid or oleyl chloride, respectively, access to amide 87 was more difficult than initially expected. In this case, removal of the Boc group of intermediate 91 followed by condensation of the free amine with the N-hydroxysuccinimidyl ester of 10-phenyldecanoic acid led to aziridine 92 due to an intramolecular cyclization instead of formation of the desired amide 87 (Scheme 6). Further attempts to open the aziridine ring with bromide allowed the isolation of intermediate 93, which after hydrogenation to remove the remaining benzyl group yielded a complex mixture of unidentified products. This result suggests that target amide 87 has a low intrinsic stability. Therefore, only compounds 85 and 86 were assessed for LPA1 activity (Table 4). The obtained results reflect the key influence of the oxygen atom, as the two studied amides displayed a dramatic loss of activity both in terms of Emax and EC50 values when compared with their ester analogues.
Scheme 6. a Synthesis of amides 85–87.

a Reagents and conditions: (a) palmitoleic acid, EDC, HOBt, DMF, rt, 18 h, 37%; (b) oleoyl chloride, Et3N, DCM, rt, 18 h, 46%; (c) TFA, DCM, rt, 18 h, 69–99%; (d) TFA, DCM, rt, 16 h, 99%; (e) N-hydroxysuccinimidyl ester of 53, Et3N, DCM, rt, 12 h, 48%; (f) MgBr2·OEt2, Et2O, rt, 4 h, 99%; (g) H2, 10% Pd/C, EtOH, rt.
Table 4.
Agonist activities of amides 85 and 86 at LPA1 receptor in comparison with their ester analogues
| Cpd | Structure | X | Emax (%)a | EC50 (μM)b |
|---|---|---|---|---|
| 85 | NH | 26 ± 1 | 5 ± 1 | |
| 69 | O | 43 ± 6 | 1.4 ± 0.6 | |
| 86 | NH | N. E.c | - | |
| 7 | O | 74 ± 14 | 6 ± 1 | |
Emax = maximal efficacy of the drug/maximal efficacy of LPA, expressed as the percentage.
For Emax > 30%, EC50 values are expressed as mean ± s.e.m, from a minimum of two independent experiments, performed in triplicate.
N.E., no effect was observed at the highest concentration of compound tested (10 μM).
In summary, the results obtained up to this moment identify compound (S)-17, with an excellent EC50 value of 0.24 μM, as the most potent within the series, being able to activate LPA1 with a slightly higher intensity (118%) than the endogenous ligand LPA. In addition, we confirmed the binding of (S)-17 to the LPA1 receptor using back-scattering interferometry (BSI),42–44 a technique which detects changes in refractive index (RI) due to variations in molecular structure, dipole moment, polarizability, conformation and solvation that occur upon binding of a ligand to a protein. Thus, BSI can be used to measure binding affinities by detecting changes in the RI of mixtures of protein and ligand after incubation to reach equilibrium. In this case, increasing concentrations of (S)-17 were incubated with a constant quantity of LPA1-expresssing membrane homogenates and the difference between the signals of protein-ligand complex and free protein solution was plotted versus ligand concentration to obtain a saturation binding curve from which the dissociation constant (KD) was obtained (Supporting Figure S1), resulting in a KD value of 19.9 nM for compound (S)-17 and of 5.6 nM for LPA, used as reference. We also carried out molecular docking studies to propose a plausible binding mode between the ligand and the receptor. The results showed that (S)-17 can interact with LPA1 in a similar manner to LPA and other known LPA1 agonists.33, 45 The phosphate moiety of (S)-17 establishes three hydrogen bonds with Arg124, Thr113, and Trp121 and the carbonyl moiety of the ligand forms an additional hydrogen bond with Gln125. Besides, (S)-17 establishes hydrophobic interactions with Tyr 102, Ile128, Leu132, Tyr202, Trp210, Leu297, and Ala300 (Figure 4).
Figure 4. Two images (0°−180° views) of the binding mode between (S)-17 and the LPA1 receptor (active conformation) as predicted by docking calculations.

Blue lines represent hydrogen bonds and grey dashed lines hydrophobic interactions. Residues marked in green are reported in the literature33, 45 to bind to other agonists. The LPA1 active state model was retrieved from the GPCRdb46, 47 (structure file provided in the Supporting Information).
Due to the high sequence similarity between LPA1–3, a selectivity profile was determined for all compounds with LPA1 agonist activity (Supporting Table 1). In addition, for compound (S)-17, we studied its selectivity against LPA4–6 receptors and also against the enzyme autotaxin (ATX), involved in the biosynthesis of LPA, and its antagonist capacity at the LPA1 receptor. Remarkably, this compound showed 10-fold selectivity versus LPA5 receptor and more than 50-fold selectivity versus any of the other analysed targets, making this compound the most potent and selective LPA1 agonist described so far. Besides, we assessed the reversibility of the binding and the stability of the compound. To determine whether derivative (S)-17 activates LPA1 in a reversible manner, we carried out a wash out experiment. The obtained results indicate that, after incubation and removal of the compound, the receptor recovers its ability for activation, thus ruling out a covalent binding between LPA1 and the compound (Supporting Figure S2). Additionally, the compound remains stable (>85%) up to 8 hours at 37 °C both in PBS and in culture media (Supporting Figure S3), making (S)-17 an excellent candidate for assessing LPA1-mediated cellular effects in vitro and for modulation of NP in vivo.
Cellular effects of (S)-17: induction of neurite retraction, cellular migration and internalization of the LPA1 receptor
We have shown that (S)-17 has the ability to bind to LPA1 and activate calcium mobilization. Several in vitro assays were performed to determine if (S)-17 has the ability to recapitulate LPA induce cellular changes. In agreement with its ability to function as a receptor agonist, (S)-17 induced neurite retraction, as indicated by a rounded cellular morphology, in a manner similar to that of LPA (Figure 5). In addition, (S)-17 induced cell migration of LPA1 overexpressing B103 cells in a transwell chamber assay (Figure 6). Finally, the ability of (S)-17 to induce LPA1 internalization was assessed in B103 neuroblastoma cells overexpressing an enhanced green fluorescent-LPA1 fusion receptor [(EGFP)-LPA1]. The results obtained clearly show that in the absence of stimulus, the receptor is localized to plasma membrane (Figure 7A), whereas in the presence of an agonist (LPA or (S)-17) the receptor is internalized, as indicated by the presence of the green fluorescence inside the cell (Figure 7B,C). Consistent with its ability to act as an agonist in vitro, (S)-17 was able to mimic other LPA mediated cellular effects including, neurite retraction, cellular migration, and LPA1 internalization. Receptor desensitization, through internalization, is of special importance because it is a mechanism of protection against LPA mediated NP that occurs through LPA1 signaling.
Figure 5. Compound (S)-17 induces neurite retraction.

B103 neuroblastoma cells overexpressing EGFP-LPA1 receptor were treated with (A, D) vehicle (0.1% fatty acid free bovine serum albumin), (B) LPA, or (C, E) (S)-17. Cells were then fixed and stained with phalloidin and DAPI, and analysed under fluorescence microscopy for visualization of LPA (green), cell morphology (red) and nuclei (blue). The number of cells with retracted neurites was counted and expressed as the percentage of the total number of cells (F). The data shown correspond to the mean±SEM of the number of cells counted in two independent experiments and three different slides per experiment. *, p<0.05; **, p<0.01 vs BSA (Student’s t test). Samples were imaged under the same conditions using a Zeiss fluorescence microscope (A-C, bars 100 μm) or Zeiss fluorescence confocal microscope (D, E, bars 10 μm).
Figure 6. Compound (S)-17 induces cellular migration.

B103 neuroblastoma cells overexpressing LPA1 receptor were seeded (2.5·105 cells) into the upper chamber of the transwell and vehicle (0.1% fatty acid free bovine serum albumin), LPA 1 μM, or (S)-17 1 μM were added in the lower chamber. After allowing the cells to migrate for 5 h, migratory cells were stained with 0.1% crystal violet solution and counted. Images shown are representative of the migratory cells in the presence of (A) vehicle, (B) LPA 1 μM, or (C) (S)-17 1 μM. (D) Data in the bar graph correspond to the mean±SEM of the number of cells counted in two independent experiments and three different transwell chambers per experiment. *, p<0.05; **, p<0.01 vs BSA (Student’s t test). Samples were imaged under the same conditions by using a Zeiss microscope (bars 50 μm).
Figure 7. Compound (S)-17 induces internalization of the LPA1 receptor.

B103 neuroblastoma cells overexpressing EGFP-LPA1 were treated with (A) vehicle (0.1% fatty acid free bovine serum albumin), (B) LPA 1 μM, or (C) (S)-17 1 μM. The cells were then fixed, stained with phalloidin and DAPI, and analysed under fluorescence microscopy for visualization of LPA1 (green), cell morphology (red) and nuclei (blue). Green fluorescence in the cytosol in (B) and (C) shows internalization of the LPA1, which, in the absence of agonist, appears almost exclusively in the cell membrane. Images shown are representative of two independent experiments and at least three different fields per experiment. All samples were imaged under the same conditions using a Zeiss fluorescence confocal microscope (bars 10 μm).
Functional desensitization induced by (S)-17
The observed internalization of the LPA1 after treatment with the agonist (S)-17 strongly suggests its associated functional desensitization. We sought to confirm this aspect in dorsal root ganglia (DRG) primary culture, since sensory neurons express LPA1 receptor and thus, represent a more relevant model than cell lines. As expected, Ca2+ microfluorometric measurements show that 10 μM LPA induces Ca2+ influx in DRG neurons (Figure 8), as detected by an increase in the Fluo-4 fluorescence.48 After the first application of LPA, neurons desensitize and the Ca2+ response is notably reduced in a second LPA application (Figure 8A). LPA produced intracellular Ca2+ changes in TRPV1-expressing nociceptors, as capsaicin also triggered a Ca2+ influx in all LPA responsive neurons, although not all capsaicin responding neurons were activated by LPA. Importantly, stimulation of LPA/capsaicin responding neurons with 40 mM KCl indicate that changes in cytosolic Ca2+ concentrations were recorded in viable, healthy nociceptors. A similar behaviour was observed for (S)-17, which under the same experimental paradigm (Figure 8B), activated the LPA1 receptor TRPV1-sensitive DRG nociceptors. Akin to LPA, repeated instillation of compound (S)-17 induced a reduction of the Ca2+ responses, consistent with the desensitization of the LPA1 receptor (see Supporting Figure S4 for representative calcium microfluorography recordings).
Figure 8. Repeated application of (S)-17 reduces the activation of DRG neurons.

Measurement of calcium-influx peak intensity elicited on cultured neonatal sensory neurons upon two repetitive applications of 10 μM LPA (A) or (S)-17 (B) and one of 500 nM capsaicin (Caps). A 10 second-pulse of 40 mM KCl was applied to distinguish neuronal viability. Data represent fluorescence expressed as mean ± SEM. First pulse was compared to the second by t-test paired values, ***p<0.001. All neurons that responded to LPA or (S)-17 were also activated by capsaicin.
To further substantiate the neuronal activity of compound (S)-17, we also investigated their effect on nociceptor excitability using multielectrode array (MEA) technology49 (see Supporting Figure S5 for the experimental protocol). For this purpose, rat nociceptors were cultured in MEA chips, stimulated with LPA or (S)-17 and capsaicin and KCl. As seen in Figure 9, a first pulse of LPA or (S)-17 provoked the firing of action potentials that were attenuated upon a second instillation of the compounds. Akin to the calcium microfluorography, LPA sensitive neurons also responded to capsaicin. Analysis of the mean spike frequency clearly demonstrates that the nociceptor desensitization was produced by the repeated instillation of LPA or (S)-17. It should be noted that LPA modified nociceptor excitability/desensitization by a direct action on the LPA1 receptor and not by sensitizing TRPV1 channels, as LPA induced nociceptor excitability was insensitive to capsazepine (see Supporting Figures S6 and S7).
Figure 9. Repeated application of (S)-17 decreases neuronal firing activity of DRG neurons.

Graphs show the 10 μM LPA (A) or (S)-17 (B) on mediated neuronal firing activity on neonatal rat DRG neurons. LPA and (S)-17 were applied in two consecutive pulses. Neurons were exposed to 500 nM capsaicin (Caps) at the end of the recording to measure TRPV1 sensitivity of the neuronal networks exposed to LPA or (S)-17. Data represent mean spike frequency (Hz) and are expressed as mean ± SEM. Number of cultures ≥2. Comparison between pulses was performed by t-test paired values. *p<0.05; ***p<0.001
Further confirmation of the in vivo desensitization of DRG neurons induced by (S)-17 was obtained assessing the effect of repeated administration of the compound on the writhing response induced by administration of 0.6% acetic acid. Animals were injected with a daily dose for five days with vehicle (3% BSA) or (S)-17 (10 mg/kg) and the acetic acid-induced writhing response was analyzed on the fifth day after 60 minutes of the first injection. The results obtained show that the total number of writhes was markedly decreased in the (S)-17-treated mice compared to the vehicle-treated group (Figure 10A), being this reduction especially remarkable in the first 10 minutes (Figure 10B). These results are consistent the LPA1 desensitization produced by the repeated administration of (S)-17, since acute single administration of (S)-17 increased the number of writhes in the first minutes after the administration (Supporting Figure S8).
Figure 10. Acetic acid-induced writhing responses were decreased by repeated (S)-17 administration.

Mice received a daily dose of vehicle (3% BSA) or (S)-17 (10 mg/kg) for 5 days. The last day, mice were injected with 0.6% acetic acid solution (AA) 60 min after the administration of (S)-17. Data show mean±SEM of writhes observed by experimenters blind to experimental conditions. *p<0.05 compared with (S)-17-treated group (Student’s t test); **p<0.0001 compared with AA group (one-way ANOVA).
Compound (S)-17 reduces NP in vivo
After confirming that (S)-17 effectively induces receptor desensitization in vitro and in vivo, we tested its efficacy in the NP in vivo model produced by spared nerve injury (SNI). This is induced by transecting two of three branches of the sciatic nerve, the tibial and common peroneal nerves. SNI mimics some of the major features observed in clinical NP, and it is characterized by the development of long-lasting mechanical hypersensitivity in the mouse hind paw. This can be quantified with an electronic Von Frey algesimeter by measuring mechanical nociceptive threshold after applying a mechanical stimulus in the plantar surface of the hind paw.50, 51 After SNI, mice were treated daily with vehicle or with 10 mg/kg (S)-17 (intraperitoneally, i.p.) and 3, 10 and 21 days post-surgery, the mechanical nociceptive threshold was assessed. As shown in Figure 11, the mechanical nociceptive threshold reached by the treated mice was significantly superior compared to vehicle-treated mice, indicating that (S)-17 protected against NP. To discard non-specific unwanted effects, we confirmed that at the selected dose, (S)-17 did not induce any alteration in the exploratory or locomotor activity (Supporting Figure S9). These results strongly support the fact that agonist stimulation of the LPA1 receptor leads to its desensitization and to attenuation of NP.
Figure 11. Compound (S)-17 significantly attenuates mechanical hypersensitivity after SNI in mice, an animal model of NP.

Effect of i.p. injection of (S)-17 (10 mg/kg) or vehicle on nociceptive behaviour over time in mice with SNI. Data are expressed as mean±SEM. n =10/group. **** p<0.0001, ** p<0.01 compared with vehicle-treated group (two-way repeated measures ANOVA with Bonferroni’s post hoc test for multiple comparisons).
CONCLUSIONS
In this work we report the most potent and selective LPA1 agonist identified to date, compound (S)-17, with Emax and EC50 values of 118% and 0.24 μM, respectively, and a KD value of 19.9 nM at the LPA1 receptor and selectivity against the LPA2 and LPA3 receptors. In agreement with this in vitro profile, (S)-17 induces characteristic LPA1-mediated cellular effects such as neuronal cell rounding and cellular migration. Importantly, this agonist also induces LPA1 internalization that leads to functional receptor desensitization. In vivo, (S)-17 shows remarkable efficacy in reducing NP pain perception. Considering that NP affects up to 10% of the western adult population and that about half of the patients do not respond to common analgesics, it is clear that new therapies are eagerly awaited for this unmet need. Our results strongly support the hypothesis that agonist stimulation of the LPA1 can lead to functional receptor antagonism and to the amelioration of NP. These results provide a new alternative for the development of novel more efficacious antinociceptive molecules with new mechanisms of action that are devoid of undesired side effects.
EXPERIMENTAL SECTION
Synthesis
Unless otherwise stated, the starting materials, reagents, and solvents were purchased as high-grade commercial products from Sigma-Aldrich, ABCR, Acros, Biotage, Fluka, Lancaster, Scharlab, or Panreac. Dichloromethane (DCM), diethyl ether and tetrahydrofuran (THF) were dried using a Pure Solv™ Micro 100 Liter solvent purification system. Triethylamine and pyridine were dried over CaH2 and distilled prior to its use. Alcohol-free chloroform was obtained by washing with water, drying over MgSO4, filtration and distillation over P2O5. Ethylenediamine was dried over 4Å molecular sieves, distilled and used immediately. All non-aqueous reactions were performed under an argon atmosphere in oven-dried glassware. Reactions under MW irradiation were performed in a Biotage Initiatior 2.5 reactor, and hydrogenation reactions were carried out in a ThalesNano H-Cube® HC 2-SS continuous-flow hydrogenation reactor using CatCart® catalyst cartridges. Analytical thin-layer chromatography (TLC) was run on Merck silica gel plates (Kieselgel 60 F-254), with detection by UV light (λ = 254 nm), ninhydrin solution, or 10% phosphomolybdic acid solution in ethanol. Flash chromatography was performed on glass column using silica gel type 60 (particle size 230–400 mesh, Merck), or on a Varian 971-FP flash purification system, using silica gel cartridges (Varian, particle size 50 μm). All compounds were obtained as oils, except for those whose melting points (m.p.) are indicated, which were solids. M.p. (uncorrected) were determined on a Stuart Scientific electrothermal apparatus. Optical rotation [α] was measured on an Anton Paar MCP 100 modular circular polarimeter using a sodium lamp (λ = 589 nm) with a 1 dm path length; concentrations are given as g/100 mL. Infrared (IR) spectra were measured on a Bruker Tensor 27 instrument equipped with a Specac ATR accessory of 5200–650 cm−1 transmission range; frequencies (ν) are expressed in cm-1. 1H-, 13C-, and 31P-NMR spectra were recorded on a Bruker Avance III 700MHz (1H, 700 MHz; 13C, 175 MHz), Bruker Avance 500MHz (1H, 500 MHz; 13C, 125 MHz; 31P, 202 MHz) or Bruker DPX 300MHz (1H, 300 MHz; 13C, 75 MHz; 31P, 121 MHz) instrument at room temperature (rt) at the Universidad Complutense de Madrid’s NMR core facility. Proton-coupled 19F-NMR spectra were recorded on a Bruker DPX 300MHz. Chemical shifts (δ) are expressed in parts per million relative to the residual solvent peak 1H and 13C nucleus (acetone-d6: δH = 2.05, δC = 29.84; CDCl3: δH = 7.26, δC = 77.16; DMSO-d6: δH = 2.50, δC = 39.52; methanol-d4: δH = 3.31, δC = 49.00), to internal phosphoric acid for 31P nucleus and to internal (trifluoromethyl)benzene for 19F nucleus; coupling constants (J) are in hertz (Hz). The following abbreviations are used to describe peak patterns when appropriate: s (singlet), d (doublet), t (triplet), q (quartet), qt (quintuplet), m (multiplet), app (apparent) and br (broad). 2D NMR experiments (H,H-COSY, HMQC and HMBC) of representative compounds were carried out to assign protons and carbons of the new structures. High resolution mass spectrometry (HRMS) was carried out on a FTMS Bruker APEX Q IV spectrometer in electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI) mode at Universidad Complutense de Madrid’s mass spectrometry core facility. For all final compounds, purity was determined by HPLC-MS, and satisfactory chromatograms confirmed a purity of at least 95% for all tested compounds. HPLC-MS analysis was performed using an Agilent 1200LC-MSD VL instrument. LC separation was achieved with a Zorbax Eclipse XDB-C18 column (5 μm, 4.6 mm x 150 mm) or Zorbax SB-C3 (5 μm, 2.1 mm x 50 mm) together with a guard column (5 μm, 4.6 mm x 12.5 mm). The gradient mobile phases consisted of A (95:5 water/methanol) and B (5:95 water/methanol) with 0.1% ammonium hydroxide and 0.1% formic acid as the solvent modifiers. MS analysis was performed with an ESI source. The capillary voltage was set to 3.0 kV and the fragmentor voltage was set at 25 eV. The drying gas temperature was 350 °C, the drying gas flow was 10 L/min, and the nebulizer pressure was 20 psi.
General procedure 1: deprotection of tert-butyl esters and alkyl di-tert-butyl phosphates
TFA (25 or 75 equiv) was added to a solution of the corresponding tert-butyl derivative (1 equiv) in anhydrous DCM (20 mL/mmol) and the reaction was stirred at rt until disappearance of the starting material. The mixture was then treated with brine and the aqueous phase was extracted with DCM. The combined organic layers were washed with water and brine, dried over Na2SO4 and filtered. The solvent was evaporated under reduced pressure to afford the corresponding final compounds 4-9, 12-16, 69-72, 78, 79, 85, 86.
General procedure 2: deprotection of dibenzyl phosphates
The corresponding dibenzyl phosphate was dissolved in absolute ethanol (0.2 mL/mg), and the solution was pumped through H-Cube® HC 2-SS reactor using a 10% Pd/C CatCart® cartridge, under full-H2 mode at a flow-rate of 1 mL/min at rt. Solvent was then removed under reduced pressure to afford the corresponding final compounds (S)-, (R)-17, 18-26, 76, 77.
[(2S)-2-Hydroxy-3-{[(9Z)-octadec-9-enoyl]oxy}propoxy]acetic acid, 4
Following general procedure 1, compound 4 was obtained from tert-butyl ester 34 (48 mg, 84 μmol) and TFA (0.16 mL, 2.10 mmol) in 52% yield. Chromatography: EtOAc/ethanol, 20:1 to 1:5. Rf: 0.11 (EtOAc/ethanol, 8:2). IR (ATR): 3301 (O-H), 1737 (C=O). 1H-NMR (CDCl3, 300 MHz): δ 0.88 (t, J = 6.6, 3H, CH3); 1.26–1.29 (m, 20H, 10CH2); 1.52–1.65 (m, 2H, CH2CH2CO); 1.93–2.05 (m, 4H, 2CH2CHalkene); 2.30 (t, J = 7.5, 2H, CH2CO); 3.32–3.60 (m, 2H, CHCH2O); 3.76–3.92 (br s, 2H, CH2CO2H); 3.95–4.19 (m, 3H, CO2CH2, CH); 5.26–5.35 (m, 2H, 2CHalkene). 13C-NMR (CDCl3, 75 MHz): δ 14.3 (CH3); 22.9, 25.0 (2CH2); 27.3, 27.4 (2CH2CHalkene); 29.2, 29.3, 29.4, 29.47, 29.48, 29.7, 29.8, 29.9, 32.1 (9CH2); 34.2 (CH2CO); 64.8 (CO2CH2); 65.8 (CH); 70.5 (CH2CO2H); 72.1 (CHCH2O); 130.0, 130.2 (2CHalkene); 173.4, 173.8 (2CO). [α]D20: −1.6 (c = 1.00, CHCl3). HRMS (ESI, m/z): calculated for C23H41O6 ([M-H]−): 413.2909, found: 413.2890. HPLC-MS retention time (min): 14.37.
(5S)-5-{[(9Z)-Octadec-9-enoyloxy]methyl}−2-oxotetrahydrofuran-3-carboxylic acid, 5
Following general procedure 1, compound 5 was obtained from di-tert-butyl ester 35 (169 mg, 0.30 mmol) and TFA (0.58 mL, 7.53 mmol) in 88% yield. Chromatography: EtOAc/ethanol, 20:1 to 1:5. Rf: 0.13 (EtOAc/ethanol, 8:2). 1H-NMR (CDCl3, 300 MHz): Mixture of diastereoisomers A:B (1:1): δ 0.88 (t, J = 7.2, 3H, CH3); 1.27–1.30 (m, 20H, 10CH2); 1.61–1.64 (m, 2H, CH2CH2CO); 1.97–2.03 (m, 4H, 2CH2CHalkene); 2.35 (dt, J = 7.7, 1.8, 2H, CH2CO); 2.37–2.41 (m, 1H, 1/2CH2CHCO2H A); 2.47–2.54 (m, 1H, 1/2CH2CHCO2H B); 2.62–2.68 (m, 1H, 1/2CH2CHCO2H B); 2.78–2.84 (m, 1H, 1/2CH2CHCO2H A); 3.71–3.76 (m, 1H, CHCO2H); 4.19 (dd, J = 12.6, 4.6, 1H, 1/2CO2CH2 A); 4.23 (dd, J = 12.5, 5.8, 1H, 1/2CO2CH2 B); 4.35–4.40 (m, 1H, 1/2CO2CH2); 4.74–4.79 (m, 1H, CO2CH2CH A); 4.89–4.94 (m, 1H, CO2CH2CH B); 5.30–5.38 (m, 2H, 2CHalkene). 13C-NMR (CDCl3, 75 MHz): Mixture of diastereoisomers A:B (1:1): δ 14.3 (CH3); 22.8 (CH2); 24.9 and 24.94 (CH2); 27.3, 27.37 (2CH2CHalkene); 27.9, 29.2, 29.22, 29.3, 29.5, 29.7, 29.8, 29.9, 31.1 (9CH2); 32.1 (CH2CHCO2H); 34.0 and 34.1 (CH2CO); 46.0 and 46.2 (CHCO2H); 64.4 and 64.9 (CO2CH2); 76.7 and 76.8 (CO2CH2CH); 129.85 and 129.8 (CHalkene); 130.17 and 130.19 (CHalkene); 171.9, 172.1 (CO2H, COlactone); 173.4 and 173.6 (CO). HRMS (ESI, m/z): calculated for C23H39O4 ([M-CO2H]−): 379.2854, found: 379.2861. HPLC-Ms retention time (min): 14.37.
(5S)-3-Fluoro-5-{[(9Z)-octadec-9-enoyloxy]methyl}−2-oxotetrahydrofuran-3-carboxylic acid, 6
Following general procedure 1, compound 6 was obtained from di-tert-butyl ester 36 (80 mg, 0.14 mmol) and TFA (0.27 mL, 3.50 mmol) without further purification in quantitative yield. IR (ATR): 3466 (O-H), 1795 (C=O), 1742 (C=O), 1163 (C-F). 1H-NMR (CDCl3, 500 MHz): Mixture of diastereoisomers A:B (1:1): δ 0.88 (t, J = 6.8, 3H, CH3); 1.21–1.37 (m, 20H, 10CH2); 1.59–1.66 (m, 2H, CH2CH2CO); 1.99–2.02 (m, 4H, 2CH2CHalkene); 2.37 (t, J = 7.5, CH2CO); 2.46–2.55 (m, 1H, 1/2CH2CF A); 2.76 (app dd, J = 26.1, 7.2, 2H, CH2CF B); 2.97–3.03 (m, 1H, 1/2CH2CF A); 4.24 (dd, J = 12.6, 4.8, 1H, 1/2CO2CH2 A); 4.29 (dd, J = 12.5, 6.0, 1H, 1/2CO2CH2 B); 4.42–4.46 (m, 1H, 1/2CO2CH2); 4.90–4.99 (m, 1H, CH); 5.31–5.38 (m, 2H, 2CHalkene). 13C-NMR (CDCl3, 125 MHz): Mixture of diastereoisomers A:B (1:1): δ 14.2 (CH3); 22.8, 24.9 (2CH2); 27.3, 27.4 (2CH2CHalkene); 29.2, 29.22, 29.3, 29.5, 29.7, 29.81, 29.84, 29.9, 32.0 (9CH2); 34.0 and 34.08 (CH2CO); 34.9 (d, J = 22.4) and 35.0 (d, J = 21.7, CH2CF); 63.6 and 63.9 (CO2CH2); 75.7 (d, J = 2.9) and 76.5 (CH); 91.7 (d, J = 197.8) and 92.1 (d, J = 200.9, CF); 129.9 (CHalkene); 130.16 and 130.18 (CHalkene); 167.5 (d, J = 23.3, CO); 167.8 (d, J = 24.7, CO); 173.8 and 173.84 (CO). HRMS (ESI, m/z): calculated for C24H38FO6 ([M-H]−): 441.2658, found 441.2667. HPLC-MS retention time (min): 21.16.
Fluoro[(2S)-2-methoxy-3-{[(9Z)-octadec-9-enoyl]oxy}propyl]propanedioic acid, 7
Following general procedure 1, compound 7 was obtained from di-tert-butyl ester 37 (10 mg, 17 μmol) and TFA (98 μL, 1.28 mmol) without further purification in 95% yield. IR (ATR): 3525 (O-H), 1742 (C=O), 1160 (C-F). 1H-NMR (methanol-d4, 700 MHz): δ 0.90 (t, J = 7.1, 3H, CH3); 1.29–1.33 (m, 20H, 10CH2); 1.61–1.63 (m, 2H, CH2CH2CO); 2.02–2.05 (m, 4H, 2CH2CHalkene), 2.24–2.30 (m, 1H, 1/2CH2CF); 2.35 (t, J = 7.4, 2H, CH2CO); 2.45–2.52 (m, 1H, 1/2CH2CF); 3.33 (s, 3H, OCH3); 3.63–3.65 (m, 1H, CH); 4.04 (dd, J = 11.7, 4.9, 1H, 1/2CO2CH2); 4.25 (dd, J = 11.7, 3.9, 1H, 1/2CO2CH2); 5.32–5.37 (m, 2H, 2CHalkene). 13C-NMR (methanol-d4, 175 MHz): δ 14.5 (CH3); 23.8, 26.0 (2CH2); 28.1 (2CH2CHalkene); 30.2 (2CH2); 30.3, 30.4, 30.5, 30.6, 30.8, 30.9, 33.1 (7CH2); 34.9 (CH2CO); 38.4 (d, J = 20.1, CH2CF); 58.1 (OCH3); 65.9 (CO2CH2); 76.0 (CH); 94.4 (d, J = 192.5, CF); 130.8, 130.9 (2CHalkene); 170.9 (br s, 2CO2H); 175.2 (CO). [α]D20: +29.5 (c = 0.20, methanol). HRMS (ESI, m/z): calculated for C25H42FO7 ([M-H]−): 473.2915, found: 473.2922. HPLC-MS retention time (min): 14.51.
(3-{[(9Z)-Octadec-9-enoyl]oxy}propyl)propanedioic acid, 8
Following general procedure 1, compound 8 was obtained from di-tert-butyl ester 38 (96 mg, 0.18 mmol) and TFA (0.34 mL, 4.45 mmol) without further purification in 90% yield. IR (ATR): 3100 (O-H), 1735 (C=O). 1H-NMR (DMSO-d6, 300 MHz): δ 0.87 (t, J = 6.7, 3H, CH3); 1.26–1.30 (m, 20H, 10CH2); 1.56–1.65 (m, 2H, CH2CH2CH(CO2H)2); 1.70–1.80 (m, 2H, CH2CH2CO); 1.93–2.07 (m, 6H, 2CH2CHalkene, CH2CH(CO2H)2); 2.30 (t, J = 7.6, 2H, CH2CO); 3.48 (t, J = 7.4, 1H, CH); 4.11 (t, J = 6.1, 2H, CO2CH2); 5.28–5.39 (m, 2H, 2CHalkene). 13C-NMR (DMSO-d6, 75 MHz): δ 14.1 (CH3); 22.7; 24.9; 25.3; 26.3 (4CH2); 27.2; 27.24 (2CH2CHalkene); 27.9; 29.1; 29.2 (3CH2); 29.3 (2CH2); 29.6; 29.7; 29.8; 31.9 (4CH2); 34.3 (CH2CO); 51.0 (CH); 63.5 (CO2CH2); 129.8; 130.0 (2CHalkene); 174.1 (2CO2H); 174.3 (CO). HRMS (ESI, m/z): calculated for C24H41O6 ([M-H]−): 425.2909, found 425.2898. HPLC-MS retention time (min): 15.18.
Fluoro(3-{[(9Z)-octadec-9-enoyl]oxy}propyl)propanedioic acid, 9
Following general procedure 1, compound 9 was obtained from di-tert-butyl ester 39 (61 mg, 0.11 mmol) and TFA (0.21 mL, 2.75 mmol) without further purification in 90% yield. IR (ATR): 1738 (C=O), 1166 (C-F). 1H-NMR (DMSO-d6, 300 MHz): δ 0.88 (t, J = 6.6, 3H, CH3); 1.18–1.39 (m, 20H, 10CH2); 1.54–1.67 (m, 2H, CH2CH2CO); 1.70–1.85 (m, 2H, CH2CH2CF); 1.93–2.04 (m, 4H, 2CH2CHalkene); 2.20–2.45 (m, 2H, CH2CF); 2.41 (t, J = 7.6, 2H, CH2CO); 4.02–4.18 (m, 2H, CO2CH2); 5.27–5.41 (m, 2H, 2CHalkene). 13C-NMR (DMSO-d6, 75 MHz): δ 14.1 (CH3); 22.7; 24.9 (2CH2); 24.93 (d, J = 2.8, CH2CH2CF); 27.2; 27.23 (2CH2CHalkene); 29.1; 29.2 (2CH2); 29.3 (2CH2); 29.5; 29.7; 29.74; 29.8 (4CH2); 31.1 (d, J = 21.6, CH2CF); 31.9 (CH2); 34.3 (CH2CO); 63.29 (CO2CH2); 129.7; 130.0 (2CHalkene); 174.7 (CO); 2CO2H and CF not observed. HRMS (ESI, m/z): calculated for C24H40FO6 ([M-H]−): 443.2814, found 443.2824. HPLC-MS retention time (min): 16.75.
{4-[(2S)-2-Hydroxy-3-{[(9Z)-octadec-9-enoyl]oxy}propoxy]phenyl}boronic acid, 10
To a solution of boronate ester 40 (50 mg, 0.09 mmol, 1 equiv) in methanol (2 mL), a 4.5 M solution of potassium hydrogen fluoride in water was added (111 μL, 0.50 mmol, 5.6 equiv) and the reaction was stirred at rt for 30 min. Then, solvent was evaporated under reduced pressure and the crude was dissolved in hot acetone and filtrated. The filtrate was concentrated under reduced pressure to afford potassium trifluorido{4-[(2S)-2-hydroxy-3-{[(9Z)-octadec-9-enoyl]oxy}propoxy]phenyl}borate in quantitative yield. 1H-NMR (methanol-d4, 300 MHz): δ 0.90 (t, J = 6.6, 3H, CH3); 1.30–1.37 (m, 20H, 10CH2); 1.60–1.64 (m, 2H, CH2CH2CO); 2.02–2.04 (m, 4H, 2CH2CHalkene); 2.36 (t, J = 7.4, 2H, CH2CO); 3.94–4.02 (m, 2H, CH2OAr); 4.09–4.28 (m, 3H, CH, CO2CH2); 5.29–5.40 (m, 2H, 2CHalkene); 6.79 (d, J = 8.0, 2H, 2CHAr); 7.42 (d, J = 8.3, 2H, 2CHAr). [α]D20: +2.4 (c = 0.9, methanol). MS (ESI, m/z): 521.3 [M-OH]−.
To a solution of the borate salt (30 mg, 54 μmol, 1 equiv) in acetonitrile (0.5 mL), water (3 μL, 0.16 mmol, 3 equiv) and trimethylsilyl chloride (20 μL, 0.16 mmol, 3 equiv) were added and the mixture was stirred for 1 h at rt. Then, a saturated aqueous solution of NaHCO3 was added, and the reaction mixture was extracted with DCM. The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. Flash chromatography of the residue (hexane/EtOAc, 10:1 to 1:1, followed by EtOAc/ethanol, 10:1 to 5:1) afforded compound 10 in 60% yield. Rf: 0.30 (hexane/EtOAc, 1:1). IR (ATR): 1740 (C=O), 1368 (B-O), 1242 (B-C). 1H-NMR (CDCl3, 300 MHz): δ 0.87 (t, J = 6.5, 3H, CH3); 1.26–1.30 (m, 20H, 10CH2); 1.63–1.67 (m, 2H, CH2CH2CO); 1.99–2.01 (m, 4H, 2CH2CHalkene); 2.38 (t, J = 7.5, 2H, CH2CO); 4.03–4.12 (m, 2H, CH2OAr); 4.24–4.37 (m, 3H, CH, CO2CH2); 5.27–5.40 (m, 2H, 2CHalkene); 7.02 (d, J = 8.5, 2H, 2CHAr); 8.16 (d, J = 8.4, 2H, 2CHAr). 13C-NMR (CDCl3, 75 MHz): δ 14.3 (CH3); 22.8, 25.1 (2CH2); 27.3, 27.4 (2CH2CHalkene); 29.2 (2CH2); 29.3 (CH2); 29.5 (2CH2); 29.7, 29.8, 29.9, 32.1 (4CH2); 34.3 (CH2CO); 65.4 (CO2CH2); 68.6 (CH2OAr); 68.8 (CH); 114.2 (2CHAr); 129.9, 130.2 (2CHalkene); 137.6 (2CHAr); 141.1 (CAr); 174.2 (CO); CB not observed. [α]D20: limited solubility. HRMS (ESI, m/z): calculated for C27H46BO6 ([M+H]+): 477.3387, found 477.3395. HPLC-MS retention time (min): 22.85.
(2S)-2-Hydroxy-3-(1H-tetrazol-5-yl)propyl (9Z)-octadec-9-enoate, 11
To a solution of intermediate 41 (64 mg, 0.18 mmol, 1 equiv) in dry DMF (0.3 mL), sodium azide (13 mg, 0.19 mmol, 1.1 equiv) and NH4Cl (12 mg, 0.23 mmol, 1.3 equiv) were added. The mixture was heated at 160°C under MW irradiation for 45 min. Then, the solvent was removed under reduced pressure and the crude was purified by flash chromatography (hexane to hexane/EtOAc, 6:4) to afford compound 11 in 18% yield. IR (ATR): 3390 (O-H, N-H), 1738 (C=O). 1H-NMR (methanol-d4, 500 MHz): δ 0.90 (t, J = 6.9, 3H, CH3); 1.29–1.33 (m, 20H, 10CH2); 1.58–1.64 (m, 2H, CH2CH2CO); 2.01–2.05 (m, 4H, 2CH2CHalkene); 2.34 (t, J = 7.4, 2H, CH2CO); 3.07 (dd, J = 14.9, 8.2, 1H, 1/2CH2CN); 3.18 (dd, J = 14.9, 4.6, 1H, 1/2CH2CN); 4.06–4.12 (m, 2H, CO2CH2); 4.17–4.22 (m, 1H, CH); 5.31–5.37 (m, 2H, 2CHalkene). 13C-NMR (methanol-d4, 125 MHz): δ 14.4 (CH3); 23.7, 25.9 (2CH2); 28.1 (2CH2CHalkene); 29.3 (CH2CN); 30.18, 30.19, 30.3, 30.33, 30.4, 30.6, 30.8, 30.84, 33.1 (9CH2); 34.8 (CH2CO); 68.2 (CO2CH2); 68.3 (CH); 130.8, 130.9 (2CHalkene); 155.5 (CN); 175.2 (CO). [α]D20: +31.7 (c = 0.09, methanol). HRMS (MALDI, m/z): calculated for C22H40N4NaO3 ([M+Na]+): 431.2998, found: 431.2978. HPLC-MS retention time (min): 19.45.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl (9Z)-tetradec-9-enoate, 12
Following general procedure 1, compound 12 was obtained from di-tert-butyl phosphate 46 (7 mg, 14 μmol) and TFA (27 μL, 0.35 mmol) without further purification in 99% yield. IR (ATR): 3375 (O-H), 1737 (C=O), 1182 (P=O), 1058 (P-O). 1H-NMR (methanol-d4, 500 MHz): δ 0.91 (t, J = 7.0, 3H, CH3); 1.29–1.30 (m, 12H, 6CH2); 1.60–1.63 (m, 2H, CH2CH2CO); 2.02–2.04 (m, 4H, 2CH2CHalkene); 2.36 (t, J = 7.4, 2H, CH2CO); 3.96–3.98 (m, 3H, CH, CH2OP); 4.07–4.17 (m, 2H, CO2CH2); 5.29–5.40 (m, 2H, 2CHalkene). 13C-NMR (methanol-d4, 125 MHz): δ 14.3 (CH3); 23.4, 26.0 (2CH2); 27.9, 28.1 (2CH2CHalkene); 30.1, 30.2, 30.3, 30.8, 33.1 (5CH2); 34.9 (CH2CO); 66.2 (CO2CH2); 67.6 (d, J = 5.2, CH2OP); 69.8 (d, J = 6.9, CH); 130.8, 130.83 (2CHalkene); 175.4 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.23. [α]D20: limited solubility. HRMS (ESI, m/z): calculated for C17H32O7P ([M-H]−): 379.2891, found: 379.2848. HPLC-MS retention time (min): 9.81.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl (9Z)-hexadec-9-enoate, 13
Following general procedure 1, compound 13 was obtained from di-tert-butyl phosphate 47 (24 mg, 46 μmol) and TFA (88 μL, 1.15 mmol) without further purification in 90% yield. IR (ATR): 3392 (O-H), 1737 (C=O), 1176 (P=O), 1057 (P-O). 1H-NMR (methanol-d4, 300 MHz): δ 0.90 (t, J = 6.7, 3H, CH3); 1.29–1.33 (m, 16H, 8CH2); 1.60–1.64 (m, 2H, CH2CH2CO); 2.00–2.04 (m, 4H, 2CH2CHalkene); 2.36 (t, J = 7.5, 2H, CH2CO); 3.92–4.04 (m, 3H, CH, CH2OP); 4.15 (ABX system, J = 11.4, 5.5, 4.2, 2H, CO2CH2); 5.29–5.40 (m, 2H, 2CHalkene). 13C-NMR (methanol-d4, 75 MHz): δ 14.5 (CH3); 23.7, 26.0 (2CH2); 28.1, 28.2 (2CH2CHalkene); 30.1 (CH2); 30.2 (2CH2); 30.3, 30.8, 30.84, 32.9 (4CH2); 34.9 (CH2CO); 66.2 (CO2CH2); 67.6 (d, J = 5.3, CH2OP); 69.8 (d, J = 8.0, CH); 130.8, 130.9 (2CHalkene); 175.4 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.79. [α]D20: limited solubility. HRMS (MALDI, m/z): calculated for C19H37NaO7P ([M+Na]+): 431.2175, found: 431.2169. HPLC-MS retention time (min): 10.06.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl (9Z)-heptadec-9-enoate, 14
Following general procedure 1, compound 14 was obtained from di-tert-butyl phosphate 48 (6 mg, 12 μmol) and TFA (22 μL, 0.30 mmol) without further purification in 99% yield. IR (ATR): 3347 (O-H), 1737 (C=O), 1652 (C=C), 1186 (P=O), 1082 (P-O). 1H-NMR (methanol-d4, 700 MHz): δ 0.90 (t, J = 6.7, 3H, CH3); 1.29–1.33 (m, 18H, 9CH2); 1.60–1.64 (m, 2H, CH2CH2CO); 2.01–2.04 (m, 4H, 2CH2CHalkene), 2.36 (t, J = 7.4, 2H, CH2CO); 3.89–3.91 (m, 2H, CH2OP); 3.95–3.98 (m, 1H, CH); 4.11 (dd, J = 11.4, 6.2, 1H, 1/2CO2CH2); 4.18 (dd, J = 11.4, 4.3, 1H, 1/2CO2CH2); 5.29–5.40 (m, 2H, 2CHalkene). 13C-NMR (methanol-d4, 175 MHz): δ 14.5 (CH3); 23.7, 26.0 (2CH2); 28.1 (2CH2CHalkene); 30.2, 30.22, 30.3, 30.34, 30.8, 30.82, 30.9, 33.1 (8CH2); 34.9 (CH2CO); 66.4 (CO2CH2); 67.2 (d, J = 5.2, CH2OP); 70.1 (d, J = 7.9, CH); 130.8, 130.9 (2CHalkene); 175.4 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 4.34. [α]D20: limited solubility. HRMS (MALDI, m/z): calculated for C20H39NaO7P ([M+Na]+): 445.2331, found: 445.2311. HPLC-MS retention time (min): 10.42.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl (9Z)-nonadec-9-enoate, 15
Following general procedure 1, compound 15 was obtained from di-tert-butyl phosphate 49 (24 mg, 43 μmol) and TFA (82 μL, 1.08 mmol) without further purification in 99% yield. IR (ATR): 3360 (O-H), 1738 (C=O), 1181 (P=O), 1060 (P-O). 1H-NMR (methanol-d4, 300 MHz): δ 0.90 (t, J = 6.6, 3H, CH3); 1.29–1.33 (m, 22H, 11CH2); 1.59–1.64 (m, 2H, CH2CH2CO); 2.02–2.04 (m, 4H, 2CH2CHalkene); 2.35 (t, J = 7.5, 2H, CH2CO); 3.91–4.05 (m, 3H, CH, CH2OP); 4.15 (ABX system, J = 11.4, 5.6, 4.3, 2H, CO2CH2); 5.29–5.40 (m, 2H, 2CHalkene). 13C-NMR (methanol-d4, 75 MHz): δ 14.4 (CH3); 23.7, 26.0 (2CH2); 28.1 (2CH2CHalkene); 30.2 (2CH2); 30.3 (2CH2); 30.5, 30.6, 30.7 (3CH2); 30.8 (2CH2); 33.1 (CH2); 34.9 (CH2CO); 66.3 (CO2CH2); 67.5 (d, J = 4.8, CH2OP); 69.9 (d, J = 7.7, CH); 130.8, 130.9 (2CHalkene); 175.4 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.88. [α]D20: limited solubility. HRMS (MALDI, m/z): calculated for C22H43NaO7P ([M+Na]+): 473.2644, found: 473.2642. HPLC-MS retention time (min): 11.30.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl (9Z)-icos-9-enoate, 16
Following general procedure 1, compound 16 was obtained from di-tert-butyl phosphate 50 (9 mg, 16 μmol) and TFA (30 μL, 0.40 mmol) without further purification in 92% yield. IR (ATR): 3350 (O-H), 1738 (C=O), 1179 (P=O), 1059 (P-O). 1H-NMR (methanol-d4, 700 MHz): δ 0.90 (t, J = 7.0, 3H, CH3); 1.29–1.33 (m, 24H, 12CH2); 1.60–1.63 (m, 2H, CH2CH2CO); 2.02–2.05 (m, 4H, 2CH2CHalkene); 2.35 (t, J = 7.5, 2H, CH2CO); 3.94 (app t, J = 5.7, 2H, CH2OP); 3.97–4.00 (m, 1H, CH); 4.11 (dd, J = 11.4, 6.0, 1H, 1/2CO2CH2); 4.18 (dd, J = 11.4, 4.3, 1H, 1/2CO2CH2); 5.32–5.37 (m, 2H, 2CHalkene). 13C-NMR (methanol-d4, 175 MHz): δ 14.5 (CH3); 23.8, 26.0 (2CH2); 28.1, 28.14 (2CH2CHalkene); 30.2, 30.3, 30.5, 30.6 (4CH2); 30.7 (2CH2); 30.77 (2CH2); 30.8 (2CH2); 33.1 (CH2); 34.9 (CH2CO); 66.1 (CO2CH2); 67.6 (d, J = 5.3, CH2OP); 69.7 (d, J = 7.6, CH); 130.8, 130.9 (2CHalkene); 175.4 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.16. [α]D20: limited solubility. HRMS (MALDI, m/z): calculated for C23H45NaO7P ([M+Na]+): 487.2801, found: 487.2798. HPLC-MS retention time (min): 12.35.
(2S)-1-Bromo-3-(phosphonooxy)propan-2-yl 10-phenyldecanoate, (S)-17
Following general procedure 2, compound (S)-17 was obtained from dibenzyl phosphate (S)-75 (50 mg, 0.08 mmol) in 99% yield. IR (ATR): 3360 (O-H), 1738 (C=O), 1199 (P=O), 1025 (P-O). 1H-NMR (methanol-d4, 500 MHz): δ 1.32 (m, 10H, 5CH2); 1.60–1.65 (m, 4H, 2CH2); 2.37 (t, J = 7.3, 2H, CH2CO); 2.59 (t, J = 7.6, 2H, PhCH2); 3.58 (dd, J = 10.9, 5.9, 1H, 1/2CH2Br); 3.66 (dd, J = 10.9, 4.9, 1H, 1/2CH2Br); 4.12–4.18 (m, 2H, CH2OP); 5.14–5.19 (m, 1H, CH); 7.11–7.17 (m, 3H, 3CHAr); 7.23 (t, J = 7.5, 2H, 2CHAr). 13C-NMR (methanol-d4, 125 MHz): δ 26.0, 30.1, 30.3, 30.31 (4CH2); 30.5 (2CH2); 30.8 (CH2Br); 32.7 (CH2); 34.9 (CH2CO); 36.9 (PhCH2); 66.3 (d, J = 5.0, CH2OP); 72.5 (d, J = 8.3, CH); 126.6 (CHAr); 129.2 (2CHAr); 129.4 (2CHAr); 144.0 (CAr); 174.3 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.10. [α]D20: +4.8 (c = 0.50, methanol). HRMS (ESI, m/z): calculated for C19H2979BrO6P ([M(79Br)-H]−: 463.0891, found: 463.0896; calculated for C19H2981BrO6P ([M(81Br)-H]−: 465.0870, found: 465.0876. HPLC-MS retention time (min): 26.05.
(2R)-1-Bromo-3-(phosphonooxy)propan-2-yl 10-phenyldecanoate, (R)-17
Following general procedure 2, compound (R)-17 was obtained from dibenzyl phosphate (R)-75 (69 mg, 0.11 mmol) in 97% yield. The spectroscopic data are in agreement with those reported for its enantiomer (S)-17. [α]D20: −4.6 (c = 0.50, methanol).
(2R)-2-Hydroxy-3-(phosphonooxy)propyl 7-phenylheptanoate, 18
Following general procedure 2, compound 18 was obtained from dibenzyl phosphate 60 (30 mg, 56 μmol) in 85% yield. IR (ATR): 1737 (C=O), 1258 (P=O), 1025 (P-O). 1H-NMR (methanol-d4, 300 MHz): δ 1.33–1.36 (m, 4H, 2CH2); 1.56–1.67 (m, 4H, 2CH2); 2.35 (t, J = 7.4, 2H, CH2CO); 2.60 (t, J = 7.5, 2H, PhCH2); 3.95–4.01 (m, 3H, CH, CH2OP); 4.10 (dd, J = 11.3, 5.4, 1H, 1/2CO2CH2); 4.17 (dd, J = 11.3, 4.3, 1H, 1/2CO2CH2); 7.10–7.17 (m, 3H, 3CHAr); 7.21–7.26 (m, 2H, 2CHAr). 13C-NMR (methanol-d4, 75 MHz): δ 25.9; 29.9; 30.0; 32.6 (4CH2); 34.8 (CH2CO); 36.8 (PhCH2); 65.9 (CO2CH2); 68.1 (d, J = 5.7, CH2OP); 69.3 (d, J = 8.3, CH); 126.6 (CHAr); 129.3 (2CHAr); 129.4 (2CHAr); 143.9 (CAr); 175.3 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.09. [α]D20: +5.3 (c = 0.47, methanol). HRMS (ESI, m/z): calculated for C16H24O7P ([M-H]−): 359.1265, found: 359.1273. HPLC-MS retention time (min): 21.72.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl 8-phenyloctanoate, 19
Following general procedure 2, compound 19 was obtained from dibenzyl phosphate 61 (63 mg, 0.11 mmol) in 80% yield. IR (ATR): 1737 (C=O), 1027 (P-O). 1H-NMR (methanol-d4, 300 MHz): δ 1.33 (app s, 6H, 3CH2); 1.58–1.60 (m, 4H, 2CH2); 2.27–2.37 (m, 2H, CH2CO); 2.59 (t, J = 7.5, 2H, PhCH2); 3.59–3.61 (m, 3H, CH, CH2OP); 3.99–4.41 (m, 2H, CO2CH2); 7.10–7.16 (m, 3H, 3CHAr); 7.21–7.26 (m, 2H, 2CHAr). 13C-NMR (methanol-d4, 75 MHz): δ 25.9, 30.07, 30.1, 30.2, 32.6 (5CH2); 34.8 (CH2CO); 36.9 (PhCH2); 65.8 (CO2CH2); 68.1 (d, J = 5.0, CH2OP); 69.3 (br s, CH); 126.6 (CHAr); 129.2 (2CHAr); 129.4 (2CHAr); 143.9 (CAr); 175.3 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.04. [α]D20: +2.8 (c = 1.58, methanol). HRMS (ESI, m/z): calculated for C17H26O7P ([M-H]−): 373.1422, found: 373.1432. HPLC-MS retention time (min): 8.07.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl 9-phenylnonanoate, 20
Following general procedure 2, compound 20 was obtained from dibenzyl phosphate 62 (90 mg, 0.16 mmol) in 99% yield. IR (ATR): 3355 (O-H), 1737 (C=O), 1259 (P=O), 1029 (P-O). 1H-NMR (methanol-d4, 300 MHz): δ 1.32 (app s, 8H, 4CH2); 1.60 (br s, 4H, 2CH2); 2.34 (t, J = 7.4, 2H, CH2CO); 2.59 (t, J = 7.8, 2H, PhCH2); 3.67–3.79 (m, 1H, CH); 3.98–4.00 (m, 2H, CH2OP); 4.07–4.21 (m, 2H, CO2CH2); 7.09–7.17 (m, 3H, 3CHAr); 7.21–7.26 (m, 2H, 2CHAr). 13C-NMR (methanol-d4, 75 MHz): δ 25.9, 30.1, 30.2, 30.3, 30.4, 32.7 (6CH2); 34.8 (CH2CO); 36.9 (PhCH2); 65.8 (CO2CH2); 68.1 (d, J = 6.0, CH2OP); 69.3 (d, J = 8.3, CH); 126.6 (CHAr); 129.2 (2CHAr); 129.4 (2CHAr); 143.9 (CAr); 175.3 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.05. [α]D20: +2.5 (c = 1.13, methanol). HRMS (ESI, m/z): calculated for C18H28O7P ([M-H]−): 387.1578, found: 387.1591. HPLC-MS retention time (min): 8.34.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl 10-phenyldecanoate, 21
Following general procedure 2, compound 21 was obtained from dibenzyl phosphate 63 (20 mg, 34 μmol) in 80% yield. IR (ATR): 1739 (C=O), 1051 (P-O). 1H-NMR (methanol-d4, 500 MHz): δ 1.29–1.32 (m, 10H, 5CH2); 1.61 (m, 4H, 2CH2); 2.35 (t, J = 7.5, 2H, CH2CO); 2.59 (t, J = 7.6, 2H, PhCH2); 3.96–4.01 (m, 3H, CH, CH2OP); 4.11 (dd, J = 11.3, 5.3, 1H, 1/2CO2CH2); 4.17 (dd, J = 11.3, 4.1, 1H, 1/2CO2CH2); 7.11–7.16 (m, 3H, 3CHAr); 7.23 (t, J = 7.5, 2H, 2CHAr). 13C-NMR (methanol-d4, 125 MHz): δ 26.0, 30.2, 30.3, 30.34 (4CH2); 30.5 (2CH2); 32.7 (CH2); 34.9 (CH2CO); 36.9 (PhCH2); 65.9 (CO2CH2); 68.1 (d, J = 5.7, CH2OP); 69.4 (d, J = 8.1, CH); 126.6 (CHAr); 129.2 (2CHAr); 129.4 (2CHAr); 144.0 (CAr); 175.4 (CO). 31P-NMR (methanol-d4, 202 MHz): δ 3.14. [α]D20: +2.3 (c = 0.40, methanol). HRMS (ESI, m/z): calculated for C19H30O7P ([M-H]−): 401.1735, found: 401.1734. HPLC-MS retention time (min): 24.37.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl 11-phenylundecanoate, 22
Following general procedure 2, compound 22 was obtained from dibenzyl phosphate 64 (5 mg, 8 μmol) in 82% yield. IR (ATR): 3387 (O-H), 1739 (C=O), 1262 (P=O), 1014 (P-O). 1H-NMR (methanol-d4, 300 MHz): δ 1.26–1.32 (m, 12H, 6CH2); 1.53–1.68 (m, 4H, 2CH2); 2.35 (t, J = 7.5, 2H, CH2CO); 2.59 (t, J = 7.7, 2H, PhCH2); 3.69–3.74 (m, 1H, CH); 3.98–4.02 (m, 2H, CH2OP); 4.08–4.16 (m, 2H, CO2CH2); 7.10–7.16 (m, 3H, 3CHAr); 7.21–7.26 (m, 2H, 2CHAr). 13C-NMR (methanol-d4, 75 MHz): δ 25.8, 30.2, 30.3, 30.4, 30.55, 30.56, 30.7, 32.8 (8CH2); 34.9 (CH2CO); 36.9 (PhCH2); 65.8 (CO2CH2); 68.1 (d, J = 5.6, CH2OP); 69.3 (d, J = 8.9, CH); 126.6 (CHAr); 129.2 (2CHAr); 129.4 (2CHAr); 144.0 (CAr); 175.3 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.07. [α]D20: +0.2 (c = 0.32, methanol). HRMS (ESI, m/z): calculated for C20H32O7P ([M-H]−): 415.1891, found: 415.1906. HPLC-MS retention time (min): 27.56.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl 15-phenylpentadecanoate, 23
Following general procedure 2, compound 23 was obtained from dibenzyl phosphate 65 (20 mg, 31 μmol) in 90% yield. IR (ATR): 1738 (C=O), 1259 (P=O), 1083 (P-O). 1H-NMR (methanol-d4, 300 MHz): δ 1.28–1.32 (m, 20H, 10CH2); 1.57–1.64 (m, 4H, 2CH2); 2.35 (t, J = 7.4, 2H, CH2CO); 2.59 (t, J = 7.5, 2H, PhCH2); 3.97–4.35 (m, 5H, CO2CH2, CH, CH2OP); 7.08–7.16 (m, 3H, 3CHAr); 7.21–7.26 (m, 2H, 2CHAr). 13C-NMR (methanol-d4, 75 MHz): δ 26.0, 30.2, 30.3, 30.4, 30.59, 30.6, 30.7 (7CH2); 30.73 (2CH2); 30.75 (2CH2); 32.8 (CH2); 34.9 (CH2CO); 36.9 (PhCH2); 64.9 (CO2CH2); 67.1 (d, J = 5.3, CH2OP); 68.3 (d, J = 8.3, CH); 126.6 (CHAr); 129.2 (2CHAr); 129.4 (2CHAr); 144.0 (CAr); 175.4 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.11. [α]D20: +4.2 (c = 0.40, methanol). HRMS (ESI, m/z): calculated for C24H40O7P ([M-H]−): 471.2517, found: 471.2520. HPLC-MS retention time (min): 33.42.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl 7-([1,1’-biphenyl]-4-yl)heptanoate, 24
Following general procedure 2, compound 24 was obtained from dibenzyl phosphate 66 (27 mg, 43 μmol) in 74% yield. IR (ATR): 1720 (C=O), 1058 (P-O). 1H-NMR (methanol-d4, 300 MHz): δ 1.36–1.40 (m, 4H, 2CH2); 1.60–1.70 (m, 4H, 2CH2); 2.36 (t, J = 7.4, 2H, CH2CO); 2.64 (t, J = 7.5, 2H, ArCH2); 3.97–4.01 (m, 3H, CH, CH2OP); 4.11 (dd, J = 11.3, 5.3, 1H, 1/2CO2CH2); 4.17 (dd, J = 11.3, 4.1, 1H, 1/2CO2CH2); 7.24 (d, J = 8.1, 2H, 2CHAr); 7.30 (t, J = 7.5, 1H, CHAr); 7.40 (t, J = 7.6, 2H, 2CHAr); 7.51 (d, J = 8.2, 2H, 2CHAr); 7.58 (d, J = 7.7, 2H, 2CHAr). 13C-NMR (methanol-d4, 125 MHz): δ 25.9; 29.9; 30.0; 32.5 (4CH2); 34.9 (CH2CO); 36.4 (ArCH2); 65.9 (CO2CH2); 68.1 (d, J = 5.4, CH2OP); 69.3 (d, J = 8.3, CH); 127.8 (2CHAr); 127.84 (2CHAr); 128.0 (CHAr); 129.8 (2CHAr); 129.9 (2CHAr); 139.9; 142.4; 143.1 (3CAr); 175.3 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.11. [α]D20: +7.6 (c = 0.67, methanol). HRMS (ESI, m/z): calculated for C22H28O7P ([M-H]−): 435.1578, found: 435.1562. HPLC-MS retention time (min): 26.61.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl 10-([1,1’-biphenyl]-4-yl)decanoate, 25
Following general procedure 2, compound 25 was obtained from dibenzyl phosphate 67 (20 mg, 30 μmol) in 85% yield. IR (ATR): 3314 (C=O), 1654 (C=O), 1015 (P-O). 1H-NMR (methanol-d4, 500 MHz,): δ 1.28–1.35 (m, 10H, 5CH2); 1.58–1.66 (m, 4H, 2CH2); 2.35 (t, J = 7.4, 2H, CH2CO); 2.64 (t, J = 7.5, 2H, ArCH2); 3.96–4.00 (m, 3H, CH, CH2OP); 4.11 (dd, J = 11.4, 5.4, 1H, 1/2CO2CH2); 4.17 (dd, J = 11.4, 4.2, 1H, 1/2CO2CH2); 7.24 (d, J = 8.1, 2H, 2CHAr); 7.29 (t, J = 7.4, 1H, CHAr); 7.40 (t, J = 7.7, 2H, 2CHAr); 7.51 (d, J = 8.2, 2H, 2CHAr); 7.58 (dd, J = 8.3, 1.1, 2H, 2CHAr). 13C-NMR (methanol-d4, 125 MHz): δ 26.0; 30.2; 30.3; 30.4; 30.5; 30.52; 32.7 (7CH2); 34.9 (CH2CO); 36.5 (ArCH2); 65.9 (CO2CH2); 68.1 (d, J = 5.6, CH2OP); 69.3 (d, J = 8.2, CH); 127.8 (2CHAr); 127.83 (2CHAr); 128.0 (CHAr); 129.8 (2CHAr); 129.9 (2CHAr); 139.9; 142.5; 143.2 (3CAr); 175.4 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 3.11. HRMS (ESI, m/z): calculated for C25H34O7P ([M-H]−): 477.2048, found: 477.2029. HPLC-MS retention time (min): 10.49.
(2R)-2-Hydroxy-3-(phosphonooxy)propyl 13-([1,1’-biphenyl]-4-yl)tridecanoate, 26
Following general procedure 2, compound 26 was obtained from dibenzyl phosphate 68 (37 mg, 53 μmol) as a white solid in 91% yield. M.p.: 95–97°C. IR (ATR): 1730 (C=O), 1258 (P=O), 1027 (P-O). 1H-NMR (DMSO-d6, 500 MHz): δ 1.23–1.29 (m, 16H, 8CH2); 1.49–1.52 (m, 2H, CH2); 1.57–1.60 (m, 2H, CH2); 2.28 (t, J = 7.4, 2H, CH2CO); 2.60 (t, J = 7.6, 2H, ArCH2); 3.35 (br s, 1H, OH); 3.73–3.76 (m, 2H, CH2OP); 3.79–3.85 (m, 1H, CH); 3.94 (dd, J = 11.2, 6.0, 1H, 1/2CO2CH2); 4.02 (dd, J = 11.2, 4.3, 1H, 1/2CO2CH2); 7.27 (d, J = 8.1, 2H, 2CHAr); 7.33 (t, J = 7.3, 1H, CHAr); 7.44 (t, J = 7.7, 2H, 2CHAr); 7.56 (d, J = 8.1, 2H, 2CHAr); 7.63 (d, J = 7.3, 2H, 2CHAr). 13C-NMR (DMSO-d6, 126 MHz): δ 26.4; 28.5; 28.6; 28.7; 28.8; 28.9 (6CH2); 28.98 (2CH2); 29.0; 30.9 (2CH2); 33.4 (CH2CO); 34.7 (ArCH2); 64.9 (CO2CH2); 66.1 (d, J = 5.3, CH2OP); 67.3 (d, J = 7.8, CH); 126.4 (2CHAr); 126.5 (2CHAr); 127.1 (CHAr); 128.85 (2CHAr); 128.87 (2CHAr); 137.5; 140.1; 141.6 (3CAr); 172.9 (CO). 31P-NMR (DMSO-d6, 121 MHz): δ 2.16. [α]D20: limited solubility. HRMS (ESI, m/z): calculated for C28H40O7P ([M-H]−): 519.2517, found: 519.2508. HPLC-MS retention time (min): 12.15.
(2S)-1-Bromo-3-(phosphonooxy)propan-2-yl 9-phenylnonanoate, 76
Following general procedure 2, compound 76 was obtained from dibenzyl phosphate 81 (249 mg, 0.39 mmol) in 80% yield. Rf: 0.51 (DCM/EtOAc, 10:1). IR (ATR): 3320 (O-H), 1021 (P-O). 1H-NMR (methanol-d4, 300 MHz): δ 1.33 (app s, 8H, 4CH2); 1.59–1.66 (m, 4H, 2CH2); 2.38 (t, J = 7.4, 2H, CH2CO); 2.60 (t, J = 7.7, 2H, PhCH2); 3.63 (ABX system, J = 11.0, 6.0, 4.9, 2H, CH2Br); 4.14 (app t, J = 5.9, 2H, CH2OP); 5.17 (qt, J = 5.2, 1H, CH); 7.10–7.17 (m, 3H, 3CHAr); 7.22–7.27 (m, 2H, 2CHAr). 13C-NMR (methanol-d4, 75 MHz): δ 24.9, 29.1, 29.2, 29.3, 29.4, 29.7 (6CH2); 31.7 (CH2Br); 33.9 (CH2CO); 35.9 (PhCH2); 65.3 (d, J = 5.1, CH2OP); 71.5 (d, J = 8.3, CH); 125.6 (CHAr); 128.2 (2CHAr); 128.4 (2CHAr); 143.0 (CAr); 173.3 (CO). 31P-NMR (methanol-d4, 121 MHz): δ 2.76. [α]D20: +0.95 (c = 0.57, methanol). HRMS (MALDI, m/z): calculated for C18H2879BrNaO6P ([M(79Br)+Na]+): 473.0705, found: 473.0681; calculated for C18H2881BrNaO6P ([M(81Br)+Na]+): 475.0684, found: 475.0666. HPLC-MS retention time (min): 9.42.
(2S)-1-Bromo-3-(phosphonooxy)propan-2-yl 11-phenylundecanoate, 77
Following general procedure 2, compound 77 was obtained from dibenzyl phosphate 82 (48 mg, 73 μmol) in 72% yield. Rf: 0.46 (DCM/EtOAc, 10:1). IR (ATR): 3336 (O-H), 1689 (C=O), 1251 (P=O). 1H-NMR (methanol-d4, 300 MHz): δ 1.26–1.36 (m, 12H, 6CH2); 1.56–1.68 (m, 4H, 2CH2); 2.37 (t, J = 7.3, 2H, CH2CO); 2.59 (t, J = 7.6, 2H, PhCH2); 3.62 (ABX system, J = 10.9, 6.0, 4.8, 2H, CH2Br); 4.13 (app t, J = 5.0, 2H, CH2OP); 5.16 (qt, J = 5.1, 1H, CH); 7.10–7.16 (m, 3H, 3CHAr); 7.21–7.26 (m, 2H, 2CHAr). 13C-NMR (methanol-d4, 75 MHz): δ 26.0, 30.1, 30.3, 30.4, 30.5, 30.55, 30.62, 30.8 (8CH2); 32.8 (CH2Br); 34.9 (CH2CO); 36.9 (PhCH2); 66.3 (d, J = 4.9, CH2OP); 72.5 (d, J = 8.8, CH); 126.6 (CHAr); 129.2 (2CHAr); 129.4 (2CHAr); 144.0 (CAr); 174.3 (CO). 31-PNMR (methanol-d4, 121 MHz): δ 2.82. [α]D20: −7.8 (c = 0.81, methanol). HRMS (MALDI, m/z): calculated for C20H32Na79BrO6P ([M(79Br)+Na]+): 501.1018, found: 501.1002; calculated for C20H32Na81BrO6P ([M(81Br)+Na]+): 503.0997, found: 503.1019. HPLC-MS retention time (min): 10.06.
(2S)-1-Bromo-3-(phosphonooxy)propan-2-yl (9Z)-hexadec-9-enoate, 78
Following general procedure 1, compound 78 was obtained from tert-butyl ester 83 (4 mg, 6.9 μmol) and TFA (40 μL, 0.51 mmol) in 93% yield. IR (ATR): 1741 (C=O), 1457 (C=C), 1015 (P-O). 1H-NMR (methanol-d4, 500 MHz): δ 0.93 (t, J = 6.8, 3H, CH3); 1.29–1.42 (m, 16H, 8CH2); 1.56–1.70 (m, 2H, CH2CH2CO); 2.03–2.06 (m, 4H, 2CH2CHalkene); 2.40 (t, J = 7.4, 2H, CH2CO); 3.62 (dd, J = 10.9, 6.1, 1H, 1/2CH2Br); 3.70 (dd, J = 10.7, 4.5, 1H, 1/2CH2Br); 4.05–4.17 (m, 2H, CH2OP); 5.13–5.24 (m, 1H, CH); 5.31–5.44 (m, 2H, 2CHalkene). 13C-NMR (methanol-d4, 125 MHz): δ 13.4 (CH3); 22.7 (CH2); 25.0, 25.1 (2CH2CHalkene); 27.1, 27.13, 27.14, 29.0, 29.2, 29.77, 29.78, 29.8, 31.9 (9CH2); 33.9 (CH2CO); 60.5 (d, J = 7.2, CH2OP); 64.9 (d, J = 5.3, CH); 129.8, 129.9 (2CHalkene); 173.4 (CO). 31P-NMR (methanol-d4, 202 MHz): δ 2.88. [α]D20: +5.9 (c = 0.09, methanol). HRMS (ESI, m/z): calculated for C19H3579BrO6P ([M(79Br)-H]−): 469.1360, found: 469.1361; calculated for C19H3581BrO6P ([M(81Br)-H]−): 471.1340, found: 471.1342. HPLC-MS retention time (min): 23.25.
(2S)-1-Bromo-3-(phosphonooxy)propan-2-yl (9Z)-octadec-9-enoate, 79
Following general procedure 1, compound 79 was obtained from di-tert-butyl phosphate 84 (5 mg, 8.17 μmol) and TFA (15 μL, 204 μmol) without further purification in 81% yield. IR (ATR): 1739 (C=O), 1666 (C=C), 1071 (P-O).1H-NMR (methanol-d4, 700 MHz): δ 0.90 (t, J = 7.0, 3H, CH3); 1.28–1.35 (m, 20H, 10CH2); 1.62–1.65 (m, 2H, CH2CH2CO); 2.02–2.05 (m, 4H, 2CH2CHalkene); 2.37 (td, J = 7.4, 3.2, 2H, CH2CO); 3.60 (dd, J = 11.0, 6.1, 1H, 1/2CH2Br); 3.68 (dd, J = 11.0, 4.6, 1H, 1/2CH2Br); 4.07–4.12 (m, 2H, CH2OP); 5.14–5.18 (m, 1H, CH); 5.32–5.37 (m, 2H, 2CHalkene). 13C-NMR (methanol-d4, 175 MHz): δ 14.5 (CH3); 23.8, 26.0 (2CH2); 28.1 (2CH2CHalkene); 30.1, 30.2, 30.3, 30.4, 30.5, 30.6, 30.8, 30.9, 31.1, 33.1 (10CH2); 35.0 (CH2CO); 66.0 (d, J = 3.1, CH2OP); 72.8 (d, J = 5.5, CH); 130.8, 130.9 (2CHalkene); 174.4 (CO). 31P-NMR (methanol-d4, 202 MHz): δ 3.36. [α]D20: +3.3 (c = 0.33, methanol). HRMS (ESI, m/z): calculated for C21H3979BrO6P ([M(79Br)-H]−): 497.1673, found: 497.1664; calculated for C21H3981BrO6P ([M(81Br)-H]−): 499.1653, found: 499.1642. HPLC-MS retention time (min): 25.58.
Fluoro[(2S)-3-{[(9Z)-hexadec-9-enoyl]amino}−2-methoxypropyl]propanedioic acid, 85
Following general procedure 1, compound 85 was obtained from tert-butyl ester 89 (10 mg, 18 μmol) and TFA (104 μL, 1.34 mmol) in 99% yield. Rf: 0.17 (hexane/EtOAc, 6:4). IR (ATR): 3273 (N-H), 1646 (C=O, broad). 1H-NMR (CDCl3, 700 MHz): δ 0.88 (t, J = 7.0, 3H, CH3); 1.25–1.34 (m, 16H, 8CH2); 1.54–1.67 (m, 2H, CH2CH2CO); 1.95–2.04 (m, 4H, 2CH2CHalkene); 2.19–2.56 (m, 4H, CH2CO, CH2CF); 3.24–3.70 (m, 6H, NHCH2, CH, OCH3); 5.30–5.36 (m, 2H, 2CHalkene). 13C-NMR (CDCl3, 175 MHz): δ 14.3 (CH3); 22.8, 26.0 (2CH2); 27.4 (2CH2CHalkene); 29.1, 29.4, 29.5 (3CH2); 29.88 (2CH2); 29.93, 31.9, 36.6 (3CH2); 36.7 (br s, CH2CF); 42.2 (br s, NHCH2); 57.6 (OCH3); 74.5 (CH); 129.8, 130.1 (2CHalkene); 169.3 (br s, 2CO2H); 175.6 (CONH); CF not observed. [α]D20: −0.5 (c = 0.14, methanol). HRMS (ESI, m/z): calculated for C23H39FNO6 ([M-H]−): 444.2767, found: 444.2784. HPLC-MS retention time (min): 29.71.
Fluoro[(2S)-2-methoxy-3-{[(9Z)-octadec-9-enoyl]amino}propyl]propanedioic acid, 86
Following general procedure 1, compound 86 was obtained from tert-butyl ester 90 (18 mg, 31 μmol) and TFA (178 μL, 2.30 mmol) in 69% yield. Rf: 0.19 (hexane/EtOAc, 6:4). IR (ATR): ν 1734 (C=O), 1648 (C=O). 1H-NMR (CDCl3, 700 MHz): δ 0.88 (t, J = 6.3, 3H, CH3); 1.25–1.26 (m, 20H, 10CH2); 1.53–1.67 (m, 2H, CH2CH2CO); 1.94–2.04 (m, 4H, 2CH2CHalkene); 2.13–2.58 (m, 4H, CH2CO; CH2CF); 3.11–3.66 (m, 6H, NHCH2, CH, OCH3); 5.27–5.40 (m, 2H, 2CHalkene). 13C-NMR (CDCl3, 175 MHz): 14.3 (CH3); 22.8; 25.9 (2CH2); 27.4 (2CH2CHalkene); 29.48; 29.5; 29.7; 29.71; 29.86; 29.9; 29.94; 32.1; 33.9; 36.5 (10CH2); 36.6 (br s, CH2CF); 41.2 (br s, NHCH2); 57.5 (OCH3); 75.3 (CH); 129.8; 130.1 (2CHalkene); 169.4 (br s, 2CO2H); 174.9 (CONH); CF not observed. [α]D20: ˗2.07 (c = 0.28, methanol). HRMS (ESI, m/z): calculated for C25H43FNO6 ([M-H]−): 472.3080, found: 472.3095. HPLC-MS retention time (min): 21.44.
Cell lines and culture
RH7777 hepatoma cells stably expressing the LPA1 receptor and their corresponding non-transfected controls were kindly provided by Prof. Gabor Tigyi (University of Tennessee Health Science Center, Memphis, Tennessee). Retrovirus expression vector (LZRS-EGFP) and Phoenix retrovirus producer cell lines were provided by Prof. Garry P. Nolan (Stanford University, Stanford, California). Collagen, poly-D-lysine, poly-L-lysine and LPA were purchased from Aldrich. Capsaicin was purchased from Sigma. Ionomycin was purchased from Cayman. All other reagents were from Gibco. All cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, 1% sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin in a 5% CO2 humidified atmosphere at 37 °C. For passage, cells were rinsed with phosphate buffered saline (PBS) and incubated with 0.125% trypsin, 0.02% EDTA solution for 2 min at 37 °C. Detached cells were resuspended in growth medium, counted if necessary and splitted onto fresh dishes.
Generation of LPA1–3 overexpressing cell lines
Cell lines stably expressing LPA1–3 receptors were generated by retroviral infection of B103 cells. Lipofectamine 2000 transfection reagent (Invitrogen) was used to transfect the Phoenix ecotropic packaging cell line with retroviral constructs expressing FLAG-tagged human LPA1–3 cDNAs and EGFP. An internal ribosomal entry site (IRES) allowed dual expression of both the LPA1–3 receptors and EGFP. Retroviral supernatant was harvested 48 h post-transfection, filtered through a 0.45 μm filter and added to B103 cells in the presence of Polybrene (Sigma). Cells were centrifuged for 90 minutes at 28°C, after which the retroviral supernatant was replaced by DMEM. B103 cells expressing high levels of LPA1–3 and high levels of EGFP were isolated by FACS using a Vantage DiVa I instrument. Control B103 cells transfected with empty vector were also obtained and used as control throughout the experiments.
Evaluation of receptor activation by Ca2+ mobilization assay
Changes in intracellular calcium levels were measured by using the fluorescent calcium sensitive dye Fluo-4 NW (Invitrogen). RH7777 cells or B103 cells were plated on poly-D-lysine or collagen-coated, respectively, black-wall clear-bottom 96-well plates (Corning) at a density of 50000 cells/well and cultured overnight. The culture medium was then replaced with Fluo-4 NW dye loading solution containing 2.5 μM of probenecid and incubated for 30 minutes at 37°C followed by an additional 30 minutes at rt. Fluorescence changes were registered in a FluoStar Optima instrument (BMG Labtech) at 525 nm using an excitation wavelength of 494 nm. Each well was monitored for 240 s. 20 μL of the test compound from a 6x stock solution in assay buffer were added after 120 s of starting the measurement. Ca2+ transient increase was quantified by calculating the difference between maximum and baseline values for each well. As positive controls, 10 μM LPA and 10 μM ionomycin were included in every experiment. At this concentration, LPA induced a response about 30–33% of the one shown by ionomycin, which is in agreement with previously described results.52 The data presented are from two to four independent experiments carried out in triplicate or quadruplicate. Dose-response curves were generated and EC50 values calculated by nonlinear regression analysis using PRISM software version 5 (GraphPad Software Inc, San Diego, CA, USA). For assessing covalent binding, after the initial stimulation with compound (S)-17, media was removed, cells washed with buffer, and, after 30 minutes, fresh compound was added and Ca2+ mobilization measured as described above.
Activity at LPA4–6 receptors
B103 naive cells were transiently transfected with pCXN2.1 vectors harboring hLPA4, hLPA5 and hLPA6, plated onto 384 well plates, and cultured overnight. The media was replaced to FreeStype 293 Expression Medium 4 h before the assay. Cells were loaded with Fluo-4 dye in the presence of 5 mM probenecid for 1 h. Ca2+ signaling was monitored with a Hamamatsu FDSS7000 instrument.
In silico experiments
Docking calculations were performed using Autodock453 (ga_num_evals = 40000000, ga_run = 100 and all the other parameters set to their default values). The LPA1 active state model was retrieved from the GPCRdb46, 47 (2019, March 11th release, downloaded 2019, October 2nd; structure file provided in the Supporting Information) and prepared for docking using pdb2pqr54, 55 with propka56, 57 protonation option at a pH of 7.4 and the peoepb force field.58 (S)-17 was modelled using RDKIT and its protonation state adjusted at pH 7.4 by ChemAxon cxcalc module. Binding mode pictures were created using PyMOL v1.8 and PLIP v1.4.4.59
Stability studies
These studies were carried out as previously described with minor modifications.60 For chemical stability, to a vial containing 998 μL of PBS pH 7.4, 2 μL of a 25 mM solution of test compound in DMSO was added and kept at rt. Remaining compound at each time point was quantified by HPLC-MS (SIM mode) using the peak area integration normalized with an internal standard. To determine the stability in cellular culture medium, 12 μL of a 50 mM solution of test compound in DMSO was added to 1188 μL of cellular culture medium pre-warmed at 37 °C. Samples were incubated at 37 °C and 200 μL aliquots were taken at the corresponding time points. Each aliquot was quenched in 200 μL of cold ACN, vortexed, incubated for 10 min in ice and centrifuged at 39000g for 10 min. Supernatants were then analyzed by HPLC-MS using SIM mode and quantification was estimated by using the peak area integration normalized with an internal standard.
Receptor internalization and neurite retraction assay
B103 neuroblastoma cells overexpressing EGFP-LPA1 receptor were plated on poly-L-lysine-coated glass coverslips in 24-well plates (5·104 cells per well) and serum-starved overnight. For the experiment, media was replaced with 1 mL fresh serum-free DMEM, supplemented with 0.1% FAF BSA, 1 μM LPA, or (S)-17. After 15 minutes, cells were fixed with 4% paraformaldehyde and permeabilized with 0.025% Triton X-100/PBS for 15 min. Immunocytochemistry experiments were carried out following experimental procedures previously described.61 Briefly, F-actin was detected with rhodamine-phalloidin (Sigma) in PBS and nuclei with DAPI (Sigma). For neurite retraction, samples were visualized on a fluorescence microscopy (Carl Zeiss). For receptor internalization, cells were analysed using confocal fluorescence microscopy (Carl Zeiss).
Migration assay
Cell migration was measured in Transwell chambers (PET membrane with 8 μm pore size, Becton Dickinson). Bottom membrane of transwells was precoated overnight at 4 °C with 10 μg/mL of collagen solution in PBS. B103 neuroblastoma cells overexpressing LPA1 receptor were serum-starved overnight, and then harvested with 0.125% trypsin containing 0.02% EDTA, washed once, counted and resuspended in serum-free DMEM. The cells were seeded into the upper chamber (2.5·105 cells per transwell), and 0.1% FAF BSA, 1 μM LPA or (S)-17 were placed in the lower chamber. Cells were allowed to migrate for 5 h at 37°C. Non-migratory cells were removed from the top filter surface with a cotton swab. Migratory cells attached to the underside of the transwells were washed with PBS, stained with 0.1% crystal violet solution and counted under a microscope.
Primary culture of sensory neurons
DRG were harvested from neonatal Wistar rats (3–5 days old). Ganglia were digested with 0.25% (w/v) collagenase (type IA) in DMEM-glutamax (Invitrogen) with 1% penicillin-streptomycin (5000 U/mL, Invitrogen) for 1 h (37 °C, 5% CO2). After digestion, rat DRG was mechanically dissociated using a glass Pasteur pipette. Single cell suspension was passed through a 100 μm cell strainer, and washed with DMEM glutamax plus 10% fetal bovine serum (FBS; Invitrogen) and 1% penicillin-streptomycin. Cells were seeded at the 30 μL of medium containing cells on MEA chambers previously coated with poly-L-lysine (8.33 μg/mL) and laminin (5 μg/mL). After 2 h, medium was replaced with DMEM glutamax, 10% FBS and 1% penicillin-streptomycin, supplemented with mouse 2.5s NGF 50 ng/mL (Promega), and 1.25 μg/mL cytosine arabinoside when required (37°C, 5% CO2). All experiments were made 48 h after cell seeding.
Microelectrode Array (MEA)
Extracellular recordings were made using multiple electrode planar arrays of 60-electrode thin MEA chips, with 30 μm diameter electrodes and, 200 μm inter-electrode spacing with an integrated reference electrode (Multichannel Systems GmbH). The electrical activity of primary sensory neuron was recorded by the MEA1060 System (Multi Channel Systems GmbH, http://www.multichannelsystems.com), and MC_Rack software version 4.3.0 at a sampling rate of 25 kHz. 15 seconds application of 10μM of LPA and its derivatives were perfused to identify evoked neuronal spikes on cultured rat DRG neurons, using continuous perfusion system (2 mL/min flux). Capsaicin (500 nM) was applied at the end of the protocol to measure TRPV1 sensitivity of the neurons which were exposed to LPA or (S)-17. Data were analyzed using MC_RACK spike sorter and Neuroexplorer Software (Nex Technologies). An evoked spike was defined when the amplitude of the neuronal electrical activity overcame a threshold set at −20 μV. The recorded signals were then processed to extract mean spike frequency.
Calcium microfluorography
DRG on coverslips loaded with 5 μM fluo-4-acetoxymethyl ester (Fluo-4AM) plus 0.02% pluronic acid (Molecular Probes, Invitrogen) in HBSS extracellular solution (in mM): 140 NaCl, 4 KCl, 1 MgCl2, 1.8 CaCl2, 5 D-glucose, and 10 HEPES, pH 7.4 for 1 h (37°C, 5% CO2) mounted in a RC-25 chamber (Harvard Apparatus), were continuously perfused with test solutions at rt. Fluorescence from individual neurons monitored through a 10x air objective (Aixiovert 200 inverted microscope, Carl Zeiss) with an ORCA-ER CCD camera (Hamamatsu Photonics). Pre-programed protocols applied via computer-controlled pinch valves (Bioscience Tools, 5 mL/min flux). Fluo-4AM excited at 500 nm and emitted fluorescence filtered at 535 nm (Lambda- 10–2-filter wheel, Sutter Instruments). Images processed with AquaCosmos package software (Hamamatsu Photonics). LPA and (S)-17 were perfused at 10 μM for 10 s. TRPV1 channel activity was evoked with 10 s-application of capsaicin at 1 μM. 10 s pulse of 40 mM KCl was applied to distinguish neuronal viability. Ionomycin was used at 5 μM. Intracellular Ca2+ increase calculated as fluorescence difference between baseline and the peak reached. An evoked response defined when intensity overcame 20% of baseline. For MEA and calcium microfluorography experiments, LPA and (S)-17 were dissolved in DMSO at a stock concentration of 10 mM and then diluted in the standard external solution to use at a final concentration of 10 μM. Capsaicin was dissolved in DMSO at a stock concentration of 10 mM and used at 500 nM final concentration.
Writhing assay and locomotor and exploratory activity
Writhing (abdominal contraction) was generated by an i.p. injection of 10 mL/kg of 0.6% acetic acid solution. Mice were randomly allocated to receive the corresponding treatment (9 animals per treatment). Exploratory and locomotor activity was assessed in the open field test (OFT). The apparatus were four open field arenas made of grey Plexiglas with the following dimensions: 40×40×30 cm (Panlab, Barcelona, Spain). Light intensity in the test room was 70 lux. Mice were habituated for 30 min before starting the test. Compound (S)-17 (1, 3 and 10 mg/kg) and vehicle (BSA 3%) were injected i.p. 30 min before placing the animal in the open field at a total volume of 10 mL/kg. Throughout the session, treatments were counterbalanced between arenas. Immobility and time spent in the center area of the test were digitally recorded (Sony DCR-SX65E) during 30 min session. Behavior was analyzed using Ethovision XT 9.0 (Noldus Information Technology, Wageningen, The Nederlands). The OFT was cleaned between mice with ethanol (30%). Compound (S)-17 was dissolved in 3% fatty acid free BSA (Sigma Aldrich) at dose 10 mg/kg and administered at volume of 10 mL/kg. Adult C57BL/6 male mice (35–40 g) were housed under controlled environment conditions (temperature: 22.5 ± 0.5 °C, humidity: 64 ± 1 %, 12/12-h light/dark cycle and free access to food and water). All experiments were carried out in compliance with the European Commission on Animal Care (EU Directive 2010/63/EU). Experimental protocols were approved by The Local Ethical Committee for Animal Research of the University of Malaga. Results are expressed as the mean±SEM. Data were analyzed by t-Student or one-way analysis of variance (ANOVA) followed, when appropriated, by the Newman-Keuls Multiple Comparisons test. A value of p<0.05 was considered as statistical significant. All statistical analyses were performed using the Graphpad Prism version 5.04 software (GraphPad Software, San Diego, CA, USA).
Spared nerve injury and treatment
All surgical procedures were approved by the Universitat Autònoma de Barcelona Animal Care Committee (CEEAH 4273) and followed the guidelines of the European Commission on Animal Care (EU Directive 2010/63/EU). Adult female C57Bl/6J mice (10–12 weeks old) were anesthetized with a mixture of ketamine (90 mg/kg) and xylazine: (10 mg/kg). An incision was made on the lateral surface of the thigh to expose the three branches of the left sciatic nerve: the sural, common peroneal, and tibial nerves. Both tibial and common peroneal nerves were ligated with a 10.0 silk suture and transected approximately at 2 mm distal to the ligation. The sural nerve was left intact. Then, the muscles and skin were sutured and the wound disinfected. Mice were left to recover on a warm pad and returned to their home cages.
Mice were randomly assigned to each treatment group. (S)-17 was diluted in 0.5% DMSO and administered daily at the dose of 10 mg/kg, starting 1 hour after the injury. Control mice received equivalent volume of the vehicle.
Mechanical nociceptive threshold
Sensibility to mechanical stimuli was measured with an electronic Von Frey algesimeter (Bioseb) before surgery and at different times after injury. Mice were placed on a wire net platform in Plexiglas chambers for approximately 15 min before the experiment for habituation. The Von Frey probe was pressed against the mid-plantar surface of the hind paw. Nociceptive response for mechanical sensitivity was expressed as the force (in grams) at which mice withdrew the hind paw in response to the mechanical stimulus. The mechanical nociceptive threshold was taken as the mean of three measurements per test with a 5 minutes interval between each measurement.
Supplementary Material
Acknowledgment
This work has been awarded with the Almirall Prize for Young Researchers from the Sociedad Española de Química Terapéutica (SEQT) to I.G.-G. This work has been supported by the Spanish Ministerio de Economía y Competitividad (MINECO, Grants SAF2016-78792-R to M.L.L.-R. and S.O.-G., SAF2015-66275-C2-1 and RTI2018-097189-B-C21 to A.V.F.-M., and SAF2016-79774-R to R.L.-V.), Wings for Life International Foundation, and Red de Terapia Celular (TERCEL) to R.L.-V, Instituto de Salud Carlos III MINECO and Regional Development Funds-European Union (ERdf-EU) (Grant PI16/01698 to F.R.F.) and National Institutes of Health (NS084398 to J.C.). The authors thank MINECO for predoctoral F.P.U. grants to I.G.-G and D.Z. RH7777 hepatoma cells stably expressing the LPA1 receptor and their corresponding non-transfected controls were kindly provided by Prof. Gabor Tigyi (University of Tennessee Health Science Center, Memphis, Tennessee). Retrovirus expression vector (LZRS-EGFP) and Phoenix retrovirus producer cell lines were provided by Prof. Garry P. Nolan (Stanford University, Stanford, California).
Abbreviations Used
- BSI
back-scattering interferometry
- DAPI
4’,6’-diamidino-2-phenylindole
- DRG
dorsal root ganglia
- EDG
endothelial differentiation gene
- EGFP
enhanced green fluorescent protein
- Emax
maximal receptor activation
- FACS
fluorescence-activated cell sorting
- FAF BSA
fatty acid free bovine serum albumin
- Fluo-4AM
fluo-4-acetoxymethyl ester
- LPA
lysophosphatidic acid
- MEA
multielectrode array
- N.E.
no effect
- NP
neuropathic pain
- OFT
open field test
- RI
refractive index
- s.e.m.
standard error of the mean
- SNI
spared nerve injury
- S1P
sphingosine 1-phosphate
- TRPV1
transient receptor potential vanilloid 1
Footnotes
Supporting Information: Representative plots of BSI signal vs ligand concentration for the determination of affinity constant (Supporting Figure S1), reversibility and stability studies (Supporting Figures S2 and S3), representative calcium microfluorography recordings on neonatal rat cultures (Supporting Figure S4), experimental protocol for microelectrode array (MEA) and representative MEA recordings (Supporting Figures S5-S7), effect of acute administration of (S)-17 in the acetic acid-induced writhing responses (Supporting Figure S8), and effect of (S)-17 in the exploratory or locomotor activity of mice (Supporting Figure S9), activity at LPA2 and LPA3 receptors for all compounds with agonist activity at the LPA1 receptor (Supporting Table S1), detailed synthetic procedures, and characterization data of all intermediates (pdf). Molecular formula strings (csv). File structure of the LPA1 receptor (pdb). This material is available free of charge via the Internet at http://pubs.acs.org.
Just Accepted
“Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
REFERENCES
- 1.Choi JW; Herr DR; Noguchi K; Yung YC; Lee CW; Mutoh T; Lin ME; Teo ST; Park KE; Mosley AN; Chun J LPA receptors: subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol 2010, 50, 157–186. [DOI] [PubMed] [Google Scholar]
- 2.Kihara Y; Maceyka M; Spiegel S; Chun J Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol 2014, 171, 3575–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaho VA; Hla T Regulation of mammalian physiology, development, and disease by the sphingosine 1-phosphate and lysophosphatidic acid receptors. Chem. Rev 2011, 111, 6299–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mutoh T; Rivera R; Chun J Insights into the pharmacological relevance of lysophospholipid receptors. Br. J. Pharmacol 2012, 165, 829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yung YC; Stoddard NC; Mirendil H; Chun J Lysophosphatidic acid signaling in the nervous system. Neuron 2015, 85, 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llona-Minguez S; Ghassemian A; Helleday T Lysophosphatidic acid receptor (LPAR) modulators: The current pharmacological toolbox. Prog. Lipid Res 2015, 58, 51–75. [DOI] [PubMed] [Google Scholar]
- 7.Yung YC; Stoddard NC; Chun J LPA receptor signaling: pharmacology, physiology, and pathophysiology. J. Lipid Res 2014, 55, 1192–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tigyi GJ; Yue J; Norman DD; Szabo E; Balogh A; Balazs L; Zhao G; Lee SC Regulation of tumor cell - microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Adv. Biol. Regul 2019, 71, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hisano Y; Hla T Bioactive lysolipids in cancer and angiogenesis. Pharmacol. Ther 2019, 193, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu YM; Nepali K; Liou JP Idiopathic pulmonary fibrosis: current status, recent progress, and emerging targets. J. Med. Chem 2017, 60, 527–553. [DOI] [PubMed] [Google Scholar]
- 11.Tager AM; LaCamera P; Shea BS; Campanella GS; Selman M; Zhao Z; Polosukhin V; Wain J; Karimi-Shah BA; Kim ND; Hart WK; Pardo A; Blackwell TS; Xu Y; Chun J; Luster AD The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med 2008, 14, 45–54. [DOI] [PubMed] [Google Scholar]
- 12.Juarez-Contreras R; Rosenbaum T; Morales-Lazaro SL Lysophosphatidic acid and ion channels as molecular mediators of pain. Front. Mol. Neurosci 2018, 11, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoue M; Rashid MH; Fujita R; Contos JJ; Chun J; Ueda H Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat. Med 2004, 10, 712–718. [DOI] [PubMed] [Google Scholar]
- 14.Lin ME; Rivera RR; Chun J Targeted deletion of LPA5 identifies novel roles for lysophosphatidic acid signaling in development of neuropathic pain. J. Biol. Chem 2012, 287, 17608–17617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoddard NC; Chun J Promising pharmacological directions in the world of lysophosphatidic acid signaling. Biomol. Ther 2015, 23, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kihara Y; Mizuno H; Chun J Lysophospholipid receptors in drug discovery. Exp. Cell Res 2015, 333, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velasco M; O’Sullivan C; Sheridan GK Lysophosphatidic acid receptors (LPARs): Potential targets for the treatment of neuropathic pain. Neuropharmacology 2017, 113, 608–617. [DOI] [PubMed] [Google Scholar]
- 18.Ueda H Lysophosphatidic acid signaling is the definitive mechanism underlying neuropathic pain. Pain 2017, 158 Suppl 1, S55–S65. [DOI] [PubMed] [Google Scholar]
- 19.Colloca L; Ludman T; Bouhassira D; Baron R; Dickenson AH; Yarnitsky D; Freeman R; Truini A; Attal N; Finnerup NB; Eccleston C; Kalso E; Bennett DL; Dworkin RH; Raja SN Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma L; Matsumoto M; Xie W; Inoue M; Ueda H Evidence for lysophosphatidic acid 1 receptor signaling in the early phase of neuropathic pain mechanisms in experiments using Ki-16425, a lysophosphatidic acid 1 receptor antagonist. J. Neurochem 2009, 109, 603–610. [DOI] [PubMed] [Google Scholar]
- 21.Ma L; Uchida H; Nagai J; Inoue M; Chun J; Aoki J; Ueda H Lysophosphatidic acid-3 receptor-mediated feed-forward production of lysophosphatidic acid: an initiator of nerve injury-induced neuropathic pain. Mol. Pain 2009, 5, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marrone MC; Morabito A; Giustizieri M; Chiurchiu V; Leuti A; Mattioli M; Marinelli S; Riganti L; Lombardi M; Murana E; Totaro A; Piomelli D; Ragozzino D; Oddi S; Maccarrone M; Verderio C; Marinelli S TRPV1 channels are critical brain inflammation detectors and neuropathic pain biomarkers in mice. Nat. Commun 2017, 8, 15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morales-Lazaro SL; Llorente I; Sierra-Ramirez F; Lopez-Romero AE; Ortiz-Renteria M; Serrano-Flores B; Simon SA; Islas LD; Rosenbaum T Inhibition of TRPV1 channels by a naturally occurring omega-9 fatty acid reduces pain and itch. Nat. Commun 2016, 7, 13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nieto-Posadas A; Picazo-Juarez G; Llorente I; Jara-Oseguera A; Morales-Lazaro S; Escalante-Alcalde D; Islas LD; Rosenbaum T Lysophosphatidic acid directly activates TRPV1 through a C-terminal binding site. Nat. Chem. Biol 2011, 8, 78–85. [DOI] [PubMed] [Google Scholar]
- 25.Brinkmann V; Billich A; Baumruker T; Heining P; Schmouder R; Francis G; Aradhye S; Burtin P Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov 2010, 9, 883–897. [DOI] [PubMed] [Google Scholar]
- 26.Huwiler A; Zangemeister-Wittke U The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharmacol. Ther 2018, 185, 34–49. [DOI] [PubMed] [Google Scholar]
- 27.Chun J; Kihara Y; Jonnalagadda D; Blaho VA Fingolimod: Lessons learned and new opportunities for treating multiple sclerosis and other disorders. Annu. Rev. Pharmacol. Toxicol 2019, 59, 149–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez-Gil IZ, D.; Vázquez-Villa H; Ortega-Gutiérrez S; López-Rodríguez ML The status of the lysophosphatidic acid receptor type 1 (LPA1R). Med. Chem. Commun 2015, 6, 13–23. [Google Scholar]
- 29.Prestwich GD; Gajewiak J; Zhang H; Xu X; Yang G; Serban M Phosphatase-resistant analogues of lysophosphatidic acid: agonists promote healing, antagonists and autotaxin inhibitors treat cancer. Biochim. Biophys. Acta 2008, 1781, 588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heise CE; Santos WL; Schreihofer AM; Heasley BH; Mukhin YV; Macdonald TL; Lynch KR Activity of 2-substituted lysophosphatidic acid (LPA) analogs at LPA receptors: discovery of a LPA1/LPA3 receptor antagonist. Mol. Pharmacol 2001, 60, 1173–1180. [DOI] [PubMed] [Google Scholar]
- 31.Santos WL; Heasley BH; Jarosz R; Carter KM; Lynch KR; Macdonald TL Synthesis and biological evaluation of phosphonic and thiophosphoric acid derivatives of lysophosphatidic acid. Bioorg. Med. Chem. Lett 2004, 14, 3473–3476. [DOI] [PubMed] [Google Scholar]
- 32.Kawanami E; Kondo Y; Shishikura J; Takahashi M; Sakamoto K; Soga T; Orita M Benzyl-12b-methyl-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizine derivatives as LPA receptor agonists. JP2008297278A 2008. [Google Scholar]
- 33.Chrencik JE; Roth CB; Terakado M; Kurata H; Omi R; Kihara Y; Warshaviak D; Nakade S; Asmar-Rovira G; Mileni M; Mizuno H; Griffith MT; Rodgers C; Han GW; Velasquez J; Chun J; Stevens RC; Hanson MA Crystal structure of antagonist bound human lysophosphatidic acid receptor 1. Cell 2015, 161, 1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parrill AL; Tigyi G Integrating the puzzle pieces: the current atomistic picture of phospholipid-G protein coupled receptor interactions. Biochim. Biophys. Acta 2013, 1831, 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ballatore C; Huryn DM; Smith AB 3rd. Carboxylic acid (bio)isosteres in drug design. ChemMedChem 2013, 8, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lassalas P; Gay B; Lasfargeas C; James MJ; Tran V; Vijayendran KG; Brunden KR; Kozlowski MC; Thomas CJ; Smith AB 3rd; Huryn DM; Ballatore C Structure property relationships of carboxylic acid isosteres. J. Med. Chem 2016, 59, 3183–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meanwell NA Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- 38.Cisneros JA; Bjorklund E; Gonzalez-Gil I; Hu Y; Canales A; Medrano FJ; Romero A; Ortega-Gutierrez S; Fowler CJ; Lopez-Rodriguez ML Structure-activity relationship of a new series of reversible dual monoacylglycerol lipase/fatty acid amide hydrolase inhibitors. J. Med. Chem 2012, 55, 824–836. [DOI] [PubMed] [Google Scholar]
- 39.Hernandez-Torres G; Cipriano M; Heden E; Bjorklund E; Canales A; Zian D; Feliu A; Mecha M; Guaza C; Fowler CJ; Ortega-Gutierrez S; Lopez-Rodriguez ML A reversible and selective inhibitor of monoacylglycerol lipase ameliorates multiple sclerosis. Angew. Chem. Int. Ed. Engl 2014, 53, 13765–13770. [DOI] [PubMed] [Google Scholar]
- 40.Orcajo-Rincón AL; Ortega-Gutiérrez S; Serrano P; Torrecillas IR; Wuthrich K; Campillo M; Pardo L; Viso A; Benhamú B; López-Rodríguez ML Development of non-peptide ligands of growth factor receptor-bound protein 2-Src homology 2 domain using molecular modeling and NMR spectroscopy. J. Med. Chem 2011, 54, 1096–1100. [DOI] [PubMed] [Google Scholar]
- 41.Troupiotis-Tsailaki A; Zachmann J; Gonzalez-Gil I; Gonzalez A; Ortega-Gutierrez S; Lopez-Rodriguez ML; Pardo L; Govaerts C Ligand chain length drives activation of lipid G protein-coupled receptors. Sci. Rep 2017, 7, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baksh MM; Kussrow AK; Mileni M; Finn MG; Bornhop DJ Label-free quantification of membrane-ligand interactions using backscattering interferometry. Nat. Biotechnol 2011, 29, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bornhop DJ; Latham JC; Kussrow A; Markov DA; Jones RD; Sorensen HS Free-solution, label-free molecular interactions studied by back-scattering interferometry. Science 2007, 317, 1732–1736. [DOI] [PubMed] [Google Scholar]
- 44.Mizuno H; Kihara Y; Kussrow A; Chen A; Ray M; Rivera R; Bornhop DJ; Chun J Lysophospholipid G protein-coupled receptor binding parameters as determined by backscattering interferometry. J. Lipid Res 2019, 60, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Omotuyi OI; Nagai J; Ueda H Lys39-Lysophosphatidate carbonyl oxygen interaction locks LPA1 N-terminal cap to the orthosteric site and partners Arg124 during receptor activation. Sci. Rep 2015, 5, 13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pandy-Szekeres G; Munk C; Tsonkov TM; Mordalski S; Harpsoe K; Hauser AS; Bojarski AJ; Gloriam DE GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–d446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. https://gpcrdb.org/structure/homology_models/lpar1_human_active.
- 48.Devesa I; Ferrandiz-Huertas C; Mathivanan S; Wolf C; Lujan R; Changeux JP; Ferrer-Montiel A AlphaCGRP is essential for algesic exocytotic mobilization of TRPV1 channels in peptidergic nociceptors. Proc. Natl. Acad. Sci. USA 2014, 111, 18345–18350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathivanan S; Devesa I; Changeux JP; Ferrer-Montiel A Bradykinin induces TRPV1 exocytotic recruitment in peptidergic nociceptors. Front. Pharmacol 2016, 7, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaggi AS; Jain V; Singh N Animal models of neuropathic pain. Fundam. Clin. Pharmacol 2011, 25, 1–28. [DOI] [PubMed] [Google Scholar]
- 51.Seltzer Z; Dubner R; Shir Y A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 1990, 43, 205–218. [DOI] [PubMed] [Google Scholar]
- 52.Hopper DW; Ragan SP; Hooks SB; Lynch KR; Macdonald TL Structure-activity relationships of lysophosphatidic acid: conformationally restricted backbone mimetics. J. Med. Chem 1999, 42, 963–970. [DOI] [PubMed] [Google Scholar]
- 53.Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dolinsky TJ; Nielsen JE; McCammon JA; Baker NA PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dolinsky TJ; Czodrowski P; Li H; Nielsen JE; Jensen JH; Klebe G; Baker NA PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sondergaard CR; Olsson MH; Rostkowski M; Jensen JH Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- 57.Li H; Robertson AD; Jensen JH Very fast empirical prediction and rationalization of protein pKa values. Proteins 2005, 61, 704–721. [DOI] [PubMed] [Google Scholar]
- 58.Czodrowski P; Dramburg I; Sotriffer CA; Klebe G Development, validation, and application of adapted PEOE charges to estimate pKa values of functional groups in protein-ligand complexes. Proteins 2006, 65, 424–437. [DOI] [PubMed] [Google Scholar]
- 59.Salentin S; Schreiber S; Haupt VJ; Adasme MF; Schroeder M PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marin-Ramos NI; Balabasquer M; Ortega-Nogales FJ; Torrecillas IR; Gil-Ordonez A; Marcos-Ramiro B; Aguilar-Garrido P; Cushman I; Romero A; Medrano FJ; Gajate C; Mollinedo F; Philips MR; Campillo M; Gallardo M; Martin-Fontecha M; Lopez-Rodriguez ML; Ortega-Gutierrez S A potent isoprenylcysteine carboxylmethyltransferase (ICMT) inhibitor improves survival in Ras-driven acute myeloid leukemia. J. Med. Chem 2019, 62, 6035–6046. [DOI] [PubMed] [Google Scholar]
- 61.Martin-Couce L; Martin-Fontecha M; Palomares O; Mestre L; Cordomi A; Hernangomez M; Palma S; Pardo L; Guaza C; Lopez-Rodriguez ML; Ortega-Gutierrez S Chemical probes for the recognition of cannabinoid receptors in native systems. Angew. Chem. Int. Ed. Engl 2012, 51, 6896–6899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.