

Key Points
Question
Can inflammation modulate lipoprotein(a)–associated cardiovascular risk during secondary prevention in optimally treated patients with high-risk vascular disease?
Findings
In a prespecified post hoc secondary analysis of the ACCELERATE trial, in patients with established vascular disease who were optimally treated, increasing lipoprotein(a) levels (assessed as either quintiles or continuous logarithmic transformed levels) during treatment were significantly associated with cardiovascular death, myocardial infarction, and stroke only in individuals with high-sensitivity C-reactive protein levels of 2 mg/L or higher during treatment, but not in those with levels less than 2 mg/L. Similar significant associations were identified in time-to-first event rates and survival curves, as well as specifically in the placebo-treated group.
Meaning
There is likely to be incremental benefit in lowering lipoprotein(a) levels in optimally treated patients with high-risk vascular disease, which appears to be optimized in patients with concomitant high-sensitivity C-reactive protein levels of 2 mg/L or more.
Abstract
Importance
Although lipoprotein(a) (Lp[a]) is a causal genetic risk factor for atherosclerotic cardiovascular disease, it remains unclear which patients with established atherosclerotic cardiovascular disease stand to benefit the most from Lp(a) lowering. Whether inflammation can modulate Lp(a)-associated cardiovascular (CV) risk during secondary prevention is unknown.
Objective
To examine whether Lp(a)-associated CV risk is modulated by systemic inflammation in optimally treated patients at high risk of CV disease.
Design, Setting, and Participants
A prespecified secondary post hoc analysis of the double-blind, multicenter randomized clinical Assessment of Clinical Effects of Cholesteryl Ester Transfer Protein Inhibition With Evacetrapib in Patients at a High Risk for Vascular Outcomes (ACCELERATE) trial was conducted between October 1, 2012, and December 31, 2013; the study was terminated October 12, 2015. The study was conducted at 543 academic and community hospitals in 36 countries among 12 092 patients at high risk of CV disease (acute coronary syndrome, stroke, peripheral arterial disease, or type 2 diabetes with coronary artery disease) with measurable Lp(a) and high-sensitivity C-reactive protein (hsCRP) levels during treatment. Statistical analysis for this post hoc analysis was performed from September 26, 2018, to March 28, 2020.
Interventions
Participants received evacetrapib, 130 mg/d, or matching placebo.
Main Outcomes and Measures
The ACCELERATE trial found no significant benefit or harm of evacetrapib on 30-month major adverse cardiovascular events (CV death, myocardial infarction [MI], stroke, coronary revascularization, or hospitalization for unstable angina). This secondary analysis evaluated rates of CV death, MI, and stroke across levels of Lp(a).
Results
High-sensitivity C-reactive protein and Lp(a) levels were measured in 10 503 patients (8135 men; 8561 white; 10 134 received concurrent statins; mean [SD] age, 64.6 [9.4] years). In fully adjusted analyses, in patients with hsCRP of 2 mg/L or more but not less than 2 mg/L, increasing quintiles of Lp(a) were significantly associated with greater rates of death, MI, and stroke (P = .006 for interaction). Each unit increase in log Lp(a) levels was associated with a 13% increased risk of CV death, nonfatal MI, or stroke only in those with hsCRP levels of 2 mg/L or more (P = .008 for interaction). There was also a significant stepwise relationship between increasing Lp(a) quintiles and time to first CV death, MI, or stroke (log-rank P < .001) when hsCRP levels were 2 mg/L or more but not less than 2 mg/L. Sensitivity analyses in the ACCELERATE placebo-treated group yielded similar significant associations exclusively in the group with hsCRP of 2 mg/L or more.
Conclusions and Relevance
Elevated Lp(a) levels during treatment are related to CV death, MI, and stroke when hsCRP levels are 2 mg/L or more but not less than 2mg/L. This finding suggests a potential benefit of lowering Lp(a) in patients with residual systemic inflammation despite receipt of optimal medical therapy.
Trial Registration
ClinicalTrials.gov Identifier: NCT01687998
This secondary analysis of the randomized clinical ACCELERATE trial examines whether lipoprotein(a)-associated cardiovascular risk is modulated by systemic inflammation in optimally treated patients at high risk for cardiovascular disease.
Introduction
Lipoprotein(a) (Lp[a]) levels are genetically mediated and considered to be causal in the development of atherosclerotic cardiovascular disease (ASCVD).1,2 Although reducing low-density lipoprotein cholesterol levels remains the dominant means of attenuating ASCVD risk, substantial residual risk despite established therapies has led to the effort to identify novel biomarkers and treatment targets.3 Lipoprotein(a) has thus emerged as a potential therapeutic target, with antisense oligonucleotides directed specifically at hepatocytes demonstrating substantial 70% to 90% reductions in circulating Lp(a) levels in early-phase clinical trials in humans.4 Despite these recent advances, it remains unclear which patient populations would benefit the most from Lp(a) reduction, and what degree of Lp(a) lowering would be required to demonstrate incremental clinical benefit despite the use of background established medical therapies.5,6
Although the inflammatory hypothesis of ASCVD was recently demonstrated in 2 large-scale multicenter randomized clinical trials (RCTs) using either a selective interleukin (IL) 1β antagonist or low-dose colchicine,7,8 its mainstream clinical application using other affordable mainstream anti-inflammatory therapies remains challenging.9 Emerging translational and clinical data suggest a synergism between the proatherosclerotic effects of Lp(a) and systemic inflammation.10,11 Accordingly, we tested the hypothesis that Lp(a)-associated ASCVD risk would be significantly modulated by residual levels of systemic inflammation in a large, contemporary optimally treated population with high-risk vascular disease.
Methods
Study Population
We undertook an exploratory post hoc analysis of the Assessment of Clinical Effects of Cholesteryl Ester Transfer Protein Inhibition With Evacetrapib in Patients at a High Risk for Vascular Outcomes (ACCELERATE) trial (study protocol in Supplement 1). The rationale, design, and results of ACCELERATE have been previously described in detail.12 Briefly, ACCELERATE was a multicenter, randomized, double-blind, placebo-controlled phase 3 trial that enrolled 12 092 patients (from October 1, 2012, to December 31, 2013) with high-risk vascular disease, defined as those with an acute coronary syndrome within the previous 30 to 365 days, cerebrovascular atherosclerotic disease, peripheral arterial disease, or type 2 diabetes with coronary artery disease (eFigure 2 in Supplement 2). Patients were randomly assigned to receive either the cholesteryl ester transfer protein inhibitor evacetrapib or placebo, in addition to standard medical therapy. The primary efficacy end point was the first occurrence of any component of the composite of death from cardiovascular causes, myocardial infarction (MI), stroke, coronary revascularization, or hospitalization for unstable angina pectoris. The data and safety monitoring board conducted an interim analysis for futility after 1363 primary composite events (81.6% of the expected 1670 events). The treatment group experienced 691 events and the placebo group experienced 672 events (hazard ratio [HR] for a composite event with evacetrapib vs placebo, 1.03; 95% CI, 0.93-1.15; P = .58). On October 12, 2015, the trial was halted for futility on the basis of these findings. The ACCELERATE investigators planned a number of prespecified analyses, including those involving Lp(a) and hsCRP.
Given that evacetrapib conferred neither benefit nor harm, the present analysis therefore pooled the total ACCELERATE trial population that had Lp(a) and high-sensitivity C-reactive protein (hsCRP) levels measured during the follow-up period. As such, 10 503 of the enrolled 12 092 patients in ACCELERATE met these criteria, forming the basis of the present analysis.
Clinical End Points
In ACCELERATE, the primary efficacy end point was the composite of death from cardiovascular causes, MI, stroke, coronary revascularization, or hospitalization for unstable angina pectoris. In the present analysis, we used the stricter major adverse cardiovascular events (MACE) definition of cardiovascular death, MI, or stroke. In ACCELERATE, Lp(a) was measured as its molar concentration in units of nanomole per liter. High-sensitivity C-reactive protein was measured by the Roche Modular Turbidimetric method, whereas Lp(a) was measured by the Randox assay, which is insensitive to isoform size. The median follow-up period for the present analysis was 28 months. The clinical end points were truncated at 915 days (30 months) owing to the study stopping early.
Statistical Analysis
Statistical analysis for this post hoc analysis was performed from September 26, 2018, to March 28, 2020. Continuous variables are reported as mean and SD values for normally distributed variables or median and interquartile range values for nonnormally distributed variables. Categorical variables are reported as frequency and percentage. Multivariable Cox proportional hazards regression modeling was used to assess the relationship between MACE and Lp(a), including interactions, while adjusting for important covariates. Hazard ratios and 95% CIs are reported. The logarithm of Lp(a) was used when examining Lp(a) as a continuous variable in the models because Lp(a) was nonnormally distributed. Kaplan-Meier curves illustrate the cumulative incidence of MACE over time. Tests of trend were performed across the Lp(a) quintiles. The number at risk at each 6-month increment is given under each plot. All P values were from 2-tailed tests and results were deemed statistically significant at P < .05. Analysis was performed using SAS, version 9.4 (SAS Institute Inc). Figures were created using SigmaPlot, version 11.0 (Systat Software Inc) and R, version 3.0.1 (R Foundation for Statistical Computing).
Results
eTable 1 in Supplement 2 describes the baseline clinical characteristics of the present population (n = 10 503) relative to the original ACCELERATE population (n = 12 092). Table 1 describes the baseline clinical and biochemical characteristics of the patient population stratified according to achieved hsCRP levels (<2 vs ≥2 mg/L). In the present population, most patients (8561 [81.5%]) were white, 6838 (65.1%) had diabetes and coronary artery disease, 10 134 (96.5%) received statin therapy (4845 of 10 373 [46.7%] received high-intensity statin therapy), and 8805 (83.8%) received aspirin. Evacetrapib was administered to 5253 (50.0%) of the present population. However, when baseline characteristics were evaluated according to mean hsCRP levels during treatment, those with hsCRP levels of 2 mg/L or more vs less than 2 mg/L had a higher mean (SD) body mass index, had a more frequent cluster of common cardiovascular risk factors (hypertension, diabetes, active smoking, greater low-density lipoprotein cholesterol levels, higher systolic blood pressure, and a greater burden of established vascular disease), and were less likely to be receiving statins.
Table 1. Baseline Patient Characteristics According to Achieved hsCRP Levels.
| Variable | No./total No. (%) | P value | |
|---|---|---|---|
| hsCRP level during treatment | |||
| <2 mg/L | ≥2 mg/L | ||
| Age, mean (SD), y | 64.5 (9.4) | 64.7 (9.4) | .28 |
| Male sex | 4602/5609 (82.0) | 3533/4894 (72.2) | <.001 |
| Race/ethnicity | |||
| White | 4412/5573 (79.2) | 4149/4869 (85.2) | <.001 |
| Black | 102/5573 (1.8) | 173/4869 (3.6) | |
| Asian | 809/5573 (14.5) | 286/4869 (5.9) | |
| Other | 250/5573 (4.5) | 261/4869 (5.4) | |
| ACS in last 30-365 d | 1757/5609 (31.3) | 1267/4894 (25.9) | <.001 |
| BMI, mean (SD) | 29.1 (4.8) | 31.7 (6.3) | <.001 |
| Hypertension | 4788/5609 (85.4) | 4388/4894 (89.7) | <.001 |
| Type 2 diabetes | 3739/5609 (66.7) | 3501/4894 (71.5) | <.001 |
| Diabetes with CAD | 3577/5609 (63.8) | 3261/4894 (66.6) | .002 |
| Prior MI | 3452/5132 (67.3) | 2802/4311 (65.0) | .02 |
| Prior PCI | 3750/5128 (73.1) | 3056/4306 (71.0) | .02 |
| Prior CABG | 1457/5128 (28.4) | 1416/4308 (32.9) | <.001 |
| High-risk PAD | 657/5609 (11.7) | 814/4894 (16.6) | <.001 |
| Current smoker | 797/5609 (14.2) | 882/4894 (18.0) | <.001 |
| Medications | |||
| Statins (any) | 5459/5609 (97.3) | 4675/4894 (95.5) | <.001 |
| Statins (high-intensity) | 2726/5537 (49.2) | 2119/4836 (43.8) | <.001 |
| Antihypertensive agents | 4789/5609 (85.4) | 4362/4894 (89.1) | <.001 |
| Aspirin | 4780/5609 (85.2) | 4025/4894 (82.2) | <.001 |
| Evacetrapib | 2640/5609 (47.1) | 2613/4894 (53.4) | <.001 |
| Lipids, mean (SD), mg/dL | |||
| LDL-C | 79.2 (26.2) | 83.7 (29.3) | <.001 |
| HDL-C | 46.1 (11.8) | 44.5 (11.7) | <.001 |
| Lp(a), median (IQR), nmol/L | 28 (11-109) | 31 (11-109) | .15 |
| hsCRP, median (IQR), mg/L | 0.9 (0.5-1.5) | 3.0 (1.6-5.5) | <.001 |
| Systolic blood pressure, mean (SD), mm Hg | 130 (16) | 132 (17) | <.001 |
Abbreviations: ACS, acute coronary syndrome; BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); CABG, coronary artery bypass grafting; CAD, coronary artery disease; HDL-C, high-density lipoprotein cholesterol; hsCRP, high-sensitivity C-reactive protein; IQR, interquartile range; LDL-C, low-density lipoprotein cholesterol; Lp(a), lipoprotein(a); MI, myocardial infarction; PAD, peripheral arterial disease; PCI, percutaneous coronary intervention.
SI conversion factor: To convert LDL-C and HDL-C to millimoles per liter, multiply by 0.0259.
eTable 2 in Supplement 2 describes the lipoprotein, hsCRP, and systolic blood pressure levels at baseline and follow-up in the present population. Considering follow-up measures, the mean low-density lipoprotein cholesterol level was 71.4 mg/dL (to convert to millimoles per liter, multiply by 0.0259), median Lp(a) level was 22.9 nmol/L, and median hsCRP level was 1.8 mg/L.
eTable 3 in Supplement 2 describes the breakdown of adjudicated clinical events in the present population. There were 714 MACE (cardiovascular death, MI, or stroke) events (6.8% of the study population).
Table 2 describes fully adjusted multivariable relationships between MACE stratified according to hsCRP and Lp(a) levels during treatment. In the overall population, higher hsCRP levels during treatment (≥2 vs <2 mg/L) were associated with a significantly greater risk of MACE (HR, 1.59; 95% CI, 1.37-1.86; P < .001), whereas higher Lp(a) levels during treatment (greater than or equal to the median vs less than the median) were not (HR, 1.03; 95% CI, 0.88-1.20; P = .72). However, when stratifying the overall population according to hsCRP levels less than 2 mg/L vs 2 mg/L or more and quintiles of Lp(a) levels, higher Lp(a) levels were significantly associated with MACE only when hsCRP levels were 2 mg/L or more (relative to quintile 1 [Q1], the HR for MACE of Q2 was 1.31 [95% CI, 0.94-1.84], of Q3 was 1.42 [95% CI, 1.02-1.98], of Q4 was 1.50 [95% CI, 1.08-2.09], and of Q5 was 1.70 [95% CI, 1.23-2.36]). In the setting of lower hsCRP levels during treatment (<2 mg/L), higher Lp(a) levels were not associated with a greater risk of clinical events. There was a significant interaction for MACE between Lp(a) quintile and hsCRP dichotomy (P = .006 for interaction). These per-quintile relationships are shown in Figure 1.
Table 2. Associations Between MACE Stratified According to Achieved hsCRP and Lp(a) Quintilesa.
| Population | Kaplan-Meier estimate, No./total No. (%) | MACE, HR (95% CI)b,c | P value |
|---|---|---|---|
| Overall hsCRP population | |||
| <2 mg/L | 289/5609 (5.5) | 1 [Reference] | NA |
| ≥2 mg/L | 425/4894 (9.6) | 1.59 (1.37-1.86) | <.001 |
| Overall Lp(a) population | |||
| <Median | 348/5251 (7.3) | 1 [Reference] | NA |
| ≥Median | 366/5252 (7.6) | 1.03 (0.88-1.20) | .72 |
| hsCRP<2 mg/L | |||
| Lp(a) | |||
| Q1d | 66/1154 (6.0) | 1 [Reference] | NA |
| Q2 | 53/1129 (4.9) | 0.82 (0.57-1.18) | .28 |
| Q3 | 60/1113 (6.1) | 0.93 (0.65-1.33) | .70 |
| Q4 | 58/1103 (5.6) | 0.90 (0.62-1.29) | .55 |
| Q5 | 52/1110 (5.0) | 0.78 (0.54-1.14) | .20 |
| hsCRP ≥2 mg/L | |||
| Lp(a) | |||
| Q1d | 59/946 (7.0) | 1 [Reference] | NA |
| Q2 | 81/972 (9.3) | 1.31 (0.94-1.84) | .12 |
| Q3 | 87/988 (10.0) | 1.42 (1.02-1.98) | .04 |
| Q4 | 94/998 (10.3) | 1.50 (1.08-2.09) | .02 |
| Q5 | 104/990 (11.4) | 1.70 (1.23-2.36) | .001 |
Abbreviations: HR, hazard ratio; hsCRP, high-sensitivity C-reactive protein; Lp(a), lipoprotein(a); MACE, major adverse cardiovascular events (cardiovascular death, myocardial infarction, or stroke); NA, not applicable; Q, quintile.
All hsCRP and Lp(a) levels are time-weighted mean values; Lp(a) is measured in units of nanomoles per liter.
Adjusted for age, sex, race/ethnicity, region, diabetes, smoking, baseline low- and high-density lipoprotein cholesterol, and randomization or treatment group.
P = .006 for interaction between Lp(a) quintile and hsCRP 2 mg/L or more vs less than 2 mg/L for MACE.
Each subsequent quintile is compared with the first quintile. Median (range) Lp(a) values per Lp(a) quintile: Q1, 8.2 (2.2-8.4); Q2, 11.1 (8.5-15.7); Q3, 22.9 (15.8-37.5); Q4, 71.7 (37.6-125.4); and Q5, 183.4 (125.5-7681).
Figure 1. Major Adverse Cardiovascular Events According to High-Sensitivity C-Reactive Protein (hsCRP) and Lipoprotein(a) (Lp[a]) Levels in a Fully Adjusted Multivariable Model.
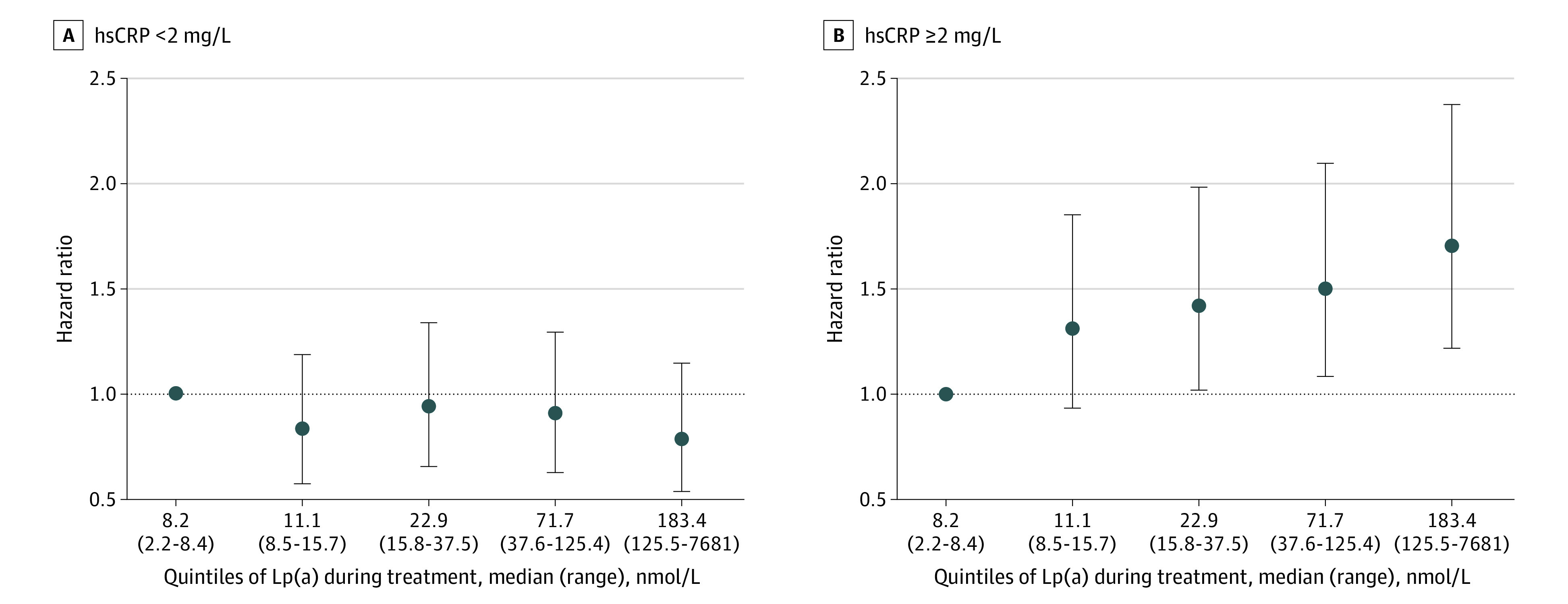
A, High-sensitivity C-reactive protein during treatment of less than 2 mg/L. B, High-sensitivity C-reactive protein during treatment of 2 mg/L or more. Points represent hazard ratios and vertical lines the 95% CIs. Major adverse cardiovascular events indicates cardiovascular death, myocardial infarction, or stroke. Quintile 1 is used as the reference group. Multivariable model adjusted for age, sex, race/ethnicity, region, diabetes, smoking, baseline low-density lipoprotein cholesterol, baseline high-density lipoprotein cholesterol, and randomized treatment group. The dashed horizontal line indicates the hazard ratio of 1. Anything with 95% CIs above this dotted line has a significant association with major adverse cardiovascular events.
Table 3 describes the associations between hsCRP and continuous Lp(a) levels during treatment with MACE in a fully adjusted multivariable model. Each unit increase in the logarithm of Lp(a) was associated with a 13% increase risk of MACE in patients with hsCRP levels of 2 mg/L or more (HR, 1.13 [95% CI, 1.05-1.22]; P = .002], whereas no significant interaction was noted in those with hsCRP levels less than 2 mg/L. There was a significant interaction for MACE between continuous Lp(a) and the hsCRP dichotomy (P = .008 for interaction).
Table 3. Associations Between MACE Stratified According to Achieved hsCRP and Continuous Lp(a) Levelsa.
| MACE | hsCRP <2 mg/L, HR (95% CI)b | P value | hsCRP ≥2 mg/L, HR (95% CI)b | P value | P value for interaction |
|---|---|---|---|---|---|
| Logarithm of Lp(a) | 0.95 (0.87-1.05) | .32 | 1.13 (1.05-1.22) | .002 | .008 |
Abbreviations: HR, hazard ratio; hsCRP, high-sensitivity C-reactive protein; Lp(a), lipoprotein(a); MACE, major adverse cardiovascular events (cardiovascular death, myocardial infarction, or stroke).
All hsCRP and Lp(a) levels are time-weighted mean values.
Adjusted for age, sex, race/ethnicity, region, diabetes, smoking, baseline low- and high-density lipoprotein cholesterol, and randomization or treatment group.
Figure 2 shows the cumulative incidence of first MACE over time stratified according to Lp(a) quintiles in the setting of hsCRP levels less than 2 mg/L (Figure 2A) or 2 mg/L or more (Figure 2B) over a 30-month period. In those with hsCRP levels less than 2 mg/L, increasing quintiles of Lp(a) levels were not associated with a higher cumulative incidence of MACE over time (P = .44 for trend). However, in those with hsCRP levels of 2 mg/L or more, increasing quintiles of Lp(a) levels were associated with a higher cumulative incidence of MACE over time in a stepwise manner (P < .001 for trend).
Figure 2. Kaplan-Meier Curves of Major Adverse Cardiovascular Events Stratified by Achieved Lipoprotein(a) (Lp[a]) Quintiles in the Setting of Achieved High-Sensitivity C-Reactive Protein (hsCRP) Levels of Less Than 2 vs 2 mg/L or More.
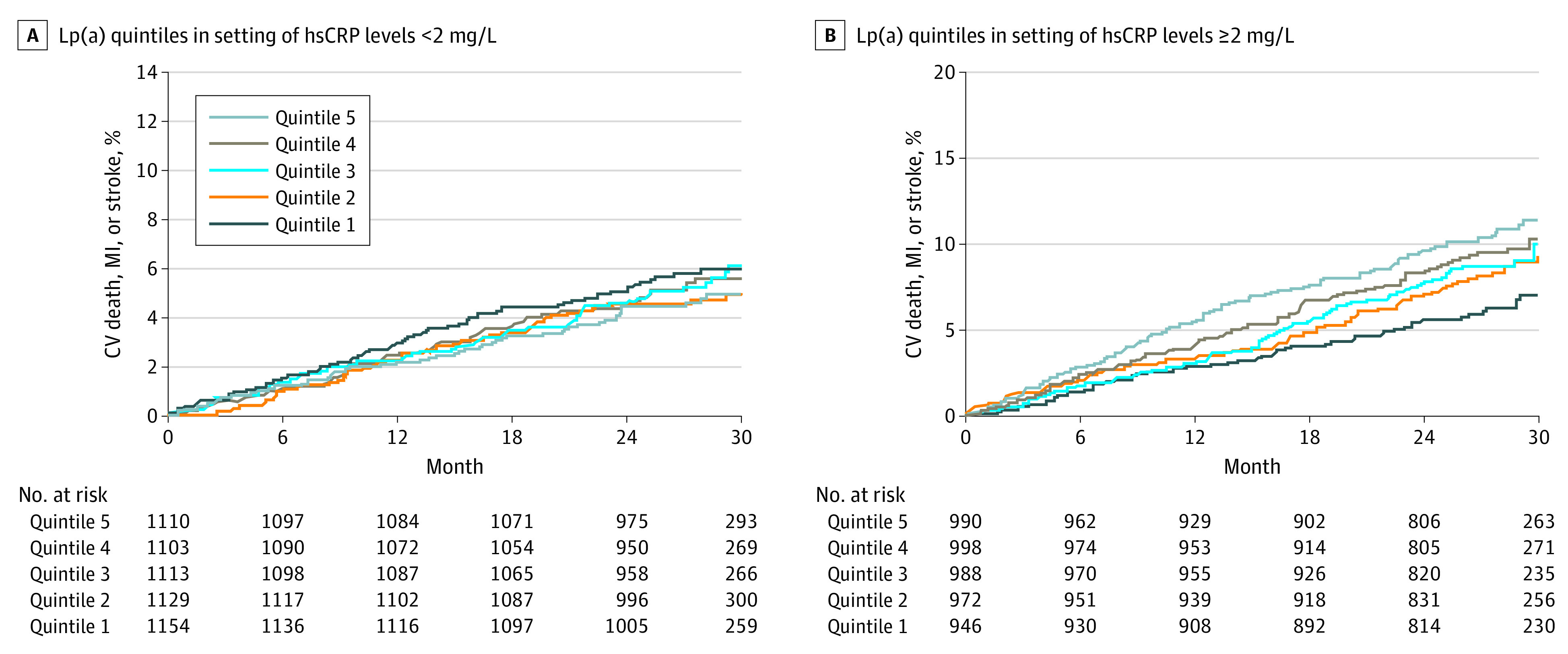
A, Test of trend across quintiles: P = .44. B, Test of trend across quintiles: P < .001. CV indicates cardiovascular; MI, myocardial infarction.
Sensitivity analyses were undertaken involving the placebo-treated group in ACCELERATE, as an attempt to validate the associations in the overall ACCELERATE population while negating the potential confounding effects of evacetrapib. eTable 4 in Supplement 2 describes Kaplan-Meier and fully adjusted multivariable relationships between MACE stratified according to hsCRP and Lp(a) levels during treatment in those who did not receive evacetrapib. Similar to what was demonstrated in the overall population, when stratifying the placebo-treated population according to hsCRP levels less than 2 vs 2 mg/L or more and quintiles of Lp(a) levels, higher Lp(a) levels were significantly associated with MACE only when hsCRP levels were 2 mg/L or more (relative to Q1, the HR for MACE of Q2 was 1.69 [95% CI, 1.02-2.79], of Q3 was 1.87 [95% CI, 1.15-3.06], of Q4 was 1.74 [95% CI, 1.06-2.84], and of Q5 was 1.91 [95% CI, 1.17-3.11]) (P = .008 for interaction between Lp[a] quintile and hsCRP dichotomy). eTable 5 in Supplement 2 describes the associations between hsCRP and continuous Lp(a) levels during treatment with MACE in a fully adjusted multivariable model in those who did not receive evacetrapib. Similar to the overall ACCELERATE population, each unit increase in logarithm of Lp(a) was associated with a 15% increased risk of MACE in patients with hsCRP levels of 2 mg/L or more (HR, 1.15 [95% CI, 1.02-1.29]), whereas no significant association was noted in those with hsCRP levels less than 2 mg/L. There was a significant interaction for MACE between continuous Lp(a) and the hsCRP dichotomy (P = .02 for interaction). eFigure 1 in Supplement 2 illustrates the cumulative incidence of MACE over time stratified according to Lp(a) quintiles in the setting of hsCRP levels less than 2 mg/L or 2 mg/L or more over a 30-month period in those who did not receive evacetrapib. In those with hsCRP levels less than 2 mg/L, increasing quintiles of Lp(a) levels were not associated with a higher cumulative incidence of MACE over time (P = .22 for trend). However, in those with hsCRP levels of 2 mg/L or more, increasing quintiles of Lp(a) levels were associated with a higher incidence of risk of MACE over time in a stepwise manner (P = .01 for trend). eTable 6 and eTable 7 in Supplement 2 describe additional sensitivity analyses using the primary MACE definition in ACCELERATE that also included coronary revascularization and hospitalization for unstable angina. Further sensitivity analyses evaluating the interplay between hsCRP and Lp(a) as continuous variables on MACE are presented in eTable 8 in Supplement 2. These data lend additional support to the significant effect of hsCRP on Lp(a)-mediated ASCVD risk (as opposed to the effect of Lp[a] on hsCRP-mediated ASCVD risk), whereby the log hsCRP × Lp(a) quintile multivariable P = .01 for interaction, whereas the Lp(a) × hsCRP quintile multivariable P = .10 for interaction.
Discussion
We demonstrate for the first time, to our knowledge, that Lp(a)-mediated ASCVD risk appears to be mediated by concomitant hsCRP levels during treatment in a large, contemporary, cohort with high-risk vascular disease ubiquitously treated with statins. In fully adjusted analyses we elucidated a stepwise association between Lp(a) levels and MACE only in individuals with greater degrees of systemic inflammation (hsCRP levels during treatment of ≥2 mg/L). No such relationship between rising Lp(a) levels and MACE was noted when achieved hsCRP levels were less than 2 mg/dL. These data shed new light on the apparent interdependence of 2 known mediators of both primary and residual ASCVD risk: Lp(a) and systemic inflammation.
Translational and epidemiologic data have outlined the causal role of Lp(a) in ASCVD development and residual ASCVD risk,13,14 and therapies specifically targeted at Lp(a) lowering were demonstrated to safely reduce Lp(a) levels by 70% to 90%.4 In anticipation of pivotal RCTs aimed to formally test the Lp(a)-ASCVD hypothesis, there remains considerable debate as to which patients stand to benefit the most from Lp(a)-lowering therapies and the magnitude of Lp(a) reductions required to produce a clinically relevant reduction in ASCVD risk. Burgess and colleagues5 used mendelian randomization techniques to speculate that absolute reductions in Lp(a) of 100 mg/dL would result in equivalent atheroprotective effects yielded from a 39-mg/dL (or 1-mmol/L) reduction of low-density lipoprotein cholesterol, whereas others have estimated Lp(a) reductions of approximately 66 mg/L to achieve similar benefit.6 The present analysis, however, suggests that a potential target population’s residual Lp(a)-mediated ASCVD risk could be further enriched by selecting individuals with concomitantly raised Lp(a) and hsCRP levels of 2 mg/dL or more. Closer inspection of the Kaplan-Meier curve data (Figure 2) of the present analysis shows that for individuals with hsCRP levels of 2 mg/dL or more, the absolute difference in 30-month rates of MACE between individuals in the fifth Lp(a) quintile (median Lp[a] level, 183 nmol/L) vs the second Lp(a) quintile (median Lp[a] 11.1 nmol/L) is 2.1% and the absolute difference in 30-month rates of MACE between individuals in the fifth Lp(a) quintile vs the first Lp(a) quintile (median Lp[a] 8.2 nmol/L) is 4.4%. Assuming a risk-continuum relationship between achieved Lp(a) levels and MACE rates,14 targeting individuals in the highest quintile with a potent Lp(a)-lowering therapy (that would lower Lp[a] levels by 70%-90%) could theoretically result in a number needed to treat of between 23 and 47 patients to prevent 1 recurrent MACE over 30 months. Similarly, closer analysis of Table 3 of the present analysis suggests that for individuals with hsCRP levels of 2 mg/dL or more, each unit increase in the logarithm of Lp(a) level confers a 13% increased risk of MACE. These data could thus prove useful for informing patient selection and planning of future RCTs designed to a priori test the Lp(a)-ASCVD hypothesis.
The inflammatory hypothesis of ASCVD risk was recently proven in the CANTOS (Canakinumab Antiinflammatory Thrombosis Outcome Study) RCT.7 Although CANTOS demonstrated its proof of concept with canakinumab significantly reducing the risk of recurrent ASCVD events, the prohibitive cost of this agent and announcement from its manufacturer not to file for a US Food and Drug Administration approval for treating ASCVD has essentially rendered it unavailable in mainstream clinical practice. The failure of methotrexate to significantly reduce recurrent ASCVD events and hsCRP levels in the CIRT (Cardiovascular Inflammation Reduction Trial) RCT9 leaves us currently without a therapeutic means of specifically reducing systemic inflammation to incrementally lower residual ASCVD risk. In parallel, targeted therapies designed to specifically lower Lp(a) levels have proven successful in early-phase clinical trials in humans.13 Given that the prevalence of increased Lp(a) levels in patients with acute coronary syndrome (such as those in CANTOS and CIRT) is approximately 30%,15 and the prevalence of a proinflammatory IL-1 genotype in populations such as those in CANTOS and CIRT is estimated at approximately 60%,11 extrapolating such data could prove useful for better identifying which patients would theoretically benefit significantly from Lp(a) lowering.
A meta-analysis of patient-level data from 7 RCTs involving statins for treating ASCVD identified a linear relationship between ASCVD risk and achieved Lp(a) levels, particularly at Lp(a) levels greater than 50 mg/dL,14 conferring a 40% increased risk for ASCVD. Closer analysis of Figure 1 in the present data suggests that in the setting of achieved hsCRP levels of 2 mg/L or more, a significant risk of ASCVD emerges at median Lp(a) molar concentrations beyond 23 nmol/L and more consistently toward 50 to 70 nmol/L, roughly equivalent to a Lp(a) particle mass of 25 to 30 mg/dL. At these Lp(a) levels, we observed a 42% to 50% increased MACE risk. Prior observations suggest that ASCVD risk for a first MACE begins to accrue beyond Lp(a) levels of 25 to 30 mg/dL.2 The present data would also suggest a similar Lp(a) cutpoint for recurrent ASCVD-related clinical events in optimally treated patients with high-risk vascular disease, although only in the context of concomitantly elevated hsCRP levels of 2 mg/dL or more. These Lp(a) levels are significantly lower than those suggested by Willeit et al14 in their meta-analysis of individuals treated with statins. Based on our findings, this observation could be a result of the synergistic effects of systemic inflammation and Lp(a)-related proatherosclerotic or prothrombotic mechanisms lowering the Lp(a) cutpoint for mediating secondary ASCVD-related events.
The present data are supported by the clinical observations of Naka and colleagues11 in the Ioannina study, who demonstrated that Lp(a)-associated ASCVD risk in patients with coronary artery disease was positively modulated by the presence of a proinflammatory IL-1 genotype, especially in those 60 years or younger. The mechanistic basis of these observations likely stems from the fact that IL-1–positive individuals produce greater tissue fluid levels of the IL-1β protein and hsCRP,16,17,18 propagating various inflammatory cytokines, thus stimulating a range of inflammatory processes. The proinflammatory effects of oxidized phospholipids bound to apolipoprotein(a) (via their immune-mediated mechanisms of promoting atherosclerosis), in addition to the range of proatherosclerotic effects of various components of the Lp(a) particle, likely renders individuals with dually elevated Lp(a) and hsCRP levels at incremental ASCVD-related risk. Although the present analysis outlines the utility of measuring hsCRP levels in an optimized secondary preventive setting to better assess Lp(a)-associated ASCVD risk, the data are also noteworthy for the lack of Lp(a)-associated ASCVD risk when hsCRP levels are less than 2 mg/dL, irrespective of the magnitude of Lp(a) elevation. The mechanistic basis of these observations warrants further study. Although the nature of Lp(a)-mediated MACE may relate more to thrombosis as opposed to atheroma progression,19 its oxidized phospholipid-based mechanisms rendering Lp(a) as proinflammatory is also likely to promote atherosclerosis.10
Limitations
This study has several limitations. Although analyses involving Lp(a) and hsCRP were initially planned by the trial steering committee, findings of the present exploratory post hoc analysis should be considered hypothesis generating until confirmed in parallel patient cohorts. The various sensitivity analyses however, revealed similar, if not stronger associations and statistical interactions between Lp(a)-mediated ASCVD risk and hsCRP levels in the placebo-treated ACCELERATE cohort, strengthening the plausibility of the present overall findings. Moreover, we used a more rigorous MACE definition of cardiovascular death, MI, or stroke than the ACCELERATE primary end point that also included coronary revascularization and hospitalization for unstable angina. Levels of Lp(a) and hsCRP during treatment (time-weighted mean), rather than baseline levels of Lp(a) and hsCRP, were used as a means of reflecting the effects of optimal medical therapies. However, it is plausible that a subsequent coronary event could have affected hsCRP levels during follow-up. Lipoprotein(a) levels in the clinical literature have tended to be reported as its particle mass in milligrams per deciliter, rather than the criterion standard molar concentration in units of nanomole per liter. It is therefore troublesome to use a single conversion factor between differing assays,20 and interpreting various Lp(a) levels and cutoffs in the present analysis are not directly interchangeable with prior data presented in milligrams per deciliter. Detailed mechanistic insights from the present analysis are lacking, as oxidized phospholipids and IL-1 levels and genotype were not measured. However, hsCRP levels represent a validated, pragmatic, and clinically useful biomarker to ascertain residual ASCVD risk. Moreover, the futility and lack of harm of evacetrapib in ACCELERATE provided a unique opportunity to test the hsCRP-Lp(a) hypothesis in a contemporary, optimized cohort of more than 10 000 patients with vascular disease, with all clinical events expertly adjudicated by an independent committee.
Conclusions
The present data demonstrate for the first time, to our knowledge, in a large cohort of optimally treated patients with high-risk vascular disease, that Lp(a)-associated ASCVD risk appears to be significantly mediated by concomitant levels of residual systemic inflammation. These findings, while shedding further insight into the mechanisms underlying Lp(a)-associated ASCVD risk, may prove useful for identifying individuals who might benefit the most from novel Lp(a)-lowering therapies.
Trial Protocol
eTable 1. Baseline Characteristics According to Achieved hsCRP Levels
eTable 2. Biochemistry and Blood Pressure Values at Baseline and Follow-up Within the Overall ACCELERATE hsCRP-Lp(a) Population
eTable 3. Clinical Event Rates Within the Overall ACCELERATE hsCRP-Lp(a) Population
eTable 4. Relationships Between MACE Stratified According to Achieved hsCRP and Lp(a) Quintiles in the Placebo Group Only
eTable 5. Associations Between MACE Stratified According to Achieved hsCRP and Continuous Lp(a) Levels in the Placebo Group Only
eTable 6. Relationships Between Primary MACE End Point Stratified According to Achieved hsCRP and Lp(a) Quintiles
eTable 7. Associations Between Primary MACE End Point Stratified According to Achieved hsCRP and Continuous Lp(a) Levels
eTable 8. Relationships Between hsCRP and Lp(a) as Continuous Variables Upon MACE
eFigure 1. Cumulative Incidence of MACE Over Time Stratified According to Lp(a) Quintiles in the Setting of hsCRP Levels <2 mg/L (A) or ≥2 mg/L (B)
eFigure 2. CONSORT Diagram
References
- 1.Clarke R, Peden JF, Hopewell JC, et al. ; PROCARDIS Consortium . Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518-2528. doi: 10.1056/NEJMoa0902604 [DOI] [PubMed] [Google Scholar]
- 2.Erqou S, Kaptoge S, Perry PL, et al. ; Emerging Risk Factors Collaboration . Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412-423. doi: 10.1001/jama.2009.1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libby P. The forgotten majority: unfinished business in cardiovascular risk reduction. J Am Coll Cardiol. 2005;46(7):1225-1228. doi: 10.1016/j.jacc.2005.07.006 [DOI] [PubMed] [Google Scholar]
- 4.Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388(10057):2239-2253. doi: 10.1016/S0140-6736(16)31009-1 [DOI] [PubMed] [Google Scholar]
- 5.Burgess S, Ference BA, Staley JR, et al. ; European Prospective Investigation Into Cancer and Nutrition–Cardiovascular Disease (EPIC-CVD) Consortium . Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a mendelian randomization analysis. JAMA Cardiol. 2018;3(7):619-627. doi: 10.1001/jamacardio.2018.1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamina C, Kronenberg F; Lp(a)-GWAS-Consortium . Estimation of the required lipoprotein(a)-lowering therapeutic effect size for reduction in coronary heart disease outcomes: a mendelian randomization analysis. JAMA Cardiol. 2019;4(6):575-579. doi: 10.1001/jamacardio.2019.1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ridker PM, Everett BM, Thuren T, et al. ; CANTOS Trial Group . Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119-1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 8.Tardif JC, Kouz S, Waters DD, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381(26):2497-2505. doi: 10.1056/NEJMoa1912388 [DOI] [PubMed] [Google Scholar]
- 9.Ridker PM, Everett BM, Pradhan A, et al. ; CIRT Investigators . Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. 2019;380(8):752-762. doi: 10.1056/NEJMoa1809798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Valk FM, Bekkering S, Kroon J, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134(8):611-624. doi: 10.1161/CIRCULATIONAHA.116.020838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naka KK, Bechlioullis A, Marini A, et al. Interleukin-1 genotypes modulate the long-term effect of lipoprotein(a) on cardiovascular events: the Ioannina Study. J Clin Lipidol. 2018;12(2):338-347. doi: 10.1016/j.jacl.2017.12.004 [DOI] [PubMed] [Google Scholar]
- 12.Lincoff AM, Nicholls SJ, Riesmeyer JS, et al. ; ACCELERATE Investigators . Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376(20):1933-1942. doi: 10.1056/NEJMoa1609581 [DOI] [PubMed] [Google Scholar]
- 13.Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692-711. doi: 10.1016/j.jacc.2016.11.042 [DOI] [PubMed] [Google Scholar]
- 14.Willeit P, Ridker PM, Nestel PJ, et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet. 2018;392(10155):1311-1320. doi: 10.1016/S0140-6736(18)31652-0 [DOI] [PubMed] [Google Scholar]
- 15.Afshar M, Pilote L, Dufresne L, Engert JC, Thanassoulis G. Lipoprotein(a) interactions with low-density lipoprotein cholesterol and other cardiovascular risk factors in premature acute coronary syndrome (ACS). J Am Heart Assoc. 2016;5(4):e003012. doi: 10.1161/JAHA.115.003012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berger P, McConnell JP, Nunn M, et al. C-reactive protein levels are influenced by common IL-1 gene variations. Cytokine. 2002;17(4):171-174. doi: 10.1006/cyto.2001.0974 [DOI] [PubMed] [Google Scholar]
- 17.Iacoviello L, Di Castelnuovo A, Gattone M, et al. ; IGIGI Investigators . Polymorphisms of the interleukin-1β gene affect the risk of myocardial infarction and ischemic stroke at young age and the response of mononuclear cells to stimulation in vitro. Arterioscler Thromb Vasc Biol. 2005;25(1):222-227. doi: 10.1161/01.ATV.0000150039.60906.02 [DOI] [PubMed] [Google Scholar]
- 18.Rogus J, Beck JD, Offenbacher S, et al. IL1B gene promoter haplotype pairs predict clinical levels of interleukin-1β and C-reactive protein. Hum Genet. 2008;123(4):387-398. doi: 10.1007/s00439-008-0488-6 [DOI] [PubMed] [Google Scholar]
- 19.Puri R, Ballantyne CM, Hoogeveen RC, et al. Lipoprotein(a) and coronary atheroma progression rates during long-term high-intensity statin therapy: insights from SATURN. Atherosclerosis. 2017;263:137-144. doi: 10.1016/j.atherosclerosis.2017.06.026 [DOI] [PubMed] [Google Scholar]
- 20.Tsimikas S, Fazio S, Viney NJ, Xia S, Witztum JL, Marcovina SM. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12(5):1313-1323. doi: 10.1016/j.jacl.2018.07.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol
eTable 1. Baseline Characteristics According to Achieved hsCRP Levels
eTable 2. Biochemistry and Blood Pressure Values at Baseline and Follow-up Within the Overall ACCELERATE hsCRP-Lp(a) Population
eTable 3. Clinical Event Rates Within the Overall ACCELERATE hsCRP-Lp(a) Population
eTable 4. Relationships Between MACE Stratified According to Achieved hsCRP and Lp(a) Quintiles in the Placebo Group Only
eTable 5. Associations Between MACE Stratified According to Achieved hsCRP and Continuous Lp(a) Levels in the Placebo Group Only
eTable 6. Relationships Between Primary MACE End Point Stratified According to Achieved hsCRP and Lp(a) Quintiles
eTable 7. Associations Between Primary MACE End Point Stratified According to Achieved hsCRP and Continuous Lp(a) Levels
eTable 8. Relationships Between hsCRP and Lp(a) as Continuous Variables Upon MACE
eFigure 1. Cumulative Incidence of MACE Over Time Stratified According to Lp(a) Quintiles in the Setting of hsCRP Levels <2 mg/L (A) or ≥2 mg/L (B)
eFigure 2. CONSORT Diagram