

Abstract
Duchenne muscular dystrophy (DMD) is caused by mutations in the gene encoding dystrophin. Prior work has shown that DMD progression can vary, depending on the genetic makeup of the patient. Several modifier alleles have been identified including LTBP4 and SPP1. We previously showed that Spp1 exacerbates the DMD phenotype in the mdx mouse model by promoting fibrosis and by skewing macrophage polarization. Here, we studied the mechanisms involved in Spp1's promotion of fibrosis by using both isolated fibroblasts and genetically modified mice. We found that Spp1 upregulates collagen expression in mdx fibroblasts by enhancing TGFβ signaling. Spp1's effects on TGFβ signaling are through induction of MMP9 expression. MMP9 is a protease that can release active TGFβ ligand from its latent complex. In support for activation of this pathway in our model, we showed that treatment of mdx fibroblasts with MMP9 inhibitor led to accumulation of the TGFβ latent complex, decreased levels of active TGFβ and reduced collagen expression. Correspondingly, we found reduced active TGFβ in Spp1−/−mdxB10 and Mmp9−/−mdxB10 muscles in vivo. Taken together with previous observations of reduced fibrosis in both models, these data suggest that Spp1 acts upstream of TGFβ to promote fibrosis in mdx muscles. We found that in the context of constitutively upregulated TGFβ signaling (such as in the mdxD2 model), ablation of Spp1 has very little effect on fibrosis. Finally, we performed proof-of-concept studies showing that postnatal pharmacological inhibition of Spp1 reduces fibrosis and improves muscle function in mdx mice.
Introduction
Mutations in DMD, the gene encoding for dystrophin protein, cause Duchenne muscular dystrophy (DMD), a disease characterized by progressive muscle deterioration and eventual cardiac and pulmonary dysfunction (1, 2). In DMD, insufficient levels of dystrophin protein cause contraction-induced damage to the sarcolemma, leading to myofiber death and inflammatory cell invasion. While muscle stem cells partially compensate for muscle cell loss, the repeated cycles of cell death and repair lead to accumulation of TGFβ, accumulation of extracellular matrix (ECM), and replacement of muscle by fat and connective tissue.
ECM deposition (i.e. fibrosis) contributes significantly to the severity and rate of DMD disease progression. Support for this hypothesis was bolstered by the identification of latent TGFβ binding protein 4 (LTBP4) as a modifier of DMD severity (3). LTBP4 belongs to a family of TGFβ binding proteins that bind to both precursor and active TGFβ ligands, regulating their release through proteolytic cleavage (4). LTBP4 was first identified as a DMD modifier through murine linkage studies, in which a 12 amino acid deletion in Ltbp4 segregated with two pathogenic features of murine muscular dystrophy (3). This “risk allele” in the DBA/2 J background correlated with higher levels of TGFβ activity, enhanced membrane permeability and increased fibrosis compared to the “protective allele” that retained the 12 amino acid sequence. This polymorphism is located in the proline rich hinge region that needs cleavage to release the sequestered TGFβ ligand. Subsequently, several independent genotype/phenotype studies identified a risk haplotype in LTBP4 that correlated with a shorter ambulatory period and more severe cardiomyopathy in DMD patients (5–7). The deleterious human haplotype involves four non-synonymous amino acids (VTTT) in LTBP4, whereas the IAAM haplotype is protective, correlating with slower disease progression. The protein produced from the protective haplotype binds more TGFβ and is associated with reduced TGFβ signaling (5, 8). Thus, TGFβ-mediated fibrosis has a strong link to DMD disease progression.
Spp1 (Osteopontin) is another modifier of dystrophinopathies that is highly upregulated with dystrophic onset and progression in mice (mdx), dogs golden retriever muscular dystrophy (GRMD) and humans (DMD) (7, 9–12). Spp1 (24) is a highly acidic, secreted protein that binds to integrins (13, 14) or CD44 (15) to modify signaling or mediate cell adhesion to the ECM (16). Correlations between Spp1 levels and dystrophic disease severity have been demonstrated in mice (9, 10), dogs (11) and humans (7, 12), which lends support for its role as a DMD modifier and potential biomarker. Ablation of Spp1 in mdx mice attenuated disease severity and was associated with reduced TGFβ and fibrosis, as well as with increased muscle regeneration and strength (10). In humans with DMD, a single nucleotide polymorphism (SNP) in the Spp1 promoter (rs28357094T > G-referred to as the “G allele”) correlated with earlier loss of ambulation and decreased grip strength (7, 12). These disease indicators also correlated with increased Spp1 mRNA expression compared to patients with the protective “T allele” (12). Not all subsequent reports reproduced these original findings; however, the studies that substantiated rs28357094T > G in Spp1 as a modifier used cohorts that were more ethnically homogeneous than those studies that did not support the original finding (17). The modifier effect was even more pronounced in patients on steroids, which may be due to a glucocorticoid-receptor binding element in the Spp1 promoter near the location of the modifier SNP (18). Thus, both LTBP4 and SPP1 are modifiers of dystrophinopathies that impact TGFβ activity and fibrosis and exacerbate disease progression.
Studies examining the relationship between Spp1 and LTBP4/TGFβ have observed a feed forward effect in the context of sarcolemmal repair of myofibers (19). Spp1 ablation correlated with reduced expression of TGFβ and, conversely, increased activation of TGFβ signaling correlated with increased Spp1. For example, muscles in mdx mice on the DBA/2 J background (carrying the LTBP4 risk allele that is associated with higher TGFβ activity) showed higher Spp1 expression than dystrophic mice expressing the protective allele (19). However, the molecular mechanisms and effects of Spp1 and TGFβ cascades are still unknown with regards to promotion of fibrosis and pathological ECM remodeling, which is the subject of this investigation.
Our previous studies demonstrated that global Spp1 ablation correlated with reduced fibrosis and decreased TGFβ in dystrophic muscle (10); however, the cell type responsible for mediating these changes was not identified. Spp1 ablation altered macrophage polarization toward M2c, but macrophage-derived TGFβ1 was unchanged, nor was there altered expression of any other macrophage-derived pro-fibrotic factor ((9) and unpublished data). Moreover, TGFβ expression was not changed with Spp1 ablation when the total leukocyte population in Spp1−/− mdx was compared to mdx muscles (Supplementary Material, Fig. S1). This result suggests that the reductions in TGFβ observed in Spp1−/− dystrophic muscles must derive from a cell type other than leukocytes.
Spp1 has been previously shown to promote fibroblast differentiation and induce upregulation of collagen I expression in pathological fibrosis associated with liver, skin and lung tissues (20–22). Here we identify resident muscle fibroblasts as an important source and target of the Spp1-TGFβ pathway and dissect the mechanism whereby Spp1 interacts with fibroblasts to promote fibrosis in the context of dystrophic muscles. We demonstrate that Spp1 promotes fibroblast expression of MMP9 and increased TGFβ processing to induce collagen expression in skeletal muscle fibroblasts. Furthermore, we show that Spp1 ablation in the setting of very high TGFβ does not improve collagen deposition and fibrosis in dystrophic muscle, thus demonstrating that Spp1 acts upstream of TGFβ to regulate fibrosis in muscular dystrophy. Postnatal pharmacological inhibition of Spp1 lowers TGFβ in skeletal muscles, decreases collagen accumulation and ameliorates disease severity. These data support Spp1 as a beneficial therapeutic target in DMD-associated fibrosis.
Results
Autocrine induction of collagen I in fibroblasts by Spp1
To interrogate Spp1's influence on fibroblasts in promotion of fibrosis in dystrophic muscles, we studied primary fibroblasts isolated from mdx Spp1−/−mdx muscles, which allowed us to examine the influence of exogenously added Spp1 on a pure population of fibroblasts, independent of Spp1 derived from other cell types. Spp1−/−mdx fibroblasts were incubated with conditioned media (CM) from Spp1−/−mdx or Spp1+/+mdx primary fibroblasts for 24 hours. Following incubation, collagen I gene expression was quantified as a marker of fibrosis, since collagen I is the main component of fibrotic ECM in dystrophic muscles. Using fibroblast-derived CM ensured that the proper cell-specific post-translational modifications would be present on Spp1 added to the cells. We observed a significant upregulation of collagen I expression in cells incubated with Spp1+/+mdx CM compared to Spp1−/−mdx CM, supporting the hypothesis that fibroblast-derived Spp1 acts in an autocrine manner to promote collagen I expression by fibroblasts (Fig. 1A). We also observed increased expression of collagen 3 (another collagen upregulated in fibrosis) but not fibronectin in cells incubated with Spp1+/+mdx CM (Supplementary Material, Fig. S2A and B).
Figure 1.

The autocrine effect of Spp1 on muscle fibroblast collagen expression is mediated by TGFβ. (A) Primary Spp1−/−mdx fibroblasts were incubated for 24 h with CM collected from either Spp1−/−mdx or Spp1+/+mdx fibroblasts, and expression of collagen I was assessed by RT-PCR. (B) Spp1 was purified from Spp1+/+mdx fibroblasts CM by immunoprecipitation for use in subsequent experiments. Non-specific beads (left panel) were used as a negative control to insure specificity of immunoprecipitation by Spp1-specific antibodies (middle panel). The concentration of immunoprecipitated Spp1 was determined by comparison with a standard curve of rSpp1 (right panel) by densitometry. (C) Immunoprecipitated (IP'd) Spp1 was incubated with Spp1−/−mdx fibroblasts, and collagen I expression was measured. (D) Expression of collagen I was significantly suppressed when Spp1−/−mdx fibroblasts were incubated with CM of Spp1+/+mdx fibroblasts, supplemented with the TGFβ inhibitor (10 μm of SB431542). (E) Twenty-four-hour incubation of Spp1−/−mdx fibroblasts with Spp1+/+mdx CM, IP'd Spp1 or rSpp1 did not upregulate expression of TGFβ gene.
To determine whether the effect of Spp1+/+CM on collagen expression was Spp1 specific, we purified Spp1 from Spp1+/+mdx CM by immunoprecipitation (Fig. 1B). Subsequently, primary Spp1−/−mdx fibroblasts were incubated with the immunoprecipitated Spp1 (using the same concentration of Spp1 as was present in Spp1+/+mdx CM, 50 ng/ml), and the effect on collagen I expression was evaluated. Surprisingly, we did not observe a similar increase in collagen expression in fibroblasts incubated with immunoprecipitated Spp1 as was observed in fibroblasts incubated with Spp1+/+mdx CM, suggesting that Spp1's effect on fibroblast secretion of ECM is indirect (Fig. 1C).
We next sought to determine whether the effect of Spp1+/+mdx CM on collagen I expression is mediated through TGFβ, a known regulator of tissue fibrosis. To answer this question, primary Spp1−/−mdx fibroblasts were incubated with Spp1+/+mdx CM in the presence or absence of a TGFβ inhibitor (10 μm of SB431542) and were subsequently assessed for collagen I expression. As shown in Figure 1D, inhibition of TGFβ in Spp1+/+mdx CM significantly reduced fibroblast collagen induction, suggesting that TGFβ mediates the effect of Spp1 on collagen expression in fibroblasts. To test whether Spp1 upregulates TGFβ mRNA, primary fibroblasts were incubated with either: 1) Spp1+/+mdx CM, 2) immunoprecipitated Spp1, or 3) commercially available recombinant Spp1 (rSpp1). After 24 h of incubation, TGFβ mRNA was assayed by quantitative PCR and none of these treatments induced TGFβ mRNA in the time frame of analysis (Fig. 1E). Thus, although the autocrine effect of Spp1 on collagen expression is mediated through a TGFβ pathway, in this experimental context, Spp1 does not accomplish this task through direct induction of TGFβ gene expression.
Spp1 is required for extracellular processing of TGFβ
We next examined whether Spp1 influences posttranslational processing of TGFβ. To address this question, CM from Spp1+/+mdx and Spp1−/−mdx fibroblasts was collected, concentrated and analyzed by Western blotting for processed TGFβ. This analysis revealed higher levels of the active form of TGFβ (25 kDa) in Spp1+/+mdx CM compared to Spp1−/−mdx CM, suggesting that Spp1 indirectly influences TGFβ cleavage and activation (i.e. processing) (Fig. 2A and B). In vivo support for this hypothesis was provided by analysis of whole muscle extracts whereby Spp1−/−mdxB10 muscles showed reduced levels of active TGFβ compared to control Spp1+/+mdxB10 muscles (Fig. 2C and D). Taken together, our results indicate that Spp1 has an indirect effect on proteolytic processing of TGFβ.
Figure 2.

Extracellular processing of TGFβ is decreased in the absence of Spp1. (A) Western blot analysis of concentrated CM from Spp1−/−mdx and Spp1+/+mdx fibroblasts revealed that levels of proteolytically processed (25 kDa) TGFβ are decreased in CM lacking Spp1. Ponceau staining demonstrates equal loading of the concentrated CM. (B) Schematic representation of the hypothesis that Spp1 is required for normal extracellular processing of TGFβ that in turn controls collagen I expression. (C) Western blot analysis of total muscle extracts from Spp1+/+mdxB10 and Spp1−/−mdxB10 mice demonstrates decreased levels of processed TGFβ in the absence of Spp1. n = 4 of each genotype. Quantitative analysis of the western blot is shown in (D). Asterisk indicates statistical significance of P < 0.05. The nitrocellulose membrane was stained with ponceau after transfer and used to assess gel loading. Molecular weight markers were run in the middle lane and used to estimate TGFβ size.
Spp1 regulates expression of MMP9 in fibroblasts
We next sought to identify whether Spp1 mediates its effects through upregulation of matrix metalloproteinases (MMPs), which have been previously associated with TGFβ processing. MMP9 is highly elevated in dystrophic mdx muscles, and a relationship between Spp1 and induction of MMP9 expression was previously established (23). Based on these observations, we tested whether Spp1 ablation and subsequent reductions in MMP9 expression could be responsible for the decreased extracellular processing of TGFβ in Spp1−/−mdx fibroblasts. Consistent with prior in vivo observations, we found that primary fibroblasts treated with rSpp1 had increased levels of MMP9 mRNA (Fig. 3A) but did not show an increased expression of MMP2 (Supplementary Material, Fig. S2C). Moreover, we observed increased levels of both intracellular and secreted MMP9 protein in Spp1+/+mdx compared to Spp1−/−mdx fibroblasts (Fig. 3B). Thus, the effects of Spp1 on fibroblasts are mediated through MMP9 and its subsequent effects on TGFβ processing to induce collagen.
Figure 3.

Spp1 regulates expression of Mmp9 that contributes to the proteolytic processing of TGFβ. (A) rSpp1 upregulates expression of Mmp9 in Spp1−/−mdx fibroblasts as assessed by quantitative RT-PCR. (B) Western blot analysis showed increased Mmp9 inside the cells and in concentrated CM from Spp1+/+mdx fibroblasts compared to Spp1−/−mdx fibroblasts. (C) rSpp1 activates Akt signaling pathway as revealed by increased phosphorylation of Thr308 (D). Expression of Mmp9 gene was significantly decreased by incubation with Akt inhibitor (HY-15431). (E) Western blot analysis of total protein lysates from Spp1−/−mdxB10 and Spp1+/+mdxB10 mice (n = 4 of each genotype) showed that Spp1 regulates Mmp9 expression in vivo; quantitative analysis of the blot is shown in (F). (G) Schematic representation of the hypothesis that Spp1 regulates expression of Mmp9, which contributes to proteolytic processing of TGFβ. Asterisk indicates statistical significance of P < 0.05; AU, arbitrary units.
It was previously demonstrated that Spp1 activates protein kinase B (AKT) signaling in hepatic stellate cells and promotes fibrogenesis in the liver (24 upregulates collagen I via integrin α(V)β (3) engagement and PI3K/pAkt/NFκB signaling). Consistent with this observation, we found that incubation with rSpp1 led to increased phosphorylation of AKT at Thr308 (Fig. 3C). Moreover, in the presence of the AKT inhibitor HY-15431 MedChemExpress (MCE), expression of MMP9 was significantly reduced, suggesting that Spp1 regulates expression of MMP9 via AKT activation (Fig. 3D). To determine whether Spp1 induces MMP9 expression in vivo, we assessed MMP9 protein levels in whole muscle lysates from Spp1−/−mdxB10 and Spp1+/+mdxB10 muscles. MMP9 was significantly reduced in the absence of Spp1 (Fig. 3E and F) supporting the hypothesis that decreased extracellular processing of TGFβ is due to reduction in MMP9 in Spp1-deficient muscles. A schematic of this hypothesis is shown in Figure 3G.
Inhibition of MMP9 reduces TGFβ extracellular processing and collagen I expression
Active TGFβ is secreted in a complex with its inactive domain (referred to as the small latent complex [SLC]). SLC binds to latent TGFβ binding protein (LTBP) to form a large latent complex (LLC) (Fig. 4A). Among the members of the LTBP family, LTBP4 is of special interest because it is preferentially expressed in skeletal and cardiac muscles and it is a known modifier of the dystrophic phenotype (3, 5). Both LTBPs and SLC can be cleaved by several extracellular proteinases including MMP9 (25).
Figure 4.
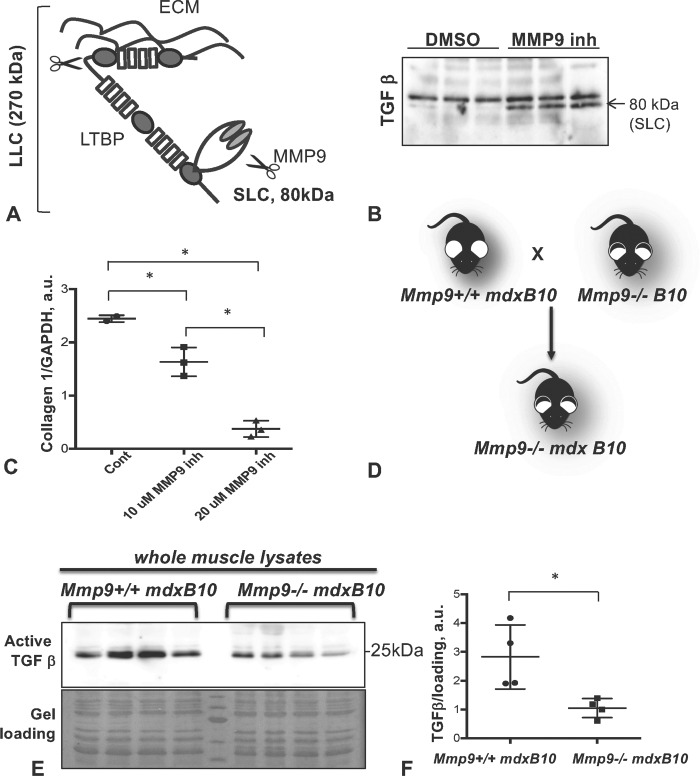
Mmp9 contributes to proteolytic processing of TGFβ precursor. (A) Schematic structure of SLC and LLC of TGFβ precursor that can both be cleaved by Mmp9. (B) CM of fibroblasts incubated with or without Mmp9 inhibitor shown in triplicate (Enzo Life Sciences) were probed with anti-TGFβ antibody. Arrow indicates 80 kDa SLC, which is accumulated in CM containing Mmp9 inhibitor. (C) Inhibition of Mmp9 activity was associated with a dose-dependent decrease in collagen I expression. (D) Schematic of a genetic cross to produce Mmp9−/−mdxB10 double mutant mice. (E) Western blot analysis of total protein lysates from Mmp9−/−mdxB10 and Mmp9+/+mdxB10 mice (n = 4 of each genotype) showed decreased active form of TGFβ (25 kDa) in Mmp9-deficient mice. Quantitative analysis of the western blot is shown in (F). Asterisk indicates statistical significance of P < 0.05; AU, arbitrary units.
To test whether MMP9 is involved in TGFβ processing, we incubated primary skeletal muscle fibroblasts with Spp1+/+mdx CM in the presence or absence of a MMP9-specific inhibitor. Inhibition of MMP9 activity led to accumulation of the SLC in CM suggesting that MMP9 regulates extracellular TGFβ activity by cleaving the SLC (Fig. 4B). Moreover, as shown in Figure 4C, inhibition of MMP9 activity reduced collagen I expression in a dose-dependent manner. Thus, these data suggest that MMP9-mediated processing and activation of TGFβ could underlie promotion of the fibrotic process in dystrophic muscles.
To determine whether these relationships exist in vivo and to confirm that MMP9 plays a role in controlling latent TGFβ processing, we compared the levels of active TGFβ in double mutant Mmp9−/−mdxB10 and Mmp9+/+mdxB10 muscles (Fig. 4D). These mice have been previously described (23), and mdxB10 lacking MMP9 were shown to have a milder dystrophic phenotype, likely due to decreased fibrosis and improved muscle regeneration (26). In agreement with these previously published data, we also showed that the levels of active TGFβ were significantly lower in Mmp9−/−mdxB10 compared to Mmp9+/+mdxB10 muscles (Fig. 4E and F), suggesting that MMP9 activity correlates with TGFβ processing in vivo. Taken together, our data suggest that the effect of Spp1 on fibrosis is mediated through induction of MMP9 and processing of latent TGFβ.
Effect of Spp1 ablation on fibrosis is diminished in the context of high TGFβ activity
To test the proposed model that Spp1 acts upstream of MMP9 to modulate TGFβ signaling and fibrosis in dystrophic muscles, we examined the effect of Spp1 ablation on the mdxD2 phenotype. Muscles of mdxD2 mice have poorly regulated TGFβ activity due to an in-frame deletion in the LTBP4 gene that is not present in the B10 background (3). This SNP generates an allelic variant of LTBP4 that is more susceptible to proteolysis, leading to increased release of TGFβ from the LLC. This model is ideal for addressing mechanisms of fibrosis in a dystrophic context, since mdxD2 mice are more severely fibrotic than mdxB10 mice (27). Our prior studies showed that genetic ablation of Spp1 on the mdxB10 background decreases fibrosis, as measured by expression of collagen I, and hydroxyproline assay (19). If TGFβ acts downstream of Spp1 to control expression of fibrotic genes, then one would expect little or no effect of Spp1 ablation in the context of unregulated TGFβ signaling.
To test this hypothesis, we generated Spp1−/− mice on the mdxD2 background (Fig. 5A) and assessed levels of active TGFβ and fibrosis. Unlike on the B10 background, the levels of active TGFβ were not different between mdxD2 and Spp1−/−mdxD2 mice (Fig. 5B and C). Moreover, lack of Spp1 in mdxD2 mice had no effect on hydroxyproline content nor on collagen I gene expression (Fig. 5D and E). Furthermore, immunostaining collagen I on cross sections did not reveal any significant differences between mdxD2 and Spp1−/−mdxD2 muscles (Fig. 5F). Thus, in the mdxB10 background, ablation of Spp1 significantly decreased active TGFβ and improved fibrosis; in contrast, lack of Spp1 in the context of unregulated TGFβ activity (as in the mdxD2 background) had only a modest effect on the level of fibrosis. These data indicate that Spp1 acts upstream of the TGFβ pathway in control of pro-fibrotic gene expression to impact ECM deposition in dystrophic muscles.
Figure 5.

Effect of Spp1 ablation in mdxD2 mice. (A) Schematic of a genetic cross to produce Spp1−/−mdxD2 mice. (B) Western blot analysis of the total protein lysates from Spp1−/−mdxD2 mice and Spp1+/+mdxD2 mice (n = 4 of each genotype). The active form of TGFβ (25 kDa) was not different between the two genotypes. Quantitative analysis of the western blot is shown in (C). Asterisk indicates statistical significance of P < 0.05; AU, arbitrary units. (D) Analysis of total collagen content in 12- and 28-week-old Spp1−/−mdxD2 mice and Spp1+/+mdxD2 mice using hydroxyproline (HYP) assay. (E) RT-PCR analysis of collagen I expression did not show a difference between Spp1−/−mdxD2 and Spp1+/+mdxD2 muscles. (F) Immunofluorescent staining using anti-collagen I antibody. No significant differences were observed between Spp1+/+mdxD2 and Spp1−/−mdxD2 muscles.
Postnatal pharmacological inhibition of Spp1 decreases active TGFβ and improves fibrosis and muscle function in mdxB10 mice
To test whether postnatal reductions of Spp1 levels can improve fibrosis and muscle function in mdxB10 mice, we used a Spp1-inhibiting compound (PTC-549) that was identified by PTC Therapeutics in a high throughput screen utilizing their GEMS™ discovery platform (28). PTC-549 efficiently reduced levels of Spp1 in both human and mouse dystrophin-deficient fibroblasts (Fig. 6A and B). Compound PTC-549 was well tolerated by mice up to 100 mg/kg, and oral administration was determined as an optimal route of delivery. Ten days of treatment of mdxB10 mice with 10 mg/kg PTC-549 resulted in a 30% reduction of Spp1 in quadriceps compared to vehicle-treated mice (Fig. 6C). Using this regimen, mdx mice were treated for 6 months and evaluated for muscle strength by wire mesh test (after 4 and 23 weeks of treatment) and then assayed for markers of fibrosis at the end of the treatment. As shown in Figures 6D and F and Fig. 7, mice treated with PTC-549 showed a significant reduction in active TGFβ levels and collagen content (assayed by hydroxyproline) after long-term treatment. Moreover, a significant improvement in muscle strength was observed after 4 weeks and up to 23 weeks of treatment. (Fig. 6G). Immunohistochemical examination of the diaphragm muscles from mice treated with PTC-549 for 6 months revealed the presence of clusters of regenerative fibers, positive for developmental myosin heavy chain (devMyHC). These data suggest that postnatal inhibition of Spp1 ameliorates fibrosis and improves muscle function in a mouse model of DMD.
Figure 6.
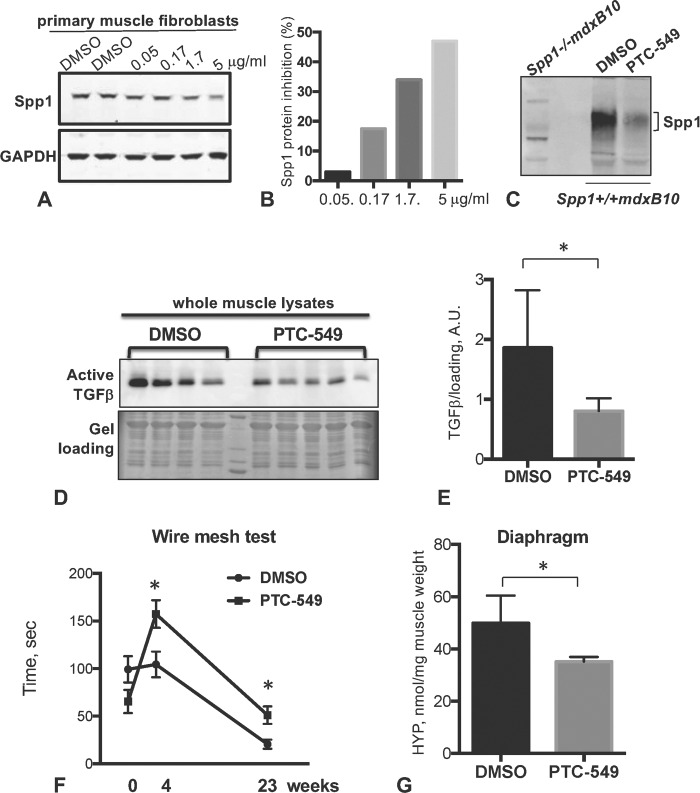
Effect of postnatal pharmacological inhibition of Spp1 in mdxB10 mice. (A) Western blot of Spp1 levels in mdxB10 primary fibroblasts that were incubated with increasing concentration of PTC-549 for 72 h. Blot was probed with anti-Spp1 antibodies (upper panel). Lower panel shows GAPDH, which was used as a loading control. (B) Quantitative analysis of the western blot shown in (A). Bars represent the percent of Spp1 inhibition relative to DMSO control. (C) Western blot of whole muscle lysates from DMSO- or PTC-549-treated mice (n = 5 per group in pooled samples) probed with anti-Spp1 antibody. First lane shows muscle lysate lacking Spp1 to ensure antibody specificity. (D) Western blot analysis of total protein lysates from mdxB10 mice treated with PTC-549 or DMSO for 6 months (n = 4 per group). The data showed decreases in the active form of TGFβ (25 kDa) in PTC-54-treated mice. Quantitative analysis of the western blot is shown in (E). (F) Wire mesh test showed that time on the wire was significantly higher for mice treated with PTC-549 at 4 weeks of treatment and up to 23 weeks (n = 8 and 9 mice per group). (G) Analysis of total collagen content after 24 weeks of treatment with PTC-549 or DMSO using hydroxyproline (HYP) assay. Asterisk indicates statistical significance of P < 0.05; AU, arbitrary units.
Figure 7.

Immunohistochemical staining of diaphragm isolated from mice treated with PTC-549 (upper panel) or DMSO (lower panel). Frozen sections of diaphragm from mice treated with PTC-549 for 6 months stained for collagen I or developmental myosin heavy chain (a marker of regenerating fibers). All images were taken at the same magnification and the same exposure.
Discussion
Excess ECM deposition or fibrosis develops in tissues undergoing chronic damage and repair, such as in the case of DMD, in which muscles sustain repeated contraction-induced sarcolemmal injury followed by muscle stem cell-mediated repair. Inflammatory cells play a necessary and important role in the process of muscle repair; thus, it is critical that signals derived from these cells appear in the lesion with the correct order and timing to facilitate removal of damaged tissue and to facilitate regeneration (29). Since dystrophic lesions arise randomly and asynchronously along a muscle fiber, the carefully orchestrated repair process is ultimately undermined, resulting in accumulation of TGFβ, incomplete repair and accretion of excess connective tissue (i.e. fibrosis). It has long been assumed that TGFβ in the tissue derives from inflammatory cells that enter the muscle; however, these studies indicate that fibroblasts are not only a fundamental target but also a pivotal source of TGFβ that contributes to muscle fibrosis. Our studies go further to demonstrate a substantial role for Spp1 on fibroblasts as a participant in this process (30).
Our prior studies revealed an overall reduction in TGFβ levels and fibrosis in muscles of genetically modified mice that were globally lacking Spp1 (10, 19). Subsequent analysis of the effects of Spp1 ablation on the immune infiltrate failed to reveal the cell source responsible for the reduced TGFβ (9). The work presented here strongly supports the concept that reduced fibrosis on Spp1 ablation is due to a change in processing of fibroblast-derived TGFβ, increased AKT signaling and MMP9 expression in dystrophic fibroblasts. We first demonstrated a relationship between fibroblast-derived Spp1 and MMP9 in cultures treated with rSpp1 and AKT inhibitors but then confirmed the in vivo relevance by assessing MMP9 levels and active TGFβ in Spp1−/−mdx mice. This analysis revealed a significant reduction in MMP9 levels in the setting of Spp1 ablation in vivo. While a relationship between Spp1 and MMP9 has been previously reported (31), our studies identified muscle fibroblasts as the site of action of this pathway. Furthermore, we and others (23, 32) observed a dramatic reduction in the concentration of processed TGFβ in Mmp9−/−mdx mice, consistent with the hypothesis that MMP9 plays a role in promotion of TGFβ activation. Mmp9 is highly elevated in dystrophic muscles and its genetic ablation improves the dystrophic phenotype (26). Upregulation of highly similar MMP2 in dystrophic muscles has also been demonstrated in mdx mice and DMD patients; however, genetic ablation of MMP2 in mdx mice had a deleterious effect on disease progression due to reduced angiogenesis and impaired muscle regeneration (33). Both MMP2 and MMP9 have been shown to cleave latent TGFβ (34); however, despite their similarities, MMP2 and MMP9 play very distinct roles in dystrophic muscles and we have demonstrated a specific role for MMP9, independent of MMP2, in promoting TGFβ processing in this context.
The identification of genetic modifiers of DMD that affect active TGFβ levels (such as LTBP4 and Spp1) and the abundant data linking TGFβ to fibrosis has lent strong support for the idea that excessive TGFβ activity and fibrosis promote DMD disease progression (3, 5, 7); however, the mechanism has not been entirely clear. One explanation is that the excess ECM deposition that occurs in fibrosis presents a physical barrier to muscle regeneration. Another is that increased matrix stiffness that is associated with fibrosis causes cellular reprogramming of myoblasts (35), leading to lost regenerative potential. Furthermore, it is well known that TGFβ can directly inhibit regeneration by blocking terminal myogenic differentiation (36); thus, TGFβ can also directly interfere with regenerative potential even in the absence of excess ECM, and these effects of TGFβ may be additive.
Studies in different muscular dystrophy patients have provided mixed results regarding a Spp1 modifier effect (37). For example, it has been reported that Spp1 does not act as a disease modulator in congenital muscular dystrophy type 1A indicating that Spp1's ability to modulate dystrophic phenotype may be context dependent (38). Moreover, Spp1 has been shown to be post-translationally modified by phosphorylation, glycosylation, sulfation, transglutamination and proteolytic cleavage (39) leading to numerous forms, each with the capacity to act on a wide range of receptors. MMPs have been shown to cleave Spp1 to expose an RGD peptide that enhances Spp1's ability to bind to integrins to affect cellular behavior (13, 14). Thus, different post-translationally modified forms of Spp1 can combine with a large number of potential receptors to regulate a diverse number of physiological and pathological processes. As an example, it has been shown that Spp1 affects sarcolemmal repair through increased TGFβ signaling and induction of Slug and Snail occupancy on the annexin promoter, leading to inhibition of annexin expression (19). Annexins are important repair-promoting proteins, and thus, their inhibition leads to reduced efficiency of membrane repair in myofibers after induced injury. This example highlights a specific interaction between Spp1 and myofibers.
Taken together with our previous work, these studies suggest that Spp1's effects on the dystrophic process are multi-faceted. On the mdxB10 background, loss of Spp1 reduces TGFβ and fibrosis and increases muscle growth and regeneration through its actions on fibroblasts and macrophages, respectively. While the current work reveals a direct relationship between Spp1 and promotion of fibrosis by fibroblasts, our prior data provide support for Spp1's modulation of macrophage polarization in mdx muscles (9, 10). M2c macrophages accumulate in dystrophic muscles and secrete growth factors essential for muscle repair. Ablation of Spp1 shifted macrophage polarization toward the M2c pro-regenerative phenotype and led to an increase in their secretion of IGF1 and LIF. Thus, Spp1 affects muscle regeneration and growth through effects on macrophage polarization toward M2c and increased levels of insulin like growth factor (IGF) and leukemia inhibitory factor (LIF) (9), while it impacts fibrosis through its effects on fibroblasts and increases in MMP9 that promotes TGFβ processing.
Our studies reveal two distinct benefits of Spp1 ablation in dystrophic muscles: 1) reduced fibrosis through decrease of both MMP9 and TGFβ activation in fibroblasts and 2) promotion of muscle growth/regeneration through skewed macrophage polarization that leads to their increased growth factor secretion (Fig. 8). Whether or not Spp1 directly affects muscle precursor cells and/or influences myogenic differentiation in dystrophic muscle remains to be determined. Recent studies using a whole muscle autograft model of muscle injury concluded that Spp1 derived from both muscle and non-muscle sources (primarily, inflammatory cells) is equally important for normal muscle recovery after acute injury; however, because these studies used whole muscle grafts, neither the essential source(s) of Spp1 nor the mechanisms of Spp1's action were determined (40). Spp1's effects may depend on its cellular origin as well as the genetic context in which it acts (acute injury in otherwise healthy muscle vs. chronic injury associated with muscular dystrophy).
Figure 8.
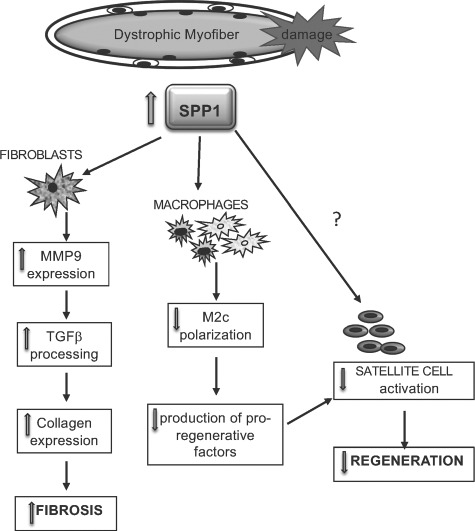
Schematic of Spp1 effects on different cell populations in dystrophic muscle. Spp1 is elevated in dystrophic muscles. Spp1 causes macrophages to polarize away from M2c and reduce their growth factor secretion of IGF1 and LIF. Spp1 also acts on fibroblasts to induce MMP9 expression, which leads to processing of TGFβ.
Since our data suggest that genetic ablation of Spp1 can ameliorate DMD disease progression in mdxB10 mice, we tested whether in vivo targeting of Spp1 is beneficial to the dystrophic phenotype, using a new compound (PTC-549) that targets Spp1. Similar to Spp1−/−mdxB10 mice (9, 10), PTC-549-treated mdxB10 mice showed significant improvements in muscle strength and fibrosis, thus providing proof of concept that post-natal ablation of Spp1 is beneficial for the dystrophic phenotype. This work opens the possibility of using pharmacological agents that target Spp1 to improve or delay fibrosis in DMD, which may also be beneficial when used in combination with gene or cell therapies.
Materials and Methods
Mice
Spp1 and Mmp9 knockout mice were obtained from Jackson laboratory. Spp−/−mdxB10 mice were generated and described previously (10). To generate Spp1−/−mdxD2 mice, Spp1−/−mdxB10 mice were bred with mdxD2 mice (The Jackson Laboratory, Bar Harbor, Maine) for over 5 generations. The following primers were used for genotyping of D2 background, forward 5' AACCGCTACCCAAACCTTCA and reverse 5' AGGCTTTCTGCCTACTCGTC. A 36 bp deletion/insertion in Ltbp4 gene was detected by genotyping PCR (3). Mmp9−/−mdxB10 mice were generated by breeding Mmp9−/− mice and mdxB10 mice. Previously published protocols were used for genotyping for mdx allele (41, 42) and for Mmp9−/− (The Jackson Laboratory).
For PTC-549 studies, Spp1−/−mdxB10 mice were housed individually during a course of treatment. In addition to regular pellets, each mouse received 1 g of peanut buffer containing PTC-549 in Dimethyl Sulfoxide (DMSO) (10 mg/kg daily) or equal volume of DMSO (control group). All animals were handled and bred according to guidelines stipulated by the Animal Research Committee at UCLA.
Cell culture
Primary skeletal muscle fibroblasts were isolated from all hindlimb muscles of three 10- to 13-day-old Spp1−/−mdxB10 mice, washed with sterile PBS and minced in 1:1 mixture of 1600 U/ml collagenase type 2 and 1.5 mg/mg dispase (Worthington Biochemical Corp., Lakewood, New Jersey) Muscle homogenate was incubated with collagenase for 30 min at 37°C with slow agitation. Once the tissue was digested, muscle homogenate was diluted with sterile PBS and passed through a 70-μm cell strainer. Cells were pelleted by centrifugation at 900g for 5 min and resuspended in growth medium (F10 Ham [Sigma], supplemented with 1% penicillin/streptomycin and 20% fetal bovine serum (FBS)). During next 2–3 passages, cells were pre-plated for 30 min to allow highly adhesive fibroblasts to attach to the plate. After 30 min, the medium with unattached cells was removed and fresh growth medium was added. To collect CM, confluent cells were incubated with serum free F10 Ham medium for 24 hours. Vivaspin 6 tubes (GE Healthcare, Pittsburgh, Pennsylvania) were used to concentrate CM for immunoprecipitation of Spp1 and for Western blotting. To inhibit MMP9 activity, cells were incubated with 10 μm or 20 μm MMP9 inhibitor I (Enzo Life Sciences, Farmingdale, NY) for 24 hours. To inhibit TGFβ, cells were incubated with 10 μm of SB431542 (Tocris) for 24 hours. To inhibit AKT, cells were incubated with 10 μm HY-15431 MedChemExpress (MCE) for 24 hours. For PTC studies, mdxB10 mouse primary fibroblasts were incubated with increasing concentrations of PTC-549 (0.05 to 5 μg/ml) for 72 hours.
Immunoprecipitation of Spp1 from concentrated CM
BrCN-Sepharose (Sigma-Aldrich, Saint Louis, Missouri) was activated by incubation with 1 mm HCl for 2 hours at 4°C. After centrifugation at 1000g for 5 min, activated beads were washed once in coupling buffer (100 mm NaHCO3, 500 mm NaCl, pH 8.3), resuspended in coupling buffer with goat anti-mouse Spp1 antibody (R&D Systems, Minneapolis, Minnesota) at 50 μg/ml and incubated overnight at 4°C. Beads were washed with coupling buffer for 30 min at room temperature and incubated with quenching buffer (100 mm Tris–HCl, pH 8.0) for 2 hours at room temperature. Concentrated CM were precleaned by incubation with BrCN-Sepharose and added to the beads with linked antibodies for overnight incubation at 4°C. After that, beads were washed three times in PBS with 0.1% Tween 20. To eluate bound proteins, beads were incubated for 5 min with elution buffer (100 mm glycine, 2.5 pH, 500 mm NaCl). Eluates were neutralized by 1:10 volume of 1 m Tris pH 8.0 and analyzed by Western blotting. Concentration of immunoprecipitated Spp1 was determined by comparing with a range of known amounts of recombinant mouse Spp1 (R&D Systems) loaded on the same gel.
Western blotting
Cell pellets were resuspended in reducing sample buffer (50 mm Tris–HCl, pH 6.8, 10% glycerol, 2% Sodium Dodecyl Sulfate (SDS), and 100 mm β-mercaptoethanol) supplemented with protease and phosphatase inhibitors (Sigma, St Louis, Missouri). DNA was shared by passing through 27G needle 10 times. The same reducing sample buffer (30 V/muscle weight) was used to homogenize whole muscle. After electrophoresis, proteins were transferred to nitrocellulose membrane. The following primary antibodies were used for Western blot analysis: anti-TGFβ (R&D), anti-MMP9 (Cell Signaling, Danvers, Massachusetts). Imaging was performed using c300 imager (Azure Biosystems, Dublin, California), and Western blot quantification was done using Image J software.
Real-time PCR
cDNA was generated using iScript Reverse Transcriptase Supermix (Bio-Rad, Hercules, CA) and was used for real-time PCR (RT-PCR)with ITaq Universal SYBR Green Supermix (Bio-Rad) according to manufacturer's instructions. All RT-PCR reactions were run in CFX Connect Real-Time PCR System (Bio-Rad). Primers for RT-PCR were selected to span intron–exon junctions (when possible) and were first tested in regular PCR amplification to ensure the production of a single band in each case. The following primer pairs were used:
GAPDH Frw 5' tccaccaccctgttgctgta and Rev 5' gacttcaacagcaactcccac.
collagen I Frw 5' gtcgcttcacctacagcac and Rev 5' caatgtccaagggagccac.
MMP9 Frw 5' gatccccagagcgtcattc and Rev 5' ccaccttgttcacctcattttg.
TGFβ1 Frw 5' gggaagcagtgcccgaaccc and Rev 5' tgggggtcagcagccggtta.
Immunohistochemistry
Frozen quadriceps cross-sections were fixed for 10 min with 4% paraformaldehyde and stained with rabbit anti-collagen I antibody (Cedarlane Laboratories, Burlington, Canada) followed by secondary anti-rabbit FITC antibody (Vector). Slides were analyzed using AxioVision Software from Zeiss.
Conflicts of Interest statement
E.W. is an employee of PTC therapeutics, which provided PTC-549 compound for these studies. The other authors have no conflicts of interest.
Funding
National Institute of Arthritis and Musculoskeletal and Skin Diseases for a Wellstone Cooperative Muscular Dystrophy Center [U54AR052646]; P30 Muscular Dystrophy Core Center [NIAMS-P30AR057230-01 to M.J.S.]; National Institutes of Health grants [RO1 AR046911 to M.J.S., R01 HL140938 to E.M.M.]; Parent Project Muscular Dystrophy (M.J.S.); Muscular Dystrophy Association [MDA Development Grant No. 479350 to M.J.S. and M.Q.].
Supplementary Material
{kind=link}
{kind=link}
References
- 1. Bushby K., Finkel R., Birnkrant D.J., Case L.E., Clemens P.R., Cripe L., Kaul A., Kinnett K., McDonald C., Pandya S. et al. (2010) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol., 9, 77–93. [DOI] [PubMed] [Google Scholar]
- 2. Bushby K., Finkel R., Birnkrant D.J., Case L.E., Clemens P.R., Cripe L., Kaul A., Kinnett K., McDonald C., Pandya S. et al. (2010) Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol., 9, 177–189. [DOI] [PubMed] [Google Scholar]
- 3. Heydemann A., Ceco E., Lim J.E., Hadhazy M., Ryder P., Moran J.L., Beier D.R., Palmer A.A. and McNally E.M. (2009) Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J. Clin. Invest., 119, 3703–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robertson I.B., Horiguchi M., Zilberberg L., Dabovic B., Hadjiolova K. and Rifkin D.B. (2015) Latent TGF-β-binding proteins. Matrix Biol., 47, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Flanigan K.M., Ceco E., Lamar K.M., Kaminoh Y., Dunn D.M., Mendell J.R., King W.M., Pestronk A., Florence J.M., Mathews K.D. et al. (2013) LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann. Neurol., 73, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bello L., Piva L., Barp A., Taglia A., Picillo E., Vasco G., Pane M., Previtali S.C., Torrente Y., Gazzerro E. et al. (2012) Importance of SPP1 genotype as a covariate in clinical trials in Duchenne muscular dystrophy. Neurology, 79, 159–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bello L., Kesari A., Gordish-Dressman H., Cnaan A., Morgenroth L.P., Punetha J., Duong T., Henricson E.K., Pegoraro E., McDonald C.M. et al. (2015) Genetic modifiers of ambulation in the cooperative international neuromuscular research group Duchenne natural history study. Ann. Neurol., 77, 684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lamar K.M., Bogdanovich S., Gardner B.B., Gao Q.Q., Miller T., Earley J.U., Hadhazy M., Vo A.H., Wren L., Molkentin J.D. et al. (2016) Overexpression of latent TGFβ binding protein 4 in muscle ameliorates muscular dystrophy through myostatin and TGFβ. PLoS Genet., 12, e1006019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Capote J., Kramerova I., Martinez L., Vetrone S., Barton E.R., Sweeney H.L., Miceli M.C. and Spencer M.J. (2016) Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J. Cell Biol., 213, 275–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vetrone S.A., Montecino-Rodriguez E., Kudryashova E., Kramerova I., Hoffman E.P., Liu S.D., Miceli M.C. and Spencer M.J. (2009) Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J. Clin. Invest., 119, 1583–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Galindo C.L., Soslow J.H., Brinkmeyer-Langford C.L., Gupte M., Smith H.M., Sengsayadeth S., Sawyer D.B., Benson D.W., Kornegay J.N. and Markham L.W. (2016) Translating golden retriever muscular dystrophy microarray findings to novel biomarkers for cardiac/skeletal muscle function in Duchenne muscular dystrophy. Pediatr. Res., 79, 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pegoraro E., Hoffman E.P., Piva L., Gavassini B.F., Cagnin S., Ermani M., Bello L., Soraru G., Pacchioni B., Bonifati M.D. et al. (2011) SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology, 76, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barry S.T., Ludbrook S.B., Murrison E. and Horgan C.M. (2000) Analysis of the alpha4beta1 integrin-osteopontin interaction. Exp. Cell Res., 258, 342–351. [DOI] [PubMed] [Google Scholar]
- 14. Barry S.T., Ludbrook S.B., Murrison E. and Horgan C.M. (2000) A regulated interaction between alpha5beta1 integrin and osteopontin. Biochem. Biophys. Res. Commun., 267, 764–769. [DOI] [PubMed] [Google Scholar]
- 15. Katagiri Y.U., Sleeman J., Fujii H., Herrlich P., Hotta H., Tanaka K., Chikuma S., Yagita H., Okumura K., Murakami M. et al. (1999) CD44 variants but not CD44s cooperate with beta1-containing integrins to permit cells to bind to osteopontin independently of arginine-glycine-aspartic acid, thereby stimulating cell motility and chemotaxis. Cancer Res., 59, 219–226. [PubMed] [Google Scholar]
- 16. Mukherjee B.B., Nemir M., Beninati S., Cordella-Miele E., Singh K., Chackalaparampil I., Shanmugam V., DeVouge M.W. and Mukherjee A.B. (1995) Interaction of osteopontin with fibronectin and other extracellular matrix molecules. Ann. N. Y. Acad. Sci., 760, 201–212. [DOI] [PubMed] [Google Scholar]
- 17. Vo A.H. and McNally E.M. (2015) Modifier genes and their effect on Duchenne muscular dystrophy. Curr. Opin. Neurol., 28, 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vianello S., Pantic B., Fusto A., Bello L., Galletta E., Borgia D., Gavassini B.F., Semplicini C., Sorarù G., Vitiello L. et al. (2017) SPP1 genotype and glucocorticoid treatment modify osteopontin expression in Duchenne muscular dystrophy cells. Hum. Mol. Genet., 26, 3342–3351. [DOI] [PubMed] [Google Scholar]
- 19. Quattrocelli M., Capote J., Ohiri J.C., Warner J.L., Vo A.H., Earley J.U., Hadhazy M., Demonbreun A.R., Spencer M.J. and McNally E.M. (2017) Genetic modifiers of muscular dystrophy act on sarcolemmal resealing and recovery from injury. PLoS Genet., 13, e1007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dong J. and Ma Q. (2017) Osteopontin enhances multi-walled carbon nanotube-triggered lung fibrosis by promoting TGF-β1 activation and myofibroblast differentiation. Part Fibre Toxicol., 14, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hunter C., Bond J., Kuo P.C., Selim M.A. and Levinson H. (2012) The role of osteopontin and osteopontin aptamer (OPN-R3) in fibroblast activity. J. Surg. Res., 176, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morimoto Y., Hirahara K., Kiuchi M., Wada T., Ichikawa T., Kanno T., Okano M., Kokubo K., Onodera A., Sakurai D. et al. (2018) Amphiregulin-producing pathogenic memory T helper 2 cells instruct eosinophils to secrete osteopontin and facilitate airway fibrosis. Immunity, 49, 134–150.e136. [DOI] [PubMed] [Google Scholar]
- 23. Dahiya S., Givvimani S., Bhatnagar S., Qipshidze N., Tyagi S.C. and Kumar A. (2011) Osteopontin-stimulated expression of matrix metalloproteinase-9 causes cardiomyopathy in the mdx model of Duchenne muscular dystrophy. J. Immunol., 187, 2723–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Urtasun R., Lopategi A., George J., Leung T.M., Lu Y., Wang X., Ge X., Fiel M.I. and Nieto N. (2012) Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin α(V)β(3) engagement and PI3K/pAkt/NFκB signaling. Hepatology, 55, 594–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Costanza B., Umelo I.A., Bellier J., Castronovo V. and Turtoi A. (2017, 6 Jan) Stromal modulators of TGF-β in cancer. J. Clin. Med., 6(1). pii: E7. doi: 10.3390/jcm6010007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H., Mittal A., Makonchuk D.Y., Bhatnagar S. and Kumar A. (2009) Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum. Mol. Genet., 18, 2584–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gordish-Dressman H., Willmann R., Dalle Pazze L., Kreibich A., van Putten M., Heydemann A., Bogdanik L., Lutz C., Davies K., Demonbreun A.R. et al. (2018) “Of mice and measures”: a project to improve how we advance Duchenne muscular dystrophy therapies to the clinic. J. Neuromuscul. Dis., 5, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bhattacharyya A., Trotta C.R. and Peltz S.W. (2007) Mining the GEMS—a novel platform technology targeting post-transcriptional control mechanisms. Drug Discov. Today, 12, 553–560. [DOI] [PubMed] [Google Scholar]
- 29. Tidball J.G. (2017) Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol., 17, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Serrano A.L. and Muñoz-Cánoves P. (2017) Fibrosis development in early-onset muscular dystrophies: mechanisms and translational implications. Semin. Cell Dev. Biol., 64, 181–190. [DOI] [PubMed] [Google Scholar]
- 31. Hindi S.M., Shin J., Ogura Y., Li H. and Kumar A. (2013) Matrix metalloproteinase-9 inhibition improves proliferation and engraftment of myogenic cells in dystrophic muscle of mdx mice. PLoS One, 8, e72121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dahiya S., Bhatnagar S., Hindi S.M., Jiang C., Paul P.K., Kuang S. and Kumar A. (2011) Elevated levels of active matrix metalloproteinase-9 cause hypertrophy in skeletal muscle of normal and dystrophin-deficient mdx mice. Hum. Mol. Genet., 20, 4345–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miyazaki D., Nakamura A., Fukushima K., Yoshida K., Takeda S. and Ikeda S. (2011) Matrix metalloproteinase-2 ablation in dystrophin-deficient mdx muscles reduces angiogenesis resulting in impaired growth of regenerated muscle fibers. Hum. Mol. Genet., 20, 1787–1799. [DOI] [PubMed] [Google Scholar]
- 34. Yu Q. and Stamenkovic I. (2000) Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev., 14, 163–176. [PMC free article] [PubMed] [Google Scholar]
- 35. Stearns-Reider K.M., D'Amore A., Beezhold K., Rothrauff B., Cavalli L., Wagner W.R., Vorp D.A., Tsamis A., Shinde S., Zhang C. et al. (2017) Aging of the skeletal muscle extracellular matrix drives a stem cell fibrogenic conversion. Aging Cell, 16, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu D., Black B.L. and Derynck R. (2001) TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev, 15, 2950–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van den Bergen J.C., Hiller M., Böhringer S., Vijfhuizen L., Ginjaar H.B., Chaouch A., Bushby K., Straub V., Scoto M., Cirak S. et al. (2015) Validation of genetic modifiers for Duchenne muscular dystrophy: a multicentre study assessing SPP1 and LTBP4 variants. J. Neurol. Neurosurg. Psychiatry, 86, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gawlik K.I., Holmberg J., Svensson M., Einerborg M., Oliveira B.M., Deierborg T. and Durbeej M. (2017) Potent pro-inflammatory and pro-fibrotic molecules, osteopontin and galectin-3, are not major disease modulators of laminin α2 chain-deficient muscular dystrophy. Sci. Rep., 7, 44059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anborgh P.H., Mutrie J.C., Tuck A.B. and Chambers A.F. (2011) Pre- and post-translational regulation of osteopontin in cancer. J. Cell Commun. Signal., 5, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wasgewatte Wijesinghe D.K., Mackie E.J. and Pagel C.N. (2019) Normal inflammation and regeneration of muscle following injury require osteopontin from both muscle and non-muscle cells. Skelet. Muscle, 9, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amalfitano A. and Chamberlain J.S. (1996) The mdx-amplification-resistant mutation system assay, a simple and rapid polymerase chain reaction-based detection of the mdx allele. Muscle Nerve, 19, 1549–1553. [DOI] [PubMed] [Google Scholar]
- 42. Liaw L., Birk D.E., Ballas C.B., Whitsitt J.S., Davidson J.M. and Hogan B.L. (1998) Altered wound healing in mice lacking a functional osteopontin gene (spp1). J. Clin. Invest., 101, 1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.