

SUMMARY
Expanded CAG repeats form stem-loop secondary structures that lead to fork stalling and collapse. Previous work has shown that these collapsed forks relocalize to nuclear pore complexes (NPCs) in late S phase in a manner dependent on replication, the nucleoporin Nup84, and the Slx5 protein, which prevents repeat fragility and instability. Here, we show that binding of the Smc5/6 complex to the collapsed fork triggers Mms21-dependent sumoylation of fork-associated DNA repair proteins, and that RPA, Rad52, and Rad59 are the key sumoylation targets that mediate relocation. The SUMO interacting motifs of Slx5 target collapsed forks to the NPC. Notably, Rad51 foci only co-localize with the repeat after it is anchored to the nuclear periphery and Rad51 exclusion from the early collapsed fork is dependent on RPA sumoylation. This pathway may provide a mechanism to constrain recombination at stalled or collapsed forks until it is required for fork restart.
Graphical Abstract
In Brief
Whalen et al. establish the mechanism by which collapsed forks caused by expanded CAG repeats relocate to the nuclear pore complex to maintain genome stability. This mechanism relies on sumoylation of repair proteins that bind the collapsed fork to facilitate relocation and constrain recombination until at the nuclear pore complex.
INTRODUCTION
The nucleus has well-defined regions; however, the various functions of these distinct areas are still not completely understood. In recent years, the role that nuclear domains play in DNA repair has been investigated. Knowing where in the nucleus the DNA is being repaired could help elucidate the mechanisms behind the repair event itself. The nuclear periphery has emerged as an important site of repair for several types of DNA damage. For example, induced double-strand breaks (DSBs) in Saccharomyces cerevisiae that lack a nearby homologous repair template (Nagai et al., 2008; Kalocsay et al., 2009; Oza et al., 2009; Horigome et al., 2014) and breaks induced in heterochromatin in Drosophila (Ryu et al., 2015; Amaral et al., 2017) both relocate to the nuclear periphery. Eroded or broken telomeres relocate from the inner nuclear membrane to the nuclear pore complex (NPC) (Khadaroo et al., 2009; Chung et al., 2015; Churikov et al., 2016). Breaks induced in the rDNA move out of the nucleolus for repair and interact with the NPC (Hauer and Gasser, 2017; Horigome et al., 2019). Less is known about the influence of the nuclear periphery in the repair of replication-associated breaks.
DNA sequences that form alternative DNA structures such as hairpins, cruciforms, triplex DNA, or G-quadruplexes are found frequently in genomes at every 10–50 kb, depending on the type (Zhao et al., 2010). CAG/CTG repeats >35 repeat units are unstable and can form hairpin structures that interfere with replication and repair, leading to repeat expansions or contractions (reviewed in Usdin et al., 2015; Polleys et al., 2017). Long tracts of CAG repeats stall replication and are prone to fork reversal (Fouché et al., 2006; Kerrest et al., 2009; Nguyen et al., 2017) and breakage (Freudenreich et al., 1998; Callahan et al., 2003). S phase delays caused by expanded CAG tracts inserted into the yeast genome require Rad52-dependent repair for recovery, suggesting that fork collapse events are occurring (Sundararajan and Freudenreich, 2011). Therefore, long CAG repeats are a useful model to study the effects of fork stalling and collapse at DNA structure-mediated barriers and the mechanisms of fork restart or repair.
Along with the physiological relevance, there are distinct differences in the nuclear localization pattern for expanded CAG repeats as compared to other types of DNA damage. In S. cerevisiae, a persistent DSB can relocate to both the NPC and the nuclear envelope protein Mps3 in both G1 and S phase, whereas eroded telomeres move from Mps3 to the NPC (Khadaroo et al., 2009; Horigome et al., 2016; Chung et al., 2015). In contrast, when a cell is not actively replicating its DNA, expanded CAG repeats can be found in the middle of the nucleus. However, during S phase, CAG-70 and CAG-130 repeats that interfere with replication relocalize to the NPC, but not the nuclear envelope. This relocation has been shown to be dependent on replication and repeat length (Su et al., 2015). These differences suggest potentially distinct pathways to relocate collapsed forks and target them to the NPC as compared to DSBs or eroded telomeres. Other conditions that create collapsed forks such as hydroxyurea+methyl methanesulfonate (HU+MMS) or longer HU treatment also cause movement to the nuclear periphery, although treatment with MMS alone or HU alone for shorter times does not lead to relocation (Nagai et al., 2008; Su et al., 2015). This suggests that fork collapse, not fork stalling due to nucleotide depletion, or post-replication gaps, is necessary for relocation. Although the exact structure provoking the relocation is not known, it could include broken forks or unbroken but altered forks, (e.g., stalled at a DNA structure, containing gaps or single-stranded DNA [ssDNA], reversed) and either an intact or uncoupled/dissociated replisome. For simplicity, we refer to both CAG repeats and collapsed forks when describing what is relocalizing to the NPC.
The pathway(s) that enable collapsed forks caused by expanded CAG repeats to migrate to the NPC remain to be understood. Su et al. (2015) showed that the SUMO-targeted ubiquitin ligase (STUbL) complex Slx5/8, which interacts with the nucleoporin Nup84 (Nagai et al., 2008), plays a key role in the relocation process. The deletion of Nup84 has been shown to cause a delay in replication fork progression (Gaillard et al., 2019). Slx5 has SUMO-interacting motifs (SIMs) that allow the complex to interact with sumoylated proteins (Xie et al., 2007, 2010), suggesting a potential mechanism for relocation. A role for the Slx5/8 STUbL complex (or its homologs) and SUMO have also been implicated in the relocation of persistent or heterochromatic DSBs and telomeres (Nagai et al., 2008; Horigome et al., 2016; Ryu et al., 2015, 2016; Churikov et al., 2016). The ubiquitin ligase Slx8 has been suggested to modify sumoylated proteins, targeting them for removal from chromatin and/or degradation, which could help facilitate fork restart at the NPC (Freudenreich and Su, 2016). The sumoylated proteins involved in the relocation of collapsed forks are not fully understood, but the recombination factor Rad52 was implicated because Rad52 associated with the CAG repeat before relocation but was no longer associated once the CAG tract was at the nuclear periphery (Su et al., 2015). The association of Rad52 with the CAG repeat at the nuclear periphery was significantly increased in slx5Δ cells, and sumoylated forms of Rad52 persisted in slx8Δ cells after fork collapse upon HU+MMS treatment (Su et al., 2015). Thus, it was hypothesized that SUMO removal could facilitate fork restart at the NPC. Preventing Rad52 sumoylation leads to increased repeat fragility and instability to the same level as the lack of Slx8. These data suggested that sumoylated Rad52 contributes to the relocation of CAG repeats and is removed through STUbLs at the NPC, allowing for fork restart or repair and therefore reduced repeat instability and fragility.
The goal of this study was to determine how collapsed forks caused by expanded CAG repeats relocalize to the NPC. Our aim was to determine whether the collapsed forks relocalize to the NPC via a SUMO-dependent mechanism, and if so, what specific sumoylated proteins facilitated the relocation. We show that mono-sumoylation of proteins by the SUMO E3 ligase Mms21 is required for collapsed fork relocation. In addition, the interaction with the SIM domains of Slx5 are required. We identified specific sumoylated repair proteins that facilitate the relocation (Rad52, Rad59, RPA, and Smc5) and found that resection by Mre11 and either the Exo1 or the Sgs1-Dna2 pathway is required to generate sufficient ssDNA for these proteins to bind. Notably, Rad51 only associates with the CAG tract after its association with the NPC and is excluded from CAG tracts in the interior of the nucleus, suggesting a regulatory mechanism for homologous recombination (HR) at collapsed forks. Rad51 exclusion was lost in a replication protein A (RPA) mutant unable to be modified by SUMO, suggesting that RPA sumoylation regulates Rad51 binding to collapsed forks. Our data support an important role for the Smc5/6 complex and its SUMO ligase subunit Mms21 (Nse2) in the sumoylation of target proteins at stalled forks to control Rad51 association and mediate relocation to the NPC. A deficiency in this process results in increased chromosome breakage, showing that proper relocation of the collapsed fork caused by CAG repeats to the NPC has a genome-protective effect.
RESULTS
Relocation of Collapsed Forks to the NPC Depends on Mono-sumoylation and Slx5-SUMO Interaction
We used the zoning assay system from Su et al. (2015) to visualize the CAG repeat in relation to the nuclear periphery. The system contains 130 CAG repeats on chromosome VI 6.4 kb away from a LacO/GFP-LacI array and Nup49-GFP to mark NPCs, allowing for the visualization of both the nuclear periphery and the CAG repeat locus in the same nucleus. A mathematical program was used to separate images of each nucleus analyzed into three equal areas to determine the frequency of CAG repeat localization in each zone. As shown in Figure 1A, zone 1 represents the nuclear periphery (where the NPC is located), zone 3 is the middle of the nucleus, and zone 2 is the area in between.
Figure 1. Relocation of Collapsed Forks to the NPC Depends on Slx5 SUMO-Interacting Motif (SIM) Domains and Mms21-Mediated Sumoylation.
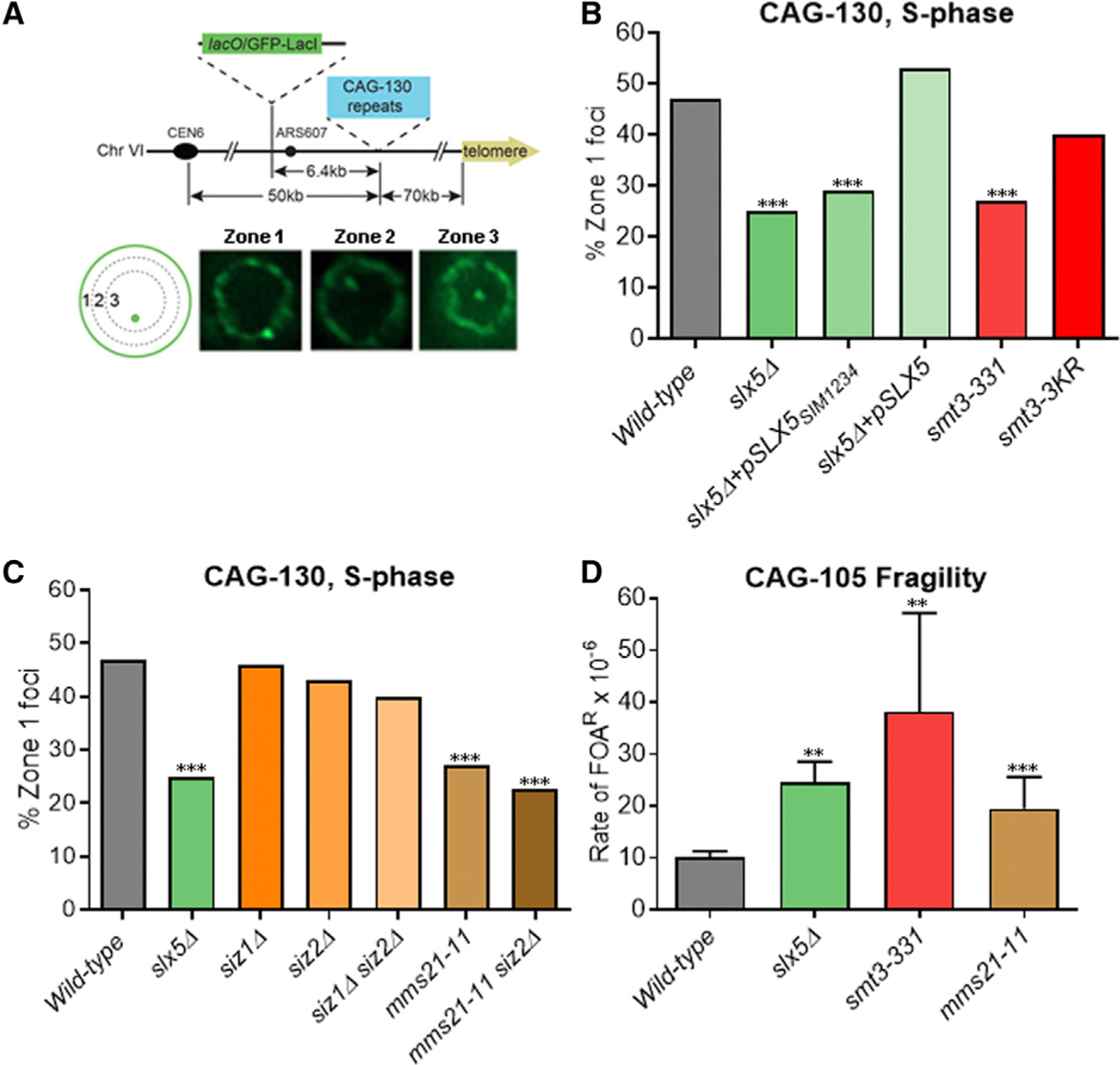
(A) Yeast chromosome VI containing integrated 130 CAG repeats (CAG-130) and a lacO array that binds the LacI-GFP protein. S phase cells were imaged, and the locations of the foci were scored into 1 of 3 zones of equal area, using the GFP-Nup49 signal to mark the nuclear periphery (NP). Representative images are shown.
(B) Percentage of zone 1 foci for CAG-130 S phase cells. Mutations in the Slx5 SIM were introduced into slx5Δ cells on a yeast centromere/autonomously replicating sequence (CEN/ARS) plasmid; mutations in the SMT3 gene coding for SUMO were integrated into the genome, replacing the WT SMT3 gene
(C) Percentage of zone 1 foci for CAG-130 S phase cells with mutation of the indicated SUMO ligases. ***p < 0.001 compared with WT by Fisher’s exact test. The number of cells analyzed per strain ranges from 150 to 403. See Table S1 for the exact number of cells analyzed, percentages, and p values. See Table S1 and Figure S6A for zoning data for individual strains.
(D) Rate of 5-fluoroorotic acid resistance (FOA)R × 10−6 calculated using the method of maximum likelihood for WT and indicated mutants in strains with YACs containing CAG-105 repeats. Percentage of end loss in FOAR colonies was confirmed by His+/−status and ranged from 92% to 98%. **p < 0.01 and ***p < 0.001 compared with WT by Student’s t test. The average of 4–6 experiments is shown. Error bars represent standard error of the mean (SEM)
See also Table S2.
Previous findings of the involvement of Slx5 in collapsed fork relocation to the NPC pointed to a SUMO-dependent mechanism (Su et al., 2015). We tested this model by examining mutations of sumoylation pathway proteins. Plasmids containing either the unaltered SLX5 gene or one in which the four SIM domains were mutated (Xie et al., 2007) were used to complement the slx5Δ strain to test whether the defect in CAG repeat relocation in slx5Δ was specifically due to its role in interacting with sumoylated proteins. The wild-type (WT) SLX5 gene complemented the relocation defect, but the SIM domain mutant did not, indicating that one or more SIM domains are required for mediating movement to the NPC (Figure 1B; Table S1). In addition, a smt3–331 mutant that lowers overall sumoylation levels (Biggins et al., 2001; Bustard et al., 2016) was found to be as defective as slx5Δ cells (Figure 1B).
Proteins can be mono- or poly-sumoylated, so we tested which was required for CAG tract relocation. The mutant smt3–3KR, which largely eliminates poly-sumoylation (Tatham et al., 2001; Bylebyl et al., 2003), showed no significant decrease in relocation (Figure 1B). Although we cannot eliminate the possibility that a low amount of SUMO modification of other Smt3 lysines occurs, the smt3–3KR results indicate that mono-sumoylation is most likely sufficient. We conclude that mono-sumoylation of a protein or proteins and the subsequent SUMO-mediated interaction with Slx5 is required for the relocation of collapsed forks.
Relocation of Collapsed Forks to the NPC Depends on Mms21-Mediated Sumoylation and Protects against Chromosome Breakage
We next tested the effects of each of the three mitotic SUMO E3 ligases (Siz1, Siz2, and Mms21 [Nse2]) to determine which was required for CAG tract relocation. Different from what was found for persistent DSBs (Horigome et al., 2016), neither Siz1 nor Siz2 deletion had a significant effect in our assay (Figure 1C). In contrast, a mutation of Mms21 that removes the C-terminal region and abolishes its SUMO ligase activity (Zhao and Blobel, 2005) had a significant defect on relocation to zone 1, similar to the Slx5 mutant (Figure 1C). We conclude that Mms21 is the primary SUMO ligase required for the relocation of the CAG repeat. It is possible that Siz2 plays a minor role due to a larger decrease in relocation in the mms21–11 siz2Δ double mutant, although this is not significant (Figure 1C). As Mms21 is mainly responsible for mono-sumoylation in both G1 and S phase, while Siz1/2 activity leads to poly-sumoylation (Horigome et al., 2016), the requirement for Mms21 and not Siz1/2 is consistent with our result that mono-sumoylation is sufficient for CAG repeat relocation. We conclude that Mms21 is responsible for sumoylation of proteins that facilitate the relocation of the collapsed fork to the NPC.
Su et al. (2015) previously showed that the deletion of Slx5 led to an increase in chromosome fragility using a yeast artificial chromosome (YAC) that contained 70 CAG repeats. We re-tested this result using a YAC that contains a second marker distal to the repeat to more accurately measure end loss rates and 105 CAG repeats to better correlate with the CAG-130 repeat in the zoning assay system (Figure S1C). Consistent with our previous finding, we observed a 2-fold increase in chromosome breakage in the slx5Δ mutant (Figure 1D). We tested smt3–331 and mms21–11 strains and both mutants also showed a significant increase in chromosome end loss (Figure 1D). Therefore, we conclude that members of the sumoylation pathway that are required for relocation also protect against chromosome breakage.
Loss of Rad52, Rad59, and RPA Sumoylation Impairs the Relocation of Expanded CAG Repeats to NPC
Su et al. (2015) showed that Rad52 was bound to the CAG repeat before relocation and removed afterward in a manner dependent on the Slx5/8 complex. Preventing sumoylation of Rad52 led to increased repeat fragility and instability to the same level as in slx8Δ cells. These data suggest that sumoylated Rad52 could be involved in the relocation of the collapsed fork and is removed through a STUbL-mediated mechanism. However, no decrease in CAG tract relocation in rad52Δ cells was observed. One possible explanation for this is that the Rad52 paralog Rad59 compensates for Rad52 functions. The deletion of both Rad52 and Rad59 resulted in a significant decrease in relocation to zone 1, although the single rad59Δ had no effect (Figure 2A). To determine whether sumoylated Rad52 and Rad59 are required for relocation, a rad52–3KR rad59–2KR double mutant was tested. This mutant prevents sumoylation of Rad52 and Rad59, but it is still expressed and functional (Silva et al., 2016). A significant decrease in relocation comparable to the full deletions was observed (Figure 2A). We conclude that sumoylated Rad52 and Rad59 facilitate the relocation of CAG repeats to the NPC.
Figure 2. Sumoylated Repair Proteins Rad52, Rad59, and RPA Facilitate the Relocation of Expanded CAG Repeats to the NPC.
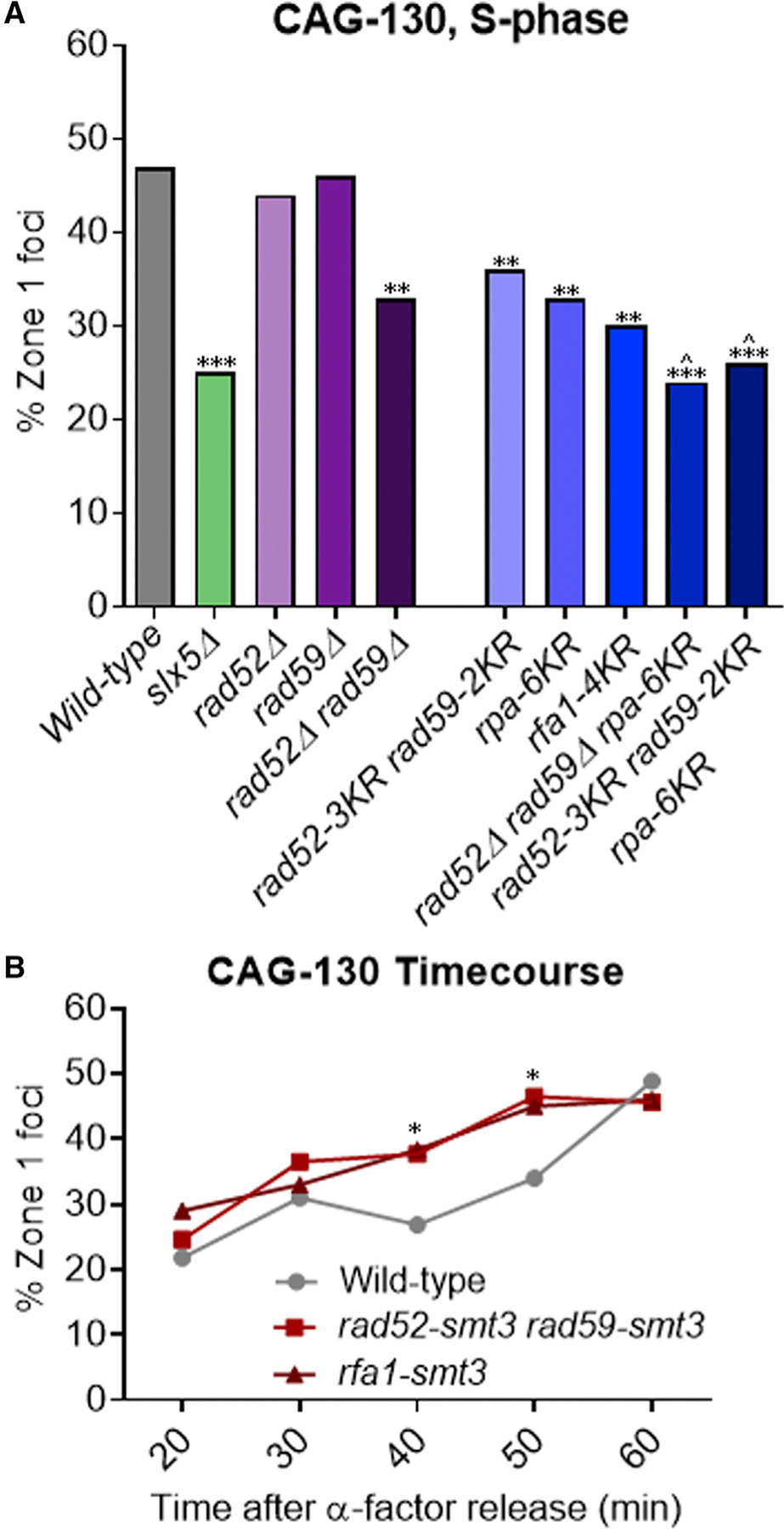
(A) Percentage of zone 1 foci for CAG-130 S phase cells in repair protein mutant strains.
(B) Percentage of zone 1 foci for CAG-130 cells at indicated time points after release from alpha factor arrest in WT and strains with rad52-smt3ΔGG rad59-smt3ΔGG (smt3ΔGG, missing the last 2 glycines of Smt3 to prevent conjugation to other proteins (Silva et al., 2016; Johnson and Blobel, 1997) and rfa1-smt3 fusion proteins (Smt3 at the C terminus of each). *p < 0.05, **p < 0.01, and ***p < 0.001 compared with WT by Fisher’s exact test. ^p < 0.05 compared to rad52–3KR rad59–2KR by Fisher’s exact test. The number of cells analyzed per strain ranges from 150 to 270.
See Tables S1 and S4 for the exact number of cells analyzed, percentages, and p values.
See Table S1 and Figure S6B for zoning data for individual strains.
RPA, which is composed of Rfa1–3 subunits, was also of interest because it binds to ssDNA at the collapsed fork and has several mono-sumoylation sites. We recently defined four Rfa1 sumoylation sites and showed that mutating these four lysines (rfa1–K170, −180, −411, and −427R or rfa1–4KR) largely eliminated Rfa1 sumoylation (Dhingra et al., 2019). We also confirmed that mutating the previously identified sumoylation sites on Rfa2 (rfa2- K199R) and Rfa3 (rfa3–K46R) (Psakhye and Jentsch, 2012) greatly reduced the sumoylation of these proteins. A strain that combined mutations of these sumoylation sites in all three subunits of RPA, rpa-6KR, showed a significant decrease in relocation comparable to rad52–3KR rad59–2KR (Figure 2A). A similar decrease was seen in the rfa1–4KR mutant (Figure 2A). It was also possible that rpa-6KR is defective in the recruitment of Rad52-Rad59, and the defect in relocation seen in rpa-6KR is an indirect effect. However, rpa-6KR did not affect Rad52-GFP foci levels in response to MMS treatment; therefore, the observed rpa-6KR defect is not due to the impairment of Rad52 recruitment to DNA lesions (Figure S1A; Table S3). We conclude that sumoylated RPA is involved in CAG repeat relocation to the NPC.
Neither of the rpa-KR mutants nor the rad52/59-KR mutant showed a reduction in relocation down to the slx5Δ level. One possibility is that sumoylation of all three repair proteins is required. The triple mutant in which the sumoylation of all three factors is eliminated showed a more severe decrease in CAG repeat relocation, similar to the defect seen in slx5Δ and significantly decreased from the rad52–3KR rad59–2KR double mutant (p = 0.03; Table S1), indicating that sumoylation of all three substrates plays a role in the relocation of collapsed forks to the NPC (Figure 2A). We conclude that sumoylated RPA along with Rad52 and Rad59 facilitates the movement of CAG repeats to the NPC.
SUMO Fusion of Rad52, Rad59, and Rfa1 Advances the Timing of CAG Repeat Relocation to NPC
To further test the roles of sumoylation of Rad52, Rad59, and Rfa1 in mediating relocation and to complement our sumoylation mutant analysis, we asked whether a gain-of-function allele in which SUMO is fused to Rad52 and Rad59 or to Rfa1 to mimic constitutive sumoylation would promote CAG repeat relocation. The rad52-smt3 rad59-smt3 strain was previously shown to have a mild sensitivity to MMS and zeocin and slightly increased protein expression levels (Silva et al., 2016). Neither the rad52-smt3 rad59-smt3 strain nor our rfa1-smt3 fusion strain, which contains a linker between Rfa1 and Smt3, exhibited sensitivity to 0.01% MMS (Figure S2). When tested in the zoning assay, we detected a non-significant increase in S phase cells in zone 1, and there was no difference from WT cells in G1 for either the rfa1-smt3 or the rad52-smt3 rad59-smt3 mutants (Figure S1B). We then assessed the timing of re-localization by examining synchronized S phase cells. Consistent with our previous Nup84 chromatin immunoprecipitation (ChIP) data (Su et al., 2015), in WT cells, the CAG-130 repeat was rarely found at the NPC in early S phase (20 min after release), but the percentage of cells in zone 1 started to increase slightly by 40 min and was maximal at 60 min (Figure 2B). In contrast, when Rad52 and Rad59 or Rfa1 are fused with SUMO, the CAG repeat relocalized to the NPC earlier than in WT cells, being significantly increased by the 40-min time point (Figure 2B; Table S4). This coincided with the time that Rad52 was detected at the CAG tract by ChIP (Su et al., 2015). Thus, the attachment of SUMO to Rad52 and Rad59 or to Rfa1 advances relocation timing, supporting our conclusion that sumoylated Rad52, Rad59, and Rfa1 contribute to the relocation of the collapsed fork to NPCs and that this is a direct effect.
Mms21 Contributes to the Sumoylation of RPA Subunits and Rad59
We next tested whether the Mms21 SUMO E3 ligase contributes to the sumoylation of Rad52, Rad59, and RPA. Biochemically measurable amounts of sumoylation of these proteins can be detected upon treatment by zeocin, which generates DNA breaks, including DSBs (Chung and Zhao, 2015). We found that zeocin, like HU+MMS, leads to an increase in zone 1 foci at ARS607 in S phase (Figure 3F). This suggests that zeocin, in addition to causing DSBs, also causes collapsed forks in S phase. This is different from MMS treatment, which does not cause relocation to the nuclear periphery. We thus examined how removing the Mms21 SUMO ligase domain (mms21–11) affects the sumoylation of RPA, Rad52, and Rad59 in zeocin-treated cells.
Figure 3. The Effects of the Mms21 SUMO E3 Mutant on RPA, Rad52, and Rad59 Sumoylation.
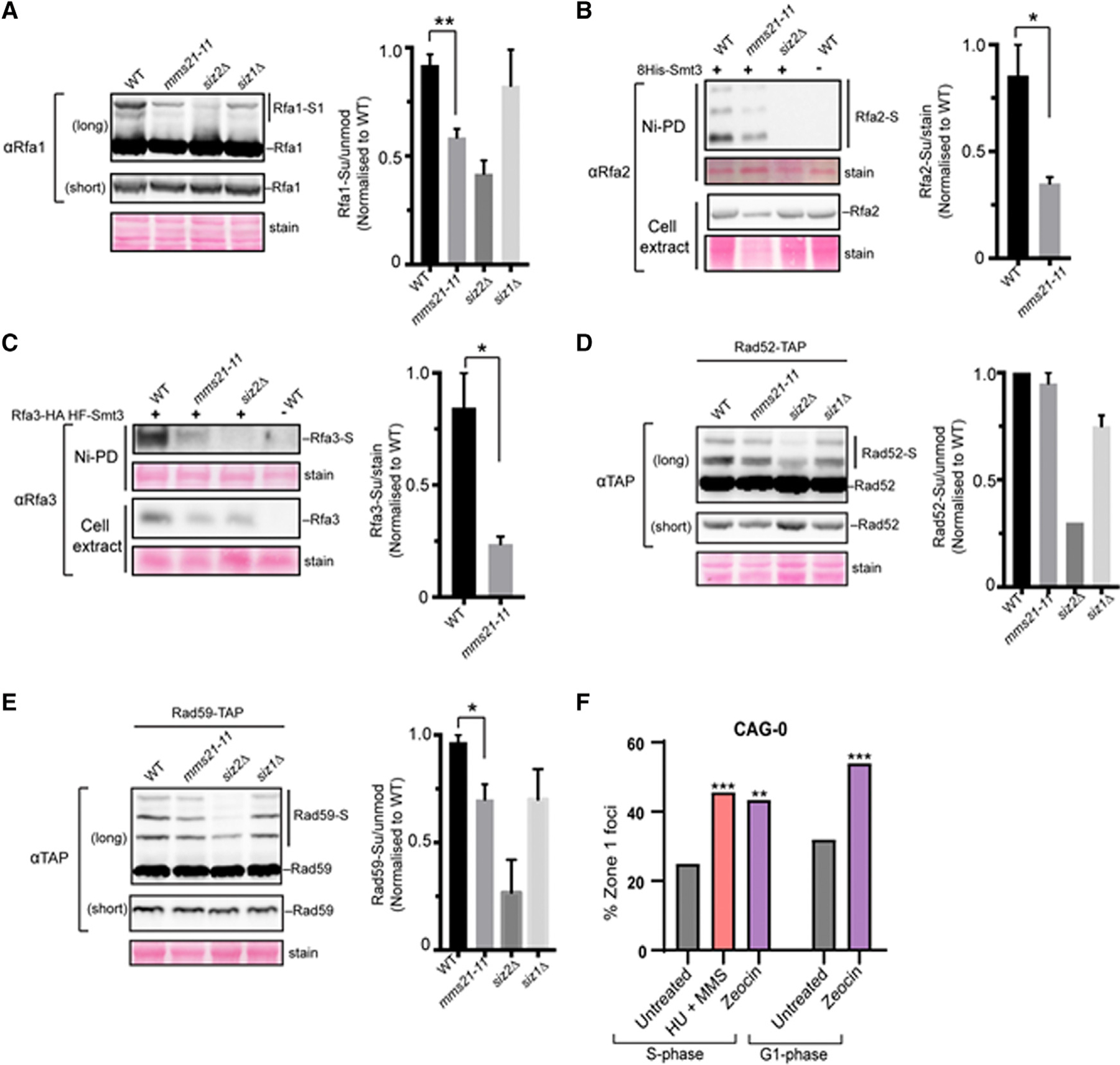
(A–E) Cell lysates of the indicated genotypes were examined by western blotting using antibodies against the tag or the protein as described in Method Details. Sumoylated forms are indicated by “S.” Note that due to the low level of sumoylation, some sumoylated proteins were not visible at the short exposure shown, but they were detected at long exposures. Plots show the relative ratios of the sumoylated forms to unmodified forms based on at least 2 independent experiments using different spore clones of the same genotype (see Method Details). Significant differences between mms2–11 and WT cells are indicated by *p < 0.05 or **p < 0.01 and based on Student’s t test. Error bars show the SEM.
(F) Percentage of zone 1 foci in S and G1 phase cells for a CAG-0 strain (containing the LacO-LacI-GFP array near ARS607, same as CAG-130 shown in Figure 1A) treated with 0.2 M HU+ 0.033% MMS or 0.3 mg/mL zeocin as compared to untreated. **p < 0.01 and ***p < 0.001 Fisher’s exact test compared to CAG-0 without drug treatment. The number of cells analyzed ranges from 92 to 159.
As shown in Figure 3, mms21–11 reduced the sumoylation of RPA subunits by up to 75% compared to WT levels and reproducibly reduced Rad59 sumoylation levels 25%, although no effect was detected for Rad52. This finding provides a link between the Mms21 SUMO E3 and substrates involved in CAG repeat relocation and suggests that RPA and Rad59 may be particularly important targets for Mms21-mediated effects. The lack of Mms21-dependent sumoylation of Rad52 could be due to a difference between zeocin-induced damage and a structure-induced replication barrier, or it could indicate a secondary role for Siz2, as was hinted at by our zoning data (Figure 1C). We hypothesize that the presence of the Smc5/6 complex and its associated Mms21 SUMO ligase at collapsed forks (Lindroos et al., 2006) provides the required proximity for sumoylation of RPA and Rad59 by Mms21, whereas Siz1 and Siz2 are recruited more readily to DSBs, a common byproduct of both zeocin and MMS-induced DNA damage. This would explain why the more relevant modification at collapsed forks is Mms21-dependent sumoylation, even though in other contexts these same repair proteins are sumoylated by Siz2 (Sacher et al., 2006; Psakhye and Jentsch, 2012). Our results support the conclusion that Mms21-dependent sumoylation of key repair proteins occurs at collapsed forks and indicate that RPA and Rad59 may be particularly important targets in this situation.
Resection Is Required for the Relocation of Collapsed Forks to the NPC
For sumoylated RPA, Rad52, and Rad59 to bind at the collapsed fork and facilitate relocation to the NPC, there needs to be processing at the fork. Therefore, we next asked whether fork resection is required for collapsed fork relocation to the NPC. We found that independently removing each of the two redundant enzymes required for longer-range resection, namely the Sgs1 helicase and the Exo1 nuclease, did not affect CAG-130 relocation to zone 1 (Figure 4A). However, the double mutant did have a significant decrease (Figure 4A). Since Exo1 and Sgs1/Dna2 are known to play redundant roles in 5′to 3′ resection (Mimitou and Symington, 2008), the requirement for removing both pathways supports a role for long-range resection. Although RPA is also involved in long-range resection, loss of Rfa1 sumoylation, which reduced CAG repeat localization at the NPC (Figure 2A), did not affect resection (Figure S3), excluding the possibility that the observed defects of Rfa1 sumoylation are due to impaired resection.
Figure 4. Long-Range Resection and the Smc5/6 Complex Are Required for the Relocation of Collapsed Forks to the NPCs.
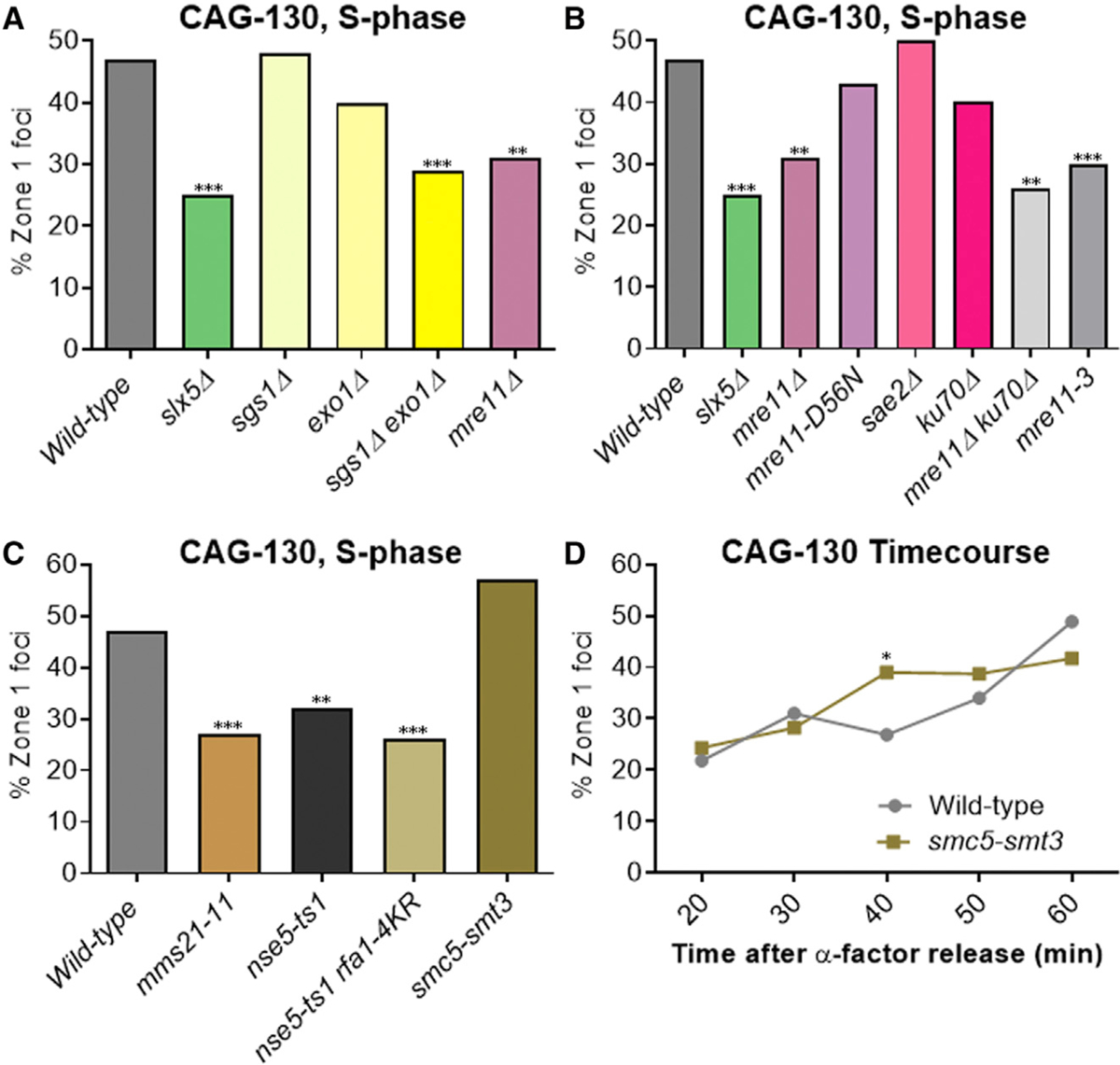
Percentage of zone 1 foci for CAG-130 S phase cells in (A) resection mutant strains, (B) mutants removing specific functions of Mre11, (C) Smc5/6 complex mutants, and (D) cells at indicated time points after release from alpha factor arrest in WT and the smc5-smt3 fusion strain (Smt3 at the C terminus). p = 0.025 for time point 40. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with WT by Fisher’s exact test. The number of cells analyzed per strain ranges from 150 to 288.
See Tables S1 and S4 for the exact number of cells analyzed, percentages, and p values.
See Table S1 and Figure S7A for zoning data for individual strains.
We further tested the Mre11 protein that acts upstream of Sgs1 and Exo1 to initiate resection. Our previous study found that cells lacking Mre11 showed a decrease in CAG-130 S phase relocation to the NPC, but the decrease did not reach significance. Here, with more cells analyzed, we now find that the decrease in mre11Δ cells is significant (Figure 4A, combined data). Mre11 has many functions and we tested several of these to determine which was required for the relocation of the collapsed fork. An endonuclease dead allele mre11-D56N (Moreau et al., 1999; Krogh et al., 2005) showed no effect, indicating that the endonuclease function of Mre11 is not required (Figure 4B). Consistent with this conclusion, deletion of Sae2, which stimulates the Mre11 endonuclease (Cannavo and Cejka, 2014), also had no effect (Figure 4B). However, the mre11–3 mutant, which impairs the exonuclease function of the protein and the resection at stalled replication forks (Bressan et al., 1998; Tittel-Elmer et al., 2009), was found to be defective in CAG repeat relocation to a comparable level as the mre11Δ (Figure 4B). This suggests that the exonuclease function of Mre11 is required for CAG repeat relocation. It has been shown that Mre11 plays a role in removing the heterodimer Ku from DNA ends to expose them to resection (Mimitou and Symington, 2010; Teixeira-Silva et al., 2017). This can also occur at reversed fork ends, which are prominent structures observed when forks encounter expanded CAG repeats (Nguyen et al., 2017) and are subjected to Exo1-mediated processing (Cotta-Ramusino et al., 2005; Lemaçon et al., 2017). However, deleting Ku70 did not rescue the mre11Δ defect in CAG repeat localization to the NPC (Figure 4B), indicating that the main role of Mre11 in this process is not to remove Ku70. We did notice a small decrease in the percentage of zone 1 foci in ku70Δ compared to WT cells, suggesting that Ku70 may play a minor or supporting role in relocation. This effect may be linked to a previous observation that Ku70 sumoylation partially requires Mms21 (Zhao and Blobel, 2005). Based on our findings, we conclude that processing the stalled or reversed fork by the Mre11 exonuclease and resection by Exo1 and Sgs1/Dna2 are required for the relocation of collapsed forks, likely by generating ssDNA for repair proteins to bind.
The Smc5/6 Complex Plays a Role in the Relocation of Expanded CAG Repeats to the NPC
We wanted to test the role of the Smc5/6 complex in the relocation of CAG repeats to the NPC for several reasons. First, the Smc5/6 complex is known to be important for rescuing collapsed forks and has been found to accumulate at replication pausing sites and collapsed forks (Ampatzidou et al., 2006; Lindroos et al., 2006; Menolfi et al., 2015). Second, the movement of CAG repeats to the NPC is dependent on sumoylation by Mms21, which is a subunit of Smc5/6. It has been proposed that the Smc5/6 complex could be a scaffold center for sumoylation in response to DNA damage (Branzei et al., 2006; Chavez et al., 2010; Bustard et al., 2016). Third, the Smc5/6 complex has been shown to be required for the relocation of DSBs to the nuclear periphery for both heterochromatic breaks in Drosophila and persistent DSBs in yeast (Ryu et al., 2015; Horigome et al., 2016). Smc5 is a SUMO target of Mms21 (Zhao and Blobel, 2005), making it an intriguing target to test for a role in relocation. The nse5-ts1 mutation exhibits reduced Smc5 sumoylation (Bustard et al., 2012); thus, it was tested here. The nse5-ts1 strain showed reduced collapsed fork relocation to the NPC, suggesting a role for Smc5 sumoylation (Figure 4C). In addition, a nse5-ts1 rfa-4KR strain was tested and showed a further decrease in zone 1 association to the level of mms21–11 (although not significant), supporting the idea that the sumoylation of Smc5 and repair proteins could both play a role in CAG tract relocation (Figure 4C). Since the nse5-ts1 mutation could affect other complex functions, including Mms21 activity, we also made a Smc5-SUMO fusion construct by fusing Smt3 onto the Smc5 C terminus. This construct did not affect growth or the MMS sensitivity of the cells (Figure S2), but it did result in a higher percentage of S phase zone 1 foci for the CAG-130 tract, although not a significant one (Figure 4C). To observe the timing of relocation, G1 cells with the Smc5-SUMO fusion construct were released into S phase and time points collected. Similar to the Rfa1- and Rad52-Rad59-SUMO fusions, the Smc5-SUMO fusion led to an earlier association of the CAG tract with the nuclear periphery (Figure 4D; Table S4). These data are consistent with the notion that the Smc5/6 complex is involved in CAG repeat relocation to the NPC in a manner that is mediated by its sumoylation.
Differential Timing of Mre11, Smc5, Rfa1, Slx5, and Rad51 Association with the CAG Repeats
To better understand the timing of repair protein association with the CAG tract, we investigated whether and when Mre11, Smc5, Rfa1, Slx5, and Rad51 interact with the repeat. To do this, we tagged each of these proteins with mCherry. An amino acid linker was added between the protein and a C-terminal mCherry tag in all of the cases (Mre11, Smc5, Slx5, and Rad51: GGSGGS, Rfa1: AAAAAAAAG). The tagged proteins were shown to be functional through MMS sensitivity assays (Figure S2). Rad51 fusions are known to affect protein function, but it was recently shown that a Rad51-GFP fusion was non-functional but also not dominant negative; the repair functionality of both an induced DSB or MMS-induced damage could be complemented by the addition of WT Rad51 protein expressed from a plasmid, and the formation and disappearance of Rad51 foci paralleled the kinetics of repair as in WT cells (Waterman et al., 2019). We tested the MMS sensitivity of the complemented Rad51-mCherry construct in our strain background and found only a slight sensitivity in line with what was reported for Rad51-GFP+ Rad51 (Waterman et al., 2019) (Figure S2). The co-localization of each protein with the CAG repeat (marked with GFP at the nearby LacO array) was assessed, and whether the co-localization was occurring at the nuclear periphery (NP) or not was scored (Figure 5A).
Figure 5. Differential Timing of Mre11, Smc5, Rfa1, Slx5, and Rad51 Association with the CAG-130 Repeat Locus Near ARS607.
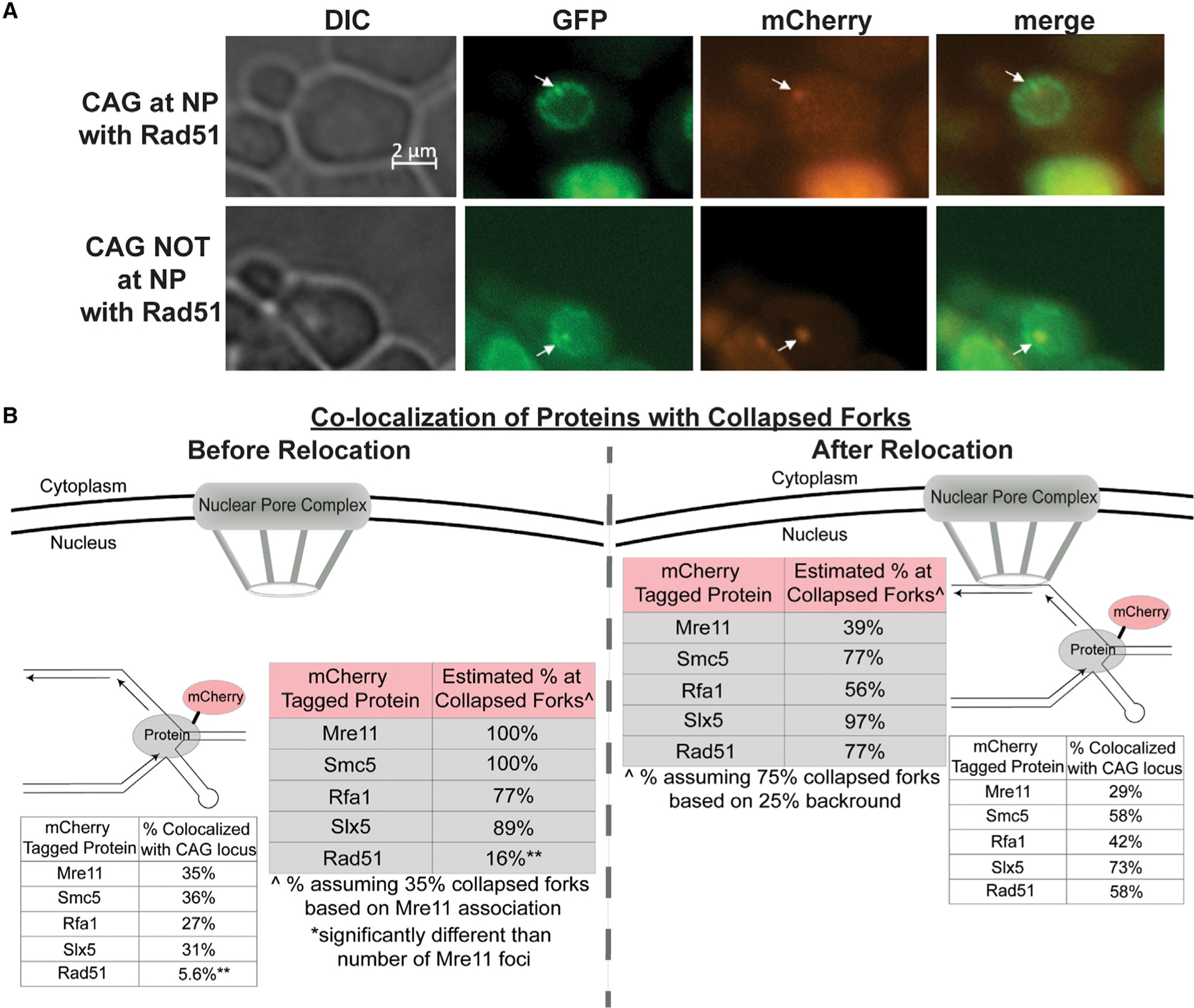
(A) Example images of the CAG locus (GFP) and Rad51 foci (mCherry) co-localization.
(B) Co-localization of mCherry-tagged Mre11, Smc5, Rfa1, Slx5, and Rad51 with the GFP-marked CAG repeat in S phase cells. The location of the CAG repeat, at the NP or not, was scored along with the co-localization. See also Table S5. The Rad51-mCherry strain contained a CEN plasmid expressing Rad51 under its own promoter as used in Waterman et al. (2019). Both the actual percentage observed and the estimated percentage at collapsed forks is shown for each tagged protein.Êstimated percentage at collapsed forks before relocation was calculated by normalizing the percentage of co-localization for each tagged protein to the percentage of co-localization of Mre11-mCherry with the CAG-130 (35%). Mre11 is used as a proxy for collapsed forks as it has been shown to promote resection and restart of collapsed forks. ^(After relocation) estimated percentage at collapsed forks at the NP was calculated by normalizing the percentage of co-localization for each tagged protein to 75%, as we see a 25% background of the locus at the NPC based on CAG-0 data (Su et al., 2015).
For before relocation data, **p < 0.01 compared with Mre11-mCherry co-localization by Fisher’s exact test. Comparisons were not done for after relocation data since it was not normalized to Mre11 occupancy.
See Table S5 for the exact number of cells analyzed, percentages, and p values.
See Figures S4 and S5 for example co-localization images of Mre11-mCherry, Smc5-mCherry, Rfa1-mCherry, Slx5-mCherry, and Rad51-mCherry.
The actual percentage of co-localization with the CAG repeat was normalized for both before and after relocation to estimate specifically the co-localization of the proteins with collapsed forks. For the data before relocation, Mre11-mCherry was used as a proxy for collapsed forks, as Mre11 has been shown to promote the resection and restart of collapsed forks (Trenz et al., 2006; Tittel-Elmer et al., 2009; Zhu et al., 2018). The data were normalized by dividing the actual co-localization percentages by 35% (the actual percentage of co-localization for Mre11-mCherry) to give an estimated percentage of each tagged protein at collapsed forks. For the data after relocation, we assumed 75% collapsed forks at the nuclear periphery, as there is a 25% background based on CAG-0 relocation data (Su et al., 2015), which was also the remaining percentage in the slx5Δ, smt3–331, and mms21–11 strains (Figure 1). The data were normalized by dividing the actual co-localization percentages by 75% to give an estimated percentage of each tagged protein at the collapsed forks. Both the normalized and non-normalized data are shown in Figure 5B.
Before relocation to the NPC, Mre11, Smc5, Rfa1, and Slx5 strongly co-localize with collapsed forks caused by the CAG-130 repeat (77%–100% of the time; normalized data) (Figure 5B; Table S5). After relocation, they all are still co-localized with the CAG tract, but to varying strengths, with Smc5 and Slx5 more strongly associated with the collapsed fork at the NPC (77% and 97%, respectively) and Mre11 and Rfa1 less strongly associated (39% and 56%, respectively) (Figure 5B; Table S5). These data provide support that the proteins identified as important for relocation are associating with the CAG repeat both before and after the movement to the NPC. In contrast, Rad51 is predominantly co-localized with the collapsed fork only when it is at the periphery (77%; normalized data) and very rarely within the internal nuclear space (16%; normalized data) (Figure 5B; Table S5). These data indicate that most Rad51 protein is recruited only once the CAG repeat is at the NPC, which is consistent with Rad51 not being required for relocation (Su et al., 2015). We conclude that Mre11, Smc5, Rfa1, and Slx5 associate with the CAG repeat before relocation, and Rad51 is primarily recruited after the repeat is at the NPC.
RPA Sumoylation Is Critical for Restraining the Association of Rad51 with the CAG Repeat before Relocating to the NPC
To further investigate the timing of Rad51 co-localization with the CAG repeat, we scored the co-localization of Rad51 with the CAG repeat locus across several time points (Figure 6A; Table S6). At the peak of CAG repeat association with the NPC (60 min), Rad51 is co-localized with the repeat at the nuclear periphery 48% of the time. For CAG tracts that are already associated with the periphery at 50 min, there is also significant Rad51 co-localization (37%), indicating that it is an early event after NPC association. However, by 70 min, a time point at which some CAG tracts are being released from the periphery, co-localization with Rad51 drops to 22% (Figure 6A). Most notably, when the CAG tract is not at the nuclear periphery, Rad51 is almost never co-localized with the repeat in S phase (0%–3% for 50- to 70-min time points) (Figure 6A; Table S6). This is striking when compared to the co-localization of the other proteins associated with the CAG tract in the inner nuclear space (Figure 5). These data provide strong support that Rad51 mostly associates with collapsed forks only after relocation to the NPC has occurred, although it is possible that a small amount of Rad51 not visible as foci could associate earlier. All repair proteins are most often found at the nuclear periphery (73%–97%; Tables S5 and S6), regardless of the interaction with the CAG repeat, pointing to the nuclear periphery being an active area for repair.
Figure 6. Rad51 Association with the CAG Repeat Is Restricted Until after Relocation to the NPCs in a Manner That Is Dependent on RPA Sumoylation.
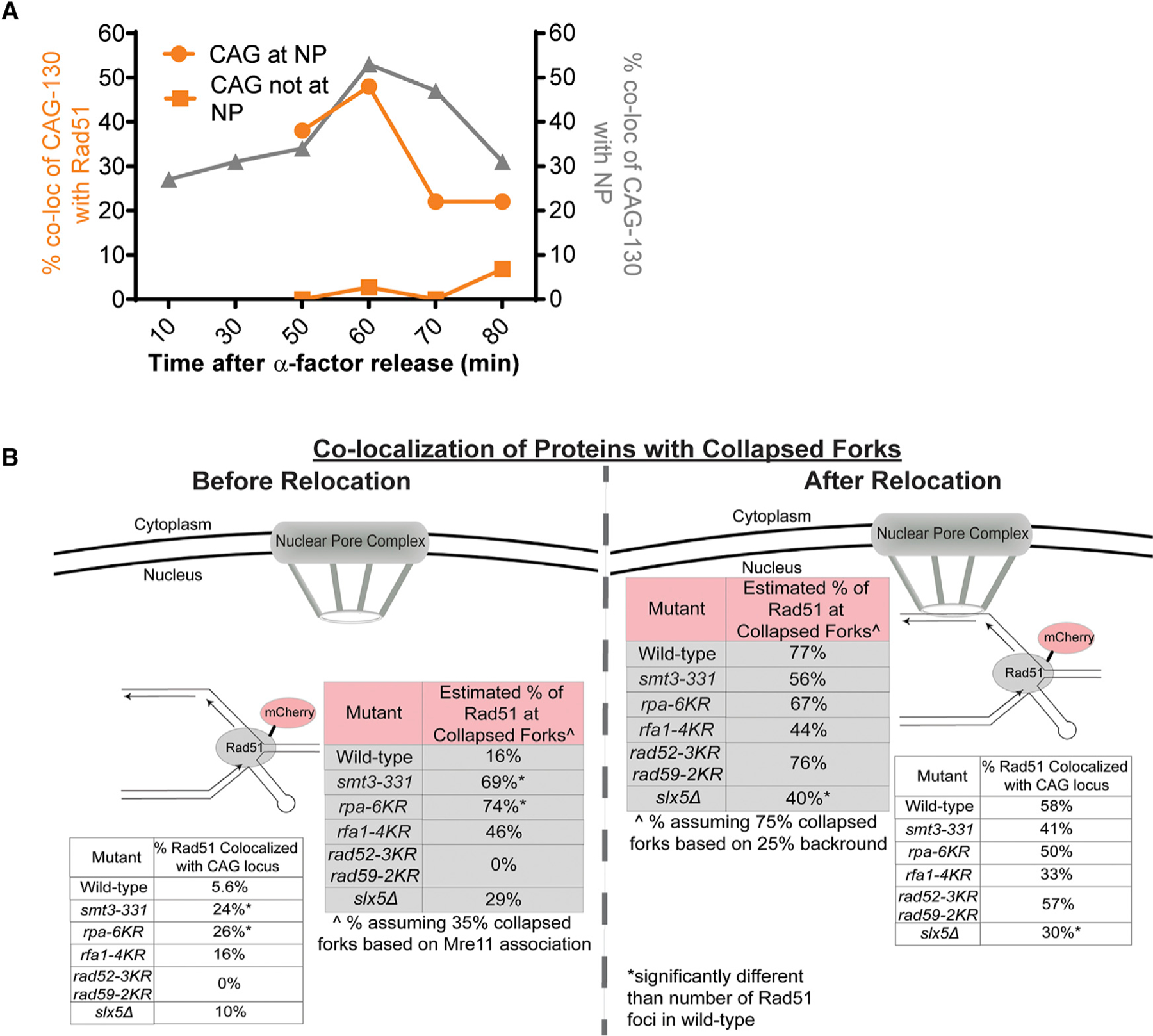
(A) Co-localization of mCherry-tagged Rad51 with the GFP-marked CAG repeat across several time points after alpha factor release. The location of the CAG repeat, at the NP or not, was scored along with the co-localization. See Table S6.
(B) Co-localization of mCherry-tagged Rad51 with the GFP-marked CAG repeat in various mutants. The location of the CAG repeat, at the NP or not, was scored along with the co-localization. See Table S7. Both the actual percentage observed and the estimated percentage at specifically collapsed forks is shown for each mutant.Êstimated percentage at collapsed forks was calculated as outlined in the Figure 5 legend. *p < 0.05 compared with number of Rad51 foci in WT cells by Fisher’s exact test.
See Tables S7 and S8 for the exact number of cells analyzed, percentages, and p values.
To assess whether sumoylation of the key targets required for relocation is needed to prevent Rad51 recruitment to the collapsed fork in the internal nuclear space, we assessed co-localization of Rad51-mCherry and CAG-130 in several mutants (rpa-4KR, rpa-6KR, smt3–331, rad52–3KR rad59–2KR, and slx5Δ). The same normalization methods were used as described for Figure 5B. In the smt3–331 mutant, in which overall sumoylation is decreased, we saw significantly more co-localization of the CAG repeat and Rad51-mCherry before relocation to the nuclear periphery occurs (69% in smt3–331 versus 16% in WT) (Figure 6B; Tables S7 and S8). Similar results were seen for rpa-6KR (74%), and both of these values were significantly different from the level of Rad51 association in the WT strain (p % 0.05). The rfa1–4KR mutant also had an increase in the co-localization of Rad51-mCherry with the CAG repeat before relocation to 46%, although the increase was not significant compared to WT (Figure 6B; Tables S7 and S8), suggesting that the remaining sites for sumoylation available on Rfa2 and Rfa3 are providing some level of interference with Rad51 binding. In contrast, the rad52–3KR rad59–2KR mutant was statistically equivalent to WT, with no Rad51 co-localizing with the CAG tract while in the internal nuclear space (Figure 6B; Tables S7 and S8). Thus, despite their role in relocation, Rad52 and Rad59 sumoylation are not playing a significant role in excluding Rad51 binding. The similar increase in Rad51 focus formation in the smt3–331 and rpa-6KR strains indicates that RPA sumoylation is primarily preventing Rad51 binding at the collapsed fork before relocation to the nuclear periphery.
After relocation, there was a marked increase in Rad51 foci co-localizing with the CAG tract in WT cells from 16% to 77%. Rad51 association with collapsed forks remained similar to that before relocation in the rpa-6KR and rfa1–4KR and smt3–331 mutants (Figure 6B; Tables S7 and S8), as expected since Rad51 was already associated due to lack of sumoylation at the fork in these backgrounds. In contrast, Rad51 foci increased in the rad52–3KR rad59–2KR mutant to 76% as in WT, showing that the modification state of Rad52 and Rad59 is not a significant factor in determining Rad51 binding levels (Figure 6B; Tables S7 and S8). However, the slx5Δ strain showed significantly less Rad51 association at the nuclear periphery (40%) compared to WT (Figure 6B; Tables S7 and S8). It is possible that since Slx5 is needed to tether the collapsed fork to the NPC, Rad51 association is disrupted when tethering is non-functional but sumoylated proteins are still present at the fork, as in this mutant.
DISCUSSION
In this study, we used long CAG repeats as a model to study the effects of fork stalling and collapse. We aimed to determine the mechanism by which forks stalled by a DNA structure relocalize to the NPC. We previously showed that the slx5Δ mutant, which has a defect in relocation to the NPC, results in increased genome instability, manifested as increased chromosome breakage at the expanded CAG tract. We further confirmed that the deletion of other members of the sumoylation pathway, which are required for relocation, also have increased chromosome breakage. Thus, a better understanding of how sumoylation is involved in relocation to the NPC will provide insight into the mechanisms of fork restart and repair. Our data indicate that the mechanism by which a fork impeded by expanded CAG repeats relocalizes to the NPC is dependent on mono-sumoylation of repair proteins by the Mms21 SUMO ligase that is associated with the Smc5/6 complex. The sumoylated proteins then interact with the SIM domains of Slx5 to mediate relocation to the NPC. Sumoylation is known to be induced in response to DNA damage, and multiple sumoylated events are often required to generate a strong effect via a so-called group effect (Sarangi and Zhao, 2015a, 2015b; García-Rodríguez et al., 2016; Zhao, 2018). Here, we show that sumoylation of repair proteins is also a critical response to replication stress and replication barriers and demonstrate a group effect of sumoylation in this context.
We identified specific sumoylated repair proteins (RPA, Rad59, and Rad52) that are required for the relocation of a collapsed fork to the NPC in late S phase. Sumoylation of all three proteins appears to be required, as the mutation of each one individually was not sufficient to completely impair relocation, but instead there was an additive effect. This result suggests that a critical level of sumoylation must be achieved for efficient relocation to the NPC. Consistently, a minimal level of resection was also important, presumably to provide a platform for RPA and Rad52/59 binding. Removal of sumoylated RPA, Rad52, and Rad59 at the same time abolished relocation, indicating that these proteins are the principal players in mediating interaction with the Slx5 SIM domains. Their sumoylation loss likely reduces the SUMO moieties at collapsed forks below a threshold required for the effective recruitment of Slx5 via its SIMs. Nonetheless, other Mms21 sumoylation targets, such as Smc5/6, the Ku complex, and Sgs1, are also known to be associated with impaired replication forks and could play a supporting role. Our data are consistent with a role for Smc5 sumoylation in this relocation process. Although we found that Sgs1 loss alone did not affect relocation, it could also play a supporting role. Sgs1 preferentially interacts with sumoylated Smc5/6 and RPA via its SIM domain, and Sgs1 sumoylation modulates its resection activity (Bermúdez-López et al., 2016; Bonner et al., 2016). Therefore, the presence of the Smc5/6 complex and subsequent RPA sumoylation at the stalled fork could promote the recruitment and sumoylation of Sgs1. This could result in the activation of resection (together with Dna2 and Exo1), promoting further RPA binding and sumoylation in a positive feedback loop. We also showed that constitutive sumoylation of several proteins (Rfa1, Rad52, Rad59, and Smc5) advanced the timing of relocation, indicating that a minimal level of sumoylation is sufficient for an increase in retention at the periphery. However, the effect is not observed until 40 min into S phase, which is when Rad52 is detected at the CAG repeat by ChIP (Su et al., 2015). This result suggests that fork collapse causes additional sumoylation, leading to more robust relocation. Our data indicate that there is a threshold of sumoylation required for efficient relocation of the collapsed fork to the NPC.
One unexpected result was that Mms21 is the main SUMO E3 ligase required for the relocation of an expanded CAG repeat, even though the Siz2 E3 sumoylates Rad52, Rad59, and RPA in response to other types of damage (Sacher et al., 2006; Psakhye and Jentsch, 2012). Our data show that Mms21 contributes significantly to the sumoylation of Rfa1, Rfa2, Rfa3, and Rad59. We hypothesize that since the Smc5/6 complex accumulates at collapsed forks (Lindroos et al., 2006), Mms21 is in close proximity to RPA and Rad52/59, favoring Mms21-dependent sumoylation. It is possible that Siz2 plays a small role—for example, targeting Rad52—as we did see a non-significant but consistent decrease in CAG-130 relocation in siz2Δ mutants. As an example, initial RPA sumoylation by Mms21 could recruit Siz2 to boost sumoylation of RPA, Rad52, and Rad59 (Chung and Zhao, 2015). Nonetheless, our results are different from what was observed at persistent DSBs, where both Mms21 and Siz2 were found to be necessary for efficient relocation to the NPC, and a role for Siz2-dependent poly-sumoylation was observed (Horigome et al., 2016). Consistent with the requirement for Mms21 and not Siz2, we also observed that mono-sumoylation was mainly responsible for CAG-130 relocation. Thus, the SUMO response at a collapsed fork differs from a persistent DSB. Furthermore, we confirm that the Slx5 SIM domain is the critical factor mediating the relocation of collapsed forks, presumably through interaction with sumoylated RPA, Rad52/59, and the Smc5/6 complex, and that Slx5 binding occurs both before and after association with the nuclear periphery. Mms21 and Smc6 mutants accumulate Rad51-dependent recombination intermediates at damaged replication forks (Branzei et al., 2006; Sollier et al., 2009), which is consistent with our model that Mms21-dependent sumoylation normally restrains recombination at stalled forks. Our data provide insight into the role of Smc5/6 at collapsed forks and link its SUMO ligase activity to its known function in fork protection.
A key observation was that Rad51 foci were mostly excluded from the CAG tract while in the internal nuclear space and only became associated at the nuclear periphery. Notably, this exclusion was dependent on sumoylation of all three RPA subunits, but not on Rad52 or Rad59 sumoylation. Therefore, RPA sumoylation inhibits Rad51 binding at the collapsed fork. Such a mechanism could serve to limit recombination at more transiently stalled forks that have not collapsed or accumulated extensive resection. There is evidence that initial Rad51 exclusion from lesions is a conserved strategy. For example, heterochromatic breaks in Drosophila move out of the chromocenter and over to the nuclear periphery, and Rad51 foci only appear once the break is at the periphery (Chiolo et al., 2011; Ryu et al., 2015). For a heterochromatic DSB, sumoylation in general was shown to prevent Rad51 recruitment in the heterochromatin (Ryu et al., 2015). We have now identified a specific sumoylation target, RPA, that is required to prevent Rad51 recruitment to collapsed forks caused by CAG repeats. In mouse cells, DSBs move to the periphery of the heterochromatin, and again, Rad51 is recruited after relocation occurs (Tsouroula et al., 2016). Similarly, in S. cerevisiae, recombination at DSBs induced in the rDNA is excluded from the nucleolus in a manner that is dependent on Rad52 SUMO modification and the Smc5/6 complex (Torres-Rosell et al., 2007). In addition, type II recombination at eroded telomeres is promoted by the relocation of the eroded telomere to NPCs (Géli and Lisby, 2015; Churikov et al., 2016). However, at a protein-mediated replication fork barrier in Schizosaccharomyces pombe, which causes a much more pronounced stall compared to a CAG-130 repeat, Rad51 is required for relocation to the NPC (K. Kramarz and S. Lambert, personal communication). We did observe a few Rad51 foci co-localized with the CAG-130 tract in the nuclear interior (in 3/94 cells) and short Rad51 filaments will not be visible as a focus (Mason et al., 2019). Thus, the subtleties of the timing and level of Rad51 binding and its effect on relocation could depend on the severity or type of fork collapse. Nonetheless, our result that sumoylation of RPA by Mms21 prevents the formation of Rad51 foci provides a mechanism to explain the previous observations of delayed Rad51 association and its dependence on Smc5/6.
Presumably, NPCs and associated components can relieve the inhibition of Rad51 binding. It is possible that upon NPC association, the Slx5/8 STUbL enzyme can ubiquitinate sumoylated proteins at the fork, leading to their extraction from chromatin and/or degradation. This in turn could facilitate Rad51 access to ssDNA. The human RNF4 protein, a Slx5/8 homolog, interacts with RPA via SIM domains and promotes RPA clearance and Rad51 accumulation at DSBs (Galanty et al., 2012). Another Slx8 homolog, RFWD3, can ubiquitinate RPA upon fork collapse induced by HU and ataxia telangiectasia and Rad3-related (ATR) inhibition and remove RPA from chromatin to facilitate the restart of collapsed forks (Elia et al., 2015). As we observed reduced levels of collapsed fork-associated RPA at the NPC compared to before relocation, a similar mechanism involving Slx5/8-mediated RPA clearance may occur in yeast at the NPC to facilitate Rad51 filament formation. This hypothesis is in line with our finding that slx5Δ both blocks collapsed fork relocation and reduces Rad51 foci levels at peripheral CAG tracts. Future studies will be needed to further test this model.
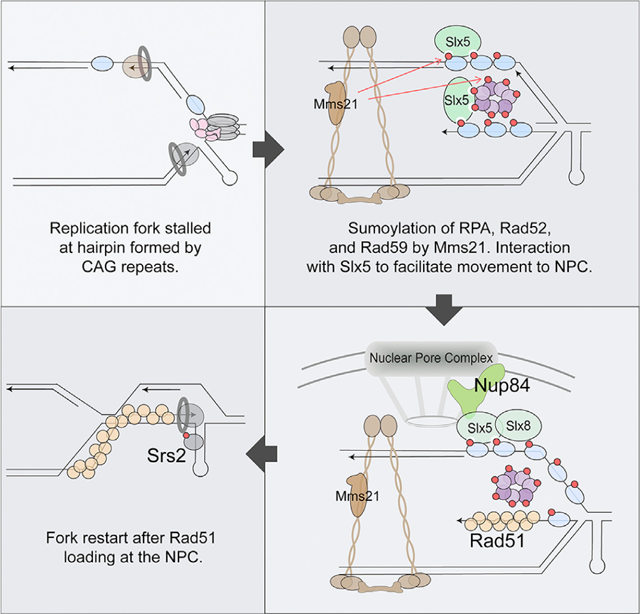
Our data support a model that a SUMO-dependent mechanism moves collapsed forks to NPCs, where Rad51 association occurs (Figure 7). Specifically, upon replication fork stalling at the CAG repeats in S phase, we envision that collapsed forks can be produced in the form of reversed or broken forks, although the exact structure that initiates the response remains to be determined. Impaired fork structures are then recognized by Mre11, which could play both a structural role (e.g., recruiting Sgs1/Dna2) and a nuclease role. Upon Mre11- and Exo1-mediated DNA resection, the produced ssDNA creates a platform for RPA binding. The Smc5/6 complex is also recruited to the collapsed fork, bringing the Mms21 SUMO E3 ligase into close proximity to sumoylate RPA and other repair factors. Sumoylated RPA disfavors Rad51 accumulation, potentially restricting recombination at this stage. These sumoylation events facilitate the relocation of the collapsed fork to the NPC, where Slx5 serves as a bridge between sumoylated proteins (via Slx5 SIMs) and Nup84. Once at the NPC, we hypothesize that Slx8 ubiquitylates the sumoylated forms of repair proteins, potentially targeting them for removal and/or degradation. Our previous results provide evidence for Slx8-mediated Rad52 degradation (Su et al., 2015); however, the details of this part of the pathway remain to be elucidated. We hypothesize that the removal of these proteins from the collapsed fork allows for Rad51 loading at the NPC, which then stimulates HR-mediated fork restart. Replication through the CTG hairpin (on the leading strand template in the construct used here) could be promoted by the Srs2 helicase, which was previously shown to unwind hairpins and promote fork restart at expanded CTG tracts (Dhar and Lahue, 2008; Anand et al., 2012; Nguyen et al., 2017), or by other D-loop-associated factors. Events at the NPC may also promote the resolution of HR intermediates or of the stalled fork and the converging fork. As CAG repeat fragility increases when sumoylation is impaired, our findings indicate that SUMO-mediated relocation of collapsed forks to the NPC is an important protective mechanism to maintain genome stability.
Figure 7. Model for the Relocation of Collapsed Forks Caused by Expanded CAG Repeats to the NPC.
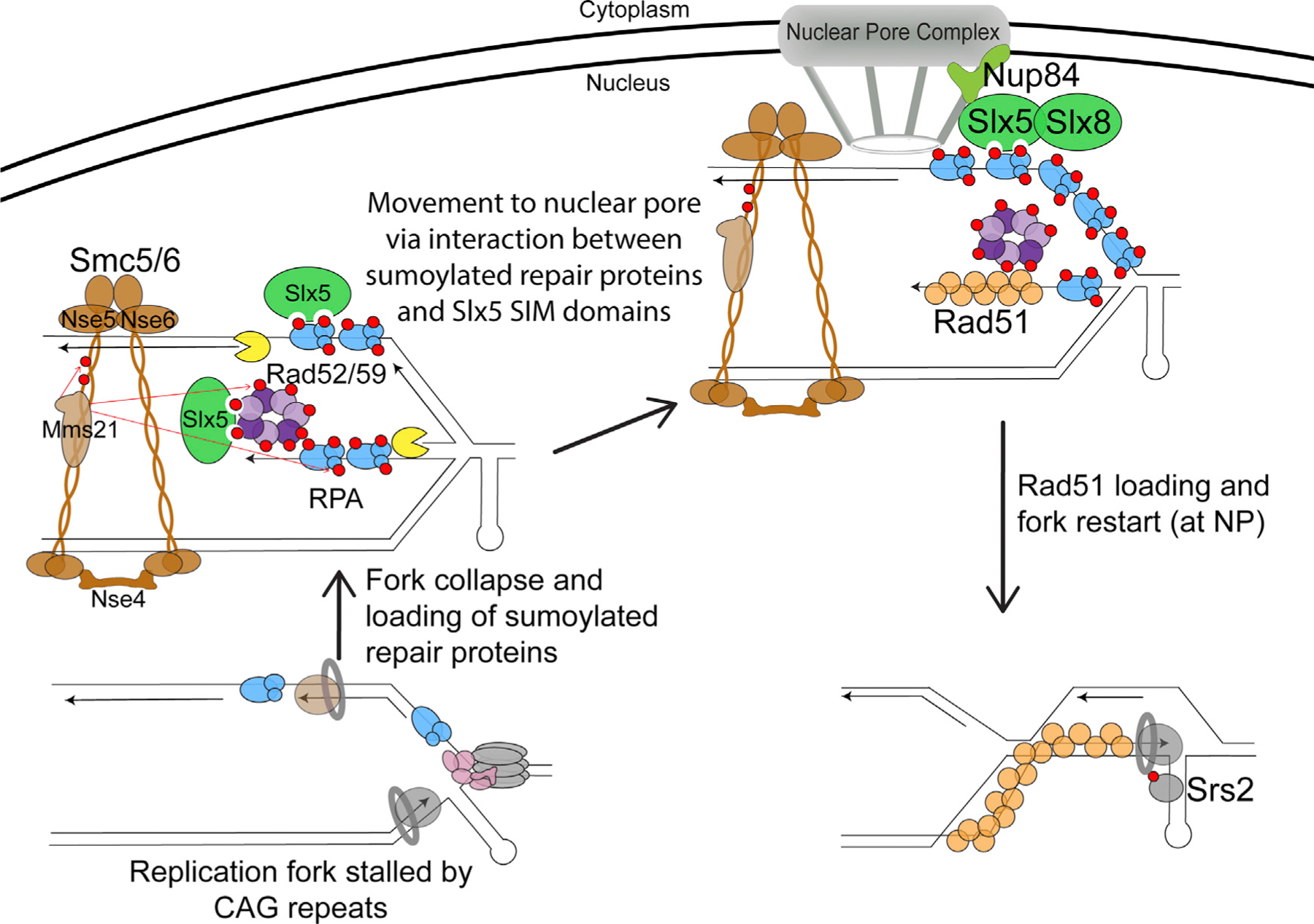
Refer to the Discussion for a detailed description of the model. Mms21-dependent sumoylation is represented by red arrows. The red circles on the proteins represent SUMO.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-FOA | US Biological | F5050 |
| 5x GC rich buffer | Empire Genomics | IDL028 |
| Alpha factor | Zymo Research | Y1001 |
| ID pol Taq polymerase | Empire Genomics | IDL007 |
| Metaphor agarose | VWR | 50184 |
| Methyl methanesulfonate (MMS) | Fisher Scientific | AC15689–0050 |
| Zeocin | Invivogen | ant-zn-05 |
| Hydroxyurea | Fisher Scientific | AC151680250 |
| NEB Thermo buffer | Fisher | B9004S |
| Paraformaldehyde | Fisher Scientific | AC41678-0250 |
| Cover glasses, high performance, square | Zeiss | 474030-9000-000 |
| 2-well concave microscope slides | VWR | 470019–020 |
| Experimental Models: Organisms/Strains | ||
| See Table S9 for a list of yeast strains used in this study. The background for the yeast strains used were W1588–4C, W303, or BY4705. | Freudenreich Lab | Strain number |
| Oligonucleotides | ||
| See Table S10 for a list of primer sequences used in this study. | Eton Bioscience Inc. | Primer name |
| Recombinant DNA | ||
| plasmid pFA6a-kanMX6 | Longtine et. al 1998 | CHF136 |
| plasmid pYM-N35 | Janke et al., 2004 | CHF397 |
| plasmid pRS415 LEU2 | Brachmann et al., 1998 | CHF188 |
| plasmid pFA6a-GFP(S65T)-kanMX6 | Longtine et al., 1998 | CHF139 |
| plasmid pFA6a-13Myc-kanMX6 | Longtine et al., 1998 | CHF145 |
| plasmid pFA6a-link-yoPA-mCherry-Kan | Addgene #44950 | CHF677 |
| Software and Algorithms | ||
| Fluctuation analysis | FLUCALC | http://flucalc.ase.tufts.edu/ |
| Image processing and analysis in Java | ImageJ | https://imagej.nih.gov/ij/ |
| Image processing and analysis in Zen | ZEN Pro/Zen Lite | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Catherine H. Freudenreich (catherine.freudenreich@tufts.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
All raw data are available in the supplemental tables
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All yeast strains were derivatives of either strain W303, BY4705, or W1588–4C, a RAD5 derivative of W303. Details of strain genotypes are listed in Table S9. Yeast strains were grown at 30°C and frozen at 80°C for long term storage.
METHOD DETAILS
Yeast strains and genetic manipulation
Standard procedures were used in cell growth and medium preparation. C.H. Freudenreich lab strains are listed in Table S9 and had either W303 or BY4705 backgrounds. Several point mutant alleles used here have been previously characterized: pSLX5SIM1234 (Xie et al., 2007), smx3–331, smt3–3KR, mms21–11 (Bustard et al., 2016), rad52–3KR, rad59–2KR (Silva et al., 2016), rpa-6KR (Dhingra et al., 2019), rad52-smt3ΔGG, rad59-smt3ΔGG (Silva et al., 2016), mre11-D56N (Moreau et al., 1999), mre11–3 (Bressan et al., 1998; Tittel-Elmer et al., 2009), and nse5-ts1 (Bustard et al., 2016). The smc5-smt3 and rfa1-smt3 mutants were generated by transformation of a SMT3-KANMX fragment such that Smt3 with a stop codon was fused to the C-terminal end of Smc5 and Rfa1 respectively. Neither the Smc5-Smt3, Rfa1-Smt3 or Smt3-mCherry fusion proteins were sensitive to MMS, indicating that protein function was not impaired (Figure S2). mCherry was fused to Mre11, Smc5, Rfa1, Slx5, and Rad51 with a linker sequence integrated between the protein and C-terminal mCherry tag (Mre11, Smc5, Slx5, and Rad51: GGSGGS, Rfa1: AAAAAAAAG). For Rad51-mCherry a plasmid expressing Rad51 under its own promoter was also transformed into the strain; the plasmid is the same one used in Waterman et al., 2019 and the complemented strain shows a slight MMS sensitivity as previously published (Figure S2). X. Zhao Lab strains used are listed in Table S9 and were isogenic to W1588–4C, a RAD5 derivative of W303 (MATa ade2–1 can1–100 ura3–1 his3–11,15 leu2–3, 112 trp1–1 rad5–535). The rfa1–4KR mutant allele used here has been previously characterized (Dhingra et al., 2019). The rfa2-KR and rfa3-KR mutant alleles were generated using the previously published sumoylation sites for Rfa2 (K199) and Rfa3 (K46) (Psakhye and Jentsch, 2012). All rfa mutant alleles were generated using the URA3-based pop-in-pop-out method as previously described (Reid et al., 2002). Mutations were generated at their endogenous loci and proteins were expressed from their own promoters, no tag or selection marker was present unless specified. All alleles were verified by sequencing. Similar to a previous report (Psakhye and Jentsch, 2012) our rfa2-KR and rfa3-KR mutants had undetectable levels of Rfa2 and Rfa3 sumoylation after treatment with MMS.
Zoning assay
Colonies were checked for 130 CAG repeats by PCR with primers flanking the repeat (CTGRev2/T720). Cells from colonies with the correct repeat length were grown to approximately 5 × 106 cells per ml in YC media. Cells were fixed with 4% paraformaldehyde and washed 3 times with 1X PBS. 1.4% YC agar was used to make agar pads on depression slides and 5uL of cells were added on top with a coverslip. Z stack images were taken using a Zeiss AX10 fluorescent microscope under 100x magnification. Step interval size was 0.15um and approximately 30 stacks were taken per field of cells. Exposure time was DIC: 100ms; GFP: 500ms. Images were deconvolved, and three-zoning criteria was used to evaluate the location of the GFP foci for S-phase cells with the ImageJ point picker program as described in Meister et al. (2010). Only the middle two-thirds of the stacks were used for analysis. S-phase cells were determined by yeast morphology using bud size criteria of 2/3rd or less the size of the mother cell as described in Hediger et al. (2004).
Rad52-GFP foci microscopy
Cells were grown overnight in YC media at 30°C, diluted to OD600 of 0.1, and grown to OD600 of 0.3. MMS was added at a concentration of 0.5%. Cultures grown at 30°C for 15 min then washed twice with sterile water. Cell pellets were resuspended in 2mL YC media and grown for 30 min. Cells were fixed with 4% paraformaldehyde. Z stack images were taken using a Zeiss AX10 fluorescent microscope under 100x magnification. Step interval size was 0.15um and approximately 30 stacks were taken per field of cells. Exposure time was DIC: 100ms; GFP: 500ms. The number of Rad52-GFP foci per S-phase cells was scored.
MMS+HU and zeocin treatment for zoning assay
Cells were grown overnight in YC media at 30°C, diluted to OD600 of 0.1, and grown again until OD600 reached 0.2. 40 uL of alpha factor mating pheromone was added to each culture and grown at 30°C for 90 min. Alpha factor was rinsed out by washing three times with sterile water. Cells were resuspended in YC media with either 0.2M HU+0.033% MMS or 0.3mg/mL zeocin and grown at 30C for 1 h. Cells were washed with sterile water, resuspended in YC media and allowed to recover for 30 min at 30C. Cultures were then processed and imaged as outlined in the zoning assay method.
Microscopy time-courses
Colonies were checked for 130 CAG repeats by PCR with primers flanking the repeat. Cells from colonies with the correct repeat length were grown overnight in YC media at 30°C, diluted to OD600 of 0.1, and grown again until OD600 reached 0.2. 40 uL of alpha factor mating pheromone was added to each culture and grown at 30°C for 90 min. Alpha factor was rinsed out by washing three times with sterile water. The cell pellets were resuspended in YC media and 20, 30, 40, 50, and 60-minute time points were taken. Each time point was processed and imaged as indicated in the Zoning Assay method.
Mre11, Smc5, Rfa1, Slx5, and Rad51 co-localization assays
Assays were set up and imaged the same as the Zoning assay previously outlined. In addition to exposure to GFP and DIC, cells were exposed to dsRed for 500ms to visualize the mCherry tagged protein of interest. Only cells with mCherry foci were scored. mCherry was added to proteins of interest such that it disrupted the stop codon and an appropriate linker sequence was added. For Rad51-mCherry a plasmid expressing Rad51 under its own promoter was also transformed into the strain as outlined in Waterman et al. (2019) and cultures were grown in YC-LEU to maintain the plasmid.
Detection of the sumoylation of Rad52, Rad59 and RPA
As sumoylated forms of proteins are dynamic and difficult to detect, in order to quantitively assess the effects of SUMO E3 mutants on the sumoylation levels of Rad52, Rad59 and RPA, we examined cells treated with 0.3 mg/ml Zeocin (Life Technologies) for 2 hr, which give rise to abundant sumoylated species of the proteins examined. For Rad52, Rad59 and Rfa1, TCA method was used to make cell lysates as described (Chung et al., 2015). Briefly, 2×108 cells were collected and lysed by bead beating in the presence of 20% TCA. The pellet was recovered by centrifugation and incubated with 1X Laemmli buffer at 95°C for 5 min. Proteins were separated on 3%–8% Tris-acetate gels (Life Technologies) followed by western blotting with antibodies against Rfa1 (gift from S. Brill) or TAP-(Peroxidase-Anti-Peroxidase, Sigma). For Rfa2 and Rfa3, the Ni-NTA pull-down method, which minimizes protein desumoylation, was followed as previously described (Bonner et al., 2016). In brief, yeast SUMO (Smt3) was tagged with either His8 or His6-Flag at its N terminus and expressed from its endogenous promoter in cells containing WT Rfa2 or HA-tagged Rfa3. 5×108 cells were har-vested and the prepared protein extract was incubated overnight at room temperature with Ni-NTA resin (QIAGEN) containing 0.05% Tween20 and 4.4 mM Imidazole. The resin was then washed and eluents were subjected to SDS-PAGE and western blotting with anti-Rfa2 antibody (gift from S.Brill) to detect sumoylated and unmodified Rfa2 or anti-HA antibody (SC-7392, Santa Cruz) to detect sumoylated and unmodified Rfa3. Accurate quantification of protein bands was achieved by scanning the western blots using a LAS-3000 luminescent image analyzer (Fujifilm) with a linear dynamic range of 104. The signal intensities of non-saturated bands were measured using ImageJ software. Signals of sumoylated Rad52, Rad59 and Rfa1 from the long exposure blots are divided by that of their unsumoylated forms from the short exposure blots, in order to use signal values with a linear range of detection. For Rfa2 and Rfa3, signals of their sumoylated forms were divided by the stain. For graphs, data are shown as mean and SEM. Statistical differences were determined using Student’s t tests.
CAG fragility assay
The CAG repeat tract was amplified from yeast colonies using primers (NewCAGFor/NewCAGRev) spanning the repeats (P1 and P2 in Figure S1C) as described in Sundararajan et al. (2010) to confirm correct tract length. Colonies were grown in YC-Leu liquid media for 6–7 cell divisions to allow breakage and were plated on FOA-Leu and YC-Leu plates. To assay fragility, colonies growing on FOA-Leu and YC-Leu were counted, and a rate of mutation was calculated using the method of maximum likelihood with the online program FLUCALC (Polleys and Freudenreich, 2018; Radchenko et al., 2018). A representative number of FOA-Leu plates for each strain were replica plated to YC-HIS plates and percent end loss was calculated. At least three independent assays were performed per strain.
Resection assay
DSB resection assays were performed as previously described (Chen et al., 2013). A DSB at the MAT a locus was introduced by galactose-inducible HO endonuclease. Cells were collected at the indicated time points after HO induction. Genomic DNA was isolated and an aliquot was subjected to XbaI and StyI digestion. Digested DNA was then subjected to native agarose gel electrophoresis, transferred to Hybond XL membrane (GE Healthcare) and hybridized with two radiolabeled DNA probes that recognize regions 0.7 kb and 3.0 kb away from the HO cut site. A Dnl4 probe was used to indicate loading. The proportion of unresected DNA at each time point was calculated as the ratio of the signal intensity at that time point to that at 30mins after HO induction and then normalized to the Dnl4 signal. The values at 30mins were set to 100%.
QUANTIFICATION AND STATISTICAL ANALYSIS
Prism software was used to calculate statistical significance. For microscopy experiments Fisher’s exact test was used. For CAG fragility assays Student’s t test was used. Error bars for CAG fragility assays are representing SEM. The number of cells analyzed, assays performed, and p values are denoted in the supplementary tables. For western blot quantifications statistical differences were determined using Student’s t tests.
Supplementary Material
Highlights.
Relocation of collapsed forks to NPCs depends on sumoylation by Mms21/Nse2
Targets of sumoylation important for relocation are RPA, Rad52, Rad59, and Smc5
Resection mediated by Mre11, Exo1, and Sgs1 is required for relocation to the NPC
RPA sumoylation prevents Rad51 binding to collapsed forks before NPC anchoring
ACKNOWLEDGMENTS
We thank Susan Gasser, Michael Lisby, Jennifer Cobb, Mark Hochstrasser, Lorraine Symington, John Petrini, and Jim Haber for sharing strains. This work was funded by National Institutes of Health (NIH) grants GM122880 (to C.H.F.) and GM080670 and GM131058 (to X.Z.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107635.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Amaral N, Ryu T, Li X, and Chiolo I (2017). Nuclear Dynamics of Heterochromatin Repair. Trends Genet. 33, 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ampatzidou E, Irmisch A, O’Connell MJ, and Murray JM (2006). Smc5/6 is required for repair at collapsed replication forks. Mol. Cell. Biol 26, 9387–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand RP, Shah KA, Niu H, Sung P, Mirkin SM, and Freudenreich CH (2012). Overcoming natural replication barriers: differential helicase requirements. Nucleic Acids Res. 40, 1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermúdez-López M, Villoria MT, Esteras M, Jarmuz A, Torres-Rosell J, Clemente-Blanco A, and Aragon L (2016). Sgs1’s roles in DNA end resection, HJ dissolution, and crossover suppression require a two-step SUMO regulation dependent on Smc5/6. Genes Dev. 30, 1339–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Bhalla N, Chang A, Smith DL, and Murray AW (2001). Genes involved in sister chromatid separation and segregation in the budding yeast Saccharomyces cerevisiae. Genetics 159, 453–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner JN, Choi K, Xue X, Torres NP, Szakal B, Wei L, Wan B, Arter M, Matos J, Sung P, et al. (2016). Smc5/6 Mediated Sumoylation of the Sgs1-Top3-Rmi1 Complex Promotes Removal of Recombination Intermediates. Cell Rep. 16, 368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, and Boeke JD (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132. [DOI] [PubMed] [Google Scholar]
- Branzei D, Sollier J, Liberi G, Zhao X, Maeda D, Seki M, Enomoto T, Ohta K, and Foiani M (2006). Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 127, 509–522. [DOI] [PubMed] [Google Scholar]
- Bressan DA, Olivares HA, Nelms BE, and Petrini JHJ (1998). Alteration of N-terminal phosphoesterase signature motifs inactivates Saccharomyces cerevisiae Mre11. Genetics 150, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustard DE, Menolfi D, Jeppsson K, Ball LG, Dewey SC, Shirahige K, Sjögren C, Branzei D, and Cobb JA (2012). During replication stress, non-SMC element 5 (NSE5) is required for Smc5/6 protein complex functionality at stalled forks. J. Biol. Chem 287, 11374–11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustard DE, Ball LG, and Cobb JA (2016). Non-Smc element 5 (Nse5) of the Smc5/6 complex interacts with SUMO pathway components. Biol. Open 5, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylebyl GR, Belichenko I, and Johnson ES (2003). The SUMO isopeptidase Ulp2 prevents accumulation of SUMO chains in yeast. J. Biol. Chem 278, 44113–44120. [DOI] [PubMed] [Google Scholar]
- Callahan JL, Andrews KJ, Zakian VA, and Freudenreich CH (2003). Mutations in yeast replication proteins that increase CAG/CTG expansions also increase repeat fragility. Mol. Cell. Biol 23, 7849–7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, and Cejka P (2014). Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125. [DOI] [PubMed] [Google Scholar]
- Chavez A, George V, Agrawal V, and Johnson FB (2010). Sumoylation and the structural maintenance of chromosomes (Smc) 5/6 complex slow senescence through recombination intermediate resolution. J. Biol. Chem 285, 11922–11930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lisby M, and Symington LS (2013). RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 50, 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, and Karpen GH (2011). Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144, 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung I, and Zhao X (2015). DNA break-induced sumoylation is enabled by collaboration between a SUMO ligase and the ssDNA-binding complex RPA. Genes Dev. 29, 1593–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DKC, Chan JNY, Strecker J, Zhang W, Ebrahimi-Ardebili S, Lu T, Abraham KJ, Durocher D, and Mekhail K (2015). Perinuclear tethers license telomeric DSBs for a broad kinesin- and NPC-dependent DNA repair process. Nat. Commun 6, 7742. [DOI] [PubMed] [Google Scholar]
- Churikov D, Charifi F, Eckert-Boulet N, Silva S, Simon MN, Lisby M, and Géli V (2016). SUMO-Dependent Relocalization of Eroded Telomeres to Nuclear Pore Complexes Controls Telomere Recombination. Cell Rep. 15, 1242–1253. [DOI] [PubMed] [Google Scholar]
- Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, and Foiani M (2005). Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell 17, 153–159. [DOI] [PubMed] [Google Scholar]
- Dhar A, and Lahue RS (2008). Rapid unwinding of triplet repeat hairpins by Srs2 helicase of Saccharomyces cerevisiae. Nucleic Acids Res. 36, 3366–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra N, Wei L, and Zhao X (2019). Replication protein A (RPA) sumoylation positively influences the DNA damage checkpoint response in yeast. J. Biol. Chem 294, 2690–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia AEH, Wang DC, Willis NA, Boardman AP, Hajdu I, Adeyemi RO, Lowry E, Gygi SP, Scully R, and Elledge SJ (2015). RFWD3-Dependent Ubiquitination of RPA Regulates Repair at Stalled Replication Forks. Mol. Cell 60, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouché N, Ozgür S, Roy D, and Griffith JD (2006). Replication fork regression in repetitive DNAs. Nucleic Acids Res. 34, 6044–6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenreich CH, and Su XA (2016). Relocalization of DNA lesions to the nuclear pore complex. FEMS Yeast Res. 16, fow095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenreich CH, Kantrow SM, and Zakian VA (1998). Expansion and length-dependent fragility of CTG repeats in yeast. Science 279, 853–856. [DOI] [PubMed] [Google Scholar]
- Gaillard H, Santos-Pereira JM, and Aguilera A (2019). The Nup84 complex coordinates the DNA damage response to warrant genome integrity. Nucleic Acids Res. 47, 4054–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanty Y, Belotserkovskaya R, Coates J, and Jackson SP (2012). RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 26, 1179–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Rodríguez N, Wong RP, and Ulrich HD (2016). Functions of ubiquitin and SUMO in DNA replication and replication stress. Front. Genet 7, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géli V, and Lisby M (2015). Recombinational DNA repair is regulated by compartmentalization of DNA lesions at the nuclear pore complex. BioEssays 37, 1287–1292. [DOI] [PubMed] [Google Scholar]
- Hauer MH, and Gasser SM (2017). Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 31, 2204–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger F, Taddei A, Neumann FR, and Gasser SM (2004). Methods for visualizing chromatin dynamics in living yeast. Methods Enzymol. 375, 345–365. [DOI] [PubMed] [Google Scholar]
- Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, and Gasser SM (2014). SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol. Cell 55, 626–639. [DOI] [PubMed] [Google Scholar]
- Horigome C, Bustard DE, Marcomini I, Delgoshaie N, Tsai-Pflugfelder M, Cobb JA, and Gasser SM (2016). PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev. 30, 931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horigome C, Unozawa E, Ooki T, and Kobayashi T (2019). Ribosomal RNA gene repeats associate with the nuclear pore complex for maintenance after DNA damage. PLoS Genet. 15, e1008103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, and Knop M (2004). A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962. [DOI] [PubMed] [Google Scholar]
- Johnson ES, and Blobel G (1997). Ybc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J. Biol. Chem 272, 267999–802. [DOI] [PubMed] [Google Scholar]
- Kalocsay M, Hiller NJ, and Jentsch S (2009). Chromosome-wide Rad51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell 33, 335–343. [DOI] [PubMed] [Google Scholar]
- Kerrest A, Anand RP, Sundararajan R, Bermejo R, Liberi G, Dujon B, Freudenreich CH, and Richard GF (2009). SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat. Struct. Mol. Biol 16, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadaroo B, Teixeira MT, Luciano P, Eckert-Boulet N, Germann SM, Simon MN, Gallina I, Abdallah P, Gilson E, Géli V, and Lisby M (2009). The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat. Cell Biol 11, 980–987. [DOI] [PubMed] [Google Scholar]
- Krogh BO, Llorente B, Lam A, and Symington LS (2005). Mutations in Mre11 phosphoesterase motif I that impair Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex stability in addition to nuclease activity. Genetics 171, 1561–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaçon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, and Vindigni A (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindroos HB, Ström L, Itoh T, Katou Y, Shirahige K, and Sjögren C (2006). Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol. Cell 22, 755–767. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Mason JM, Chan YL, Weichselbaum RW, and Bishop DK (2019). Non-enzymatic roles of human RAD51 at stalled replication forks. Nat. Commun 10, 4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister P, Gehlen LR, Varela E, Kalck V, and Gasser SM (2010). Visualizing yeast chromosomes and nuclear architecture. Methods Enzymol. 470, 535–567. [DOI] [PubMed] [Google Scholar]
- Menolfi D, Delamarre A, Lengronne A, Pasero P, and Branzei D (2015). Essential Roles of the Smc5/6 Complex in Replication through Natural Pausing Sites and Endogenous DNA Damage Tolerance. Mol. Cell 60, 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, and Symington LS (2008). Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, and Symington LS (2010). Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 29, 3358–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Ferguson JR, and Symington LS (1999). The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell. Biol 19, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, and Krogan NJ (2008). Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 322, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JHG, Viterbo D, Anand RP, Verra L, Sloan L, Richard GF, and Freudenreich CH (2017). Differential requirement of Srs2 helicase and Rad51 displacement activities in replication of hairpin-forming CAG/CTG repeats. Nucleic Acids Res. 45, 4519–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza P, Jaspersen SL, Miele A, Dekker J, and Peterson CL (2009). Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 23, 912–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleys EJ, and Freudenreich CH (2018). Methods to study repeat fragility and instability in Saccharomyces cerevisiae. Methods Mol. Biol 1672, 403–419. [DOI] [PubMed] [Google Scholar]
- Polleys EJ, House NCM, and Freudenreich CH (2017). Role of recombination and replication fork restart in repeat instability. DNA Repair (Amst.) 56, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psakhye I, and Jentsch S (2012). Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151, 807–820. [DOI] [PubMed] [Google Scholar]
- Radchenko EA, McGinty RJ, Aksenova AY, Neil AJ, and Mirkin SM (2018). Quantitative analysis of the rates for repeat-mediated genome instability in a yeast experimental system. Methods Mol. Biol 1672, 421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid RJ, Lisby M, and Rothstein R (2002). Cloning-free genome alterations in Saccharomyces cerevisiae using adaptamer-mediated PCR. Methods Enzymol. 350, 258–277. [DOI] [PubMed] [Google Scholar]
- Ryu T, Spatola B, Delabaere L, Bowlin K, Hopp H, Kunitake R, Karpen GH, and Chiolo I (2015). Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol 17, 1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu T, Bonner MR, and Chiolo I (2016). Cervantes and Quijote protect heterochromatin from aberrant recombination and lead the way to the nuclear periphery. Nucleus 7, 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher M, Pfander B, Hoege C, and Jentsch S (2006). Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol 8, 1284–1290. [DOI] [PubMed] [Google Scholar]
- Sarangi P, and Zhao X (2015a). SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem. Sci 40, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarangi P, and Zhao X (2015b). Erratum to: SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem. Sci 40, 183–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva S, Altmannova V, Eckert-Boulet N, Kolesar P, Gallina I, Hang L, Chung I, Arneric M, Zhao X, Buron LD, et al. (2016). SUMOylation of Rad52-Rad59 synergistically change the outcome of mitotic recombination. DNA Repair (Amst.) 42, 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Driscoll R, Castellucci F, Foiani M, Jackson SP, and Branzei D (2009). The Saccharomyces cerevisiae Esc2 and Smc5–6 proteins promote sister chromatid junction-mediated intra-S repair. Mol. Biol. Cell 20, 1671–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su XA, Dion V, Gasser SM, and Freudenreich CH (2015). Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Genes Dev. 29, 1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan R, and Freudenreich CH (2011). Expanded CAG/CTG repeat DNA induces a checkpoint response that impacts cell proliferation in Saccharomyces cerevisiae. PLoS Genet. 7, e1001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararajan R, Gellon L, Zunder RM, and Freudenreich CH (2010). Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae. Genetics 184, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatham MH, Jaffray E, Vaughan OA, Desterro JMP, Botting CH, Naismith JH, and Hay RT (2001). Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem 276, 35368–35374. [DOI] [PubMed] [Google Scholar]
- Teixeira-Silva A, Ait Saada A, Hardy J, Iraqui I, Nocente MC, Fréon K, and Lambert SAE (2017). The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun 8, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittel-Elmer M, Alabert C, Pasero P, and Cobb JA (2009). The MRX complex stabilizes the replisome independently of the S phase checkpoint during replication stress. EMBO J. 28, 1142–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Rosell J, Sunjevaric I, De Piccoli G, Sacher M, Eckert-Boulet N, Reid R, Jentsch S, Rothstein R, Aragón L, and Lisby M (2007). The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol 9, 923–931. [DOI] [PubMed] [Google Scholar]
- Trenz K, Smith E, Smith S, and Costanzo V (2006). ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 25, 1764–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsouroula K, Furst A, Rogier M, Heyer V, Maglott-Roth A, Ferrand A, Reina-San-Martin B, and Soutoglou E (2016). Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 63, 293–305. [DOI] [PubMed] [Google Scholar]
- Usdin K, House NCM, and Freudenreich CH (2015). Repeat instability during DNA repair: insights from model systems. Crit. Rev. Biochem. Mol. Biol 50, 142–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman DP, Zhou F, Li K, Lee CS, Tsabar M, Eapen VV, Mazzella A, and Haber JE (2019). Live cell monitoring of double strand breaks in S. cerevisiae. PLoS Genet. 15, e1008001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Kerscher O, Kroetz MB, McConchie HF, Sung P, and Hochstrasser M (2007). The yeast Hex3.Slx8 heterodimer is a ubiquitin ligase stimulated by substrate sumoylation. J. Biol. Chem 282, 34176–34184. [DOI] [PubMed] [Google Scholar]
- Xie Y, Rubenstein EM, Matt T, and Hochstrasser M (2010). SUMO-independent in vivo activity of a SUMO-targeted ubiquitin ligase toward a short-lived transcription factor. Genes Dev. 24, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X (2018). SUMO-mediated regulation of nuclear functions and signaling processes. Mol. Cell 71, 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, and Blobel G (2005). A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc. Natl. Acad. Sci. USA 102, 4777–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Bacolla A, Wang G, and Vasquez KM (2010). Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci 67, 43–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Zhao H, Limbo O, and Russell P (2018). Mre11 complex links sister chromatids to promote repair of a collapsed replication fork. Proc. Natl. Acad. Sci. USA 115, 8793–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are available in the supplemental tables