

Abstract
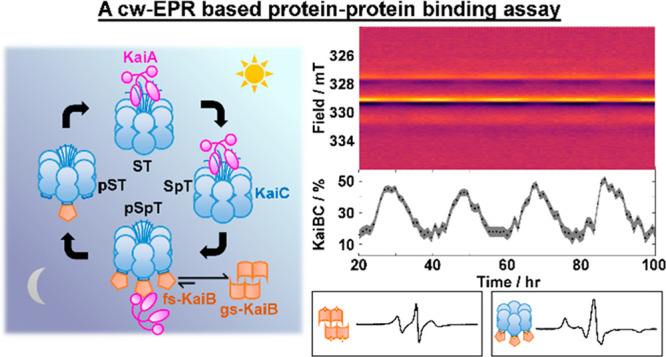
The cyanobacterial circadian clock in Synechococcus elongatus consists of three proteins, KaiA, KaiB, and KaiC. KaiA and KaiB rhythmically interact with KaiC to generate stable oscillations of KaiC phosphorylation with a period of 24 h. The observation of stable circadian oscillations when the three clock proteins are reconstituted and combined in vitro makes it an ideal system for understanding its underlying molecular mechanisms and circadian clocks in general. These oscillations were historically monitored in vitro by gel electrophoresis of reaction mixtures based on the differing electrophoretic mobilities between various phosphostates of KaiC. As the KaiC phospho-distribution represents only one facet of the oscillations, orthogonal tools are necessary to explore other interactions to generate a full description of the system. However, previous biochemical assays are discontinuous or qualitative. To circumvent these limitations, we developed a spin-labeled KaiB mutant that can differentiate KaiC-bound KaiB from free KaiB using continuous-wave electron paramagnetic resonance spectroscopy that is minimally sensitive to KaiA. Similar to wild-type (WT-KaiB), this labeled mutant, in combination with KaiA, sustains robust circadian rhythms of KaiC phosphorylation. This labeled mutant is hence a functional surrogate of WT-KaiB and thus participates in and reports on autonomous macroscopic circadian rhythms generated by mixtures that include KaiA, KaiC, and ATP. Quantitative kinetics could be extracted with improved precision and time resolution. We describe design principles, data analysis, and limitations of this quantitative binding assay and discuss future research necessary to overcome these challenges.
The circadian clock of the cyanobacterium Synechococcus elongatus regulates global gene expression1 and provides a selective growth advantage.2 The fact that transcriptional and translational oscillations (TTO) are unnecessary to drive posttranslational oscillations (PTOs)3 sets this clock apart from circadian clocks from other organisms.4 Indeed, in vitro reconstitution of the core oscillator proteins, KaiA, KaiB, and KaiC (Figure 1a), generates an autonomous macroscopic circadian rhythm of sequential KaiC phosphorylation–dephosphorylation at residues Ser-431 (S431) and Thr-432 (T432) in the presence of ATP5,6 mirroring in vivo oscillations,7 making the Kai system a unique model system for studying circadian clock systems in general.
Figure 1.
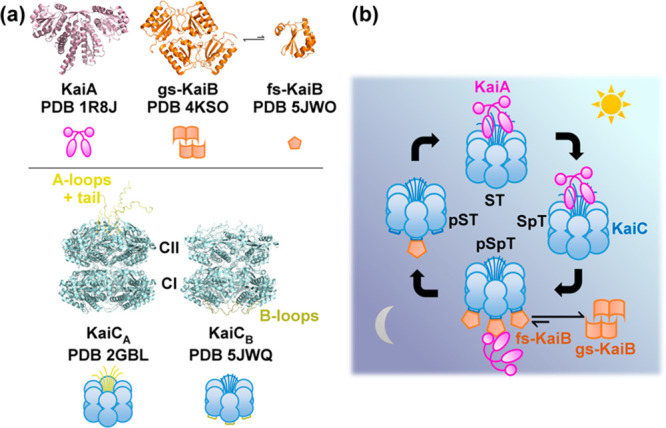
Overview of the core Kai oscillator. (a) Representative crystal structures and simplified representations of isolated clock components KaiA (magenta), KaiB [orange, ground state (gs) vs fold-switched (fs) states], and KaiC (blue, KaiA-binding competent state KaiCA vs KaiB-binding competent state KaiCB). The crystal structure of KaiCB (Protein Data Bank entry 5JWQ) was obtained from the KaiC homologue in Thermosynechococcus elongatus. All other crystal structures were obtained from S. elongatus. For KaiC, the KaiA-binding A-loops and tail (yellow) and KaiB-binding B-loops (olive) are highlighted in both the crystal structures and simplified representations. (b) Model of events governing circadian oscillations in the Kai clock. Input and output pathways are not shown.
KaiC belongs to the AAA+ ATPase family and consists of two homologous domains, CI and CII (Figure 1a).8 CI possesses slow ATPase activity,9−11 with its activity correlated with the frequencies of both TTO and PTO.10 On the other hand, CII contains two phosphorylation sites, S431 and T432, that undergo ordered phosphorylation as S/T → S/pT → pS/pT → pS/T → S/T (p, phosphorylated).5,6 This order is dictated by phosphorylation-dependent CII ring dynamics12 and CI–CII ring stacking interactions.13 The autophosphatase activity of CII is atypical in that ATP is regenerated from ADP and pS431 or pT432 as an intermediate prior to hydrolysis.14
In the absence of KaiA or KaiB, KaiC autophosphatase activity dominates, leading to an unphosphorylated KaiCST resting state.5 To drive autokinase activity in KaiC, KaiA, a domain-swapped homodimer (Figure 1a),15 interacts with the C-terminal residues (“A-loops” and tail) of KaiC during the day (Figure 1a,b)16,17 as a KaiAC complex. This KaiA–KaiC interaction promotes nucleotide exchange in KaiC and consequently autokinase activity in CII.18,19 As CII progresses from S/T to pS/pT, corresponding to the day-to-night transition, the CII ring tightens, withdrawing its A-loops,12 which decreases the binding affinity between KaiA and KaiC20 and suppresses the autokinase activity in KaiC. This decrease in affinity is critical to maintaining macroscopic phase coherence.20 Concurrently, the CII and CI rings of KaiC stack13 such that CI adopts a posthydrolysis conformation that exposes its “B-loops” to KaiB binding (Figure 1b),10,21 a step that marks the beginning of clock resetting.22 The interchanging competences for KaiA and KaiB binding by two conformations of KaiC23 are designated here as KaiCA and KaiCB, respectively. Curiously, the conformation of KaiB that can bind to CI in this KaiBC complex differs from KaiB as crystallized in that the former has a thioredoxin fold and is termed fold-switched KaiB (fs-KaiB),21,24 whereas the latter possesses an uncommon tetrameric state and is termed ground-state KaiB [gs-KaiB (Figure 1a)].25,26 The tetramer and monomer have been shown to interconvert reversibly under native conditions,27 whereas the mechanism of fold switching within monomerized KaiB remains elusive. On fold switching, a β-strand (β2) together with a newly formed α-helix (α3) in fs-KaiB is capable of KaiA binding, thereby stabilizing KaiA in an “autoinhibited” conformation where the CII-binding motif of KaiA is blocked.21,28 The KaiB–KaiCpS(p)T binary complex can thus recruit KaiA to form a ternary KaiABC nighttime complex,21,28 suppressing further KaiC phosphorylation and forming a negative feedback loop. Subsequent autodephosphorylation of KaiCpS(p)T during the night enables resetting of KaiC to KaiCST and prepares the proteins for the next day. As the Kai system consists of discrete molecules, intrinsic stochastic noise can lead to desynchronization of the population.29 To combat this, populations of KaiC synchronize via intersubunit allosteric interactions,23,30 phosphorylation-dependent KaiA–KaiC binding affinities,20,31 and monomer shuffling.32,33
Biochemical circadian rhythms of the Kai clock were first revealed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) in which KaiC phosphostates in SDS-quenched reaction samples were quantitatively resolved.5−7 As KaiC phosphodistribution represented only one facet of the Kai oscillator, coimmunoprecipitation assays5,23 and native mass spectrometry27 were developed in parallel in attempt to quantify relevant protein–protein interactions over the oscillations. However, the discontinuous nature of these assays meant that multiple samples had to be prepared to provide time-domain information. More recently, fluorescence correlation spectroscopy (FCS) and fluorescence polarization/anisotropy (FA) have been employed to study KaiB binding by fluorophore tagging, producing tagged proteins using either cell free synthesis34,35 or site-directed mutagenesis.24,36 Fluorescence assays are an improvement to previous discontinuous methods as the fluorescence signals could be continuously monitored provided the fluorophore tags are not photobleached. However, the myriad of stoichiometries existing in Kai reaction mixtures, tetrameric26 and monomeric KaiB24 and (sub)stoichiometric KaiBC and KaiABC complexes,20,27,28 invalidate models that assume two species with differing molecular weights. Thus, fluorescence signals via either FCS or FA are not directly comparable to coimmunoprecipitation results, limiting their usefulness. Here, we introduce site-directed spin labeling–electron paramagnetic resonance (SDSL–EPR)37 to probe real-time evolution of KaiB–KaiC interactions. SDSL–EPR provides an alternative to fluorescence assays by providing comparable time resolution while avoiding ambiguities in stoichiometry inherent to fluorescence-based methods. Furthermore, interaction specificity is maintained, allowing quantitative binding information comparable to coimmunoprecipitation or native-PAGE. In the following section, we discuss in detail the sample preparation and data analysis necessary to obtain reproducible results. Then, we describe the design principles to site-directed spin labeling. Once the theoretical background has been laid, we implement cw-EPR to the Kai system and demonstrate that cw-EPR gives complementary information to existing methods. Finally, we outline the limitations of this quantitative binding assay and discuss future research and instrumentation necessary to overcome these challenges.
Materials and Methods
Cloning, Protein Expression, Purification, and Fluorescence/Spin Labeling
All genes were cloned into pET-28b using the NdeI/HindIII sites for production of SUMO fusion proteins. Details of the cloning protocol have been described previously.12,13 Proteins were expressed in Escherichia coli BL21(DE3) (Novagen) and purified by Ni-NTA affinity chromatography and size-exclusion chromatography as described previously.12,13 6-Iodoacetamidofluorescein (6IAF) labeling of KaiB was performed according to a previous report.24 A modified protocol for 3-iodoacetamido-PROXYL (3IAP) labeling of KaiB was performed as follows. A 5-fold excess of tris(2-carboxyethyl)phosphine (TCEP) was added to a purified aliquot of unlabeled KaiB (typically 100 μM) and incubated at room temperature for 15 min in labeling buffer [20 mM Tris and 150 mM NaCl (pH 8.0)], followed by overnight incubation of a 10-fold excess of 3IAP in the dark at 4 °C using a 200 mM 3IAP stock solution in DMSO. The difference in concentrations between the spin-label and TCEP is critical to a high labeling efficiency; TCEP is known to directly react with iodoacetamide-based fluorescent dyes38 and is inferred to do the same to 3IAP, decreasing its effective concentration. After labeling, excess 3IAP was removed with a HiPrep 26/10 desalting column (GE Healthcare Life Sciences). The sample was then concentrated using Amicon ultrafiltration stirred cells fitted with 10 kDa cutoff membranes.
Protein Characterization
The concentrations of stock protein solutions were determined via a Bradford assay using bovine serum albumin (Thermo Fisher). All concentrations refer to the monomer unless otherwise stated. The labeling efficiencies of all spin-labeled KaiB samples were determined by electrospray ionization high-resolution liquid chromatography mass spectrometry [ESI-HR-LCMS (Figure S1)] using an LTQ Orbitrap XL mass spectrometer equipped with an electrospray ionization source (ThermoFisher, San Jose, CA) operating in positive ion mode. Fifty microliter portions of spin-labeled KaiB samples (400 μg/mL) were desalted using Zeba Spin Desalting Columns (ThermoFisher). LCMS was performed on a C18 column eluted with gradient of 0.1% formic acid in water to 0.1% formic acid in acetonitrile. The LCMS data were then analyzed using MagTran.39 The labeling efficiency was determined by fitting the transformed spectrum with Gaussian lines and determining the peak areas of both labeled and unlabeled proteins. Samples that were not at least 95% labeled on the basis of LCMS peak areas were relabeled by repeating the labeling protocol described above.
Sample Preparations for In Vitro Kai Protein Reactions
All reactions were performed at 30 °C in reaction buffer [20 mM Tris, 150 mM NaCl (pH 8.0), 0.5 mM EDTA, 5 mM MgCl2, and 1 mM ATP]. The in vitro 1× oscillator was carried out using 1.2 μM KaiA, 3.5 μM KaiB, and 3.5 μM KaiC. Fluorescence samples were prepared as 100 μL samples with additional 50 nM KaiB-K25C-6IAF included as described previously.36 For real-time cw-EPR characterization, 10–40 μL samples were prepared by replacing WT-KaiB with spin-labeled KaiB. KaiCS431E,T432E (KaiCEE) phosphomimetic reactions were performed with a 1× or 5× protein concentration without supplementing extra ATP. KaiB was added to KaiCEE and incubated for 7 h (5×) or 1 day (1×), after which KaiA was added to the KaiB/KaiCEE reaction mixture to a final Kai protein concentration of 4.6× (5×) or 0.9× (1×) relative to the 1× oscillator. Native-PAGE samples were prepared as 200 μL reaction mixtures at a 1× protein concentration and incubated at 30 °C overnight. Twenty microliters of the sample was then removed at predefined time points and mixed with 20 μL of 2× native-PAGE buffer [62.5 mM Tris, 20% (w/v) glycerol, 0.01% (w/v) bromophenol blue, 1 mM ATP, and 5 mM MgCl2 (pH 6.8)] to give final KaiB and KaiCEE concentrations of 1.75 μM. The samples were frozen in a −80 °C freezer and then thawed over ice for 3 min prior to loading.
Native-PAGE
Thawed samples were electrophoresed at 60 V for 30 min followed by 140 V for 135 min using hand-cast 6% Tris-glycine mini gels with a 4% stacking gel in ATP-supplemented native-PAGE running buffer (25 mM Tris, 192 mM glycine, 1 mM ATP, and 5 mM MgCl2) in an electrophoresis cell surrounded by ice–water. Gels were incubated in 0.5% SDS in 7.5% acetic acid, stained in SYPRO Orange (Fisher Scientific, Waltham, MA), and imaged using a BioSpectrum AC Imaging System (UVP, Upland, CA). Image contrast adjustment and densitometry were performed on ImageJ (National Institutes of Health). Formation of the KaiB–KaiCEE complex was estimated by normalizing the BCEE complex band intensity against the sum of band intensities of the BCEE complex and free KaiCEE (Figure S2).
Fluorescence Anisotropy (FA) Measurements
FA measurements of 1× oscillators (Figure S3) and phosphomimetic reactions (Figure S13) using 6IAF-labeled samples were performed at 30 °C on a BMG Clariostar plate reader (BMG LABTECH, Cary, NC) with an excitation wavelength of 492 nm and an emission wavelength of 530 nm.
EPR Spectroscopy
X-Band cw-EPR (9.2 GHz) spectra were recorded using a Bruker ECS 106 spectrometer equipped with an SHQE cavity. In vitro reaction spectra were recorded at 30 °C using 2 G modulation at a 100 kHz modulation frequency with a conversion time of 40 ms and a time constant of 20 ms. Scan times were approximately 7.5 min per spectrum. Samples were loaded into custom sample tubes by sealing one end of glass pipets (10 μL, Fisher Scientific) or rectangular miniature hollow tubing (20–40 μL, VitroCom, Mountain Lakes, NJ) to ensure a high quality factor (Q) and to suppress sample heating. For near-rigid limit samples for determination of g and A, the spin-labeled samples were mixed with WT-KaiB and equilibrated overnight followed by dilution to a final concentration of 17.5 μM labeled KaiB, 157.5 μM WT-KaiB, and 20% (w/w) sucrose (20 Bx), 100 μL (4 mm inside diameter, Wilmad, Vineland, NJ), and the spectra were recorded at −40 °C and 1 G modulation. Temperature control was achieved using an FTS XR401 Air-jet Crystal Cooler (SP Scientific, Stone Ridge, NY). Real-time temperature monitoring in the sample cavity was achieved using an external type T thermocouple (Omega Inc.) connected to a Universal Thermocouple Connector Direct USB to PC Connection (Omega Inc.) on a separate computer. The thermocouple readings inside the EPR sample tube and Air-jet outlet were consistent, ruling out microwave heating of the sample. The microwave frequency was continuously monitored externally via an EIP 578B frequency counter (National Instruments, Santa Clara, CA) connected to a separate computer.
EPR Data Preprocessing
Prior to data analysis, the EPR data were (1) microwave frequency drift corrected and (2) background subtracted as follows. Unlike previous spin labeling and spin trapping studies in which spectral acquisition is usually complete within hours, the Kai clock oscillates over a duration of days. The length of the acquisition process enables extensive averaging yet poses an additional challenge that the resonance microwave frequency of the resonance cavity can drift over time due to electronic imperfections, cavity and/or ambient-temperature changes, and sample evaporation. For a given spin system, this microwave frequency drift is manifested as a shift in the spectrum predicted by considering the Hamiltonian (H̑).
| 1a |
where μB is the Bohr magneton, B is the incident magnetic field, and g is the g factor. S and I are the electron and nuclear spin quantum numbers, respectively, and the energy states are coupled by an anisotropic hyperfine matrix A that is incompletely averaged by motion. For a single nitroxide component with an isotropic rotational correlation time (τc), a modest simulated frequency drift of 0.5 MHz over a 4 day period (Figure S4a) could lead to observable differences between the spectra at 0 h and 96 h (Figure S4b). Because similar levels of microwave frequency drift were observed in our experiments (Figure S4a), a correction (Figure S4c,d) was applied by either multiplying the magnetic field of each spectrum by the ratio of their microwave frequencies (eq 1b) or adding a linear term proportional to their frequency difference (eq 1c) followed by interpolation using MATLAB interpolation routine interp1:
| 1b |
| 1c |
Both correction schemes reduced the difference spectra to the noise level. The estimated error due to microwave frequency correction is 2 orders of magnitude smaller than the magnetic field intervals used (see the Supplementary Text for details). While microwave frequency correction is theoretically impossible as the Zeeman term of the spin Hamiltonian is linearly dependent on frequency whereas hyperfine is not and the g and A tensors are transition-dependent, with this difference forming the basis of multifrequency EPR,40 we found this protocol to be a reasonable compromise to allow quantitative analysis over extended experimental durations.
To perform background subtraction, the background spectrum (Bbg) was first acquired using sample cells containing reaction buffer only. Then, the background spectrum was subtracted from the data (Figure S5). As the cw-EPR signal intensity is directly proportional to the resonator quality factor (Q),41 intersample variations in Q could lead to variations in intensity in both the signal of interest originating from the spin-label and the resonator cavity background (Figure S5a,b). The signal intensity could also fluctuate due to microwave frequency detuning. Thus, the magnitude of background subtraction (w) applied was determined individually for each spectrum Y by considering only magnetic field positions that are not within the span of the nitroxide spectrum (null window, Figure S5b). This background subtraction is hence a linear regression problem to determine the weight of the background (w):
| 2 |
Once w is determined, the correction is applied to the entire spectrum to recover the background and baseline-corrected spectrum (Figure S5c).
Determination of EPR Spin System Parameters
Simulations and least-squares fitting (LSQ) were carried out with EasySpin toolbox (version 4.5.5) and Optimization Toolbox in MATLAB (Mathworks Inc., Natick, MA) using the routine chili for slow-motion cw-EPR spectra by solving the stochastic Liouville equation with rotational eigenfunctions as a basis.42 The feasibility of changes in the rotational correlation time (τc) in distinguishing spin components was illustrated by simulating slow-motion spectra over isotropic τc values from 0.01 to 100 ns (Figure 2b and Table S1).
Figure 2.
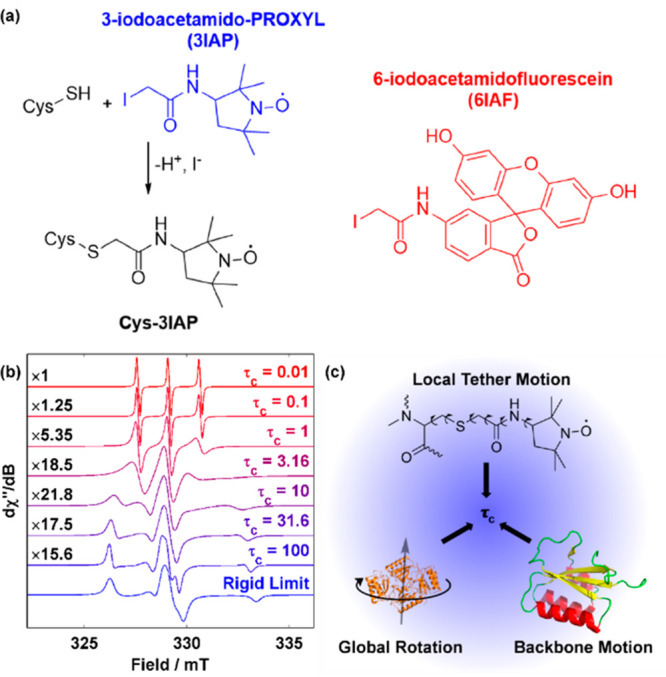
cw-EPR is a strategy for detecting and quantifying intermediates in the Kai clock. (a) Spin labeling reaction used to attach a nitroxide probe onto proteins of interest showing the spin-label 3-iodoacetamido-PROXYL (3IAP, blue) used in this work. The fluorophore 6-iodoacetamido-fluorescein (6IAF, red), also used in this work for fluorescence assays, is also shown for comparison. (b) Simulated nitroxide spectra as a function of mobility normalized by the maximum transition intensity. The scaling factor is shown to the left of each trace, whereas the isotropic rotational correlation time (τc in nanosecond) is shown at the right. Simulation details can be found in Table S1. (c) Three types of motions contribute to the experimentally observed rotational correlation time (τc), which reflects the mobility of the probe.
The best fits [Popt (Table S2)] for spin system parameters (P) were determined by interfacing the EasySpin functions pepper (to determine g and A tensors for frozen samples) or chili (to determine rotational correlation times for ambient-temperature samples)42 with MATLAB nonlinear least-squares (NLLS) routine lsqnonlin using the objective function obj(spectrum, P) defined below:
| 3a |
For frozen samples, P(pepper) consists of rhombic g and A tensors as well as Gaussian and Lorentzian line widths, totaling eight parameters (Figure S8a). For ambient-temperature N19C3IAP and its corresponding KaiBCEE samples, P(chili) consists of a rotational correlation time (a = 1, 2, or 3) parameters (Figures S8b,c and S9). The use of lsqnonlin in place of Easyspin native NLLS function esfit42 led to (i) reduced computational time, (ii) allowed user-defined lower and upper bounds for spin parameters as opposed to the [center ± vary] scheme, and (iii) enabled calculation of confidence intervals of spin parameters via the Jacobian matrix using the MATLAB function nlparci. We note that the Jacobian matrix-derived uncertainty measured only the depth or flatness of the local minimum in the objective function landscape with respect to P and thus could underestimate its uncertainty due to insufficient modeling or correlations in P. An alternative scheme was implemented via case resampling bootstrapping by magnetic field bootstrapping. In this scheme, the spectrum was first fitted as described above to obtain the best fit for Popt. Then, 200 bootstrap spectra were generated via randomly selecting magnetic field positions with replacement and performing NLLS using only the magnetic field positions specified and Popt as a guess. The resultant fits P* were then used to estimate the uncertainty in P. The (1–2α) confidence interval ([L, U]) of P will then have the form
| 3b |
where P(0.01γ)* refers to the γth quantile of P*.
To facilitate ambient-temperature simulations, the g and A tensors were first determined in the near-rigid limit at −40 °C in a 20% (w/w) sucrose (20 Bx) solution using the EasySpin function pepper (Figure S8a) with their uncertainties estimated as described above. The 30 °C spectra were then fitted with fixed g and A tensors with reduced line widths while floating their rotational correlation time(s). For both free and KaiC-bound KaiB, their spectra were simulated with isotropic, axial, and rhombic rotational correlation times in that order (Figure 3f and Figure S8b,c). For selectivity testing of KaiB with respect to direct KaiB–KaiA binding (Figure S9), spectral subtraction was employed by subtracting the spectrum of free KaiB from the reaction spectrum of a 10-fold excess of KaiA and the resultant spectrum was simulated as described above (also see the Supplementary Text). The necessity of rotational anisotropy in simulations was tested via an F-test and AIC computed using the formulas below:
| 4a |
| 4b |
where NRMSD refers to the normalized root-mean-square deviation and is the square root of the residual sum of squares (RSS), normalized by the sum of squares of the background-corrected spectra. For AIC model selection, the quantity of adjustable parameters is equal to k + 2 due to the contribution from isotropic rotation and noise. The AIC is calculated by
| 4c |
where the log-likelihood ln(L̑) is computed by
| 4d |
Due to the possibility for the slight temperature- and local environment-dependent nature of g and A, the spin parameters were further floated within small constraints to better fit the data and compared with the results derived from floating only the rotational correlation time (Figure S8b,civ). It was observed that significantly better fits were obtained by floating g and A, but the values obtained deviated from those of typical nitroxide spin-labels in aqueous solutions determined from multifrequency studies40 and thus were not further considered.
Figure 3.

KaiB-N19C-3IAP (N19C3IAP) reports KaiB–KaiC binding. (a) Crystal structure of tetrameric KaiB [Protein Data Bank (PDB) entry 4KSO] with N19 highlighted in red. (b) Crystal structures of KaiB6C6 (PDB entry 5JWQ, chains E and F) and KaiA2BCI (PDB entry 5JWR, chains A, B, E, and F) with N20 (PDB entries 5JWQ and 5JWR, T. elongatus) highlighted in red. Residue numbers of KaiB in PDB entries 5JWQ and 5JWR are offset from those of S. elongatus by +1. Color code: orange, KaiB; sky blue, KaiC; magenta, KaiA. Pictorial representations of the location of N19(N20) where 3IAP is installed are shown below the respective PDB codes. (c and d) cw-EPR spectra of N19C3IAP (5×, 17.5 μM) incubated with KaiCEE (17.5 μM) for 7 h and then spiked with KaiA (6 μM). Panel c shows a surface plot, whereas panel d shows a stacked plot at selected times. The red vertical arrow in panel d indicates B = 326 mT. (e) Reproduction of the experimental spectrum of N19C3IAP with KaiCEE and KaiA (7 h spectrum, lilac as in panel d)) with experimental spectra of free N19C3IAP (red) and N19C3IAP incubated with a 10-fold excess of KaiCEE (12 h, orange). (f) Overlay of spectral simulations (black) of free (red) and KaiCEE-bound (orange) N19C3IAP using rhombic rotational correlation times. See Table S2 for simulation parameters.
Quantitative Weight Fitting for N19C3IAP
The microwave frequency drift-corrected real-time cw-EPR data Y1024×n were simulated by varying the weights (wij) of the r components as represented by the matrix multiplication in eq 5, where B1024×r corresponds to the spectra of the r components and wij corresponds to the weight of the ith species in the jth spectrum (i = 1–r; j = 1–n):
| 5 |
where ε represents the residual of the fit. The background-corrected spectra of 35 μM spin-labeled KaiB in reaction buffer and 17.5 μM KaiB (5×) in equilibrium with a 10-fold excess of KaiCEE were taken as Bfree,unscaled and Bbound, respectively. In general, two spectra may differ in total intensity due to differences in concentration/volume due to experimental design and/or pipetting error, sample alignment, Q factor, or other unexplained factors. The Bfree,unscaled and Bbound obtained thus cannot be directly used for quantification and must be scaled or normalized. Double integration-based normalization was avoided due to known numerical instability and susceptibility to noise (see Figure S10a).43 Instead, the spectra were normalized by using the real-time kinetic data of KaiB in the presence of a 10-fold excess of KaiCEE. The real-time data can be written as
| 6 |
where θ is the scaling factor to scale Bfree. Bbound is set to be the mean of all spectra in the window t = 12–24 h where there is complete binding. Under these conditions, wt tends toward unity as binding goes to completion as the spectrum is dominated by Bbound. The equation implies that wt and θ(1 – wt) can be solved via linear regression (Figure S10b). As Bfree,unscaled is not scaled, the weight coefficients do not add up to unity. θ can then be solved via linear regression by plotting θ(1 – wt) against 1 – wt (Figure S10c). Bfree is then scaled by θ (Figure S10d).
In addition to Bfree and Bbound, background spectrum Bbg and constant/linear correction terms Bb0 and Bb1 were included. Thus, for a cw-EPR spectrum Yj that contains two components KaiBfree and KaiBbound, the equation is overdetermined (1024 equations, 5 unknowns). The corresponding normal equation is
| 7a |
The shape of each component can be found in Figure S11c. The weights are reported as ratios to total nitroxide content (aij).
| 7b |
We noted that simulations of spectra from technical replicates using experimental spectra of pure KaiB and KaiBCEE as basis spectra (B) occasionally led to slight magnetic field offsets (“B-shift”) that could not be accounted for even after correcting for changes in microwave frequency. We reason that these differences were due to (1) minor variations in magnetic shielding between experiments, (2) removing and reinserting the variable-temperature Dewar, and/or (3) ambient-temperature drift leading to minor magnetic field output. To determine the variation in “B-shift” over time, 20 μM 4-hydroxy-TEMPO (TEMPOL) was used as a spin standard (Figure S6a). It was found that the spectrum drifted horizontally over a 3 day period even after accounting for microwave frequency drift (Figure S4a,b). This drift was correlated to ambient-temperature changes and had a period of ∼24 h but with a lag of 5 h (Figure S6b), hinting that the origin of this time-dependent “B-shift” was potentially due to periodic drift in (1) magnet temperature, (2) expansion or contraction near the magnet, and/or (3) circuit load on the electrical outlets supplying the magnet, all of which can result in variations in magnetic field output.
Because minor shifts in the magnetic field abscissa directly impact spin quantification (Figure S6c), the experimental spectra were shifted horizontally (B′) to account for this difference. The discrete form of this shift can be written as a matrix operator P below:
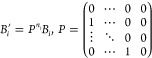 |
8a |
As the optimal shift (in millitesla) is unlikely to be an integer multiple of the magnetic field unit, the magnitude of the shift ni may take any real value s and is achieved via interpolation. Hence, quantitative cw-EPR at any time t becomes a nonlinear problem of 1024 equations and r + 1 unknowns:
| 8b |
Hence, simultaneously solving for wt and st yields the weights of each component and the “B-shift” provided that the spectra of the components were known and “B-shift” corrected. However, eq 5 did not explicitly take “B-shift” into account. Accordingly, eq 5 is modified to the following:
| 9 |
where Yt and st are the spectrum and “B-shift” magnitude at time t, respectively, wit is the weight of the ith component at time t, and si is the component-dependent shift. The term si accounts for the intersample “B-shift” contribution due to changes in sample placement and thus effective magnetic field experienced at the sample, whereas st accounts for intrasample “B-shift” due to fluctuations in magnetic field output that was found to correlate with ambient temperature (Figure S6b). Nonlinear least squares is used to solve for wit, si, and st simultaneously by considering data at multiple time points (tn) using the components Bfree,unscaled, Bbound, and Bbg. This results in 1024 × n equations and 3n (wit) + (3 – 1)(si) + n(st) unknowns with initial guesses for wij obtained via linear regression and si and st set to zero. Only r – 1 components require a non-zero si as si and st co-vary and si=1 is hence defined to be zero. The fitted wit is then used to compute θ (eq 6) for scaling, whereas si is used to shift the B components for further quantitative analysis (eq 8b).
Uncertainty Analysis in Quantitative Real-Time cw-EPR Kai Reactions
There are three contributions to the observed variance in the data: (1) spin model/spectral line shape uncertainty, (2) noise-induced uncertainty, and (3) other random errors, including pipet errors and protein batch-to-batch variability.
(1) Spin Model/Spectral Line Shape Uncertainty (σmodel). Although experimental spectra were used for fitting of all real-time spectra, giving rise to well-defined line shapes, the necessity of scaling Bfree relative to Bbound introduces error in the form of uncertainty in scaling factor θ. As θ is determined via linear regression (eq 6), the confidence interval in θ can be readily obtained by standard linear regression formulas and implemented by MATLAB function regress.
(2) Noise-Induced Uncertainty (σS/N). Real data are noisy and thus give rise to uncertainty in quantification. The weights of Bfree and Bbound are simultaneously determined with “B-shift” via nonlinear regression (see eq 8b). Uncertainty can thus be estimated via the Jacobian matrix and MATLAB function nlparci.
(3) Other Random Errors (σr). Other random errors include protein batch-to-batch variations, pipetting error, and other unknown sources of error. This error can be observed when comparing results obtained between technical replicates.
For phosphomimetic reactions, the overall uncertainty is then computed as
| 10a |
The modeling or line shape uncertainty error is intrinsic to the data processing. On the other hand, performing multiple technical replicates simultaneously measures σr and σS/N.
For oscillator reactions, direct averaging of multiple replicates can lead to cancellations in oscillations due to minor variations in period and/or phasing. Thus, calculation of the inter-replicate standard error of the mean (SEM) was not performed. Instead, the error reported does not contain inter-replicate uncertainty:
| 10b |
Uncertainty estimates from the Jacobian matrix obtained from nonlinear least-squares correspond to weights (w) to be converted to fractions (a). This propagation is done via
| 11 |
where wi in eq 11 corresponds to the weight of component i in the spectrum and ai corresponds to the ratio of component i to the overall concentration. Notice that the first term is common for all values of i and j whereas the second term is a correction term arising from the i = j case in the summation. The final 95% confidence interval (CI) is calculated by
| 12 |
where a̅ is the best estimate for a and t0.975,df and z are t and z values, respectively, for the 95% confidence interval, where df refers to the degrees of freedom in the evaluation of θ.
In experiments in which the binding velocity was desired to be computed (see Figures S11 and S12), a Savitzky–Golay filter was applied via Easyspin function savgol to simultaneously smooth and differentiate the binding kinetic curve using seven-point window and quadratic polynomials.
Circadian Oscillation Rhythm Period and Waveform Analysis
The extracted KaiB binding kinetics were further analyzed by curve fitting to mFourfit44 (MFF) by modeling the kinetic trace f(t) as a sum of cosines as shown below using lsqnonlin as the interface:
| 13a |
where the constant C, amplitude Aj, phase angle φj, and period T are adjustable parameters and the residual term ε is minimized. The quantity of cosines used can be adjusted by changing q. The number of adjustable parameters is thus equal to 3 + 2q. To prevent overfitting, the corresponding AIC was computed at each q by
| 13b |
Confidence intervals in amplitude and period were determined with nlparci. To ascertain the period of oscillation, the maximum entropy spectral analysis (MESA)45 and Lomb–Scargle periodogram46 were computed and compared with the period obtained via MFF (Figure S15b,c).
Results
cw-EPR Strategies for Quantifying Complex Mixtures
As cw-EPR spectra of mixtures are additive, quantitative kinetics can be extracted from real-time cw-EPR experiments with spin-labeled proteins if the spectra of underlying components are known. Thus, successful cw-EPR quantification is contingent upon design and/or discovery of spectroscopic handles that respond to relevant interactions. Because none of the Kai proteins contain paramagnetic centers necessary for direct cw-EPR characterization, a spin-label in the form of nitroxide can be introduced via bioconjugation to provide this handle. This is most conveniently done via site-directed mutagenesis to cysteine followed by labeling with a cysteine-reactive reagent that bears the nitroxide moiety (Figure 2a). In this regard, KaiB is an ideal candidate for spin labeling due to its lack of native cysteine residues as compared to KaiA (six cysteines per monomer) or KaiC (three cysteines per monomer).
There are multiple strategies for designing a labeling site such that the nitroxide spectroscopic handle will respond to binding once installed on the protein of interest by exploiting various spin-based phenomena. One such strategy is to install spin-labels that are close to one another in a multimeric protein, resulting in spin exchange47 and/or dipolar broadening48 that broadens the spectra and thus differentiates monomeric versus multimeric species. Another strategy that is exploited in this work is the mobility dependence of the width of the nitroxide spectrum. Unlike the free nitroxide molecule in solution that tumbles freely, a protein-bound nitroxide moiety exhibits a reduced level of global tumbling. Motions originating from the connecting tether and protein backbone dominate. As the motion slows from rapid tumbling toward the rigid limit, the 14N hyperfine anisotropy is no longer averaged out by the motion and the cw-EPR spectrum broadens (Figure 2b). This mobility as quantified by the rotational correlation time (τc) of a spin-labeled residue is a combination of local and global motions (Figure 2c), changes in which are directly reflected in cw-EPR spectra (Figure 2b). Thus, a successful spin labeling site should lead to either (a) a nitroxide tether that becomes motionally restricted or (b) a change in secondary structure on protein–protein binding. In theory, the global protein tumbling motion (Figure 2c) should contribute to experimentally observed mobility as predicted by the Stokes–Einstein relation (SE, τcSE):
| 14 |
where η is the dynamic viscosity of the buffer (assumed to be equal to that of water at 30 °C), V is the estimated volume of the crystal structures, ν̅p (= 0.73 cm3 g–1) is the partial specific volume, Mp is the molecular weight of the protein, kB is the Boltzmann constant, and T is the temperature in kelvin. In practice, τcSE often underestimates global rotation by a factor of 2 due to the negligible level of hydration.49 This puts a lower limit on the molecular mass of the protein in question as nitroxide spectra stemming from spin-labeled proteins smaller than this limit will be influenced by both local mobility and global rotation. The parallel contributions from both local and global motions complicate interpretations as spectral changes can no longer be attributed to only local binding events.
Site Design and Other Practical Considerations in SDSL–cw-EPR for Probing KaiB–KaiC Binding
Like fluorescence anisotropy (FA),36 the success of EPR spin labeling-based assays is dependent on the labeling site. In the former, site design is guided by two potentially conflicting principles that both local rotational effects (“propeller effects”) and binding affinity interference should be minimized.50 In the latter, it is precisely the changes in local rotational motion that enable a meaningful binding assay as reflected by nitroxide line shape changes while also taking advantage of the smaller footprint of a nitroxide moiety compared to common fluorophores (Figure 2a). To that end, we chose KaiB-N19 (Figure 3a) as the mutation site for studying KaiB–KaiC binding without interference from KaiA. N19 is located at the N-terminal cap of KaiB-α1 and is close to the KaiB-CI binding interface in published crystal structures (Figure 3a).21 However, it is pointed away from the KaiB–KaiA binding interface in the ternary KaiABC complex (Figure 3b).21 Hence, we hypothesized that the N19C mutant of KaiB when spin-labeled will report a decrease in flexibility on KaiB–KaiC binding but that further binding to KaiA will not report the ternary KaiABC complex as a spectroscopically distinct species.
To generate the spin-labeled KaiB-N19C-3IAP construct (N19C3IAP hereafter), a 5-fold excess of TCEP was first added to unlabeled KaiB to reduce preformed disulfide bonds that prevent cysteine-iodoacetamide bioconjugation. Then, a 10-fold excess of 3IAP was added. The labeling efficiency can be checked by intact mass spectrometry (Figure S1). As the EPR signal intensity varies with the quantity of spins in a sample, the labeling reaction should be repeated on samples with low labeling efficiencies to drive the labeling reaction toward completion to provide an optimal signal-to-noise (S/N) ratio. The more commonly used spin-labels S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methylmethanesulfonothioate (R1) and 4-maleimido-TEMPO (4MT) were avoided because they release free PROXYL via disulfide exchange when encountering free thiols51−53 and undergo maleimide hydrolysis to the more flexible succinamic acid ester (SAT),54,55 respectively, rendering them unsuitable for functional assays that span multiple days.
Despite the enhanced reactivity between N19C3IAP and KaiCEE as compared to WT-KaiB as revealed by native-PAGE (Figure S2) on mutagenesis and spin labeling, wild-type KaiB could be completely replaced by N19C3IAP without significantly altering the period of the Kai oscillator based on our previously developed FA assay using 6IAF-labeled KaiB-K25C as a fluorescence reporter [K25C6IAF (Figure S3)].36 Our spin-labeled KaiB mutants are thus robust functional surrogates for wild-type KaiB and therefore suitable as reporters of the Kai clock.
The Spin-Labeled Construct Provides a Selective Response to KaiB–KaiC Binding
To test our hypothesis that N19C3IAP provides a spectroscopic handle for quantifying KaiB–KaiC binding without interference due to ternary KaiABC complex formation, N19C3IAP was first reacted with a stoichiometric amount of KaiCEE and then the mixture was spiked with KaiA after 7 h (Figure 3c,d). KaiCEE, a KaiCpSpT phosphomimetic, was chosen as the KaiC surrogate as S431 phosphorylation was shown to be necessary for KaiBC complex formation, and KaiCEE outperformed KaiCEA in binding to KaiB.23 As spectra from the same sample were collected at 7.5 min intervals over 24 h, 192 spectra were recorded and displayed as a surface plot (Figure 3c). Comparison between cw-EPR spectra recorded immediately after mixing N19C3IAP and KaiCEE versus that of a sample that had been incubated for 7 h (Figure 3d and Figure S7) revealed the formation of a slowly tumbling species in the latter as indicated by the appearance of a peak at 326 mT that is identified as originating from KaiB–KaiC interactions. The intensity of this peak reached a maximum in approximately 6 h (Figure 3c), in agreement with native-PAGE (Figure S2). When the sample was spiked with KaiA, the intensity of the slowly tumbling species decreased marginally but no new spectroscopically distinct species were visually discernible (Figure S7a). Furthermore, the real-time KaiB–KaiCEE–KaiA reaction spectra could be satisfactorily reproduced as a linear combination of the spectrum of pure N19C3IAP and that of N19C3IAP in a 10-fold excess of KaiCEE (Figure 3e and Figure S7b). This implies that cw-EPR of N19C3IAP does not distinguish between various KaiA2jBiC6 complexes (i = 1–6, and j = 0–i),27,28,56 and thus, the spectra derived from N19C3IAP and KaiC can be used as a readout reflecting the fraction of KaiC-bound KaiB independent of KaiA forming a complex with KaiBC.
To investigate the origin of the observed equivalence of cw-EPR spectra of N19C3IAP in KaiABC complexes of various stoichiometries, spectral simulations were applied to analyze the rotational motion of the KaiB-bound spin-labels. The best fits for free and KaiCEE-bound N19C3IAP are shown in Figure 3f. The g and A tensors determined at the near-rigid limit at −40 °C using 20% (w/w) sucrose (Figure S8a and Table S2) are highly comparable to those obtained via W-band cw-EPR (94 GHz) of 3IAP57 as well as multifrequency studies of the related spin-label R1.40 Although the Brownian-motion model oversimplifies the spin-labeled systems in question as global rotation, backbone, and local tether motions are all known to contribute to rotational diffusion (Figure 2c), experimental spectra of both free and KaiCEE-bound N19C3IAP acquired at 30 °C could be satisfactorily simulated via such treatment and hence justify its application. The effective isotropic correlation times (τciso,eff) of both forms of N19C3IAP are shorter than their Stokes–Einstein (SE) estimates [τcSE (Figure S8b,c and Tables S2 and S3)]. Similar observations have been demonstrated with 3IAP-labeled IDPs57 and R1-labeled lysozyme.40 As typical rotational correlation times estimated by SE often underestimate τciso by a factor of 2 for globular proteins,49 our simulations imply that the combined motions of the nitroxide tethers and protein backbone in both free and KaiC-bound KaiB exhibit high flexibility as the tethered nitroxide probe moves faster than global rotational motion. The experimentally observed τceff is thus attributed to the combined motions of the nitroxide tether and protein backbone and explains the independence of the spectral signature to N19C3IAP–KaiC binding stoichiometry.
For the sake of completeness, we also reacted N19C3IAP with KaiA in the absence of KaiC to determine its spectral sensitivity to direct KaiA–KaiB interactions (Figure S9). It has previously been reported that KaiA could bind to WT-KaiB in trace quantities in the form of KaiA2B1 based on native mass spectrometry.28 Separately, mutations that destabilize tetrameric gs-KaiB via either the G88A mutation24 or C-terminal deletions20 have been shown to enhance direct KaiA–KaiB interactions. On the basis of these observations, it is deduced that KaiB likely adopts the fs-KaiB conformation in the KaiA2B1 complex. To determine if N19C3IAP responds to direct KaiA–KaiB interactions, cw-EPR spectra of 17.5 μM N19C3IAP, corresponding to 5× in vitro oscillator, in the presence of varying concentrations of KaiA were compared. The cw-EPR spectrum in the presence of 6 μM KaiA (5×) could not be discerned from that of N19C3IAP alone (Figure S9a). There are three possibilities to this observation. (a) KaiB and KaiA do not interact at all. (b) They interact, but X-band cw-EPR of N19C3IAP treats the KaiA-bound KaiB population as spectroscopically equivalent to free KaiB. (c) They interact to give a spectroscopically distinct species, but the limited S/N ratio renders detection impossible. The last scenario may be attributed to (i) weak interactions at the physiologically relevant KaiA:KaiB ratio (1:3) and/or (ii) the high degree of cw-EPR spectral similarity between free and KaiA-bound N19C3IAP due to insignificant changes in local mobility. In an attempt to distinguish the three scenarios and drive KaiA–KaiB interactions, we increased the KaiA:KaiB ratio to 10:1 and observed subtle changes in the h–1 transition region as well as slight broadening in the central transition (Figure S9b), ruling out possibilities (a) and (b). Subtraction and simulation studies indicate that this motional component constitutes at least 7% of the total signal under excess KaiA loading (Figure S9c, Table S4, and the Supplementary Text). Because this ratio is unphysiological as ratios of 1:329 to 1:3022 have been reported in vivo, little insight into the biological relevance of direct KaiA–KaiB interactions can be gained. Nevertheless, the selectivity of the spectroscopic response of N19C3IAP to KaiB–KaiC interactions is established.
Quantitative Interpretation of KaiB–KaiC Binding
Because N19C3IAP is the only EPR-active species in the experiments described above, the fraction of KaiB in KaiC-bound KaiB relative to total KaiB is equal to its contribution (weight) normalized by the total weight of the spectrum. It was found above that the N19C3IAP reaction spectra could be explained by two components, free and KaiC-bound KaiB (Figure 3e). The former was readily obtained by acquiring the spectrum of N19C3IAP alone, whereas the latter was obtained by adding it to a 10-fold excess of KaiCEE (Figure S7b). Quantitative kinetics with N19C3IAP could thus be extracted using these two spectra as basis spectra.
We first applied this strategy to analyze the KaiB–KaiCEE binding data to obtain a binding progression curve. The extracted kinetics exhibited sigmoidal kinetics (Figure 4a and Figures S11 and S12). Qualitatively, the sigmoidal kinetics observed in our experiments rules out the possibility of single-step KaiB–KaiC binding and is consistent with previous studies that indicate binding cooperativity among KaiB subunits27 as well as gating via KaiB fold switching and/or CI ATPase activity.24 To quantify binding latency, we determined the time taken to reach the maximum rate of binding (tv,max) to be 0.9 ± 0.2 h [SEM; n = 6 (Figure 4b and Figures S11 and S12)]. Compared to native-PAGE (Figure S2), the nondestructive nature of ambient-temperature cw-EPR allows kinetics to be determined using a single sample. This contrasts with native-PAGE in which the sample is adulterated by the loading buffer and consumed during electrophoresis, rendering the need to prepare multiple samples flash frozen at predetermined times. The improved time resolution of cw-EPR over native-PAGE allows identification of sigmoidal kinetics in N19C3IAP–KaiCEE binding that is not observed in native-PAGE.
Figure 4.
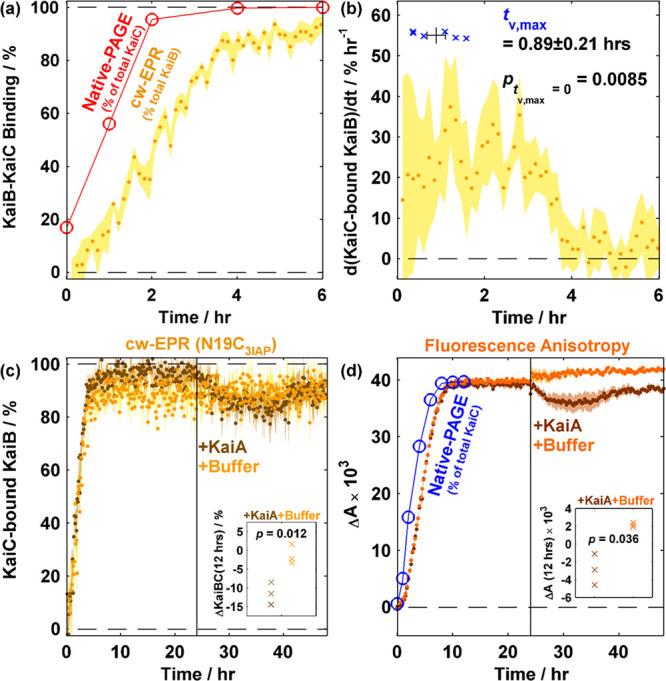
cw-EPR is an orthogonal method for quantitatively assaying KaiB–KaiC binding as illustrated by KaiB–KaiCEE binding followed by KaiA spiking at 24 h. (a) Overlay of N19C3IAP–KaiCEE binding kinetics obtained via native-PAGE (red empty circles) and cw-EPR (yellow filled circles) from 0 to 6 h. (b) Rate of formation of KaiC-bound KaiB (yellow circles) and time at maximum binding velocity (tv,max, blue crosses). For panels a and b, the shaded area (cw-EPR) and error bars (tv,max) indicate SEM (n = 6). (c) Overlay of N19C3IAP–KaiCEE cw-EPR binding kinetics followed by spiking with KaiA (brown) or buffer (yellow) at 24 h. Shaded areas show the SEM (n = 3). (d) Overlay of WT-KaiB–KaiCEE fluorescence anisotropy-based binding kinetics using 50 nM KaiB-K25C-6IAF (K25C6IAF) as a fluorescence probe with KaiA (brown) or buffer (orange) spiking at 24 h. Shaded areas show the SEM (n = 3). Insets in panels c and d show two-tailed t-tests comparing the effects of KaiA vs buffer spiking after 12 h of spiking. cw-EPR data were binned in 2 h bins prior to performing the t-test.
Curiously, an apparent offset of approximately 1 h was observed between kinetics derived from cw-EPR versus native-PAGE (Figure 4a). As KaiC (58 kDa) is larger than KaiB (11 kDa), the native-PAGE band intensity is likely dominated by KaiC in KaiBC complexes. Furthermore, native-PAGE electrophoretic mobility is a function of protein conformation. We interpret the discrepancy in native-PAGE and either cw-EPR or FA (Figure 4d and Figure S13, vide infra) as originating from the former being sensitive to KaiC conformational changes in KaiB binding. This discrepancy is consistent with the multistep nature of KaiB–KaiC binding based on native mass spectrometric identification of substoichiometric KaiBC complexes.27 However, the possibility of in-gel KaiB–KaiC binding during electrophoresis cannot be ruled out.
We further analyzed the KaiB–KaiCEE binding data when the steady-state mixtures were subjected to KaiA spiking (Figure 4c and Figures S11 and S12). On the basis of previous studies, the addition of KaiA to an equilibrated mixture of KaiB and KaiCEE would lead to a ternary mixture of KaiB-containing species: KaiB, KaiBCEE, and KaiABCEE.20 As demonstrated above, cw-EPR of N19C3IAP is insensitive to KaiABC complexes due to the lack of formation of spectroscopically distinct species at physiologically relevant concentrations. Thus, the slight increase in the mobile component’s intensity after KaiA was added to the preequilibrated mixture of N19C3IAP and KaiCEE provides unequivocal evidence that KaiA can antagonize KaiBCEE interactions slowly (Figures 3d and 4c and Figures S11 and S12). On the contrary, interpretation of the K25C6IAF FA data (Figure 4d and Figure S13) is complicated by the presence of at least three KaiB-containing species even under the assumption that KaiBC complexes of all stoichiometries (KaiBiC6, where i = 1–6) have identical fluorescence anisotropies. Because there is little theoretical basis to assume that K25C6IAF in the ternary KaiABCEE complex possesses an FA identical to that of the binary KaiBCEE complex, the use of FA to quantify KaiB–KaiCEE binding in the presence of KaiA is akin to solving an underdetermined system of equations. Nevertheless, if these assumptions hold, we anticipate that formation of the ternary complex would marginally increase the observed anisotropy whereas KaiB–KaiC interaction antagonization should reduce the anisotropy. The observed decrease in anisotropy could thus be interpreted qualitatively as KaiBCEE complex antagonization, a fortuitous conclusion that agrees with cw-EPR-derived kinetics (Figure 4c and Figures S11 and S12). This KaiA spiking experiment highlights the potential ambiguity in extracting kinetics from single-wavelength FA data. On the other hand, cw-EPR benefits from the use of the magnetic field as a second abscissa, offering the possibility of statistically testing the quantity of components via rank analysis58 and quantifying more than two components if warranted. Thus, cw-EPR of spin-labeled KaiB offers an orthogonal and quantitative method for probing KaiB–KaiC binding while eliminating interference due to ambiguity in variable Kai protein stoichiometries.
Application to Oscillator Reactions
Equipped with the knowledge that N19C3IAP probes KaiB–KaiC binding, we measured the time dependence of KaiB binding in the in vitro oscillator using real-time cw-EPR (Figure 5 and Figures S14–S16) using N19C3IAP as a spin-labeled surrogate for WT-KaiB.
Figure 5.
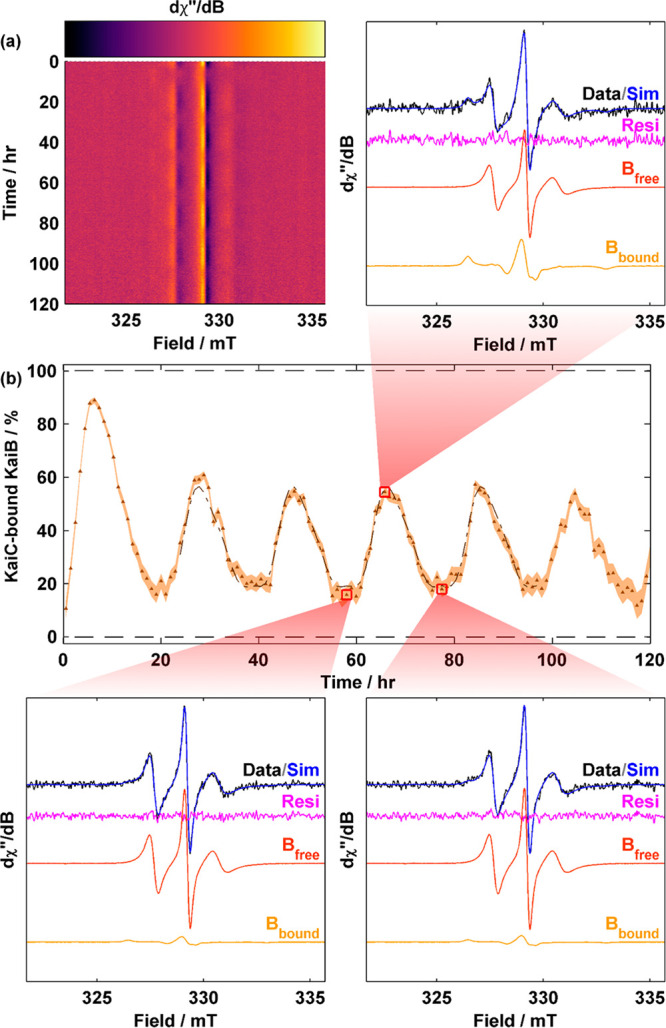
Observation of KaiB–KaiC binding and dissociation throughout the Kai oscillations via cw-EPR. (a) Surface plot of cw-EPR spectra of N19C3IAP 1× oscillator (3.5 μM N19C3IAP, 3.5 μM KaiC, and 1.2 μM KaiA). (b) cw-EPR-based kinetics of KaiC-bound KaiB as a function of time. Data were binned in 1 h bins. The shaded area indicates the 95% CI. A fit to the sum of cosines is overlaid as a dashed–dotted line (see Table S5). Selected spectra at three time points (red squares) with their respective fit are displayed.
The cw-EPR spectra of N19C3IAP in the reconstituted oscillator demonstrated periodic time dependence as seen in height oscillations of the central (h0) transition at B0 = 329.1 mT as well as broader features corresponding to h–1 and h+1 transitions that are out of phase with the h0 transition (Figure 5a and Figure S14a). The periodic spectral variations indicate periodic KaiB binding–unbinding events. Although individual spectra at a 1× concentration were very noisy, reasonable fits using experimental spectra of pure KaiB and KaiBCEE were still possible after binning (Figure 5b and Figure S14b) and enabled quantitative determination of binding progression with high reproducibility (Figure S15). Using a sum of cosines via mFourfit44 (Figure S16), we determined the phenomenological amplitude of oscillation to be 41.7 ± 3.4% [SEM; n = 4 (Figure 5b, Figure S15, and Table S5], slightly smaller than 50% as determined by FCS by Goda et al.34 We reasoned that the minor discrepancy is due to a smaller translational diffusion coefficient of the ternary KaiABC complex compared to KaiBC that leads to amplitude overestimation in a two-component model. However, the difference in reactivity between WT-KaiB and N19C3IAP revealed by native-PAGE (Figure S2) may also contribute to the difference in amplitudes. The overall shape of the binding curve is consistent with previous literature34 that revealed that (i) KaiB binding occurs over a shorter time span than KaiB unbinding and (ii) the duration of minimally bound KaiB (trough) is wider than the duration of maximally bound KaiB (peak) (Figure 5b, Figure S16, and Table S5). We note that a two-component model for describing FCS data is likely inappropriate as FCS is based on the translational diffusion time, which is in turn dependent on the molecular weights of various KaiABC complexes with various stoichiometries, and previous native mass spectrometry results27 point toward this fact. On the other hand, for sufficiently large proteins where global rotational motion is slow, cw-EPR is dependent on only the local nitroxide tether and protein backbone motion. Thus, a two-component model is appropriate for N19C3IAP (Figure 3e).
Discussion
Implications of cw-EPR Results for the Current Model of the Kai Oscillator
The discovery of N19C3IAP as a KaiB–KaiC interaction selective probe allowed binding kinetics to be observed in real time. The improved time resolution and theoretical support for selectivity offer an orthogonal window into the understanding of the Kai system.
Our cw-EPR and FA data suggest that the KaiB–KaiCEE interaction is weakly antagonized by KaiA transiently (Figure 4c,d). As pointed out above, the existence of more than two KaiB-containing species prevents interpretation of K25C6IAF FA as KaiB–KaiC binding when KaiA is present. Nevertheless, the consistency between these two techniques at differing labeling sites implies that this transient KaiA antagonization is unlikely to be a probe or technique-based artifact. The observed antagonization is surprising given that KaiA is known to be sequestered during the night state as a ternary KaiABC complex when S431 is phosphorylated.20 As KaiA was observed to impact KaiB–KaiC binding in a phospho-state-dependent manner as determined by immunoprecipitation SDS–PAGE,23 with an increasing level of KaiA leading to a nonlinear response in KaiCpSpT–KaiB binding, the real-time observations presented in this study were not unreasonable. KaiA has been shown to act as a nucleotide exchange factor for KaiC,18,19 the latter of which has only been crystallized in conjunction with KaiB in a post-ATP hydrolysis state.21 We speculate that this transient KaiA-induced KaiBC antagonization is due to interactions between KaiA and A-loops on CII. This interaction weakens the CII ring integrity, which in turn weakens CII–CI ring stacking interactions.13 As ring stacking stabilizes the posthydrolysis state of CI, weakened stacking promotes the prehydrolytic state of CI, which destabilizes KaiBC interactions. This antagonization occurs over 12 h, but the KaiBC population recovers on longer time scales as the spiked KaiA is sequestered into a KaiABC complex. This observation is consistent with exclusive activities of CI and CII rings that are reciprocally regulated.23 Further experiments are necessary to evaluate the plausibility and molecular mechanism of this pathway and its relevance in clock resetting.
Limitations of cw-EPR as an Analytical Technique for Studying Protein–Protein Interactions
Prior to this work, SDSL–EPR has been used to determine the secondary structure59 and oligomeric state47 of membrane proteins as well as folding of both globular60 and intrinsically disordered proteins.53 Our current work illustrates that cw-EPR can be an analytical technique for studying protein–protein interactions in real time. However, there are two limitations that can limit the usefulness of cw-EPR in quantification and generalization to other systems to be discussed below.
(1) Site Design for Spin Labeling. Unlike FA where the fluorescence probe can be attached to surface residues or either terminus of the protein of interest,50 the requirement for a change in the local environment (Figure 2c) for a corresponding spectral change in the nitroxide cw-EPR spectrum poses a unique problem. Researchers are often limited by existing crystal structures and biochemical intuition in designing mutants for probe conjugation while minimizing perturbations to biochemical reactivity. In this work, we showed that N19C3IAP was selective with respect to KaiB–KaiC interactions but the mutagenesis and labeling also enhanced KaiB’s reactivity (Figure S2). SDSL is thus not necessarily a conservative mutation, and orthogonal functional assays should be employed to test the spin-labeled constructs’ reactivity. Nevertheless, considering the potential of SDSL–EPR in resolving multiple species in real time by using magnetic field as the second abscissa, its application could be extended if this barrier in site design is lowered. We envision that future researchers should be able to design SDSL–EPR probe sites based on molecular dynamics (MD) simulations.61−65 To that end, multiple strategies have been implemented to directly compute the cw-EPR spectra of spin-labeled proteins from MD simulations.62−65 However, screening of multiple sites via a full MD simulation can be computationally demanding. Alternatively, rotamer libraries have previously been computed for multiple nitroxide spin-labels as coarse-grain approximations to the true dynamics of the nitroxide moiety to aid in designing probe sites for favorable distance measurements.61,66 Computed within the rotamer library-based calculations are metrics such as nitroxide partition function Z, which measures the tightness of the site and root-mean-square deviation of the midpoint of the N–O group from the mean position, both of which might be correlated with nitroxide mobility. As only a single biologically active mutant was described in this work, there are insufficient data to correlate our results with MD or rotamer library predictions. Nevertheless, we anticipate that such a correlation can be established in the future with additional data and increasingly efficient schemes in computing nitroxide molecular dynamics.
(2) Sensitivity and Throughput. A second limitation to developing an SDSL–EPR-based biochemical assay is the limited sensitivity and throughput of cw-EPR. SDSL–cw-EPR at ambient temperatures has previously been performed with proteins at high concentrations40 (∼1 mM) to allow a sufficient S/N ratio. For practical biological assays, the protein concentration is often kept low to mimic physiological conditions and reduce the level of aggregation. This trade-off in sample concentration renders a low S/N ratio and ambiguity in spectral interpretation. In the context of the Kai clock, increasing the concentration of the Kai oscillator can lead to an increased level of ATP consumption and premature failure of the oscillator due to ATP depletion.67 This contrasts with routine fluorescence assays in which nanomolar probe concentrations coupled with optical plate readers provide decent S/N ratios with high throughput. The S/N ratio limitation highlights the urgency for innovative detection schemes to improve cw-EPR sensitivity. In that respect, we anticipate that rapid-scan EPR68 coupled with imaging69 can satisfy both requirements of increased S/N ratio and throughput at physiological concentrations.
Acknowledgments
The authors thank Drs. William Jewell and Armann Andaya for their help in acquiring LCMS data. The authors thank Carrie L. Partch for valuable insights and G. Rao, L. Tao, A. R. Balo, and M. A. Nesbit for critical reading/helpful discussion.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00279.
Tables S1–S5, Figures S1–S16, and Supplementary Text (PDF)
Accession Codes
S. elongatus (PCC 7942) KaiC, UnitProt entry Q79PF4; S. elongatus (PCC 7942) KaiB, UnitProt entry Q79PF5; S. elongatus (PCC 7942) KaiA, UnitProt entry Q79PF6.
Author Present Address
∇ Y.-G.C.: Monash Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia.
This work was supported by grants from the U.S. National Science Foundation (NSF-MCB-1615752 to R.D.B. and NSF-HRD-1547848 to CREST, the Center for Cellular and Biomolecular Machines at the University of California, Merced, CA), the National Institutes of Health (GM107521 to A.L.), and the Army Research Office (W911NF-17-1-0434 to A.L.).
The authors declare no competing financial interest.
Supplementary Material
References
- Liu Y.; Tsinoremas N. F.; Johnson C. H.; Lebedeva N. V.; Golden S. S.; Ishiura M.; Kondo T. (1995) Circadian orchestration of gene expression in cyanobacteria. Genes Dev. 9, 1469–1478. 10.1101/gad.9.12.1469. [DOI] [PubMed] [Google Scholar]
- Ouyang Y.; Andersson C. R.; Kondo T.; Golden S. S.; Johnson C. H. (1998) Resonating circadian clocks enhance fitness in cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 95, 8660–8664. 10.1073/pnas.95.15.8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita J.; Nakajima M.; Kondo T.; Iwasaki H. (2005) No transcription-translation feedback in circadian rhythm of KaiC phosphorylation. Science 307, 251–254. 10.1126/science.1102540. [DOI] [PubMed] [Google Scholar]
- Young M. W.; Kay S. A. (2001) Time zones: a comparative genetics of circadian clocks. Nat. Rev. Genet. 2, 702–715. 10.1038/35088576. [DOI] [PubMed] [Google Scholar]
- Rust M. J.; Markson J. S.; Lane W. S.; Fisher D. S.; O’Shea E. K. (2007) Ordered phosphorylation governs oscillation of a three-protein circadian clock. Science 318, 809–812. 10.1126/science.1148596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiwaki T.; Satomi Y.; Kitayama Y.; Terauchi K.; Kiyohara R.; Takao T.; Kondo T. (2007) A sequential program of dual phosphorylation of KaiC as a basis for circadian rhythm in cyanobacteria. EMBO J. 26, 4029–4037. 10.1038/sj.emboj.7601832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M.; Imai K.; Ito H.; Nishiwaki T.; Murayama Y.; Iwasaki H.; Oyama T.; Kondo T. (2005) Reconstitution of circadian oscillation of cyanobacterial KaiC phosphorylation in vitro. Science 308, 414–415. 10.1126/science.1108451. [DOI] [PubMed] [Google Scholar]
- Wang J. (2004) Nucleotide-dependent domain motions within rings of the RecA/AAA(+) superfamily. J. Struct. Biol. 148, 259–267. 10.1016/j.jsb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Phong C.; Markson J. S.; Wilhoite C. M.; Rust M. J. (2013) Robust and tunable circadian rhythms from differentially sensitive catalytic domains. Proc. Natl. Acad. Sci. U. S. A. 110, 1124–1129. 10.1073/pnas.1212113110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe J.; Hiyama T.; Mukaiyama A.; Son S.; Mori T.; Saito S.; Osako M.; Wolanin J.; Yamashita E.; Kondo T.; Akiyama S. (2015) Atomic-scale origins of slowness in the cyanobacterial circadian clock. Science 349, 312–316. 10.1126/science.1261040. [DOI] [PubMed] [Google Scholar]
- Murakami R.; Miyake A.; Iwase R.; Hayashi F.; Uzumaki T.; Ishiura M. (2008) ATPase activity and its temperature compensation of the cyanobacterial clock protein KaiC. Genes Cells 13, 387–395. 10.1111/j.1365-2443.2008.01174.x. [DOI] [PubMed] [Google Scholar]
- Chang Y. G.; Kuo N. W.; Tseng R.; LiWang A. (2011) Flexibility of the C-terminal, or CII, ring of KaiC governs the rhythm of the circadian clock of cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 108, 14431–14436. 10.1073/pnas.1104221108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. G.; Tseng R.; Kuo N. W.; LiWang A. (2012) Rhythmic ring-ring stacking drives the circadian oscillator clockwise. Proc. Natl. Acad. Sci. U. S. A. 109, 16847–16851. 10.1073/pnas.1211508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiwaki T.; Kondo T. (2012) Circadian autodephosphorylation of cyanobacterial clock protein KaiC occurs via formation of ATP as intermediate. J. Biol. Chem. 287, 18030–18035. 10.1074/jbc.M112.350660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S.; Vakonakis I.; Ioerger T. R.; LiWang A. C.; Sacchettini J. C. (2004) Crystal structure of circadian clock protein KaiA from Synechococcus elongatus. J. Biol. Chem. 279, 20511–20518. 10.1074/jbc.M400077200. [DOI] [PubMed] [Google Scholar]
- Kim Y. I.; Dong G.; Carruthers C. W.; Golden S. S.; LiWang A. (2008) The day/night switch in KaiC, a central oscillator component of the circadian clock of cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 105, 12825–12830. 10.1073/pnas.0800526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayek R.; Egli M. (2015) Protein-Protein Interactions in the Cyanobacterial Circadian Clock: Structure of KaiA Dimer in Complex with C-Terminal KaiC Peptides at 2.8 angstrom Resolution. Biochemistry 54, 4575–4578. 10.1021/acs.biochem.5b00694. [DOI] [PubMed] [Google Scholar]
- Nishiwaki-Ohkawa T.; Kitayama Y.; Ochiai E.; Kondo T. (2014) Exchange of ADP with ATP in the CII ATPase domain promotes autophosphorylation of cyanobacterial clock protein KaiC. Proc. Natl. Acad. Sci. U. S. A. 111, 4455–4460. 10.1073/pnas.1319353111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L.; Vani B. P.; Thiede E. H.; Rust M. J.; Dinner A. R. (2018) Molecular dynamics simulations of nucleotide release from the circadian clock protein KaiC reveal atomic-resolution functional insights. Proc. Natl. Acad. Sci. U. S. A. 115, E11475–E11484. 10.1073/pnas.1812555115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng R.; Chang Y. G.; Bravo I.; Latham R.; Chaudhary A.; Kuo N. W.; LiWang A. (2014) Cooperative KaiA-KaiB-KaiC interactions affect KaiB/SasA competition in the circadian clock of cyanobacteria. J. Mol. Biol. 426, 389–402. 10.1016/j.jmb.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng R.; Goularte N. F.; Chavan A.; Luu J.; Cohen S. E.; Chang Y. G.; Heisler J.; Li S.; Michael A. K.; Tripathi S.; Golden S. S.; LiWang A.; Partch C. L. (2017) Structural basis of the day-night transition in a bacterial circadian clock. Science 355, 1174–1180. 10.1126/science.aag2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitayama Y.; Iwasaki H.; Nishiwaki T.; Kondo T. (2003) KaiB functions as an attenuator of KaiC phosphorylation in the cyanobacterial circadian clock system. EMBO J. 22, 2127–2134. 10.1093/emboj/cdg212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.; Chew J.; Chockanathan U.; Rust M. J. (2014) Mixtures of opposing phosphorylations within hexamers precisely time feedback in the cyanobacterial circadian clock. Proc. Natl. Acad. Sci. U. S. A. 111, E3937–E3945. 10.1073/pnas.1408692111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. G.; Cohen S. E.; Phong C.; Myers W. K.; Kim Y. I.; Tseng R.; Lin J.; Zhang L.; Boyd J. S.; Lee Y.; Kang S.; Lee D.; Li S.; Britt R. D.; Rust M. J.; Golden S. S.; LiWang A. (2015) A protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science 349, 324–328. 10.1126/science.1260031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal S. A.; Pattanayek R.; Williams D. R.; Mori T.; Qin X. M.; Johnson C. H.; Egli M.; Stewart P. L. (2013) CryoEM and Molecular Dynamics of the Circadian KaiB-KaiC Complex Indicates That KaiB Monomers Interact with KaiC and Block ATP Binding Clefts. J. Mol. Biol. 425, 3311–3324. 10.1016/j.jmb.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase R.; Imada K.; Hayashi F.; Uzumaki T.; Morishita M.; Onai K.; Furukawa Y.; Namba K.; Ishiura M. (2005) Functionally important substructures of circadian clock protein KaiB in a unique tetramer complex. J. Biol. Chem. 280, 43141–43149. 10.1074/jbc.M503360200. [DOI] [PubMed] [Google Scholar]
- Snijder J.; Burnley R. J.; Wiegard A.; Melquiond A. S. J.; Bonvin A. M. J. J.; Axmann I. M.; Heck A. J. R. (2014) Insight into cyanobacterial circadian timing from structural details of the KaiB-KaiC interaction. Proc. Natl. Acad. Sci. U. S. A. 111, 1379–1384. 10.1073/pnas.1314326111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder J.; Schuller J.; Wiegard A.; Lossl P.; Schmelling N.; Axmann I.; Plitzko J.; Forster F.; Heck A. (2017) Structures of the cyanobacterial circadian oscillator frozen in a fully assembled state. Science 355, 1181–1184. 10.1126/science.aag3218. [DOI] [PubMed] [Google Scholar]
- Chew J.; Leypunskiy E.; Lin J.; Murugan A.; Rust M. J. (2018) High protein copy number is required to suppress stochasticity in the cyanobacterial circadian clock. Nat. Commun. 9, 3004. 10.1038/s41467-018-05109-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitayama Y.; Nishiwaki-Ohkawa T.; Sugisawa Y.; Kondo T. (2013) KaiC intersubunit communication facilitates robustness of circadian rhythms in cyanobacteria. Nat. Commun. 4, 2897. 10.1038/ncomms3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zon J. S.; Lubensky D. K.; Altena P. R.; ten Wolde P. R. (2007) An allosteric model of circadian KaiC phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 104, 7420–7425. 10.1073/pnas.0608665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H.; Kageyama H.; Mutsuda M.; Nakajima M.; Oyama T.; Kondo T. (2007) Autonomous synchronization of the circadian KaiC phosphorylation rhythm. Nat. Struct. Mol. Biol. 14, 1084–1088. 10.1038/nsmb1312. [DOI] [PubMed] [Google Scholar]
- Emberly E.; Wingreen N. (2006) Hourglass model for a protein-based circadian oscillator. Phys. Rev. Lett. 96, 038303. 10.1103/PhysRevLett.96.038303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda K.; Ito H.; Kondo T.; Oyama T. (2012) Fluorescence correlation spectroscopy to monitor Kai protein-based circadian oscillations in real time. J. Biol. Chem. 287, 3241–3248. 10.1074/jbc.M111.265777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda K.; Kondo T.; Oyama T. (2014) Effects of adenylates on the circadian interaction of KaiB with the KaiC complex in the reconstituted cyanobacterial Kai protein oscillator. Biosci., Biotechnol., Biochem. 78, 1833–1838. 10.1080/09168451.2014.940833. [DOI] [PubMed] [Google Scholar]
- Heisler J.; Chavan A.; Chang Y. G.; LiWang A. (2019) Real-Time In Vitro Fluorescence Anisotropy of the Cyanobacterial Circadian Clock. Methods Protoc. 2, 42. 10.3390/mps2020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berliner L. J., Ed. (1976) Spin Labeling: Theory and Applications, Academic Press, New York. [Google Scholar]
- Tyagarajan K.; Pretzer E.; Wiktorowicz J. E. (2003) Thiol-reactive dyes for fluorescence labeling of proteomic samples. Electrophoresis 24, 2348–2358. 10.1002/elps.200305478. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Marshall A. G. (1998) A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 9, 225–233. 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Fleissner M. R.; Tipikin D. S.; Liang Z.; Moscicki J. K.; Earle K. A.; Hubbell W. L.; Freed J. H. (2010) Multifrequency electron spin resonance study of the dynamics of spin labeled T4 lysozyme. J. Phys. Chem. B 114, 5503–5521. 10.1021/jp910606h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton G. R., Eaton S. S., Barr D. P., and Weber R. T. (2010) Quantitative EPR: A Practitioners Guide, Springer, Vienna. [Google Scholar]
- Stoll S.; Schweiger A. (2006) Easy Spin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55. 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Czoch R. (1996) Quantitative EPR — Sensitivity to experimental conditions and optimal setting of recording parameters. Appl. Magn. Reson. 10, 293–317. 10.1007/BF03163115. [DOI] [Google Scholar]
- Edwards K. D.; Akman O. E.; Knox K.; Lumsden P. J.; Thomson A. W.; Brown P. E.; Pokhilko A.; Kozma-Bognar L.; Nagy F.; Rand D. A.; Millar A. J. (2010) Quantitative analysis of regulatory flexibility under changing environmental conditions. Mol. Syst. Biol. 6, 424. 10.1038/msb.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrodale I.; Erickson R. E. (1980) Algorithms for least-squares linear prediction and maximum entropy spectral analysis—Part I: Theory. Geophysics 45, 420–432. 10.1190/1.1441091. [DOI] [Google Scholar]
- Lomb N. R. (1976) Least-squares frequency analysis of unequally spaced data. Astrophys. Space Sci. 39, 447–462. 10.1007/BF00648343. [DOI] [Google Scholar]
- Langen R.; Isas J. M.; Luecke H.; Haigler H. T.; Hubbell W. L. (1998) Membrane-mediated assembly of annexins studied by site-directed spin labeling. J. Biol. Chem. 273, 22453–22457. 10.1074/jbc.273.35.22453. [DOI] [PubMed] [Google Scholar]
- Altenbach C.; Oh K. J.; Trabanino R. J.; Hideg K.; Hubbell W. L. (2001) Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry 40, 15471–15482. 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- Halle B.; Davidovic M. (2003) Biomolecular hydration: from water dynamics to hydrodynamics. Proc. Natl. Acad. Sci. U. S. A. 100, 12135–12140. 10.1073/pnas.2033320100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerke N. J. (2009) Fluorescence Polarization (FP) Assays for Monitoring Peptide-Protein or Nucleic Acid–Protein Binding. Curr. Protoc. Chem. Biol. 1, 1–15. 10.1002/9780470559277.ch090102. [DOI] [PubMed] [Google Scholar]
- Mutoh R.; Mino H.; Murakami R.; Uzumaki T.; Takabayashi A.; Ishii K.; Ishiura M. (2010) Direct interaction between KaiA and KaiB revealed by a site-directed spin labeling electron spin resonance analysis. Genes Cells 15, 269–280. 10.1111/j.1365-2443.2009.01377.x. [DOI] [PubMed] [Google Scholar]
- Erales J.; Lorenzi M.; Lebrun R.; Fournel A.; Etienne E.; Courcelle C.; Guigliarelli B.; Gontero B.; Belle V. (2009) A new function of GAPDH from Chlamydomonas reinhardtii: a thiol-disulfide exchange reaction with CP12. Biochemistry 48, 6034–6040. 10.1021/bi900569h. [DOI] [PubMed] [Google Scholar]
- Pirman N. L.; Milshteyn E.; Galiano L.; Hewlett J. C.; Fanucci G. E. (2011) Characterization of the disordered-to-α-helical transition of IA3 by SDSL-EPR spectroscopy. Protein Sci. 20, 150–159. 10.1002/pro.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine S. D.; Reid R.; Robinson L.; Ashley G. W.; Santi D. V. (2015) Long-term stabilization of maleimide-thiol conjugates. Bioconjugate Chem. 26, 145–152. 10.1021/bc5005262. [DOI] [PubMed] [Google Scholar]
- Mutoh R.; Mino H.; Murakami R.; Uzumaki T.; Ishiura M. (2011) Thermodynamically Induced Conformational Changes of the Cyanobacterial Circadian Clock Protein KaiB. Appl. Magn. Reson. 40, 525–534. 10.1007/s00723-011-0228-2. [DOI] [Google Scholar]
- Murakami R.; Yunoki Y.; Ishii K.; Terauchi K.; Uchiyama S.; Yagi H.; Kato K. (2019) Cooperative Binding of KaiB to the KaiC Hexamer Ensures Accurate Circadian Clock Oscillation in Cyanobacteria. Int. J. Mol. Sci. 20, 4550. 10.3390/ijms20184550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L.; Liu Z.; Kaur P.; Esquiaqui J. M.; Hunter R. I.; Hill S.; Smith G. M.; Fanucci G. E. (2016) Toward increased concentration sensitivity for continuous wave EPR investigations of spin-labeled biological macromolecules at high fields. J. Magn. Reson. 265, 188–196. 10.1016/j.jmr.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Wax M.; Kailath T. (1985) Detection of signals by information theoretic criteria. IEEE Trans. Acoust., Speech, Signal Process. 33, 387–392. 10.1109/TASSP.1985.1164557. [DOI] [Google Scholar]
- Altenbach C.; Yang K.; Farrens D. L.; Farahbakhsh Z. T.; Khorana H. G.; Hubbell W. L. (1996) Structural features and light-dependent changes in the cytoplasmic interhelical E-F loop region of rhodopsin: a site-directed spin-labeling study. Biochemistry 35, 12470–12478. 10.1021/bi960849l. [DOI] [PubMed] [Google Scholar]
- Hubbell W. L.; Mchaourab H. S.; Altenbach C.; Lietzow M. A. (1996) Watching proteins move using site-directed spin labeling. Structure 4, 779–783. 10.1016/S0969-2126(96)00085-8. [DOI] [PubMed] [Google Scholar]
- Polyhach Y.; Bordignon E.; Jeschke G. (2011) Rotamer libraries of spin labelled cysteines for protein studies. Phys. Chem. Chem. Phys. 13, 2356–2366. 10.1039/C0CP01865A. [DOI] [PubMed] [Google Scholar]
- DeSensi S. C.; Rangel D. P.; Beth A. H.; Lybrand T. P.; Hustedt E. J. (2008) Simulation of nitroxide electron paramagnetic resonance spectra from brownian trajectories and molecular dynamics simulations. Biophys. J. 94, 3798–3809. 10.1529/biophysj.107.125419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P. D.; Svensson B.; Thomas D. D.; Stoll S. (2019) Trajectory-Based Simulation of EPR Spectra: Models of Rotational Motion for Spin Labels on Proteins. J. Phys. Chem. B 123, 10131–10141. 10.1021/acs.jpcb.9b02693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budil D. E.; Sale K. L.; Khairy K. A.; Fajer P. G. (2006) Calculating slow-motional electron paramagnetic resonance spectra from molecular dynamics using a diffusion operator approach. J. Phys. Chem. A 110, 3703–3713. 10.1021/jp054738k. [DOI] [PubMed] [Google Scholar]
- Robinson B. H.; Slutsky L. J.; Auteri F. P. (1992) Direct simulation of continuous wave electron paramagnetic resonance spectra from Brownian dynamics trajectories. J. Chem. Phys. 96, 2609–2616. 10.1063/1.462869. [DOI] [Google Scholar]
- Jeschke G. (2018) MMM: A toolbox for integrative structure modeling. Protein Sci. 27, 76–85. 10.1002/pro.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi K.; Kitayama Y.; Nishiwaki T.; Miwa K.; Murayama Y.; Oyama T.; Kondo T. (2007) ATPase activity of KaiC determines the basic timing for circadian clock of cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 104, 16377–16381. 10.1073/pnas.0706292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. G.; Rosen G. M.; Tseitlin M.; Symmes B.; Eaton S. S.; Eaton G. R. (2013) Use of rapid-scan EPR to improve detection sensitivity for spin-trapped radicals. Biophys. J. 105, 338–342. 10.1016/j.bpj.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton S. S.; Shi Y.; Woodcock L.; Buchanan L. A.; McPeak J.; Quine R. W.; Rinard G. A.; Epel B.; Halpern H. J.; Eaton G. R. (2017) Rapid-scan EPR imaging. J. Magn. Reson. 280, 140–148. 10.1016/j.jmr.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.