

Abstract
LRRK2 has been implicated in endolysosomal function and likely plays a central role in idiopathic Parkinson’s disease (iPD). In iPD, dopaminergic neurons within the substantia nigra are characterized by increased LRRK2 kinase activity, endolysosomal deficits, and accumulation of autophagic vesicles with incompletely degraded substrates, including α-synuclein. Although LRRK2 has been implicated in endolysosomal and autophagic function, it remains unclear whether inhibition of LRRK2 kinase activity can prevent endolysosomal deficits or reduce dopaminergic neurodegeneration. In this study, we characterized the endolysosomal and autophagic defects in surviving dopaminergic neurons of iPD patient brain tissue. We next showed that these defects could be reproduced reliably in vivo using the rotenone model of iPD. Results suggested that there was impaired endosomal maturation, resulting in lysosomal dysfunction and deficits in protein degradation. A highly selective, brain-penetrant LRRK2 kinase inhibitor not only improved apparent endosomal maturation and lysosomal function, but also prevented rotenone-induced neurodegeneration in vivo. The fact that a LRRK2 kinase inhibitor was capable of preventing the neuropathological and endolysosomal abnormalities observed in human iPD suggests that LRRK2 inhibitors may have broad therapeutic utility in iPD, not only in those who carry a LRRK2 mutation.
1. Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized, in part, by the loss of dopaminergic neurons in the substantia nigra and accumulation of cytoplasmic proteinaceous inclusions called Lewy bodies and Lewy neurites. Insoluble α-synuclein is a major component of these inclusions (Spillantini et al., 1997), but other proteins such as ubiquitin and p62 are also present (Watanabe et al., 2012). Although the underlying mechanisms leading to the pathogenesis of PD are not fully understood, accumulating evidence suggests that endolysosomal dysfunction contributes to pathological accumulation of α-synuclein and may underlie disease progression.
Mutations in GBA1 and LRRK2 are the most common genetic risk factors for idiopathic PD (iPD). However, recent work demonstrated that deficits in the lysosomal hydrolase glucocerebrosidase (GCase) activity are also found in the brains of iPD patients who do not harbor a GBA1 mutation (Rocha et al., 2015). In neurons, LRRK2 is a low-abundance protein, but we recently demonstrated that LRRK2 kinase activity is increased in the substantia nigra dopamine neurons of iPD patients, and this sustained LRRK2 kinase activity had pathological consequences (Di Maio et al., 2018). The mechanisms that cause deficits in GCase or increases in LRRK2 kinase activity in iPD are unclear. GCase is a lysosomal hydrolase, whose only known function is to hydrolyze the plasma membrane glycolipids, glucosylceramide (GluCer) and glucosylsphingosine (GluSph). In contrast, LRRK2 localizes to vesicular structures, including endosomes, autophagosomes and lysosomes, and it appears to be involved in vesicular trafficking and autophagy-lysosomal degradation through phosphorylation of various Ras Analog in Brain (Rab) GTPases (Watanabe et al., 2012; Rocha et al., 2015).
Lysosomes are the terminal compartment for the major autophagic and endocytic pathways of degradation (Nixon et al., 2008) and contain essential proteases and hydrolases for degradation. In macroautophagy, substrates are enclosed in double membrane-bound autophagosomes, which can fuse with either late endosomes to form amphisomes, or with lysosomes to form autolysosomes. Substrates targeted for degradation through the endocytic pathway are first endocytosed from the plasma membrane and sorted into early, then late endosomes, and finally lysosomes, where degradation occurs. Deficits in either the endocytic pathway or macroautophagy can promote accumulation of soluble α-synuclein oligomers and may be central to iPD progression (Lee et al., 2004; Mak et al., 2010; Rideout et al., 2004).
The endolysosomal pathway uses a series of cargo vesicles to internalize nutrients and recycle and degrade receptors (via lysosomes). Rab GTPase proteins tightly regulate trafficking of these vesicles from early to late endosomes and then to lysosomes. LRRK2 phosphorylates several of the Rab GTPases that regulate endolysosomal vesicle trafficking (MacLeod et al., 2013; Steger et al., 2017). LRRK2-induced phosphorylation of Rab5 or Rab10 inhibits their function by preventing binding to Rab GDP-dissociation inhibitor factors necessary for membrane delivery and recycling. As such, it is possible that prolonged LRRK2 kinase activity leads to general disruption of maturation from early to late endosomes. If so, this may result in lysosomal impairment because lysosomes rely on dynamic fusion events with late endosomes to maintain their function. Recent findings of deficits in the lysosomal hydrolases, α-galactosidase A and GCase, in brains of iPD patients compared to controls (Rocha et al., 2015; Alcalay et al., 2018), provide support for the idea that lysosomes are defective in iPD.
Abnormalities in endolysosomal health and vesicular trafficking are also implicated in the pathogenesis of neurodegenerative diseases other than iPD (Neefjes and van der Kant, 2014). Accumulation of swollen early endosomes, labeled by Rab5 immunoreactivity, is one of earliest pathological disturbances observed in Alzheimer’s disease (Nixon, 2005), but it is unclear if early endosomes are similarly altered in iPD brains. Although normal function of Rab5-positive early endosomes is critical for endocytosis of α-synuclein – and dysfunction therein may lead to formation intracytoplasmic inclusions in vitro (Sung et al., 2001) – the status of Rab5 in iPD remain to be defined. It remains a possibility that early endosomal accumulation, due to impaired maturation or defective trafficking, may be one of the fundamental mechanisms underlying neurodegenerative diseases, including iPD.
LRRK2 has been implicated in vesicle trafficking and endolysosomal function and it likely plays a central role in iPD pathogenesis; however, it is not clear whether inhibition of LRRK2 kinase activity can prevent endolysosomal deficits or reduce dopaminergic neurodegeneration. In this study, we used postmortem human brain specimens to characterize in more detail the endolysosomal and autophagic defects in surviving dopaminergic neurons. Next, we showed that these defects could be reproduced reliably in adult male rats, using the rotenone model of PD. Finally, we showed that these defects could be prevented by inhibiting LRRK2 kinase activity, ultimately resulting in protection of nigrostriatal neurons.
2. Results
2.1. Endolysosomal deficits in postmortem substantia nigra dopaminergic neurons in iPD
As a general strategy, we used compartment-selective markers and quantitative confocal immunofluorescence to assess the status of early endosomes, late endosomes and lysosomes. Markers of early endosomes included Rab5, as well as transferrin, which is normally recycled from the extracellular space through early endosomes and then released from the cell. Late endosomes contain both mannose-6-phosphate receptor (M6PR) and Lamp1, so subcellular colocalization of these proteins was used to define this compartment. Lamp1-positive puncta that did not colocalize with M6PR were identified as lysosomes.
We found that the number of punctae per cell marked by the early endosomal Rab protein, Rab5, was markedly increased in nigral dopaminergic neurons in iPD brains compared to age-matched controls (P < .001, unpaired, two-tailed t-test). Moreover, the size of each puncta was significantly larger in iPD compared to control tissue (P < .01, unpaired, two-tailed t-test; Fig. 1a–b). These results suggest that early endosomal maturation and/or trafficking may be disrupted. If so, we would expect to see resultant alterations in early endosomal cargoes, such as transferrin.
Fig. 1.
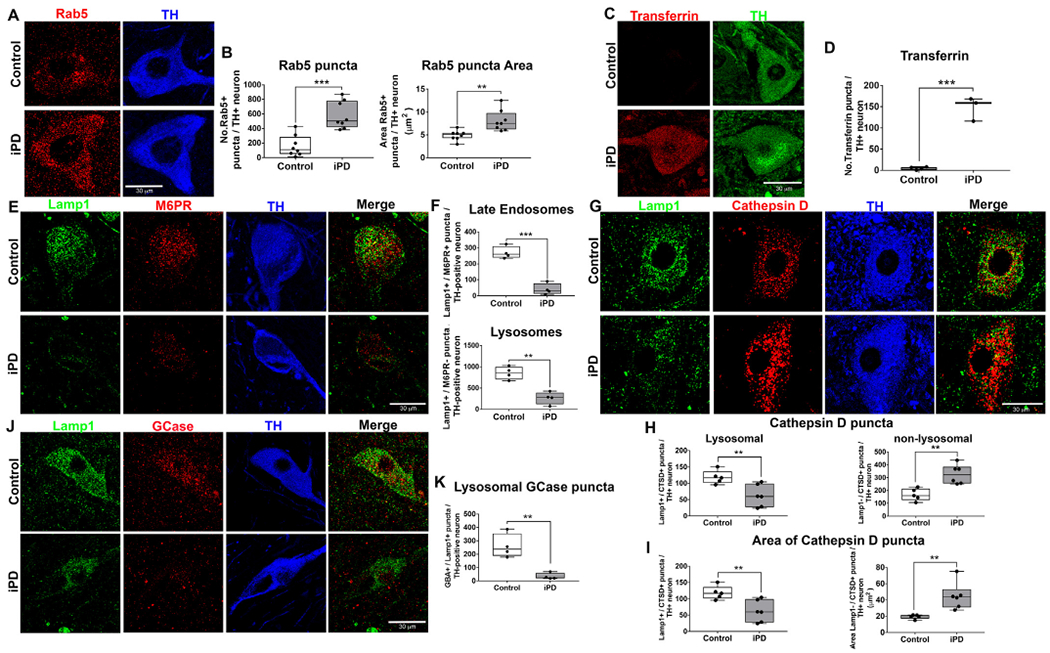
Endolysosomal deficits in human iPD postmortem brain tissue. Paraffin embedded tissue sections of the substantia nigra from age-matched control human brains and individuals with iPD. Immunoreactivity of the early endosomal protein Rab5 in surviving nigral dopaminergic neurons of iPD patients in comparison to age-matched controls (A). Quantification of the average number and size of each Rab5 puncta per dopaminergic neuron in the substantia nigra; n = 8/grp (B). Representative photomicrographs of transferrin in surviving nigral dopaminergic neurons of iPD patients in comparison to age-matched controls (C). Quantification of the average number transferrin puncta per dopaminergic neuron in the substantia nigra; n = 4/grp (D). The number of late endosomes and lysosomes are assessed using specific markers: lysosomal-associated membrane protein 1 (Lamp1) and mannose-6-phosphate receptor (M6PR). The number of late endosomes (Lamp1+ and M6PR+) and lysosomes (Lamp+ and M6PR−) in iPD patient dopaminergic neurons compared with age-matched controls (E). Quantification for average number of late endosomes and lysosomes per dopaminergic neuron in the substantia nigra; n = 4/grp (F). As shown, representative photomicrographs of lysosomal (Lamp+ and cathepsin D +) and non-lysosomal (Lamp− and cathepsin D+) cathepsin D immunoreactivity in surviving nigral dopaminergic neurons of iPD patients in comparison to age-matched controls (G). Quantification for the average number of cathepsin D puncta per dopaminergic neuron in the substantia nigra of iPD and age-matched controls; n = 5/grp (H). Quantification for the average size of each cathepsin D puncta per dopaminergic neuron in the substantia nigra of iPD and age-matched controls; n = 5/grp (I). Representative photomicrographs of lysosomal GCase (GCase+ and Lamp1+) immunoreactivity in surviving nigral dopaminergic neurons of iPD patients in comparison to age-matched controls (J). Quantification of the average number and size of each lysosomal GCase puncta per dopaminergic neuron in the substantia nigra; n = 4/grp (K). Data are analyzed using unpaired t-test ** p < .01, *** p < .001 graphs are expressed as mean ± SEM. Symbols represent individual brains.
Transferrin receptor 1 (TfR1) facilitates the transport of iron across the plasma membrane and its ligand, transferrin, readily binds to extracellular iron, allowing the TfR1 / iron-bound transferrin complex to be internalized by receptor-mediated endocytosis and trafficked to early endosomes. The slightly acidic pH of early endosomes (pH = 6.0) releases iron, allowing it to leave the endosome, whereas transferrin and TfR1 normally recycle back to the plasma membrane via recycling endosomes. Both iron and transferrin accumulate in the dopaminergic neurons of the substantia nigra in PD patients (Mastroberardino et al., 2009; Oakley et al., 2007). In this context, we measured transferrin-positive punctae in the nigral dopaminergic neurons from iPD patients and age-matched controls. We found that the number of transferrin-labeled punctae was significantly increased in the surviving dopaminergic neurons in iPD patients relative to age-matched controls (P < .001, unpaired, two-tailed t-test; Fig. 1c–d). Thus, the transferrin results are congruent with the abnormalities seen in Rab5-labeled early endosomes and, together, they suggest impairment of early endosomal function.
We recently demonstrated that Lamp1 is significantly decreased in substantia nigra dopaminergic neurons in iPD brains, further suggesting endolysosomal deficits (Di Maio et al., 2018). However, Lamp1-positive vesicles include both late endosomes and lysosomes, which are functionally distinct and are differentially vulnerable in various disease states (Lie and Nixon, 2019). Therefore, it was unclear if the reduction in Lamp1 we found in iPD represents late endosomal or lysosomal impairment, or both. Lysosomes are single membrane-bound organelles characterized by a highly acidic lumen, enrichment of acid hydrolases and a lack of M6PR (Saftig and Klumperman, 2009; Lie and Nixon, 2018), whereas late endosomes are multivesicular bodies that contain intraluminal vesicles and are enriched with both Lamp1 and M6PR. Our analysis of iPD brain tissue showed that the average number of late endosomal (M6PR+ / Lamp1 +) punctae was significantly depleted in nigral dopaminergic neurons in iPD compared to age-matched controls (P < .0001, unpaired, two-tailed t-test). Similarly, the average number of lysosomal (M6PR− / Lamp1+) punctae per dopamine neuron was reduced in iPD brains compared to age-matched controls (P < .01, unpaired, two-tailed t-test; Fig. 1e–f).
The reduction of Lamp1 + punctae in the remaining dopaminergic neurons was accompanied by a decrease in lysosomal proteins, including the aspartyl protease, cathepsin D, and the hydrolase, GCase. The average number of lysosomal cathepsin D punctae (Lamp1 + / cathepsin D + ) was significantly decreased in dopaminergic neurons in iPD (P < .01, unpaired, two-tailed t-test). We also found that the average size of each Lamp1 + / cathepsin D + puncta was smaller in iPD dopaminergic neurons (P < .01, unpaired, two-tailed t-test). In contrast, the number of Lamp1−/ cathepsin D+ puncta was significantly increased in dopaminergic neurons of iPD brains (P < .01, unpaired, two-tailed t-test; Fig. 1g–h), and the average size of each Lamp1− / cathepsin D+ puncta was significantly larger in the iPD dopamine neurons (P < .01, unpaired, two-tailed t-test; Fig. 1i). This observed decrease in the number and size of lysosomal cathepsin D punctae, concomitant with an increase in non-lysosomal localization of the protease, may be an indication of impaired structural integrity of lysosomes in iPD.
We and others previously reported that GCase activity is reduced in brains of people with iPD (Rocha et al., 2015; Gegg et al., 2012). Based on these reports, we also measured the number of GCase puncta in nigral dopaminergic neurons. The number of lysosomal GCase puncta (GCase+ / Lamp1+) was significantly reduced in the iPD brains, providing further support for lysosomal dysfunction in iPD neurons (P < .01, unpaired, two-tailed t-test; Fig. 1j–k).
2.2. Rotenone caused neuropathological alteration of nigrostriatal neurons
Rats treated with rotenone for 9–14 days until they reach a behavioral endpoint of severe parkinsonism and reproduce many features of iPD, including endolysosomal defects, accumulation of α-synuclein, and nigrostriatal degeneration (Di Maio et al., 2018; Cannon et al., 2009). In the current study, in addition to treating a group of rats until endpoint was reached (endpoint dosing), another group of animals received only 5 daily doses of rotenone (acute dosing). Histopathological analysis revealed that rats dosed with acute rotenone (5 doses) did not have a loss of tyrosine hydroxylase (TH)-positive terminals in the dorsal striatum, or of dopamine neurons in the substantia nigra. Thus, this regime was considered “pre-degenerative”. However, rats treated to endpoint had a 35% loss TH-positive terminals in the striatum (P < .001, unpaired, two-tailed t-test; Fig. 2b–c), which corresponded to a 45% loss of TH- and Nissl-positive dopaminergic neuronal cell bodies in the substantia nigra (P < .0001, unpaired, two-tailed t-test; Fig. 2d–e).
Fig. 2.
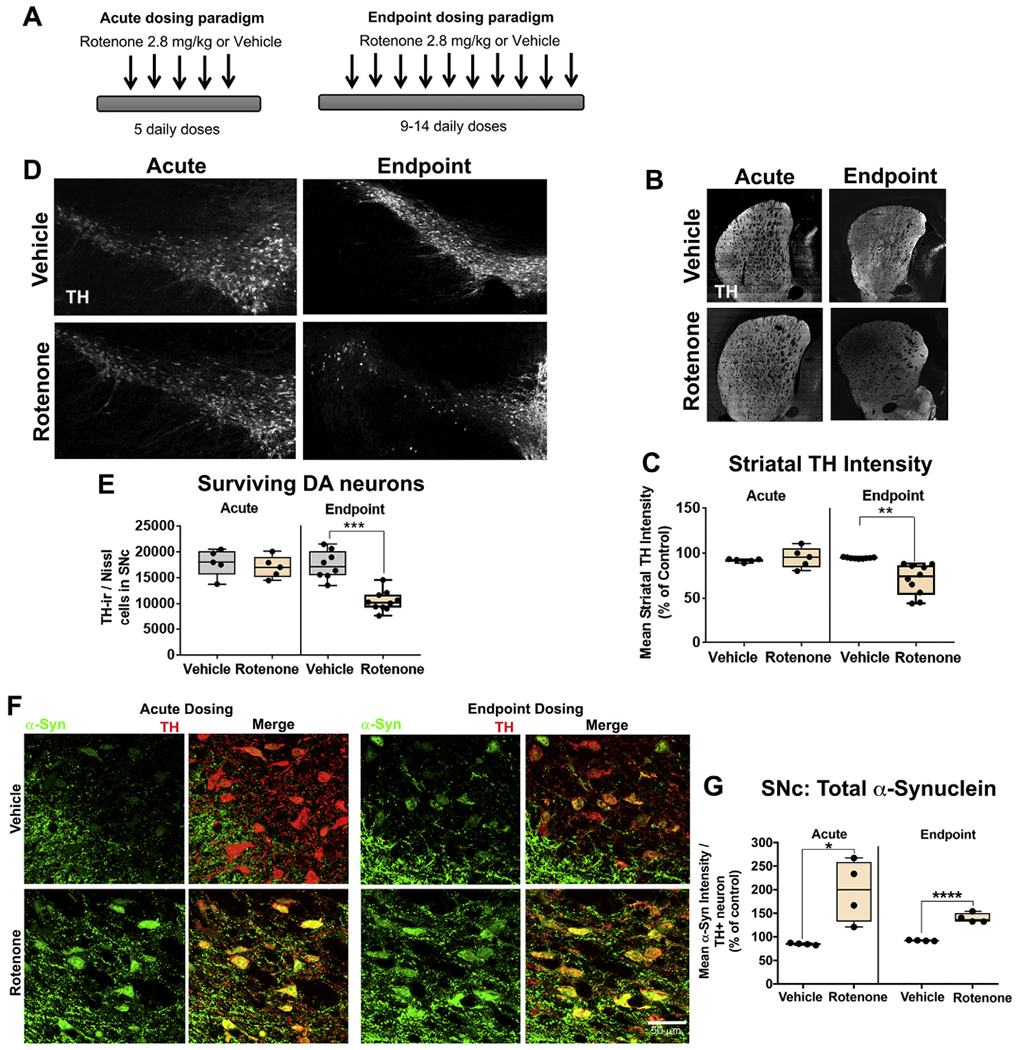
Rotenone causes α-synuclein accumulation prior to dopaminergic degeneration. Schematic of acute and endpoint dosing paradigms (A). Nigrostriatal dopaminergic degeneration was assessed by immunohistochemistry using specific markers; tyrosine hydrolase (TH) for dopaminergic neurons and fluorescent Nissl (Neurotrace) for all neurons of aged rats treated with 5 doses (acute) of rotenone or 9–14 doses (endpoint) of rotenone. As shown, representative photomicrographs of striatal dopaminergic terminals of rats treated with 5 doses (acute) of rotenone and 9–14 doses (endpoint) of rotenone (B). Quantification of striatal dopaminergic terminals loss in rats dosed with acute or endpoint rotenone; n = 5–8/grp (C). Representative photomicrographs of dopaminergic neuronal survival in the substantia nigra of rats treated with 5 doses (acute) of rotenone and 9–14 doses (endpoint) of rotenone (D). Quantification of the total number of dopaminergic neurons in the substantia nigra pars compacta of rats treated with 5 doses (acute) of rotenone and 9–14 doses (endpoint) of rotenone using a semi-automatic stereology protocol (E). Total endogenous α-synuclein levels in dopaminergic neurons in the substantia nigra following either acute or endpoint rotenone treatment (F). Quantification of the average α-synuclein intensity per nigral dopaminergic neuron of rats dosed with either acute or endpoint rotenone; n = 5–8/grp (G). Data are analyzed using unpaired t-test ** p < .01, *** p < .001 graphs are expressed as mean ± SEM. Symbols represent individual brains.
α-Synuclein accumulation is a major hallmark of iPD, and this can be reproduced by rotenone, which has been shown to cause accumulation of endogenous wildtype α-synuclein in dopaminergic neurons (Cannon et al., 2009). We measured total α-synuclein levels in nigral dopaminergic neurons by measuring the intensity of α-synuclein immunofluorescence. We found that α-synuclein significantly increased in dopaminergic neurons of rats treated with acute rotenone (P < .05, unpaired, two-tailed t-test; Fig. 2f–g) as well as with endpoint rotenone (P < .0001, unpaired, two-tailed t-test; Fig. 2f–g). These data demonstrate that, in rotenone-treated rats, α-synuclein protein levels rise in the dopaminergic neurons prior to the onset of frank neurodegeneration. Moreover, if proper endolysosomal function is required for normal α-synuclein homeostasis, these results also suggest that endolysosomal impairment occurs early in the pathogenic cascade.
2.3. Rotenone impaired endogenous GCase and altered levels of glycolipids
We next examined whether GCase activity is reduced in substantia nigra after rotenone treatment and, if so, whether this occurred early or late in pathogenesis. After acute dosing of rotenone or vehicle for 5 days, the substantia nigra was dissected and homogenized, and a fluorometric assay was used to measure GCase activity. We found that acute, pre-degenerative rotenone dosing caused a ~25% reduction in endogenous GCase activity in the substantia nigra compared to vehicle treated rats (P < .01, unpaired, two-tailed t-test, Fig. 3a). To characterize the impact of rotenone on glycolipid metabolism, we measured glucosylsphingosine (GluSph), glucosylceramide (GluCer), as well as ceramide using tandem mass spectrometry in samples from striatum, cortex, and hippocampus of rats dosed with acute rotenone. (Substantia nigra samples were not available as they had been consumed in the activity assay.) Consistent with the GCase activity assay, acute rotenone dosing caused an accumulation of GluSph in the striatum (P < .01, unpaired, two-tailed t-test) and hippocampus (P = .06, unpaired, two-tailed t-test; Fig. 3d). Neither GluCer levels nor ceramide levels changed in rats dosed with acute rotenone (Fig. 3d). To determine whether the loss of GCase activity was reflected in a loss GCase protein levels, we measured lysosomal GCase (GCase+ / Lamp1+) punctae in nigral dopaminergic neurons and unexpectedly found an increase in the number of lysosomal GCase puncta (P < .05, unpaired, two-tailed t-test; Fig. 3b–c).
Fig. 3.
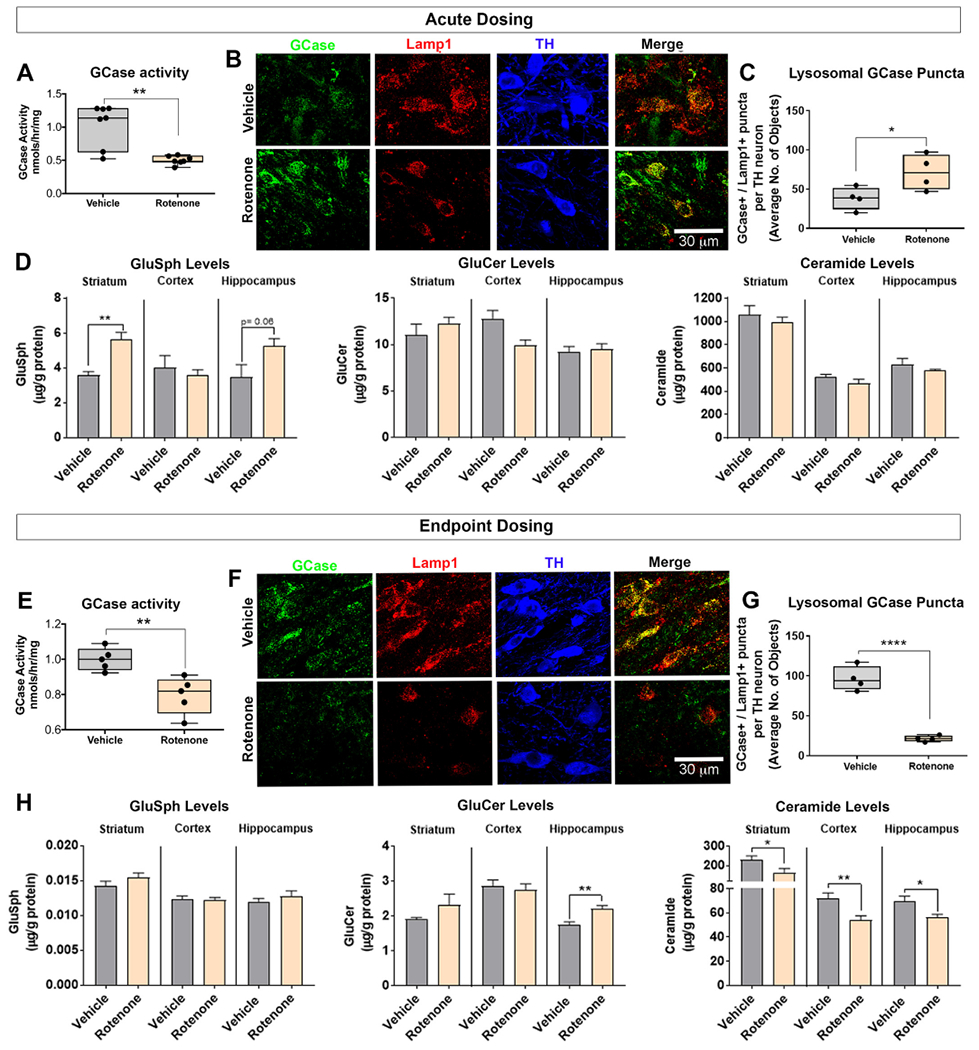
Rotenone impaired endogenous GCase and altered levels of glycolipids. GCase activity was measured in the substantia nigra using lOmM 4-Mu-β-d-glucopyranoside substrate activity assay in rats dosed with vehicle or acute rotenone (A). As shown, representative photomicrographs of lysosomal GCase (Lamp1+ and GCase+ ) puncta in nigral dopaminergic neurons in rats dosed with vehicle or acute rotenone (B). Quantification of the average number of lysosomal GCase puncta per nigral dopaminergic neuron in rats dosed with vehicle or acute rotenone; n = 4/grp (C). GluSph, GluCer and ceramide levels were measured using tandem mass spectrometry in the striatum, cortex and hippocampus in rats dosed with either vehicle or acute rotenone; n = 5/grp (D). GCase activity was measured in the substantia nigra using 10 mM 4-Mu-β-d-glucopyranoside substrate activity assay in rats dosed with vehicle or endpoint rotenone; n = 8/grp (E). Immunoreactivity of lysosomal GCase (Lamp1+ and GCase+) puncta in nigral dopaminergic neurons in rats dosed with vehicle or endpoint rotenone (F). Quantification of the average number of lysosomal GCase puncta per nigral dopaminergic neuron in rats dosed with vehicle or endpoint rotenone; n = 8/grp (G). GluSph, GluCer and ceramide levels were measured using tandem mass spectrometry in the striatum, cortex and hippocampus in rats dosed with either vehicle or endpoint rotenone; n = 8/grp (H). Data are analyzed using unpaired t-test ** p < .01, *** p < .001 graphs are expressed as mean ± SEM. Symbols represent individual brains.
Another cohort of rats was treated with rotenone or vehicle for 9–14 days (endpoint dosing). We first measured GCase activity in the substantia nigra of these rats and found that endpoint rotenone dosing caused a ~25% reduction in endogenous GCase activity (P < .01, unpaired, two-tailed t-test; Fig. 3e). To further characterize rotenone induced GCase related deficits, we measured GluSph, GluCer, as well as the downstream product, ceramide, in samples of striatum, cortex, and hippocampus. Endpoint rotenone dosing caused an accumulation of GluCer in the striatum (P = .06, unpaired, two-tailed t-test) and hippocampus (P < .01, unpaired, two-tailed t-test) and a significant decrease in ceramide levels in the striatum (P < .05, unpaired, two-tailed t-test), cortex (P < .01, unpaired, two-tailed t-test) and hippocampus (P < .05, unpaired, two-tailed t-test; Fig. 3h). Next, we examined whether this decrease in GCase activity corresponded to a change in GCase punctae. In contrast to rats dosed with acute rotenone, endpoint rotenone dosing caused a significant reduction in lysosomal GCase (GCase+ / Lamp1+) punctae in nigral dopaminergic neurons (P < .0001, unpaired, two-tailed t-test; Fig. 3f–g).
2.4. Rotenone mimicked iPD by causing widespread endolysosomal dysfunction in rats
To determine whether the rotenone model can cause the same endolysosomal deficits as observed in iPD postmortem brains (Fig. 1), rats received either acute or endpoint rotenone dosing. We found that the average number of lysosomal (M6PR− / Lamp1+) punctae was also significantly reduced in dopaminergic neurons of rats dosed with either acute (P < .0001, unpaired, two-tailed t-test) or endpoint rotenone (P < .01, unpaired, two-tailed t-test) compared to vehicle treated rats (Fig. 4a–b). Similarly, the number of late endosomal (M6PR+ / Lamp1+) punctae was significantly depleted within nigral dopaminergic neurons of rats dosed with acute rotenone (P < .01, unpaired, two-tailed t-test) and endpoint rotenone (P < .001, unpaired, two-tailed t-test) in comparison to vehicle treated rats (Fig. 4a, c).
Fig. 4.
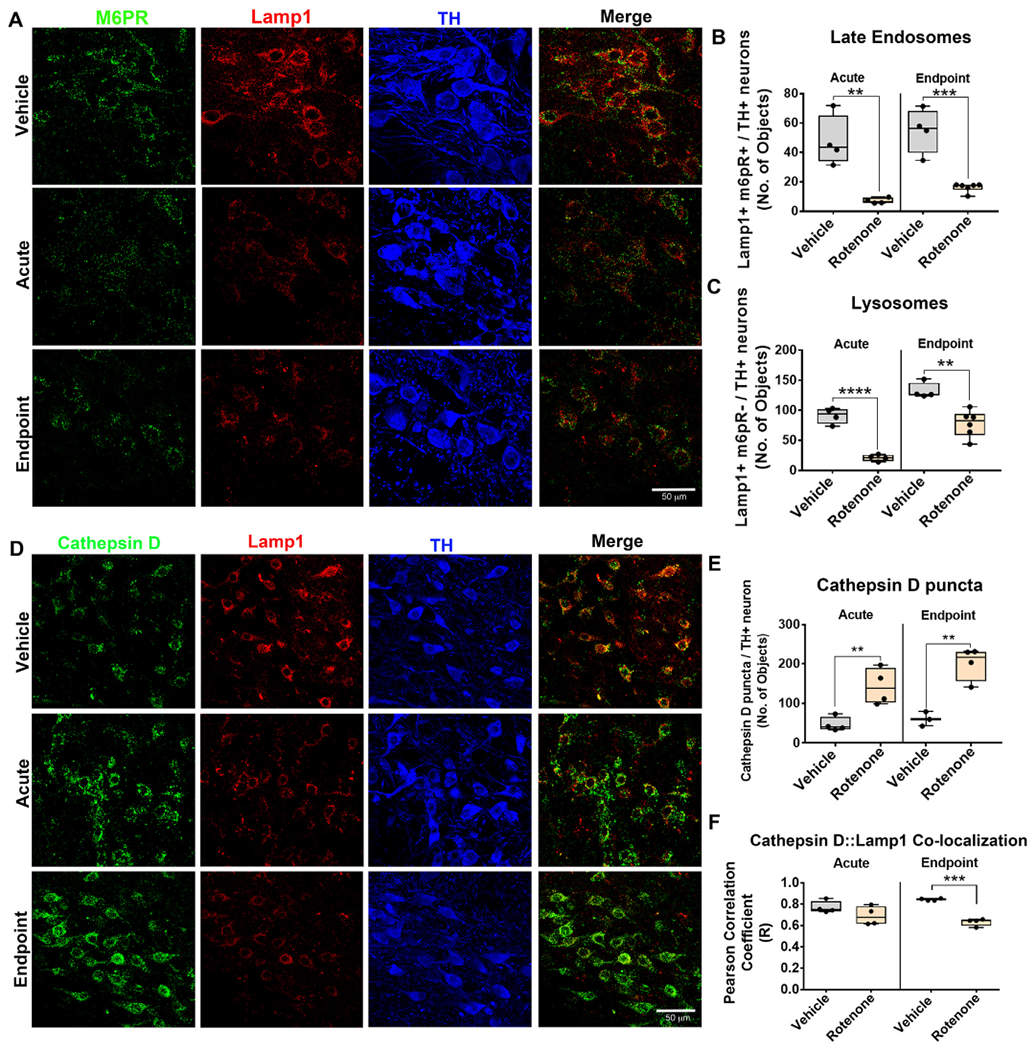
Rotenone caused endolysosomal deficits in rats. The number of late endosomes and lysosomes in nigral dopaminergic neurons were assessed by immunohistochemistry using Lamp1 and M6PR for late endosomes and lysosomes and TH for dopaminergic neurons. The number of late endosomes (Lamp 1+, M6PR+) and lysosomes (Lamp 1+, M6PR−) in nigral dopaminergic neurons of rats dosed with vehicle, acute or endpoint rotenone (A). Quantification for the average number of late endosomal and lysosomal puncta in nigral dopaminergic neurons in rats dosed with acute and endpoint rotenone; n = 4/grp (B–C). Lysosomal and non-lysosomal cathepsin D immunoreactivity was assessed in nigral dopaminergic neurons using specific markers. The number of lysosomal cathepsin D (Lamp 1+, cathepsin D+) and non-lysosomal cathepsin D (Lamp 1−, cathepsin D+) in nigral dopaminergic neurons of rats dosed with vehicle, acute or endpoint rotenone (D). Quantification for average number of cathepsin D puncta per nigral dopaminergic neuron; n = 4/grp (E). Quantification for the average number of lysosomal cathepsin D puncta per dopaminergic neuron assessed using Pearson correlation coefficient (F). Data are analyzed using unpaired t-test ** p < .01, *** p < .001, **** p < .001 graphs are expressed as mean ± SEM. Symbols represent individual brains.
Cathepsin D is a lysosomal protease known for degrading α-synuclein in the lysosome (Cullen et al., 2009). Given that rotenone causes endogenous α-synuclein to accumulate (Fig. 2), one might expect rotenone to cause changes in cathepsin D levels or subcellular localization. We found that the average number of cathepsin D punctae was significantly increased within dopaminergic neurons of rats dosed with either acute (P < .01, unpaired, two-tailed t-test) or endpoint (P < .01, unpaired, two-tailed t-test) rotenone compared to vehicle treated rats (Fig. 4d–e). To determine whether the accumulation of cathepsin D occurred within lysosomes we measured the pixel-by-pixel Pearson correlation coefficient between Lamp1 and cathepsin D. With acute rotenone dosing, cathepsin D colocalized with Lamp1 in dopaminergic neurons. However, the colocalization (Pearson correlation coefficient) between Lamp1 and cathepsin D decreased significantly in the rats dosed with endpoint rotenone (P < .001, unpaired, two-tailed t-test; Fig. 4f). Accumulation of cathepsin D punctae negative for Lamp1 was consistent with our data collected in human iPD tissue (Fig. 1).
The accumulation of Rab5-positive, enlarged early endosomes, we found in iPD (Fig. 1) reproduces key aspects of the early endosomal phenotype found in AD (Cataldo et al., 2000), and likely has downstream consequences in other compartments, including lysosomes. Here, as in the human iPD brains, we found that Rab5-positive punctae (early endosomes) increase in substantia nigra dopaminergic neurons in rats dosed with either acute (P < .05, unpaired, two-tailed t-test) or endpoint (P < .001, unpaired, two-tailed t-test) rotenone in comparison to vehicle treated rats (Fig. 5a–b).
Fig. 5.
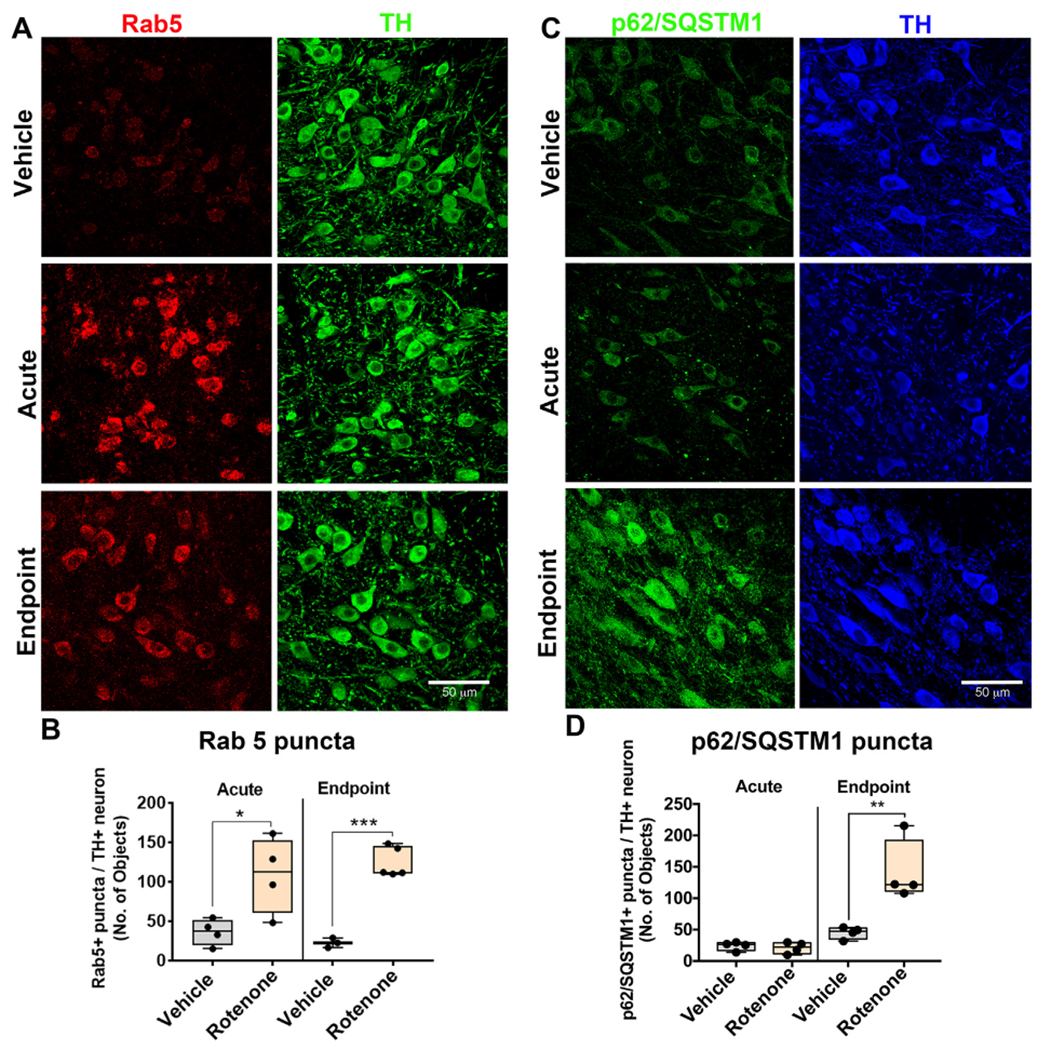
Rotenone causes accumulation of early endosomes prior to deficits in macroautophagy in rats. The number of early endosomes in dopaminergic neurons were assessed by immunohistochemistry using the early endosomal specific marker Rab5 and the dopaminergic specific marker TH in aged rats. Representative photomicrographs of Rab5 (Red) in nigral TH+ (Green) neurons in rats dosed with vehicle, acute or endpoint rotenone (A). The quantification for the average number of Rab5 puncta per dopaminergic neuron; n = 4/grp (B). The number of autophagosomes in dopaminergic neurons were assessed by immunohistochemistry using the p62/SQSTM1 and TH (C). The quantification for the average number of p62/SQSTM1 puncta per dopaminergic neuron; n = 4/grp (D). Data are analyzed using unpaired t-test * p < .05, ** p < .001, *** p < .001 graphs are expressed as mean ± SEM. Symbols represent individual brains. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
This initial accumulation of early endosomes may reflect impairment of maturation or trafficking of early endosomes and may initiate the downstream endolysosomal deficits caused by rotenone. Eventually, such endolyososmal deficits would be expected to impact macroautophagy. Consistent with the idea that endolysosomal defects precede dysfunctional macroautophagy, only rats dosed with endpoint rotenone displayed a significant accumulation of p62/SQSTM1-positive autophagosomes (P < .01, unpaired, two-tailed t-test) in comparison to vehicle treated rats (Fig. 5c–d).
2.5. LRRK2 kinase inhibition prevented rotenone-induced endolysosomal defects
We previously reported that endogenous wildtype LRRK2 kinase activity is significantly increased in nigral dopaminergic neurons in iPD (Di Maio et al., 2018) and this can be reproduced in rotenone-treated rats. Furthermore, we provided preliminary results suggesting the LRRK2 activation was accompanied by endolysosomal dysfunction in the remaining dopaminergic neurons. Additional experiments showed that with acute rotenone dosing, co-treatment with a brain-penetrant LRRK2 kinase inhibitor, PF-360 (Pfizer), was able to prevent early endolysosomal dysfunction (Di Maio et al., 2018). Therefore, in the current study, we assessed whether blocking LRRK2 kinase activity with PF-360 could improve lysosomal health, promote degradation of synuclein and prevent nigral dopaminergic neurodegeneration. For these experiments, rats were dosed with rotenone or its vehicle, with or without co-treatment with PF-360 or its vehicle, until each rotenone + vehicle rat reached its behavioral endpoint.
In this cohort of animals, endpoint rotenone caused a significant decrease in the number of lysosomes (F1,13 = 4.686, p < .05, two-way ANOVA) and late endosomes (F1,13 = 15.06, p < .001, two-way ANOVA) in the surviving dopaminergic neurons (Fig. 6a–c). Co-treatment with the LRRK2 kinase inhibitor, PF-360, prevented the rotenone-induced loss of lysosomes (F1,13 = 6.385, p < .05, two-way ANOVA) and late endosomes (F1,12 = 7.461, p < .05, two-way ANOVA) (Fig. 6b–c). Similarly, PF-360 also prevented the rotenone-induced decrease in lysosomal GCase puncta (F1,12 = 11.76, p < .01, two-way ANOVA) in dopaminergic neurons (Fig. 6d–e).
Fig. 6.
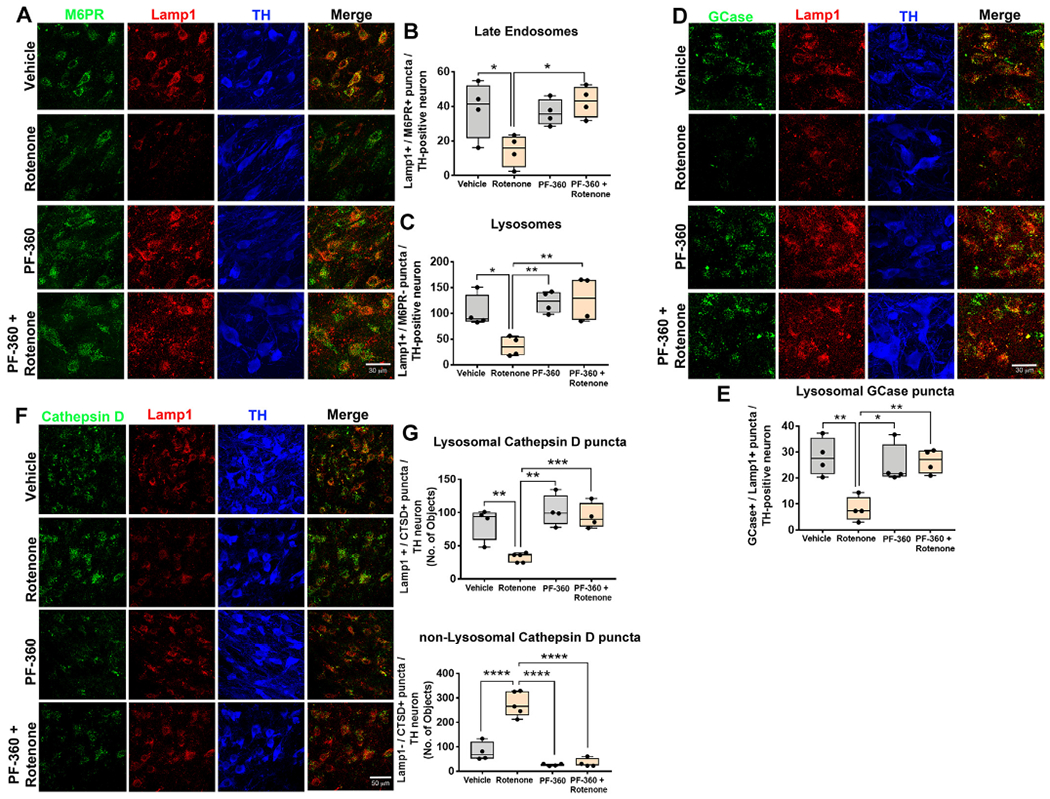
LRRK2 kinase inhibition prevented rotenone-induced endolysosomal deficits in rats. Endolysosomal deficits were assessed by immunohistochemistry in rats dosed with endpoint rotenone in the presence or absence of the LRRK2 kinase inhibitor PF-360. As shown, late endosomal (Lamp1+ and M6PR+) and lysosomal (Lamp1+ and M6PR−) puncta in rats treated with vehicle, PF-360 alone, endpoint rotenone alone or rotenone + PF-360. TH used as a marker of dopaminergic neurons. Representative photomicrographs for late endosomes and lysosomes in nigral dopaminergic neurons using M6PR (Green), Lamp1 (Red) and TH (Blue) (A). Quantification for the number of late endosomal and lysosomal puncta per nigral dopaminergic neuron; n = 4/grp (B–C). Lysosomal GCase (Lamp1+ and GCase+) in nigral dopaminergic neurons assessed by immunohistochemistry in rats dosed with endpoint rotenone in the presence or absence of the LRRK2 kinase inhibitor PF-360. As shown, representative photomicrographs of GCase (Green), Lamp1 (Red) and TH (Blue) in rats treated with vehicle, PF-360 alone, endpoint rotenone alone or rotenone + PF-360 (D). Quantification for the number of lysosomal GCase puncta per nigral dopaminergic neuron; n = 4/grp (E). Lysosomal and non-lysosomal cathepsin D immunoreactivity assessed in nigral dopaminergic neurons using specific markers. Representative photomicrographs for lysosomal cathepsin D (cathepsin D+ , Lamp 1+) and non-lysosomal (cathepsin D+ , Lamp 1−) puncta in nigral dopaminergic neurons using cathepsin D (Green), Lamp1 (Red) and TH (Blue) (F). Quantification for average number of cathepsin D puncta per nigral dopaminergic neuron; n = 4/grp (G). Data are analyzed using two-way ANOVA and Tukey post hoc analysis. * p < .05, ** p < .01, graphs are expressed as mean ± SEM. Symbols represent individual brains. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Endpoint rotenone caused a reduction in the average number of lysosomal cathepsin D puncta in the surviving dopaminergic neurons (F1,13 = 10.47, p < .01, two-way ANOVA) and accumulation of cytoplasmic cathepsin D (F1,13 = 35.85, p < .0001, two-way ANOVA). PF-360 prevented the rotenone-induced decrease in lysosomal cathepsin D puncta (F1,13 = 5.5, p < .05, two-way ANOVA) and the accumulation of cytoplasmic cathepsin D (F1,13 = 30.67, p < .0001, two-way ANOVA; Fig. 6 f–g). These data suggest that LRRK2 inhibition may preserve lysosomal integrity.
LRRK2 localizes to vesicular structures including endosomes and autophagosomes and is involved in vesicular trafficking and autophagic-lysosomal degradation through phosphorylation of various Rab GTPases, including Rab10 (Alessi and Sammler, 2018; Kalogeropulou et al., 2018). Because Rab10 is phosphorylated at Thr73 exclusively by LRRK2 (Lis et al., 2018), measurement of pThr73-Rab10 can serve as a surrogate marker of LRRK2 kinase activity. As we reported previously (Di Maio et al., 2018), endpoint rotenone dosing caused an increase of pThr73-Rab10 in surviving dopaminergic neurons (F1,13 = 23.23 p < .01, two-way ANOVA). Co-treatment with PF-360 prevented the accumulation of pThr73-Rab10 (F1,13 = 20.62 p < .01, two-way ANOVA), indicating that PF-360 blocked LRRK2 kinase activation (Fig. 7a–b).
Fig. 7.
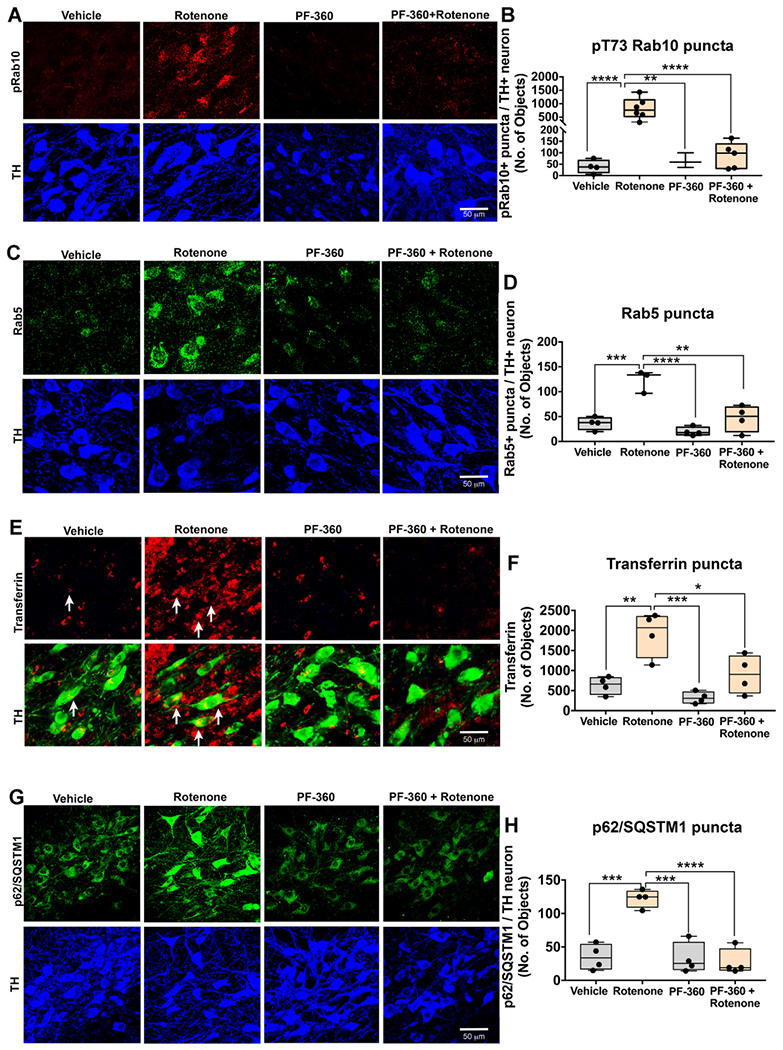
LRRK2 kinase inhibition prevents rotenone-induced protein trafficking deficits in rats. As shown, phosphorylated (T73) Rab10, and Rab5, p62/SQSTM1 and Transferrin puncta were assessed by immunohistochemistry in rats dosed with endpoint rotenone in the presence or absence of the LRRK2 kinase inhibitor PF-360. As shown, representative photomicrographs for pRab10 (Red) puncta in rats treated with vehicle, PF-360 alone, endpoint rotenone alone or rotenone + PF-360. TH (Blue), a marker of dopaminergic neurons (A). Quantification for average number of pRab10 puncta per nigral dopaminergic neuron; n = 4–6/grp (B). As shown, representative photomicrographs for Rab5 (Green) puncta and TH (Blue) in rats treated with vehicle, PF-360 alone, endpoint rotenone alone or rotenone + PF-360 (C). Quantification for average number of Rab5 puncta per nigral dopaminergic neuron; n = 4/grp (D). Representative photomicrographs for Transferrin (Red) and TH (Green) in nigral dopaminergic neurons of rats treated with vehicle, PF-360 alone, endpoint rotenone alone or rotenone + PF-360 (E). Quantification for average number of Transferrin puncta per nigral dopaminergic neuron; n = 4/grp (F). Representative photomicrographs for p62/SQSTM1 puncta (Green) and TH (Blue) per nigral dopaminergic neurons of rats treated with vehicle, PF-360 alone, endpoint rotenone alone or rotenone + PF-360 (G). Quantification for average number of p62/SQSTM1 puncta per nigral dopaminergic neuron; n = 4/grp (H). Data are analyzed using two-way ANOVA and Tukey post hoc analysis. * p < .05, ** p < .01, *** p < .001, **** p < .001 graphs are expressed as mean ± SEM. Symbols represent individual brains. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Endpoint rotenone caused aberrant accumulation of Rab5-positive early endosomes (F1,11 = 34.16 p < .0001, two-way ANOVA) and the LRRK2 inhibitor, PF-360, prevented this accumulation (F1,11 = 9.648 p < .0001, two-way ANOVA; Fig. 7 c–d). The early endosomal cargo, transferrin, accumulated in nigral dopaminergic neurons of rats dosed with endpoint rotenone (F1,12 = 61.11 p < .0001, two-way ANOVA), and this was prevented by LRRK2 kinase inhibition (F1,11 = 15.58 p < .05, two-way ANOVA; Fig. 7 e–f).
LRRK2 interacts with and phosphorylates the macroautophagy signaling adapter p62/SQSTM1 (Kalogeropulou et al., 2018). Therefore, we investigated the role of LRRK2 in p62/SQSTM1 accumulation in rats dosed with rotenone. Endpoint rotenone dosing caused an accumulation of p62/SQSTM1 punctae in the surviving dopaminergic neurons (F1,12 = 22.97 p < .01, two-way ANOVA) and PF-360 prevented this increase (F1,12 = 32.4 p < .001, two-way ANOVA; Fig. 7 g–h).
2.6. LRRK2 kinase inhibition prevented rotenone-induced α-symiclein accumulation and neurodegeneration
This study and others have shown that rotenone causes accumulation of endogenous α-synuclein within dopaminergic neurons (Cannon et al., 2009; De Miranda et al., 2019). Because PF-360 preserves endolysosomal function and the autophagic receptor, p62/SQSTM1, it was of interest to determine whether it would have the downstream effect of preventing α-synuclein accumulation. We found that the LRRK2 kinase inhibitor, PF-360 prevented rotenone-induced accumulation of total α-synuclein in the nigral dopaminergic neurons (F1,14 = 15.12 p < .05, two-way ANOVA; Fig. 8a,c). Next, we looked at whether a LRRK2 inhibitor can prevent accumulation of rotenone-induced phosphorylated Serine 129 (pSer129) α-synuclein. We found that rotenone caused an increase pSer129-α-synuclein and treatment with PF-360 prevented this accumulation in the nigral dopaminergic neurons (F1,14 = 14.15 p < .001, two-way ANOVA; Fig. 8b,d).
Fig. 8.
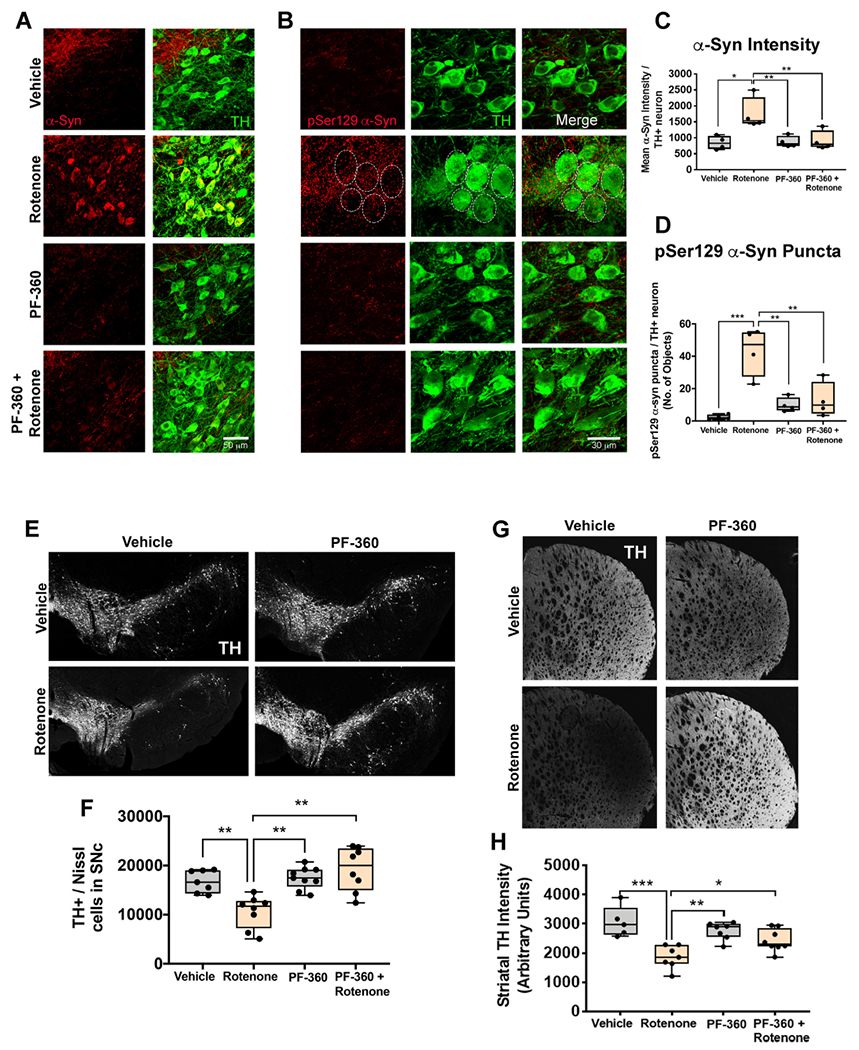
LRRK2 kinase inhibition prevented rotenone-induced dopaminergic degeneration and α-synuclein accumulation in rats. Total endogenous α-synuclein levels was assessed in surviving dopaminergic neurons in the substantia nigra in rats treated with endpoint rotenone in the presence or absence of PF-360. Representative photomicrographs of endogenous α-synuclein (Red) in dopaminergic neurons (Green) (A). Endogenous pSer129 α-synuclein levels was assessed in surviving dopaminergic neurons in the substantia nigra in rats treated with endpoint rotenone in the presence or absence of PF-360. Representative photomicrographs of pSer129 α-synuclein (Red) in dopaminergic neurons (Green) (B). Quantification of the average total α-synuclein intensity per nigral dopaminergic neuron; n = 4/grp (C). Quantification of pSer129 α-synuclein puncta per nigral dopaminergic neuron; n = 4/grp (D). Nigrostriatal dopaminergic degeneration was assessed in aged rats treated with endpoint rotenone in the presence or absence of the LRRK2 kinase inhibitor, PF-360 using specific markers; TH for dopaminergic neurons and fluorescent Nissl (Neurotrace) for all neurons. Representative photomicrographs of surviving dopaminergic neurons in the substantia nigra of rats treated with endpoint rotenone in the presence or absence of PF-360 (E). Quantification of the total number of nigral dopaminergic neurons using a semi-automatic stereology protocol; n = 8/grp (F). Photomicrographs of dopaminergic terminals in the striatum of rats treated with endpoint rotenone in the presence or absence of PF-360 using a TH specific antibody (G). Quantification of the average intensity values for dopaminergic terminals using TH; n = 5–8/grp (H). Data are analyzed using two-way ANOVA and Tukey post hoc analysis. *p < .05, ** p < .01, *** p < .001 graphs are expressed as mean ± SEM. Symbols represent individual brain. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Pathological accumulation of α-synuclein has been strongly implicated in dysfunction and death of neurons in iPD. Given that PF-360 treatment prevented α-synuclein buildup, we examined the status of nigrostriatal dopamine neurons. Histopathological analysis revealed that rats dosed with endpoint rotenone had a significant loss of TH- and Nissl-positive dopaminergic neuronal cell bodies within the substantia nigra (F1,29 = 11.92 p < .01, two-way ANOVA), which was prevented by treatment with PF-360 (F1,29 = 15.81 p < .01, two-way ANOVA; Fig. 8e–f). This loss of substantia nigral cell bodies corresponded to loss of striatal TH+ fibers (F1,23 = 24.57 p < .0001, two-way ANOVA), which was also prevented by PF-360 (F1,23 = 8.099 p < .01, two-way ANOVA; Fig. 8g–h).
3. Discussion
Although many neuronal cell types are affected in iPD, the substantia nigra dopamine neurons are the best studied. Among other findings, affected neurons in iPD are characterized by increased LRRK2 kinase activity, endolysosomal deficits and accumulation of autophagic vesicles with incompletely degraded substrates, including α-synuclein (Di Maio et al., 2018; Chu et al., 2009; Dehay et al., 2010). In this study, we used postmortem human brain specimens to characterize in more detail the endolysosomal and autophagic defects in surviving dopamine neurons in iPD. Next, we showed that these defects could be reproduced reliably in the rat using rotenone. Finally, we showed that the defects observed in human specimens, which can be reproduced in rats, can be prevented effectively by inhibiting LRRK2 kinase activity – and this results in protection of nigrostriatal neurons. Thus, this study provides compelling support for LRRK2 kinase as a therapeutic target for individuals with PD, independent of LRRK2 mutation status.
The formation of Lewy bodies and Lewy neurites in surviving dopaminergic neurons is a major pathological feature of the human disease. We previously reported that acute rotenone dosing caused an accumulation of pSer129-α-synuclein, and treatment with PF-360 prevented this accumulation (Di Maio et al., 2016). We now extend these findings by showing that rats dosed with endpoint rotenone also had accumulation of both endogenous total and pSer129-α-synuclein levels in nigrostriatal dopamine neurons – and co-treatment with PF-360 prevented this accumulation. Although the mechanism by which α-synuclein accumulates in response to rotenone is unclear, the accumulation of α-synuclein does not occur at the transcript level (Sherer et al., 2002). As oligomeric and pSer129-α-synuclein are thought to be degraded by autophagy (Kalogeropulou et al., 2018; Lis et al., 2018), it is possible that deficits in the endolysosomal pathway contribute to the observed accumulation α-synuclein. Moreover, we previously demonstrated that the chaperone-mediated autophagy (CMA) marker Lamp2A and endolysosomal marker Lamp1, were dramatically lost after acute rotenone treatment, and preserved by co-treatment with PF-360 (Di Maio et al., 2016). Our data suggests that rotenone causes an early and profound impairment in autophagy-lysosomal degradation. The observed changes in the endo-lysosomal pathway likely contribute to endogenous α-synuclein accumulation either directly or indirectly. Moreover, inhibition of LRRK2 kinase activity using PF-360 not only prevented endolysosomal impairment but also stopped the accumulation of α-synuclein caused by rotenone.
Dopaminergic neurons are highly vulnerable to oxidant stress and rotenone is known to cause accumulation of reactive oxygen species (ROS). This enhanced sensitivity to oxidative stress may in part be due to dopamine itself, which can auto-oxidize and accumulate in the cytosol following rotenone treatment. The highly reactive dopamine-quinone so formed can lead to further to mitochondrial dysfunction and ROS production and oxidative damage, as reviewed by Hastings (Hastings, 2009). Dopamine and its oxidation products can lead to deficits in GCase activity and lysosomal dysfunction (Burbulla et al., 2017). Both rotenone and elevated α-synuclein levels can increase the formation of ROS and activate LRRK2 (Di Maio et al., 2018). As rotenone causes an increase in LRRK2 kinase activity and LRRK2 is associated with increases in oxidative stress (Di Maio et al., 2018; Heo et al., 2010), it raises the possibility that components of the endolysosomal pathway may be damaged by oxidation and/or nitration leading to further accumulation of α-synuclein. Moreover, rotenone-induced activation of LRRK2 kinase activity leads to phosphorylation of its Rab GTPase substrates which, in turn, may interfere with their key roles in vesicle trafficking and endolysosomal function. Additionally, mitochondrial inhibition by rotenone has been shown to cause deficits in lysosomal biogenesis. In fact, rotenone can cause a decrease in mRNA of several degradative lysosomal enzyme genes, most likely because of oxidant stress (Fernandez-Mosquera et al., 2017). More in-depth mechanistic studies are needed to elucidate the underlying mechanism linking rotenone to endolysosomal deficits. Nonetheless, the rotenone model recapitulates core pathogenic and pathological features of iPD, which makes it a highly valuable tool to study underlying mechanisms of iPD.
Abnormalities of endocytosis and of early endosomes may be of fundamental importance and occur early in the pathogenic cascade of neurodegenerative diseases. For example, it has been reported that swollen Rab5-positive early endosomes accumulate in neurons very early in sporadic Alzheimer’s disease, even before there is deposition of amyloid beta (Nixon, 2005). Similarly, we observed accumulation of swollen Rab5-positive early endosomes in dopaminergic neurons in iPD; however, our studies of end-stage iPD human brain tissue could not tell us how early this occurs in the disease process. On the other hand, in rats treated with 5 days of rotenone (pre-degenerative, acute dosing), there was already Rab5 accumulation before any evidence of cell loss. At this early time point, we also observed concomitant accumulation of α-synuclein. α-Synuclein accumulation may contribute to the observed accumulation of Rab5 within the dopaminergic neurons. This is supported by recent data demonstrating that a transgenic Alzheimer’s disease mouse model that causes accumulation of α-synuclein also resulted in an accumulation of Rab5, which was normalized when α-synuclein was knocked out of these transgenic mice (Fang et al., 2017). Alternatively, because Rab5 is a LRRK2 kinase substrate, and LRRK2 is significantly activated in iPD and the rotenone model, Rab5 phosphorylation may be central to the early endosomal dysfunction we found. LRRK2-induced phosphorylation of Rab GTPases inhibits their function by preventing binding to Rab GDP-dissociation inhibitor factors necessary for membrane delivery and recycling. As such, it is possible that prolonged LRRK2 kinase activity leads to general disruption of endosomal maturation or trafficking.
The defect in early endosomes was also reflected in the abnormal elevation of transferrin levels. An iron transport protein, transferrin is an important cargo of early endosomes. It is normally trafficked from the cell surface, bound to TfR1, by endocytosis into early endosomes, wherein iron is released and TfR1 and transferrin are recycled to the cell surface and extracellular space. The marked accumulation of transferrin-laden early endosomes in iPD and the rotenone model further suggests defects in early endosomal trafficking; it may also play a central role in the pathological iron accumulation that characterizes the parkinsonian substantia nigra.
Early endosomes are the first cargo vesicle in the endolysosomal pathway and as such, if there were a problem with maturation or trafficking of early endosomes to late endosomes, overall lysosomal cell content and function would be impacted. Such a scenario (Fig. 9) is consistent with our study in which we found early and prolonged increases in abnormal early endosomes, along with a depletion of late endosomes and lysosomes. Only at endpoint rotenone dosing did we observe accumulation of the macroautophagy signaling adapter p62/SQSTM1, which may indicate that autophagic defects are secondary to endolysosomal dysfunction. That is, autophagosomes (marked by p62/SQSTM1) may accumulate if there are insufficient functional lysosomes with which to fuse.
Fig. 9.
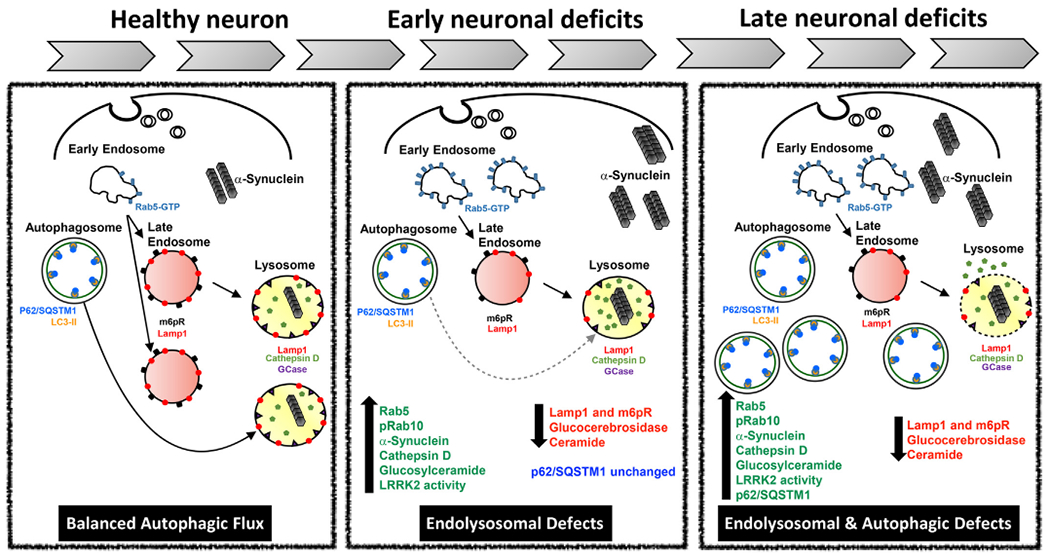
Left panel: Balanced autophagic flux. Rab5-positive early endosomes are the initial sorting station of the endocytic pathway. Normally, endocytic cargoes are either recycled to the cell surface or degraded via lysosomes. Rab5-positive early endosomes mature into late endosomes through a process called ‘Rab conversion’. Late endosomes, containing cargo from early endosomes, acquire newly synthesized lysosomal hydrolases and continue to mature and may eventually fuse with lysosomes. The resultant endolysosome matures into a classical highly acidic lysosome containing Lamp1 and the hydrolases and proteases necessary for degradation. Autophagosomes, the major cargo vesicle for macroautophagy, also traffic cargo to the lysosome for degradation, thereby forming an autolysosome. Under normal cellular homeostasis, endolysosomal function and autophagic flux are ‘balanced’.
Middle panel: Acute rotenone dosing causes selective endolysosomal defects. Rotenone causes oxidative stress and activates LRRK2, and the resultant increase in LRRK2 kinase activity leads to phosphorylation of Rab10 and accumulation of swollen, Rab5-positive early endosomes. The apparent failure of Rab5-positive early endosomes to mature properly decreases the number of late endosomes and lysosomes, which likely impairs degradation and likely explains the accumulation of α-synuclein. At this stage, autophagosomes, labeled with p62 are unaffected.
Right panel: Combined endolysosomal and autophagic deficits in endpoint rotenone and iPD. With continued cellular stress (mitochondrial impairment and LRRK2 activation), the endolysosomal deficits persist and worsen, and there are eventually fewer lysosomes with which autophagosome can fuse. As a result, p62-positive autophagosomes accumulate. Additionally, the finding of extra-lysosomal cathepsin D suggests that lysosomal integrity may be compromised. At this stage, there is ongoing neurodegeneration and depletion of dopamine neurons.
In addition to the apparent depletion of late endosomal and lysosomal punctae, seen in both iPD brains and rotenone rats, rotenone caused early and sustained loss of GCase activity, which was associated with altered glycolipid metabolism. Transient accumulation of GluSph in the striatum and hippocampus may be directly related to the reduction in GCase activity or it may be caused by an overall disruption in lysosomal glycolipid metabolism. Previous reports showed a decrease GCase activity and accumulation of GluCer in postmortem iPD brains (Rocha et al., 2015; Chiasserini et al., 2015). Based on our histological studies looking at endolysosomal proteins in nigral dopaminergic neurons of iPD patients, and others reporting a reduction in both Lamp1 and cathepsin D in nigral dopaminergic neurons of iPD patients that contained α-synuclein inclusions (Chu et al., 2009), the reduction in GCase activity may be a result of generalized lysosomal dysfunction. Along these lines, we observed cathepsin D accumulation outside of the lysosomes, which may indicate a disruption of lysosomal integrity. However, to fully elucidate the mechanism responsible for the observed accumulation of non-lysosomal cathepsin D and how detrimental it is to neuronal survival will require further exploration. As α-synuclein is degraded by cathepsin D in the lysosome under acidic conditions, (Cullen et ah, 2009), a reduction of lysosomal cathepsin D would impair effective degradation of α-synuclein. In this context, it is noteworthy that we observed an increase in endogenous α-synuclein and pSer129-α-synuclein in nigral dopamine neurons in rotenone-treated rats.
LRRK2, which has been implicated in vesicle trafficking, endolysosomal function and autophagy, is activated in nigral dopamine neurons in iPD and in rotenone-treated rats. Therefore, it was of interest to determine whether inhibition of LRJRK2 kinase activity could impact the endolysosomal defects seen in the rotenone model. For this purpose, we used a highly selective, brain-penetrant inhibitor, PF-360, at a dose that results in a pharmacokinetic profile in which an IC90 concentration in brain is achieved for 15 h daily and an IC50 concentration is achieved for a full 24 h. Rab10 is a known LRRK2 substrate and is involved in vesicle trafficking from the endoplasmic reticulum to the golgi. Enhanced LRRK2 kinase activity is associated with an increase in phosphorylation of Rab10. Therefore, to assess target engagement (LRRK2 kinase inhibition) we measured Rab10 phosphorylation and found that PF-360 effectively reduced pThr73-Rab10 levels. We also found that treatment with PF-360 prevented the rotenone-induced increase in Rab5- and transferrin-positive early endosomes seen in iPD dopaminergic neurons, suggesting that maturation/trafficking of early endosomes was restored. Consistent with this interpretation, the number of late endosomes and lysosomes was normalized by LRRK2 kinase inhibition. Interestingly, GCase levels and cathepsin D levels and their respective subcellular localization were preserved by PF-360 treatment. The preservation of endolysosomal function was associated with restoration of autophagic function, as indicated by normal p62/SQSTM1 levels and baseline levels of intracellular α-synuclein. Most importantly, treatment with PF-360 prevented rotenone-induced nigrostriatal dopaminergic degeneration.
In conclusion, our data suggests that increased LRRK2 kinase activity impairs endosomal maturation or trafficking, resulting in lysosomal dysfunction and deficits in proteostasis. Pharmacological inhibition of LRRK2 kinase activity improves endosomal maturation and lysosomal function and is neuroprotective in a model of PD. The fact that a LRRK2 kinase inhibitor is capable of preventing the neuropathological and endolysosomal abnormalities documented in human iPD suggests that LRRK2 inhibitors may have broad therapeutic utility in iPD, not only LRRK2 mutation carriers.
4. Materials and methods
4.1. Experimental design
Retired breeder male Lewis rats aged 8–10 months were purchased from Envigo. Rats were single housed and used for experimental procedures. Animals were housed in standard conditions in a dark/light cycle of 12 h, with ad libitum access to food and water. Rats were randomly assigned to each experimental group.
4.2. Drug treatment
Rats received a single daily interperitoneal (i.p.) injection of 2.8 mg/kg of rotenone resuspended in 2% DMSO, 98% Miglyol 812 N as previously described (Cannon et al., 2009). A cohort of rats (acute dosing) received 5 daily doses of rotenone and euthanized 24 h following the last injection. A second cohort of rats (endpoint dosing) received 9–14 doses of rotenone. These endpoint rats received daily rotenone dosing until each rat reached their behavioral endpoint. Endpoint for each animal is defined as a rat’s inability to successfully perform the postural instability test or 25% body mass was lost as previously described (Cannon et al., 2009). Animals were euthanized using 0.3 mg/kg pentobarbital, followed by transcardial perfusion.
To test the therapeutic potential of LRRK2 inhibitors in our rotenone model. Rats were treated with rotenone until endpoint in the presence or absence of the LRRK2 inhibitor PF-360 (Pfizer). Rats were dosed with PF-360 (10 mg/kg p.o. twice daily) by oral gavage, as previously described (Di Maio et al., 2018). This PF-360 dosing regimen results in a pharmacokinetic profile in which an IC90 concentration in brain is achieved for 15 h daily and an IC50 concentration is achieved for a full 24 h.
4.3. Human brain tissue
Paraffin embedded post-mortem brain tissue from male and female neurologically unaffected patients (healthy subject controls) and idiopathic PD-patient brain tissue was provided by the University of Pittsburgh Pathology Department. All PD cases met a pathological diagnosis of PD made by the brain bank, which was based on the extent of neuronal (pigment) loss in the substantia nigra and locus coeruleus (0–4), and Braak staging (0–6). Postmortem brains from healthy subject controls and PD-patients were closely matched for age, sex, and postmortem interval. See supplemental Table S1 for a full description of patient demographics.
4.4. Immunohistochemistry and antibodies
Animals were terminally anaesthetized with sodium pentobarbital and perfused transcardially with 25 ml phosphate buffered 0.9% saline (PBS). Rats used for histological analyses were also perfused with 4% paraformaldehyde in phosphate buffer. Brains were removed and post-fixed in 4% paraformaldehyde for 24-h before placing them 30% sucrose. 35 pm thick free-floating sections were collected using a microtome and stored in antifreeze at −20 °C until use. See Table 1. for a list of antibodies used for each analysis. For each experiment, all tissues were stained at the same time, using the same primary and secondary antibodies.
Table 1.
Antibody list.
| Antibody | Catalogue No. | Vendor | Concentration |
|---|---|---|---|
| Rabbit anti-Rab5 | Ab109534 | Abcam | Human tissue – 1:100 |
| Rabbit anti-Rab5 | NB120–13253 | Novus Biologicals | Rat tissue – 1:500 |
| Rabbit anti-Transferrin | 82,411 | Abcam | Human tissue - 1:250 Rat tissue – 1:500 |
| Rabbit anti-pThr73 Rab10 | Ab230261 | Abcam | Rat tissue – 1:500 |
| Rabbit anti-Lamp1 | ab24170 | Abcam | Human tissue - 1:250 Rat tissue – 1:500 |
| Mouse anti-M6PR | ab2733 | Abcam | Human tissue - 1:250 Rat tissue – 1:500 |
| Mouse anti-GCase | ab55080 | Abcam | Human tissue - 1:100 Rat tissue – 1:500 |
| Goat anti-Cathepsin D | sc-6487 | Santa Cruz | Human tissue - 1:250 Rat tissue – 1:500 |
| Mouse anti-α-Synuclein | 610,787 | BD Transduction | Rat tissue – 1:1000 |
| Sheep anti-TH | AB1542 | Millipore | Human tissue - 1:250 Rat tissue – 1:2000 |
| Mouse anti-p62/SQSTM1 | H00008878-M01 | Abnova | Rat tissue – 1:500 |
| Rabbit anti-pSer129 α-synuclein | GTX50222 | GeneTex | Rat tissue – 1:500 |
4.5. Confocal microscopy and imaging parameters
Brain sections (35μm) were stored at −20 °C in cryoprotectant until use. Tissue sections were stained free-floating and mounted onto Superfrost Plus glass slides (Fisher, 12–550-15). Fluorescent images were taken using an Olympus BX61 confocal microscope and Fluoview 1000 software (Melville, NY). A negative control (secondary antibody only) slide was prepared for each experiment to subtract the background fluorescence. Imaging parameters were monitored to ensure that images were above background level and below pixel saturation. For quantitative comparisons between groups, all imaging parameters (e.g. laser power, exposure, and pinhole) were held constant across specimens. Confocal images were captured using either a UPlansApo 60×/1.35 oil or 100×/1.40 oil objective len. A minimum of 3 images per tissue section and 3 tissue sections per animal were used for each analysis. Each image contained approximately 6–20 neurons per image; therefore, 54–180 neurons per animal were analyzed. Depending on the variability of each stain, 4–8 animals were analyzed for each confocal experiment.
Confocal images were analyzed using Nikon NIS-Elements Advanced Research software (Version 4.5, Nikon, Melville, NY). Standard threshold parameters were set up for each analysis and used to identify each puncta or ‘objects’. The number objects (# of objects) were collected within each TH-positive cell body within the substantia nigra and averaged across each image and animal.
4.6. Striatal terminal intensity
Serial striatal brain sections (every 6th section) were stained for TH and analyzed using near-infrared imaging for density of dopamine neuron terminals (LiCor). Region of interest (ROI) were drawn around the dorsal lateral region of the striatum and the average intensity for each striatal section was recorded using Odyssey software (V3.0).
4.7. Stereology
The total number of surviving dopaminergic neurons in the substantia nigra was determined using an adapted protocol from Tapias et al. (2013), employing an unbiased, automated stereological system. Briefly, 1 in every 6th nigral tissue section were stained for TH and counterstained with NeuroTrace Dye (640; Life Technologies). Tissue sections were scanned and imaged using an Olympus microscope equipped with 20× objective high N.A. plan fluor/apochromat objective. Twenty times magnification was used to generate montage imaging of the ventral midbrain, for which the entire substantia nigra was analyzed per image using anatomical region of interest boundaries. Images were processed using Nikon NIS-Elements software, and quantitative analysis was performed on fluorescent images colocalizing TH and Nissl-positive stains.
4.8. Ghtcocerebrosidase activity
GCase activity was measured in the substantia nigra using a previously described protocol with some modifications (Rocha et al., 2015). Substantia nigra was microdissected, frozen and stored at −80 °C until use. Nigral sections were homogenized in 250 μL of TE buffer + sucrose (10 mM Tris-HCL, 320 mM sucrose, and 5 mM EDTA) containing Halt Protease Inhibitor Cocktail (78,430; Thermo Fisher Scientific). Samples were incubated with 5 mM 4-methylumbelliferyl (4-Mu)-β-D-glucopyranoside substrate (M3633; Sigma) diluted in a citric acid sodium phosphate buffer at pH 5. Using special optics 96-well plate (3615; Corning) 10 μL of sample was added to 75 μL glucopyranoside substrate for 60 min at 37 °C on a plate shaker. The reaction was terminated using 200 μL of 0.3 M glycine/0.2 M sodium carbonate at pH 10.7. GCase activity was analyzed using a Molecular Device SPEC-TRAmax plate reader using Softmax Pro software. The SpectraMax Plus machine set with Ex 360/Em 460 with auto cutoff, PMT setting at low. A standard curve was generated using 4-Mu sodium salt (M1508; Sigma). GCase enzymatic activity was extrapolated from a 4-Mu (Sigma, USA) standard curve and normalized to protein content in each sample as determined using a BCA kit (Thermo Scientific Pierce, USA).
4.9. Extraction of lipids from brain tissues
Frozen brain regions of interest were supplied for glycosphingolipid analyses. Brain tissue sample wet weights were obtained, and tissue homogenates were normalized across samples by addition of MeoH:H2O (1:1) to generate an equivalent 1:1 wet weight to volume ratio. Homogenization was performed at 200 oscillations/min for 2 min using a QIAGEN TissueLyser II bead mill (QIAGEN Inc.-USA, Germantown, MD). Lipid extraction performed at room temperature with 50 μL homogenates in 96 well plates by addition of 150 μL of MeOH spiked with d3 glucosylceramide C16:0 and 13C6-glucosylsphingosine (Matreya) at 200 ng/mL and 4 ng/mL respectively. Plates were briefly vortexed and spun down prior to addition of 200 μL Acetone:MeOH (1:1). Plates were shaken for 15 min at 1500 rpm, briefly spun down and resuspended with 100 μL of H2O before final centrifugation at 10, 000 xg. Supernatants (250 μL × 2) were transferred to new plates containing 200 μL MeOH:H2O (1:1) per well and mixed prior to sample preconcentration via C18 solid phase extraction (isolute C18, Biotage AB, Uppsala, Sweden) for ceramide/glucosylceramide measures or strong cation exchange (Oasis MCX, Waters Corp. Inc., Milford, MA) for glucosylsphingosine analyses as previously reported (Hamler et al., 2017). Eluates were evaporated to dryness under gentle N2 gas as above and reconstituted in 50 μL DMSO and 200 μL mobile phase B liquid chromatography buffer (95% acetonitrile, 2.5% MeOH, 2.0% H2O, 0.5% formic acid and 5 mM ammoinium formate). Sample sets were processed on the same day. LC-MS/MS analyses immediately followed sample processing steps.
4.10. LC-MS/MS quanti!cation of glucosylceramides and glucosylsphingosine
Multiple reaction monitoring targeted LC-MS/MS quantification of selected glycosphingolipids was performed using a Waters Acquity UPLC (Waters Corp., Inc.) and SCIEX 4500 triple quadrupole mass spectrometer (AB Sciex LLC, Framingham, MA) in positive ion electrospray mode. Hydrophilic Interaction Liquid Chromatography (HILIC) was carried out using a HALO HILIC 2.7 mm column (Advanced Materials Technology, Inc., Wilmington, DE) and 10 min normal phase LC gradient to separate brain derived glucosylceramides from abundant galactosylceramide isomeric species. Mobile phase A consisted of 0.1% formic acid in H2O; mobile phase B is described above. Transitions for selected endogenous glucosylceramide chain length variants were as follows: C16:0 m/z 700.5 > 264.2, C18:0 m/z 728.6 > 264.2, C20:0 m/z 756.6 > 264.2, C22:0 m/z 784.6 > 264.2, C23:0 m/z 798.6 > 264.2, C24:1 m/z 810.6 > 264.2, C24:0 m/z 812.6 > 264.2 and d3 glucosylceramide C16:0 reference standard m/z transition was 703.5 > 264.2. Linear calibration curves using d5 labeled glucosylceramide Cl8:0 standard (Avanti Polar Lipids, Inc., Alabaster, Alabama; m/z 733.6 > 269.4) were used to estimate concentrations of each of the selected glucosylceramide isoforms; total glucosylceramide values were represented as the sum of the C16:0 through C24:0 isoforms. Glucosylsphingosine was monitored as a single analyte (m/z 462.4 > 282.1), and concentrations were determined using linear calibration curves of glucosylsphingosine and 13C6-glucosylsphingosine (m/z 467.4 > 282.1) standards (Matreya). Analyst Multiquant software (AB Sciex) was used to obtain peak area and linear curve fit information.
4.11. Statistics
Experimental results collected using human postmortem tissue was obtained from 4 to 8 control or iPD patients. Experimental results collected using rat tissue was obtained from 4 to 8 rats per experimental condition. Data expressed as mean values ± standard error of the mean (SEM). For simple comparison between two experimental conditions, two-tailed unpaired t-test were used to determine statistical significance. For multiple comparisons, two-way analysis of variance (ANOVA) were used. If overall significant, additional Bonferroni post-hoc tests were used for multiple pairwise comparisons. Statistical significance between treatment groups is represented in each figure as *p < .05, **p < ,01,***p < ,001,****p < .0001. Statistical outliers from each data set were determined using GraphPad Gubbs’ test (also referred to as extreme studentized deviate) with significance level α = 0.05.
4.12. Study approval
All animal procedures were performed in accordance with National Institute of Health guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) at University of Pittsburgh.
Supplementary Material
Acknowledgments
This work was supported by research grants from the National Institutes of Health (NS100744, R21ES027470, NS095387, AG005133), the American Parkinson Disease Association, the Parkinson’s Foundation, the Michael J Fox Foundation, the Blechman Foundation, and the friends and family of Sean Logan. The brain bank has received support from the University of Pittsburgh Brain Institute. The LRRK2 inhibitor, PF-360, was a generous gift from Pfizer.
Footnotes
Declaration of Competing Interest
The authors have declared that no conflict of interest exists.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2019.104626.
References
- Alcalay RN, Wolf P, Levy OA, Kang UJ, Waters C, Fahn S, et al. , 2018. Alpha galactosidase a activity in Parkinson’s disease. Neurobiol. Dis 112, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Sammler E, 2018. LRRK2 kinase in Parkinson’s disease. Science. 360 (6384), 36–37. [DOI] [PubMed] [Google Scholar]
- Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, et al. , 2017. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science. 357 (6357), 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT, 2009. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis 34 (2), 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA, 2000. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 157 (1), 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiasserini D, Paciotti S, Eusebi P, Persichetti E, Tasegian A, Kurzawa-Akanbi M, et al. , 2015. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH, 2009. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol. Dis 35 (3), 385–398. [DOI] [PubMed] [Google Scholar]
- Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, et al. , 2009. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miranda BR, Fazzari M, Rocha EM, Castro S, Greenamyre JT, 2019. Sex differences in rotenone sensitivity reflect the male-to-female ratio in human Parkinson’s disease incidence. Toxicol. Sci 170, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, et al. , 2010. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci 30 (37), 12535–12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, et al. , 2016. Alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med. 8 (342), 342ra78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, et al. , 2018. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med 10 (451). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F, Yang W,, Florio JB, Rockenstein E, Spencer B, Orain XM, et al. , 2017. Synuclein impairs trafficking and signaling of BDNF in a mouse model of Parkinson’s disease. Sci. Rep 7 (1), 3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Mosquera L, Diogo CV, Yambire KF, Santos GL, Luna Sanchez M, Benit P, et al. , 2017. Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci. Rep 7, 45076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, et al. , 2012. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol 72 (3), 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamler R, Brignol N, Clark SW, Morrison S, Dungan LB, Chang HH, et al. , 2017. Glucosylceramide and glucosylsphingosine quantitation by liquid chromatographytandem mass spectrometry to enable in vivo preclinical studies of neuronopathic Gaucher disease. Anal. Chem 89 (16), 8288–8295. [DOI] [PubMed] [Google Scholar]
- Hastings TG, 2009. The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson’s disease. J. Bioenerg. Biomembr 41 (6), 469–472. [DOI] [PubMed] [Google Scholar]
- Heo HY, Park JM, Kim CH, Han BS, Kim KS, Seol W, 2010. LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp. Cell Res. 316 (4), 649–656. [DOI] [PubMed] [Google Scholar]
- Kalogeropulou AF, Zhao J, Bolliger MF, Memou A, Narasimha S, Molitor TP, et al. , 2018. P62/SQSTM1 is a novel leucine-rich repeat kinase 2 (LRRK2) substrate that enhances neuronal toxicity. Biochem. J 475 (7), 1271–1293. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ, 2004. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci 24 (8), 1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie PPY, Nixon RA, 2018. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis 122, 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie PPY, Nixon RA, 2019. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis 122, 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis P, Burel S, Steger M, Mann M, Brown F, Diez F, et al. , 2018. Development of phospho-specific Rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson’s disease kinase. Biochem. J 475 (1), 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, et al. , 2013. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 77 (3), 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA, 2010. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem 285 (18), 13621–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroberardino PG, Hoffman EK, Horowitz MP, Betarbet R, Taylor G, Cheng D, et al. , 2009. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiol. Dis 34 (3), 417–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neefjes J, van der Kant R, 2014. Stuck in traffic: an emerging theme in diseases of the nervous system. Trends Neurosci. 37 (2), 66–76. [DOI] [PubMed] [Google Scholar]
- Nixon RA, 2005. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging 26 (3), 373–382. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang DS, Lee JH, 2008. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy. 4 (5), 590–599. [DOI] [PubMed] [Google Scholar]
- Oakley AE, Collingwood JF, Dobson J, Love G, Perrott HR, Edwardson JA, et al. , 2007. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology. 68 (21), 1820–1825. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Dietrich P, Wang Q, Dauer WT, 2004. Stefanis L. alpha-synuclein is required for the fibrillar nature of ubiquitinated inclusions induced by proteasomal inhibition in primary neurons. J. Biol. Chem 279 (45), 46915–46920. [DOI] [PubMed] [Google Scholar]
- Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P, et al. , 2015. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann Clin Transl Neurol. 2 (4), 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P, Klumperman J, 2009. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 10 (9), 623–635. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, et al. , 2002. An in vitro model of Parkinson’s disease: linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci 22 (16), 7006–7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M, 1997. Alpha-synuclein in Lewy bodies. Nature. 388 (6645), 839–840. [DOI] [PubMed] [Google Scholar]
- Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, et al. , 2017.Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JY, Kim J, Paik SR, Park JH, Ahn YS, Chung KC, 2001. Induction of neuronal cell death by Rab5A-dependent endocytosis of alpha-synuclein. J. Biol. Chem 276 (29), 27441–27448. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Tatebe H, Taguchi K, Endo Y, Tokuda T, Mizuno T, et al. , 2012. p62/SQSTM1-dependent autophagy of Lewy body-like alpha-synuclein inclusions. PLoS One 7 (12), e52868. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.