

Abstract
The novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has led to an ongoing pandemic of coronavirus disease (COVID-19), which started in 2019. This is a member of Coronaviridae family in the genus Betacoronavirus, which also includes SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV). The angiotensin-converting enzyme 2 (ACE2) is the functional receptor for SARS-CoV and SARS-CoV-2 to enter the host cells. In particular, the interaction of viral spike proteins with ACE2 is a critical step in the viral replication cycle. The receptor-binding domain of the viral spike proteins and ACE2 have several cysteine residues. In this study, the role of thiol–disulfide balance on the interactions between SARS-CoV/CoV-2 spike proteins and ACE2 was investigated using molecular dynamics simulations. The study revealed that the binding affinity was significantly impaired when all of the disulfide bonds of both ACE2 and SARS-CoV/CoV-2 spike proteins were reduced to thiol groups. The impact on the binding affinity was less severe when the disulfide bridges of only one of the binding partners were reduced to thiols. This computational finding possibly provides a molecular basis for the differential COVID-19 cellular recognition due to the oxidative stress.
1. Introduction
The novel coronavirus known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or simply COVID-19 is the seventh member of the coronavirus family.1 The other two viruses in this family that infect humans are severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV). These are positive-sense, single-strand enveloped RNA viruses. The coronavirus particles contain four main structural proteins: the spike, membrane, envelope, and nucleocapsid.1,2 The spike protein protrudes from the envelope of the virion and consists of two subunits: a receptor-binding domain (RBD) that interacts with the receptor proteins of host cells and a second subunit that facilitates fusion of the viral membrane into the host cell membrane. Recent studies showed that the RBD of spike proteins of SARS-CoV and SARS-CoV-2 interact with angiotensin-converting enzyme 2 (ACE2). ACE2 belongs to the membrane-bound carboxydipeptidase family. It is attached to the outer surfaces of cells and is widely distributed in the human body. In particular, higher expression of ACE2 is observed in organs such as small intestine, colon, kidney, and heart, while ACE2 expression is comparatively lower in the liver and lungs.3,4
The role of oxidative stress on the binding of viral proteins on the host cell surface receptors is a relatively underexplored area of biomedical research.5−10 Previous studies have indicated that the entry of viral glycoprotein is impacted by thiol–disulfide balance on the cell surface.5,7−9,11,12 Any perturbations in the thiol−disulfide equilibrium are also found to deter the entry of viruses into their target cells.5 The first step of the viral entry involves binding of the viral envelop protein onto a cellular receptor. This is followed by endocytosis, after which conformational changes of the viral protein help the induction of the viral protein into the endosomal membrane, finally releasing the viral content into the cell. These conformational changes are mediated by pH changes as well as the conversion of disulfide into the thiol group of the viral spike protein.7 Several cell surface oxidoreductases9 regulate the thiol–disulfide exchange, responsible for conformational changes of viral proteins needed for virus entry into host cells.
In the backdrop of a significant mortality rate for SARS-CoV-2 (hereinafter referred to as CoV-2) infection, it is important to know if the thiol–disulfide balance plays any role in the binding of the spike glycoprotein onto the host cell receptor protein ACE2. A recent study with the spike glycoprotein of SARS-CoV (hereinafter referred to as CoV) has exhibited a complete redox insensitivity;7 despite the reduction of all disulfide bridges of CoV to thiols, its binding to ACE2 remained unchanged.7 However, this study did not probe the redox sensitivity of the ACE2 receptor. Thus, in the present study, we computationally investigated the redox state of both partners (ACE2 and CoV/CoV-2) on their binding affinities. The structure of CoV13 and CoV-214,15 complexed with ACE2 are known, and the noncovalent interactions at the protein–protein interface16 have been reported recently. Using these reported structures, molecular dynamics simulations and electrostatic field calculations were performed to explore the impact of thiol–disulfide balance on CoV/CoV-2 and ACE2 binding affinities. The structural and dynamical changes due to the alteration in the redox states of cysteines in the interacting proteins were analyzed, and their effects on binding free energies were studied.
2. Results and Discussion
The molecular basis of the binding of spike proteins to ACE2 is known from X-ray crystallographic (SARS-CoV)13 and cryo-electron microscopic (SARS-CoV-2)16 studies. The sequence alignment of CoV and CoV-2 spike proteins showed a high sequence identity (>75%), indicating that their binding to ACE2 receptors will be similar (Figure 1). In both bound structures, the RBD of CoV and CoV-2 is found to be complexed with ACE2 (Figure 2). Both ACE2 and CoV-2 possess four disulfide bridges, whereas the CoV subunit has only two disulfide linkages (Table 1 and Figure 2a,b). Two large helices of ACE2 form a curved surface (Figure 2, illustrated by the dashed curved line) that interacts with the concave region of CoV or CoV-2 subunit.
Figure 1.

Sequence alignment (generated by Clustal Omega35) between the receptor-binding domain of SARS-CoV and SARS-CoV-2 proteins. “*” represents the identical residues, “:” and “.” represent strongly and weakly similar residues, respectively, and gap represents dissimilar residues. The cysteine residues are highlighted in yellow.
Figure 2.

Structures of protein complexes: (a) SARS-CoV···ACE2 and (b) SARS-CoV-2···ACE2. All of the disulfide bridges between cysteine residues are shown in green van der Waals (vdW) spheres and thiol groups in cyan licorice.
Table 1. Sequences of the Receptor-Binding Domain of SARS-CoV and SARS-CoV-2 Proteinsa.

The cysteine residues are highlighted in yellow and disulfide bonds are shown using black solid lines. The start and end residues in the crystal structures are numbered and highlighted in red. The gray highlighted residues are missing in the crystal structure.
2.1. Structural Changes along the Trajectory
In all cases, the simulation started with an equilibrated structure that was obtained after minimizing the neutralized solvated protein complex built from the experimentally determined structures. The evolution of the protein structure along the molecular dynamics (MD) trajectory was monitored by calculating the root-mean-square deviation (RMSD) of each structure from the starting structure as a frame of reference following the standard procedure.17 Briefly, during the MD simulation, the protein coordinates were recorded for every 10 ps interval and an RMSD of each frame was calculated from the average root-mean-square displacement of backbone Cα atoms with respect to the initial structure. Then, the RMSD values, averaged over conformations stored during 1 ns time, were plotted against the simulated time (Figure 3). Compared to the starting structure, only a moderate backbone fluctuation was noted in all protein complexes during 20 ns simulations, and the maximum of RMSD was in the range of 2.0–3.8 Å (Table 2). The evolution of dynamics was smooth, and its stability was demonstrated by the standard deviation of the computed RMSDs, which was less than 0.3 Å. Taken together, the results of the simulations showed no unexpected structural deformation of the SARS-CoV/CoV-2···ACE2 complex in both, reduced (thiol groups) and oxidized (disulfide bonds) states (Table 2).
Figure 3.
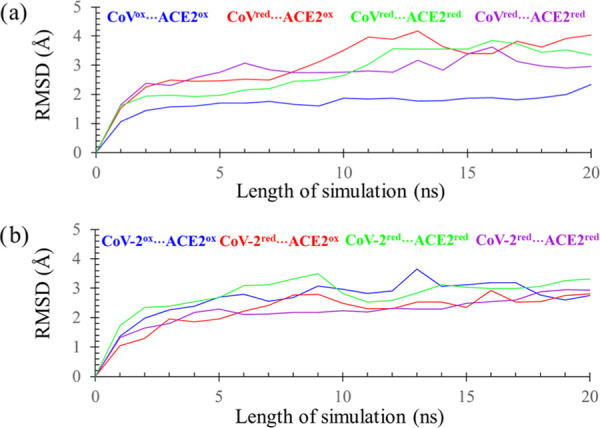
RMSDs, averaged over 1 ns MD simulation, of the protein complexes of ACE2 and SARS-CoV or SARS-CoV-2. Disulfide-containing proteins are referred to as oxidized with a shorthand notation of “ox”, while thiol variants are denoted with “red” notation.
Table 2. Evolution of the Structure of the Protein Complexes for the Oxidized and Reduced Variants Observed during 20 ns Molecular Dynamics Simulationsa.
| protein systems | number of disulfide moieties remaining | disulfide bridges present | RMSD (Å) | fluctuation (Å) |
|---|---|---|---|---|
| CoVox···ACE2ox | 6 | CoV: C366:::C419, C467:::C474, ACE2: C133:::C141, C344:::C361, C366:::C419, C530:::C542 | 2.0 | 0.2 |
| CoVred···ACE2ox | 4 | ACE2: C133:::C141, C344:::C361, C366:::C419, C530:::C542 | 3.8 | 0.3 |
| CoVox···ACE2red | 2 | CoV: C366:::C419, C467:::C474 | 3.1 | 0.3 |
| CoVred···ACE2red | 0 | none | 3.6 | 0.2 |
| CoV-2ox···ACE2ox | 8 | CoV-2: C336:::C361, C379:::C432, C391:::C525, C480:::C488, ACE2: C133:::C141, C344:::C361, C366:::C419, C530:::C542 | ||
| CoV-2red···ACE2ox | 4 | ACE2: C133:::C141, C344:::C361, C366:::C419, C530:::C542 | 2.7 | 0.2 |
| CoV-2ox···ACE2red | 4 | CoV-2: C336:::C361, C379:::C432, C391:::C525, C480:::C488 | 2.8 | 0.2 |
| CoV-2red···ACE2red | 0 | none | 3.1 | 0.2 |
For each protein system, the “RMSD” column contains the average of the last 5 ns RMSD, and the “fluctuation” column represents the standard deviation computed based on the starting structure as a reference.
2.2. Thermal Fluctuations due to the Cleavage of Disulfide Bridges
The MD simulation results indicated that the flexibility of different structural elements of the interacting proteins altered upon the cleavage of disulfide bridges (oxidized state) producing sulfydryl (thiol) groups (reduced state). For each complex, the impact of the reduction of the disulfide bridges was probed by calculating residue-level root-mean-square fluctuations (RMSF) of Cα atoms with respect to the average structure from its MD simulation trajectory. Changes in the backbone flexibility due to complete reduction of all disulfides to thiols of both interacting proteins are shown in Figure 4a,b, where the backbone is color-coded to indicate the magnitude of thermal fluctuations. Larger and smaller backbone fluctuations are shown in red and blue, respectively, whereas the medium fluctuation regions are shown in green. The backbone fluctuation analysis demonstrated that the cleavage of disulfide bridges indeed affected the flexibility of the local regions as they became more dynamic (Figure 4b). A significant alteration in backbone flexibility was noticed at the interface of CoV and ACE2 subunits when both proteins are in the fully reduced states, i.e., all disulfides are changed to sulfydryl groups. As it is evident from Figure 4b, the helix (residues 52–64) of ACE2 became very mobile, due to the rupture of the nearby C344:::C361 disulfide. The second region belongs to the flexible loop consisting of residues 463–474 of the CoV subunit (residues 478–489 of the CoV-2 subunit), which interacts with the N-terminal segment of the ACE2. The cleavage of the disulfides C467:::C474 in CoV and C480:::C488 in CoV-2 resulted in higher thermal fluctuation.
Figure 4.
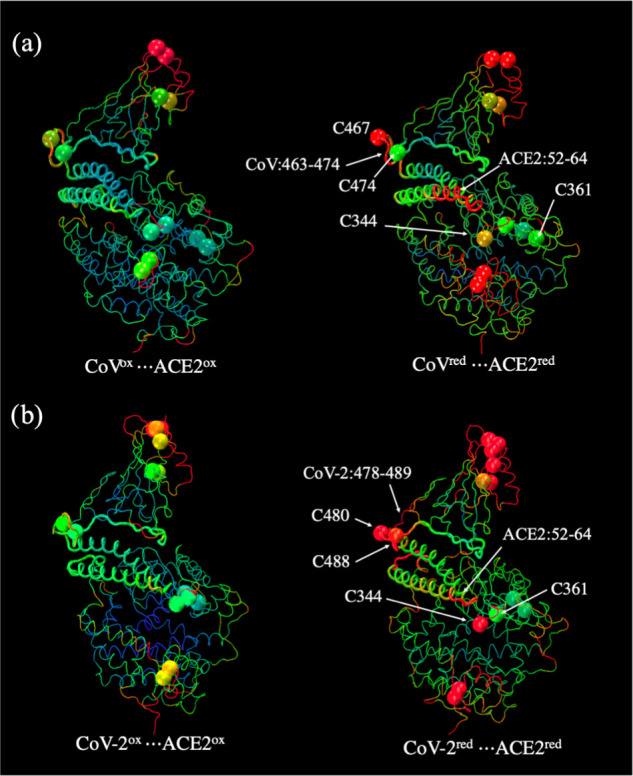
Change in residue-level RMSFs of Cα atoms in the oxidized and reduced protein complexes: (a) SARS-CoV···ACE2 and (b) SARS-CoV-2···ACE2. The cysteine residues, for each protein system (labeled in Figure 2), are highlighted as vdW spheres. The backbone as well as the cysteine residues are color-coded, large fluctuations are shown in red, and small fluctuations are in blue. The medium-scale fluctuations are shown in green.
2.3. Binding Study
The computed Gibbs binding free energy (ΔbindG°) of protein complexes (Table 3) demonstrated that the binding of CoV or CoV-2 with ACE2 occurs because the attractive electrostatic interactions between the individual subunits prevail over the desolvation due to the complexation. The complex formation results in desolvation from proteins’ surfaces at the protein–protein interface, thereby producing a positive ΔΔsolvGcorr for all protein systems (Table 3). For the formation of a stable protein–protein complex, this energy has to be counterbalanced by the negative electrostatic interactions, ΔCoulG, between the interacting proteins.
Table 3. Various Components of the Gibbs Free Energy of Binding Calculated by Implicit Solvation and Adaptive-Basis Poisson–Boltzmann Solver (APBS)a.
| protein systems | Coulombic interaction free energy ΔCoulG | corrected solvation free energy difference ΔΔsolvGcorr | Gibbs free energy of binding ΔbindG° |
|---|---|---|---|
| CoVox···ACE2ox | –564.5 | 557.2 | –7.3b |
| CoVred···ACE2ox | –673.0 | 661.8 | –11.2 |
| CoVox···ACE2red | –696.6 | 693.1 | –3.4 |
| CoVred···ACE2red | –528.4 | 580.4 | 52.0 |
| CoV-2ox···ACE2ox | –659.6 | 648.2 | –10.4c |
| CoV-2red···ACE2ox | –638.2 | 632.2 | –6.0 |
| CoV-2ox···ACE2red | –531.9 | 528.1 | –3.8 |
| CoV-2red···ACE2red | –655.3 | 712.6 | 57.3 |
All energies are expressed in kcal/mol. An estimated uncertainty of 1–3 kcal/mol was determined for the computed Gibbs free energy.
An experimental value of −10.3 kcal/mol was obtained for SARS-CoV by Lan et al.34
When the disulfides of the RBD of CoV and CoV-2 were reduced to thiols, the impact of reduction on the binding of these two proteins with ACE2 was different. The computed ΔbindG° for CoVox···ACE2ox and CoVred···ACE2ox are −7.3 and −11.2 kcal/mol, respectively (Table 3), indicating a slightly tighter binding of the reduced CoV RBD to ACE2. The experimentally known Kd value for the CoVox···ACE2ox system is 325 nM,14 which corresponds to a ΔbindG° value of −8.9 kcal/mol. Therefore, the computationally determined ΔbindG° for CoVox···ACE2ox binding is comparable to the experimental value. Although there is no experimentally determined Kd available for the CoVred···ACE2ox system, the binding assay with CoVred RBD was found to have no redox sensitivity for its binding to ACE2.7 The small favorability of CoVred···ACE2ox binding, as observed from the computation, indicates that the reduction of CoV did not have much impact on its binding affinity for ACE2 and could explain the experimentally observed redox insensitivity.7 In the case of CoV-2, its binding with ACE2 became less favorable when all of the disulfides of RBD were reduced to thiols. The computed Gibbs binding free energies were increased by ∼4.5 kcal/mol for the reduced CoV-2; ΔbindGo of CoV-2ox···ACE2ox and CoV-2red···ACE2ox are −10.4 and −6.0 kcal/mol, respectively.
In contrast, the reduction of disulfides of ACE2 impaired the binding significantly for both CoV and CoV-2 proteins. The Gibbs binding free energies of viral spike proteins CoV and CoV-2 with ACE2red, namely, CoVox···ACE2red and CoV-2ox···ACE2red are −3.4 and −3.8 kcal/mol, respectively. These ΔbindG° values are ∼4 to 6 kcal/mol more positive than what were observed for the oxidized forms of ACE2 (i.e., CoVox···ACE2ox and CoV-2ox···ACE2ox in Table 3). However, when all disulfides in CoV/CoV-2 as well as ACE2 were reduced to thiols, the binding became thermodynamically unfavorable. In both cases, the binding free energies have positive values; ΔbindG° of CoVred···ACE2red and CoV-2red···ACE2red are 52.0 and 57.3 kcal/mol, respectively. These results indicate that the binding will be severely impacted, when the disulfides of both interacting proteins are converted to thiols. This finding is potentially significant as it indicates that the cleavage of disulfide bridges in ACE2 has a significant destabilizing effect on the spike protein binding.
2.4. Molecular Basis of the Impaired Binding
The conjoined two helices of ACE2 (residues 19–53 and residues 56–94) form a complementary shape (Figure 5) that fits into the concave-shaped loop-β-sheet-loop motif (residues 460–490 of CoV and residues 475–505 of CoV-2) of RBD of viral spike proteins. The loss of disulfide bridges in ACE2 had a strong impact on the junction of the two conjoined helices (Figure 5, CoVred···ACE2red). This region is close to the disulfide bridge C344:::C361, and its cleavage has a significant impact on the shape complementarity at the protein–protein interface. Similarly, the loss of C467:::C474 disulfide in CoV (C480:::C488 in CoV-2) resulted in a significant conformational change in the loop region, which was displaced away from the protein–protein interface by ∼6 Å, indicating a reduced binding interaction as confirmed by the binding free energy calculations. This conformational change as well as the alteration in the backbone flexibility (Figure 4) must have resulted in impaired binding of CoV/CoV-2 with ACE2.
Figure 5.
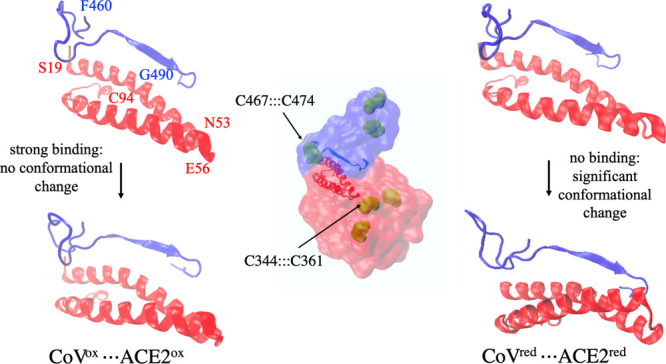
Comparison of the conformational change at the protein–protein interface in CoVox···ACE2ox (left) and CoVred···ACE2red (right). CoV and ACE2 subunits in the complex are shown in blue and red colors, respectively, at the center. The structural motif containing the two helices of ACE2 and a β sheet of the CoV (or CoV-2) was monitored before (top) and after (bottom) 20 ns MD simulation. The left and right panels show the difference in conformational changes in the oxidized form (CoVox···ACE2ox) and reduced form (CoVred···ACE2red), respectively.
3. Conclusions
The study found that the reduction of all disulfides into sulfydryl groups completely impairs the binding of SARS-CoV/CoV-2 spike protein to ACE2. This is evident from the positive Gibbs energy of binding (ΔbindG°) obtained for both CoVred···ACE2red and CoV-2red···ACE2red complexes. When the disulfides of only ACE2 were reduced to sulfydryl groups, the binding becomes weaker, as ΔbindG° becomes less negative by 4–6 kcal/mol with respect to the fully oxidized systems. On the other hand, the reduction of disulfides of the RBD of CoV-2 has comparatively less effect on ΔbindG°, while the reduction of disulfides in CoV RBD does not impact the binding with ACE2. This finding is consistent with the observed redox insensitivity of the binding between CoV and ACE2.7
The redox environment of cell surface receptors is regulated by the thiol–disulfide equilibrium in the extracellular region.12,18 This is maintained by glutathione transporters,19 a number of oxidoreductases12 including protein disulfide isomerase,8 and several redox switches.12 Under oxidative stress, the extracellular environment becomes oxidation-prone resulting in more disulfide formation on protein surfaces.12 Therefore, under severe oxidative stress, the cell surface receptor ACE2 and RBD of the intruding viral spike protein are likely to be present in its oxidized form having predominantly disulfide linkages. This computational study shows that under oxidative stress, the lack of a reducing environment would result in significantly favorable binding of the viral protein on the cell surface ACE2. In terms of energetics, this computational study demonstrates that the oxidized form of proteins with disulfide bridges would cause more than 50 kcal/mol of decrease in Gibbs binding free energy. Furthermore, ACE2, which the viral spike proteins latch on to, is known to be a key player in the remedial of oxidative stress.20 Binding of the viral protein will prevent the key catalytic function of ACE2 of converting angiotensin 2 (a strong activator of oxidative stress) to angiotensin 1–7 thereby creating a vicious circle of enhanced viral attack. In summary, the present study demonstrates that the absence of or reduced oxidative stress would have a significant beneficial effect during the early stage of viral infection by preventing viral protein binding on the host cells.
4. Methodology
4.1. Computational Setup
Setting up of protein systems and all structural manipulations were carried out using visual molecular dynamics (VMD).21 Disulfide groups were modified to thiols during the setting up of structures using standard VMD scripts. Molecular optimization and dynamics (MD) simulations were carried out using nanoscale molecular dynamics (NAMD) package using CHARMM36 force field.22−26 During MD simulations, electrostatic energy calculations were carried out using the particle mesh Ewald method.27 Backbone root-mean-square-deviation (RMSD) calculations were performed using VMD. Protein–protein interactions were studied using adaptive-basis Poisson–Boltzmann solver (APBS).28 Electrostatic field calculations were performed using PDB 2PQR program suit.29
4.2. Molecular Dynamics Simulations
All simulations were performed using the structure of ACE2 bound SARS-CoV (PDB entry: 3D0G)13 and SARS-CoV-2 (PDB entry: 6M0J).15 In all simulations, set up of protein complex systems was carried out following protocols used in the previous studies from this lab.17,30 Briefly, hydrogens were added using the HBUILD module of CHARMM. Ionic amino acid residues were maintained in a protonation state corresponding to pH 7. The protonation state of histidine residues was determined by computing the pKa using the Propka option of PDB 2PQR. The protein structures were explicitly solvated with water of the TIP3P model and neutralized by adding with 24 sodium atoms. The prepared solvated protein complexes were of dimensions 92 Å × 132 Å × 104 Å for SARS-CoV and 94 Å × 144 Å × 100 Å for SARS-CoV-2 systems.21
All systems were subjected to equilibration for 50 ps. Following equilibration, 20 ns MD simulations were performed for each system. The velocities and positions of atoms during dynamics were calculated using the velocity Verlet integration algorithm31 using a time step of 2.0 fs. All simulations were run with NAMD implementation of Langevin dynamics32 at a constant temperature of 298 K using periodic boundary conditions. To model, nonbonding interactions, a switching function was turned on with a “switchdist” of 10 Å, a cutoff of 14 Å, and a “pairlistdist” of 16 Å.
4.3. Binding Free Energy Calculations
Gibbs free energies of binding between the ACE2 and SARS-CoV or CoV-2 proteins were calculated using APBS, a standardized method of a treecode-accelerated boundary integral Poisson–Boltzmann (TABI-PB) equation solver.33 In this method, the protein surface is triangulated and electrostatic surface potentials are computed. The discretization of surface potentials is utilized to compute the net energy due to solvation as well as electrostatic interactions between the two protein subunits, as outlined in the thermodynamic scheme (Scheme 1). Following Scheme 1, the free energy of binding of the two protein fragments in water can be expressed as a sum of two components (eq 1)
| 1 |
where ΔCoulG represents the Coulombic (electrostatic) interactions between the proteins occurring at the protein–protein interface (Scheme 1) and ΔΔsolvG is the difference of the solvation energies between the complex and the corresponding free proteins
| 2 |
However, the solvation calculation used only part of the entire spike protein as well as the ACE2; therefore, ΔbindGTABI-PB was calibrated by correcting ΔΔsolvG using experimentally known binding free energy of ACE2···CoV-2
| 3 |
where Kd = 37 nM is the experimental dissociation constant.16
Scheme 1.

The corrected free energy of the solvation
| 4 |
Using ΔGcorr and eq 4, the corrected binding free energy, ΔbindG°, of all protein complexes is expressed by
| 5 |
As shown in eq 4, the combination of the last two terms in eq 5 is equal to ΔΔsolvGcorr. Therefore, eq 5 can be simplified as
| 6 |
Acknowledgments
We acknowledge computational support from the Blugold Super Computing Cluster (BGSC) of the University of Wisconsin—Eau Claire.
Glossary
Abbreviations Used
- ACE
angiotensin-converting enzyme
- CoV
coronavirus
- MD
molecular dynamics
- RBD
receptor-binding domain
- TABI-PB
treecode-accelerated boundary integral Poisson–Boltzmann
This work was supported in part by the National Institute of Health (grant number 1R15GM117510-01 to S.H. and S.B.).
The authors declare no competing financial interest.
References
- Schoeman D.; Fielding B. C. Coronavirus envelope protein: current knowledge. Virol. J. 2019, 16, 69 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr A. R.; Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Guo Y.; Pan Y.; Zhao Z. J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020, 525, 135–140. 10.1016/j.bbrc.2020.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Zhong L.; Deng J.; Peng J.; Dan H.; Zeng X.; Li T.; Chen Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8 10.1038/s41368-020-0074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryser H. J.; Levy E. M.; Mandel R.; DiSciullo G. J. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 4559–4563. 10.1073/pnas.91.10.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina A.; Hanley T. M.; Mandel R.; Trahey M.; Broder C. C.; Viglianti G. A.; Ryser H. J. Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and prevent HIV-1 entry. J. Biol. Chem. 2002, 277, 50579–50588. 10.1074/jbc.M204547200. [DOI] [PubMed] [Google Scholar]
- Lavillette D.; Barbouche R.; Yao Y.; Boson B.; Cosset F. L.; Jones I. M.; Fenouillet E. Significant redox insensitivity of the functions of the SARS-CoV spike glycoprotein: comparison with HIV envelope. J. Biol. Chem. 2006, 281, 9200–9204. 10.1074/jbc.M512529200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S.; Chen H. H.; Correia C.; Dai H.; Witt T. A.; Kleppe L. S.; Burnett J. C. Jr.; Simari R. D. Cell surface protein disulfide isomerase regulates natriuretic peptide generation of cyclic guanosine monophosphate. PLoS One 2014, 9, e112986 10.1371/journal.pone.0112986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys L.; Balzarini J. The role of cellular oxidoreductases in viral entry and virus infection-associated oxidative stress: potential therapeutic applications. Expert Opin. Ther. Targets 2016, 20, 123–143. 10.1517/14728222.2015.1068760. [DOI] [PubMed] [Google Scholar]
- Camini F. C.; da Silva Caetano C. C.; Almeida L. T.; de Brito Magalhães C. L. Implications of oxidative stress on viral pathogenesis. Arch. Virol. 2017, 162, 907–917. 10.1007/s00705-016-3187-y. [DOI] [PubMed] [Google Scholar]
- Fenouillet E.; Barbouche R.; Jones I. M. Cell entry by enveloped viruses: redox considerations for HIV and SARS-coronavirus. Antioxid. Redox Signaling 2007, 9, 1009–1034. 10.1089/ars.2007.1639. [DOI] [PubMed] [Google Scholar]
- Yi M. C.; Khosla C. Thiol-Disulfide Exchange Reactions in the Mammalian Extracellular Environment. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 197–222. 10.1146/annurev-chembioeng-080615-033553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J. Virol. 2008, 82, 6984–6991. 10.1128/JVI.00442-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D.; Wang N.; Corbett K. S.; Goldsmith J. A.; Hsieh C. L.; Abiona O.; Graham B. S.; McLellan J. S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zhang Y.; Wu L.; Niu S.; Song C.; Zhang Z.; Lu G.; Qiao C.; Hu Y.; Yuen K. Y.; Wang Q.; Zhou H.; Yan J.; Qi J. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R.; Zhang Y.; Li Y.; Xia L.; Guo Y.; Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams L. M.; Andrews R. J.; Hu Q. H.; Schmit H. L.; Hati S.; Bhattacharyya S. Crowder-Induced Conformational Ensemble Shift in Escherichia coli Prolyl-tRNA Synthetase. Biophys. J. 2019, 117, 1269–1284. 10.1016/j.bpj.2019.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel T. J.; Weerapana E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1600391 10.1002/pmic.201600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N.; Krance S. M.; Marchan R.; Hammond C. L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Aspects Med. 2009, 30, 13–28. 10.1016/j.mam.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H.; Suda S.; Bindom S.; Feng Y.; Gurley S. B.; Seth D.; Navar L. G.; Lazartigues E. ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS One 2011, 6, e22682 10.1371/journal.pone.0022682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Guvench O.; Hatcher E.; Venable R. M.; Pastor R. W.; MacKerell A. D. CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J. Chem. Theory Comput. 2009, 5, 2353–2370. 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman E. P.; Guvench O.; MacKerell A. D. Jr. CHARMM additive all-atom force field for glycosidic linkages in carbohydrates involving furanoses. J. Phys. Chem. B 2010, 114, 12981–12994. 10.1021/jp105758h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O.; Mallajosyula S. S.; Raman E. P.; Hatcher E.; Vanommeslaeghe K.; Foster T. J.; Jamison F. W. II; Mackerell A. D. Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. 10.1021/ct200328p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E. M.; Mittal J.; Feig M.; MacKerell A. D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallajosyula S. S.; Guvench O.; Hatcher E.; Mackerell A. D. Jr. CHARMM Additive All-Atom Force Field for Phosphate and Sulfate Linked to Carbohydrates. J. Chem. Theory Comput. 2012, 8, 759–776. 10.1021/ct200792v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; et al. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. 10.1063/1.470117. [DOI] [Google Scholar]
- Jurrus E.; Engel D.; Star K.; Monson K.; Brandi J.; Felberg L. E.; Brookes D. H.; Wilson L.; Chen J.; Liles K.; Chun M.; Li P.; Gohara D. W.; Dolinsky T.; Konecny R.; Koes D. R.; Nielsen J. E.; Head-Gordon T.; Geng W.; Krasny R.; Wei G. W.; Holst M. J.; McCammon J. A.; Baker N. A. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. 10.1002/pro.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky T. J.; Czodrowski P.; Li H.; Nielsen J. E.; Jensen J. H.; Klebe G.; Baker N. A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. M.; Sanford B. L.; Strom A. M.; Tadayon S. N.; Lehman B. P.; Zirbes A. M.; Bhattacharyya S.; Musier-Forsyth K.; Hati S. Multiple pathways promote dynamical coupling between catalytic domains in Escherichia coli prolyl-tRNA synthetase. Biochemistry 2013, 52, 4399–4412. 10.1021/bi400079h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlet L. Computer “experiments” on classic fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967, 159, 98–103. 10.1103/PhysRev.159.98. [DOI] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng W.; Krasny R. A treecode-accelerated boundary integral Poisson–Boltzmann solver for electrostatics of solvated biomolecules. J. Comput. Phys. 2013, 247, 62–78. 10.1016/j.jcp.2013.03.056. [DOI] [Google Scholar]
- Lan J.; Ge J.; Yu J.; Shan S.; Zhou H.; Fan S.; Zhang Q.; Shi X.; Wang Q.; Zhang L.; Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Madeira F.; Park Y. M.; Lee J.; Buso N.; Gur T.; Madhusoodanan N.; Basutkar P.; Tivey A. R. N.; Potter S. C.; Finn R. D.; Lopez R. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]