

Abstract
Traumatic brain injury (TBI) is a leading cause of death and disability worldwide. TBI can have a long-term impact on the quality of life for survivors of all ages. However, there remains no approved treatment that improves outcomes following TBI, which is partially due to poor delivery of therapies into the brain. Therefore, there is a significant unmet need to develop more effective delivery strategies that increase the accumulation and retention of potentially efficacious treatments in the injured brain. Recent work has revealed that nanoparticles (NPs) may offer a promising approach for site-specific delivery; however, a detailed understanding of the specific NP properties that promote brain accumulation and retention are still being developed. Multimodal imaging plays a vital role in the understanding of physicochemical properties that initiate the uptake and accumulation of NPs in the brain at both high spatial (e.g., fluorescence imaging) and temporal (e.g., magnetic resonance imaging, MRI) frequency. However, many NP systems that are currently used in TBI only provide contrast in a single imaging modality limiting the imaging data that can be obtained, and those that offer multimodal imaging capabilities have complicated multistep synthesis methods. Therefore, the goal of this work was to develop an ultrasmall NP with simple fabrication capable of multimodal imaging. Here, we describe the development, characterization, accumulation, and retention of poly(ethylene glycol) (PEG)-coated europium–gadolinium (Eu–Gd) mixed magnetic NPs (MNPs) in a controlled cortical impact mouse model of TBI. We find that these NPs having an ultrasmall core size of 2 nm and a small hydrodynamic size of 13.5 nm can be detected in both fluorescence and MR imaging modalities and rapidly accumulate and are retained in injured brain parenchyma. These NPs should allow for further testing of NP physicochemical properties that promote accumulation and retention in TBI and other disease models.
1. Introduction
Traumatic brain injury (TBI) results from a trauma that causes sudden damage to the brain. TBI causes approximately 2.87 million annual reported deaths, hospitalizations, and emergency room visits in the United States alone.1 Primary injury occurs as a direct result of mechanical trauma that results in the displacement and/or physical contusioning of the brain leading to intracranial hematomas, skull fractures, lacerations, contusions, and penetrating wounds. Secondary brain injury arises from the complications of the primary injury including necrotic cell death, oxidative stress, and damage to the blood–brain barrier (BBB) and leads to the secondary spread of damage into the surrounding brain, which can lead to progressive neurodegenerative diseases.2−4 Secondary injury continues in the hours, days, and even years following the primary injury.5−8 Therefore, this secondary injury cascade is the target for therapy development to help minimize the spread of damage.9−11 Several clinical trials have attempted to minimize secondary processes that cause additional damage following TBI. Unfortunately, no strategy has been successful so far in humans. At present, only limited supportive care therapies are available to TBI patients.
Nanoparticles (NPs) can play a significant role in the imaging and treatment of TBI.12,13 The improvement of NPs for therapeutic uses has been continuing for decades.14 Several nanomaterials have been studied for TBI, and they are mostly antioxidant NPs, which contain functional groups that can deactivate reactive oxygen species (ROS).15−19 For example, superoxide dismutase (SOD)-coated NPs were synthesized to minimize the oxidative stress-mediated neuronal damage,17 carbon particles were used as antioxidant treatment for TBI,16,20 and thioether polymer and core-cross-linked NPs have been shown to reduce post-traumatic neurodegeneration, neuroinflammation, and improve the outcome.15,19 However, the physicochemical properties of NPs that promote accumulation and retention in the injured brain are still being developed.21 While NP properties and behaviors have been widely studied in the context of cancer, knowledge on how these properties affect their ability to accumulate and be retained in TBI are still in the early stages. The size of a NP and administration time following the injury seem to play a major role in accumulation and retention,22−24 but there is still a wide range of NP properties that remain unstudied such as surface charge, shape, and surface chemistry including possible active targeting mechanisms. An NP-based tool that will allow for imaging through multiple imaging modalities would provide a framework for these properties to be studied. We have recently developed an MR imaging method to assess the accumulation and retention kinetics of various NPs in TBI.22 However, an NP system with a simple synthesis scheme that can be used for both MR and fluorescence imaging is still needed to achieve both high temporal and high spatial resolution imaging of the NP behavior in TBI.
Mixed lanthanide oxide magnetic NPs (MNPs) are desirable because they have both inherent paramagnetic and fluorescent properties, which are significantly useful for dual imaging.25 Mixed MNPs have an advantage over heterojunction, core–shell, and dye-coated NPs because of their robustness, stability, compactness, and compositionally controlled synthesis procedure. Furthermore, magnetic and fluorescent properties are minimally affected by surface coating due to the compactness of 4f orbitals close to the nucleus.26 Additionally, a small particle diameter enhances fluorescence intensities because the reduced excitation migration to quenching sites is proportional to the particle diameter.27,28 Biocompatible ligand-coated MNPs provide a promising option to gain insight into ideal NP properties as their size, shape, and surface properties can be tuned. Furthermore, MNPs offer the ability to be imaged through multiple imaging modalities to provide both high temporal and spatial resolution imaging to identify uptake and retention kinetics as well as cellular distribution to gain a better understanding of brain delivery.29 Here, we describe, for the first time, the development and use of mixed lanthanide oxide MNPs containing europium (Eu) for fluorescence imaging and gadolinium (Gd) for MR imaging in TBI. Looking forward, these MNPs could be coated with different length poly(ethylene glycol) (PEGs) to alter their hydrodynamic size, heterobifunctional PEGs (e.g., carboxyl and amine) to alter the surface charge, and functionalized with targeting agents to better study how physicochemical properties affect accumulation and retention in TBI.
2. Results and Discussion
It is required to coat the NPs with a biocompatible and water-soluble ligand for biomedical applications. The addition of PEG to NP surfaces reduces the reticuloendothelial system (RES) uptake and increases circulation time.30−32 PEG coating also shields the surface from aggregation, and the solubility in serum increases because of repeating the hydrophilic ethylene glycol units.33,34 PEG diacid was used in this work. The high-resolution transmission electron microscopy (HRTEM) images of PEG diacid-coated Eu–Gd NPs are shown in Figure 1a. The average particle diameter is around 2.1 nm verifying their ultrasmall core size. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was also measured to validate the presence of both Eu and Gd in the NPs. As HAADF-STEM is correlated to the atomic mass, we were able to determine the presence of Eu, Gd, and O in the mixed Eu–Gd NP system (Figure 1b). From these measurements and inductively coupled plasma mass spectrometry (ICP-MS), we determined that the final ratio of Eu and Gd in the NPs was the same as the feed ratio. We synthesized Eu–Gd (25–75%), Eu–Gd (50–50%), and Eu–Gd (75–25%) NPs. From dynamic light scattering (DLS) measurement, the average hydrodynamic size of the nanoparticle was 13.5 nm (Figure 1c), and ζ-potential was −25.1 mV (Figure 1d) in Dulbecco’s phosphate-buffered saline (DPBS) (pH 7.4). Surface modification of nano-drug delivery systems is the most common strategy for controlling the opsonization process and thus sustain the systems for a longer period in the bloodstream.35 The negative ζ-potential provides increased passivation by preventing adsorption by negatively charged serum proteins, reduced agglomeration, and promoting distribution into the tissue.36 The PEG diacid coating was confirmed after Fourier transform-infrared (FT-IR) spectroscopy. We compared PEG diacid-coated Eu–Gd NPs with free PEG diacid and observed that the C=O stretch was red-shifted by ∼140 cm–1 from that (=1736 cm–1) of a free PEG diacid (Figure 1e), confirming the attachment of the −COOH group to the NPs as commonly observed in metal oxide NPs coated with the −COOH group containing ligands.37,38 The broad peak at around 3400 cm–1 corresponds to the −OH groups.
Figure 1.
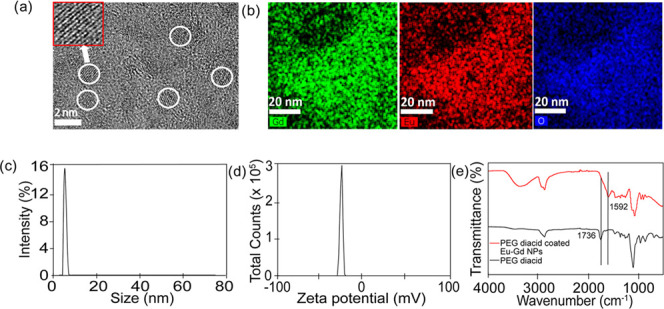
(a) HRTEM images of the PEG diacid-coated ultrasmall Eu–Gd nanoparticles. The corresponding circle shows the diameter of the nanoparticle, which is 2.1 nm (n = 71). (b) High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and element mapping [Gd (green), Eu (red), O (blue)] of Eu–Gd NPs. (c) From DLS, the average size of the nanoparticle is 13.5 nm. (d) ζ-potential value is −25.1 mM for the Eu–Gd nanoparticles. (e) FT-IR absorption spectra of (I) free PEG diacid (black line), (II) PEG diacid-coated ultrasmall Eu–Gd nanoparticles (red line).
To determine the fluorescence properties of the NPs, we generated an excitation–emission matrix. We investigated excitation from 250 to 700 nm and corresponding emission wavelengths (300 to 700 nm). From the two-dimensional excitation–emission (2D Ex–Em) matrix, a maximum fluorescent signal for the synthesized Eu–Gd NPs was observed using excitation and emission wavelengths of 365 and 450 nm, respectively (Figure 2a). We also investigated the fluorescence of different compositions of Eu–Gd NPs to identify the maximum intensity containing Eu–Gd NPs. We found that Eu–Gd (75–25%) showed the maximum fluorescence intensity (Figure 2b), which is expected because Eu is responsible for the majority of the fluorescence in the NPs.
Figure 2.
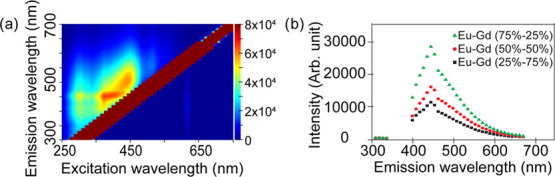
(a) 2D Excitation–emission matrix of Eu–Gd nanoparticles. (b) Fluorescence intensity in different compositions of Eu–Gd NPs.
We also tested the fluorescence lifetime of the NPs since many lanthanides including Eu39,40 are commonly used in fluorescence lifetime imaging studies.41 However, the fluorescence lifetime of the NPs of 1.6 ns (Table 1) is very close to the lifetime of cell autofluorescence42 of around 2 ns,43,44 and is not extended as observed with other Eu-based imaging agents.39,40 This is likely because the extended lifetime afforded by Eu is a result of chelation to specific ligands that enhance the fluorescence lifetime of Eu, which were not on the NP. Therefore, different polymeric coatings for these NPs are likely needed to take advantage of fluorescence lifetime imaging capabilities. Furthermore, other lanthanides could be included to tune the photophysical properties of the MNPs to achieve desired luminescent lifetimes41 or upconversions45 for enhanced brightness and depth of imaging.46
Table 1. Properties of NPs.
| name | size (nm) | ζ-Potential | R1 relaxivity (s–1 mM–1) at 9.4 T | quantum yield, ϕ | molar extinction coefficient, ε (M–1 cm–1) | fluorescence lifetime (ns) |
|---|---|---|---|---|---|---|
| Eu–Gd | 13.5 | –25.1 | 4.7697 | 0.33 | 2.15 × 103 | 1.6 |
To determine the magnetic resonance imaging (MRI) enhancing properties of the NPs, both R1 and R2 were measured at 9.4 T and plotted as a function of Ln (Ln = Gd and Eu) concentration. Three different ratios of the Eu–Gd sample were measured and are shown in Figure 3a. The highest relaxivities were observed for Eu–Gd (25–75%), which was expected as Gd is responsible for the majority of MRI contrast in the NPs. Longitudinal (r1) water proton relaxivities were estimated from the corresponding slopes, giving a value of 7.11 s–1 mM–1 for Eu–Gd (25–75%). The r1 relaxivities were 4.77 and 1.66 s–1 mM–1 for Eu–Gd (50–50%) and Eu–Gd (75–25%), respectively. Transverse (r2) water proton relaxivities were estimated from the corresponding slopes, giving a value of 187.14 s–1 mM–1 for Eu–Gd (25–75%). The r2 relaxivities were 90.16 and 78.66 s–1 mM–1 for Eu–Gd (50–50%) and Eu–Gd (75–25%), respectively. T1 and T2 map images (Figure 3a,b) show clear dose-dependent contrast enhancement. Eu–Gd (50–50%) had the lowest r2/r1 value of 18.9, indicating its greater function as a T1 contrast agent as compared to the other ratios. This, combined with the moderate fluorescence at this ratio, we chose Eu–Gd (50–50%) for the in vivo experiment.
Figure 3.
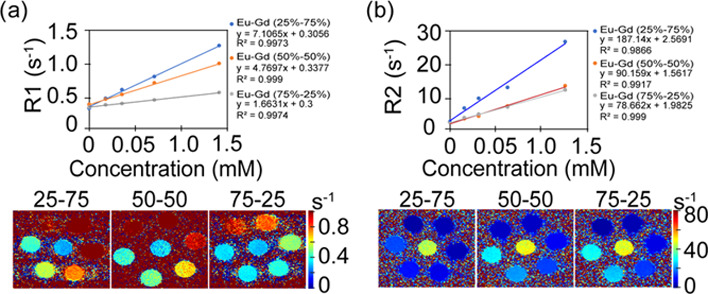
(a) R1 relaxivity and T1 map images, (b) R2 relaxivity and T2 map images of Eu–Gd nanoparticles. Both relaxivity and map images are changing with concentration variations of NPs.
To determine if NPs were able to accumulate and be retained in the injured brain, we utilized a controlled cortical impact (CCI) mouse model of TBI because of its high reproducibility and characterization within the literature.47 We used Eu–Gd (50–50%) for in vivo experiments because of their combined fluorescence and magnetic characteristics. Following catheter placement into the tail veins of the mice and precontrast MRI scans, NPs (100 μL of 0.5 mM) were injected, followed by a 100 μL chaser of phosphate-buffered saline (PBS) to ensure that all NPs were injected. We monitored NP uptake and retention kinetics through the measurement of Ktrans, a permeation coefficient that reflects the movement of a NP from the blood to the tissue, which we have recently shown provides a robust measurement of NP uptake and retention kinetics in TBI.22 A major advantage of Ktrans modeling over other MRI methods for NP tracking such as ΔR1 or ΔR2 mapping is that these other MRI methods cannot distinguish the NP in the blood from the NP in the tissue parenchyma.22 We found that the NPs rapidly accumulated and were retained in the injured region of the brain, as shown by higher Ktrans in the injury than in the surrounding brain (Figure 4). The focal mean Ktrans value for Eu–Gd NP was 0.202 min–1. The contralateral mean Ktrans value of 0.00122 min–1 for NP was significantly lower (p < 0.001). Eu–Gd NPs showed significantly high uptake in the focal injury than in the contralateral brain, as expected with previous results assessing the uptake of NP-based contrast agents into the brain following TBI. This distinction in accumulation kinetics between focal and contralateral regions illustrates the spatial variability of the BBB dysfunction at early time points following TBI, with much higher disruption occurring at the site of the injury. This result also further supports the use of NP-based therapeutics for passive targeting of the focal injury in TBI as the NPs can rapidly accumulate in the injury and be retained for a significantly longer time than small molecules.19,22
Figure 4.
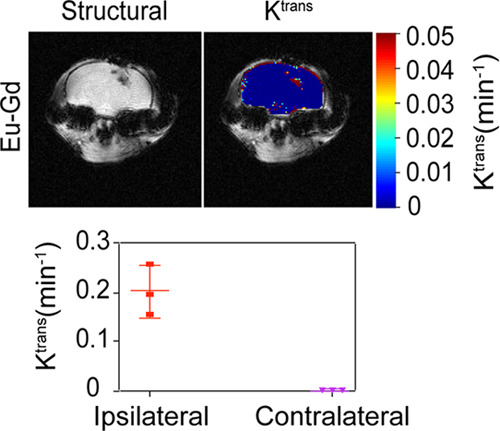
NP uptake and retention kinetics in TBI. Ktrans maps showing Eu–Gd NP accumulation and retention localization with respect to damage, a plot of average Ktrans for all mice.
To determine how NP uptake and retention kinetics compare to blood kinetics, we assessed blood half-life parameters from the MR image series used for Ktrans measurements. Eu–Gd NPs showed the elimination profiles (Table 2) in vivo with a t1/2,elim value of 26.53 min and a t1/2,dist value of 1.84 min. This rapid distribution phase and relatively short elimination half-life combined with our Ktrans measurements that show the transfer of NPs from the blood into tissue suggest that the NPs rapidly distribute into the injured region of the brain and are retained there while NPs are quickly removed from circulation likely through kidney clearance because of their small size. This reduces the likelihood that NPs are first taken up into circulating monocytes or peripheral macrophages before subsequently being trafficked into the injury, and that NPs are directly accumulating in the injury. This is further supported by the evidence that first peripheral immune cells that enter the injury are neutrophils at 24–48 h postinjury.48,49 The small size of these NPs allowed for rapid kidney clearance to minimize potential deleterious off-target effects as compared to larger NPs with significantly longer circulation times. While the area under the curve (AUC) pharmacokinetic parameter is often used as an indicator of exposure of the target tissue to small-molecule drugs, and rapidly clearing ultrasmall NPs do not provide a very large AUC, these ultrasmall NPs allow for prolonged exposure in the target tissue because of their significantly reduced diffusivity as compared to small-molecule drugs. Therefore, the NPs are retained in the target tissue for extended periods as compared to the elimination from blood, as we observed from our Ktrans measurements (Figure 4), which are nearly 10-fold higher than the small-molecule Gd–diethylenetriaminepentaacetic acid (DTPA), from our previous22 results. In this way, ultrasmall NPs provide the benefit of prolonged target tissue exposure combined with short circulation time that minimizes exposure to off-target tissues.
Table 2. Pharmacokinetic Parameters of NPsa.
| NP type | distribution half-life (t1/2,dist) (min) | Kel,dist (min–1) | elimination half-life (t1/2,elim) (min) | Kel,elim (min–1) | area under concentration–time curve (AUC) (min mM) | refs |
|---|---|---|---|---|---|---|
| Eu–Gd | 1.84 ± 0.62 | 0.409 ± 0.15 | 26.53 ± 4.95 | 0.027 ± 0.0045 | 76.6 ± 8.2 | this work |
| Gd–DTPA | 4.01 ± 2.47 | 0.23 ± 0.11 | 15.70 ± 8.50 | 0.056 ± 0.023 | 6.6 ± 3.8 | (22) |
Data shown are averages ± standard deviations.
The overall rationale behind mixed Eu–Gd NPs was the ability to visualize NP uptake kinetics using MRI because of the contrast proved by Gd followed by fluorescence imaging of tissue distribution because of the fluorescence signal provided by Eu. This is important for better understanding the brain delivery.29 We observed the signal from Eu–Gd NPs in brain slices using an A4 filter that has an excitation wavelength at 360 ± 20 nm and emission wavelength at 470 ± 20 nm, which fits perfectly with the excitation and emission wavelength of Eu–Gd NP. The images (Figure 5) were taken in the ipsilateral cortex since the ipsilateral cortex had the highest Ktrans value, where the NPs were observed to accumulate and be retained. From a fluorescence microscope, we observed that the Eu–Gd NPs were found in the brain tissue and outside of the blood vessel (Figure 5), whereas no signal was observed for a control mouse injured brain that received no NP injection. This further supports the idea that the NPs accumulate across the disrupted BBB and are retained in the brain parenchyma.
Figure 5.
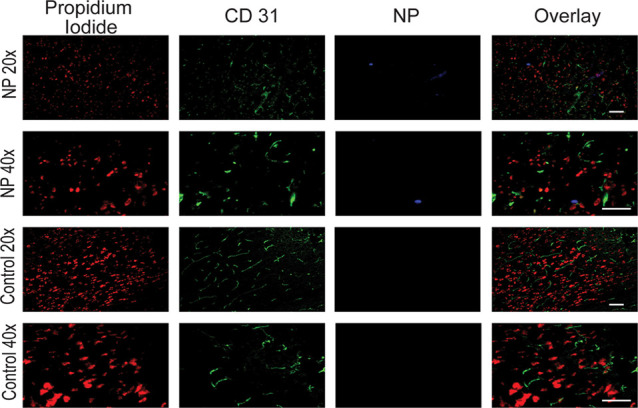
Fluorescence microscopy imaging of the ipsilateral cortex at 1 h after NP injection in Eu–Gd NP-treated CCI mice and mice that did not receive NPs with 20× and 40× objective lens magnification. Red is nucleus staining with propidium iodide, green is CD31 immunostaining of blood vessel, and blue is Eu–Gd NP. Scale bar is 10 μm.
3. Conclusions
We developed ultrasmall Eu–Gd NPs as a multimodal contrast agent and tool for studying NP behavior in TBI. In this work, we showed the influence of the reaction composition on the magnetic relaxivity and fluorescence properties to find an ideal combination of T1 contrast enhancement and fluorescence. In a CCI mouse model of TBI, we found rapid accumulation and retention of NPs within the injury using MRI analysis of Ktrans. Additionally, the ultrasmall size of the NPs allowed for rapid kidney clearance and relatively small AUC pharmacokinetic parameter to reduce off-target exposure. Moreover, the cellular localization of the NPs was determined by fluorescence imaging to generate high spatial resolution information on tissue and cellular distribution. Therefore, Eu–Gd NPs can be used for combined MRI and fluorescence imaging to find optimal NP properties that improve delivery to and especially retention in TBI.
4. Experimental Section
4.1. Materials
Gadolinium chloride hydrate (GdCl3·xH2O, 99.9%), europium(III) nitrate hydrate (Eu(NO3)3·5H2O, 99.9%), triethylene glycol (TEG, 99%), sodium hydroxide (NaOH), poly(ethylene glycol) diacid (Mn, 600), ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC, ≥98%), N-hydroxysuccinimide (NHS, 98%), and molecular weight cutoff dialysis membranes (Flot-A-Lyzer, 20 kDa) were purchased from Sigma-Aldrich. Deionized water in the experiments was obtained using a Millipore water purification system. All other chemicals and solvents used in this work were of high-performance liquid chromatography (HPLC) grade.
4.2. Synthesis of Eu–Gd NPs
Eu(NO3)3·5H2O (1 mmol) and GdCl3·xH2O (1 mmol) were added into 30 mL of triethylene glycol in a 100 mL three-necked flask. The mixture was heated to 80 °C and magnetically stirred until the two precursors were completely dissolved in the solvent. Then, 6 mmol NaOH was added and stirring was continued for 4 h at 180 °C. To coat the hydrophobic MNPs, 4 mmol PEG diacid was added and the reaction was continued with stirring for 12 h at 150 °C. After completely cooling, the synthesized nanoparticles were washed three times using deionized water.
4.3. Characterization
A high-voltage transmission electron microscope (TEM) (Tecnai Osiris TM, 200 kV) was used to measure particle diameters of PEG diacid-coated ultrasmall Eu–Gd NPs. A copper grid (PELCO mesh size 400, TED PELLA, INC.) covered with an amorphous carbon membrane was placed onto a filter paper. Then, a sample solution diluted in triple-distilled water was dropped over the copper grid using a micropipette (Eppendorf, 2–20 μL). Core diameter measurements from 71 MNPs were averaged to determine the average core size. Dynamic light scattering (DLS) studies of the NPs were conducted using a Malvern Instruments Zetasizer Nano series instrument. Solutions of the NPs were prepared in DPBS (pH 7.4) at a concentration of 0.05 mM. The resulting solutions were filtered with 0.22 μm filters before the measurement. The concentration of Eu and Gd was determined using an inductively coupled plasma mass spectrometer (ICP-MS) (Agilent 7500 cx). To determine this, ∼1 mL of the NP solution was taken out and treated with HNO3 to dissolve nanoparticles in solution completely. A Fourier transform-infrared (FT-IR) spectrometer (Nicolet AVATAR 380 FT-IR) was used to verify the surface coating. To record the FT-IR absorption spectrum (400–4000 cm–1), the powder sample was prepared.
4.4. Fluorescence Characterization
A microplate reader (Synergy H1, BioTek) was used to record fluorescence spectra of PEG diacid-coated ultrasmall Eu–Gd nanoparticles dispersed in ethanol. To find out the maximum fluorescence intensity, excitation wavelengths were used from 250 to 700 nm and emission wavelengths from 300 to 700 nm. After that, fluorescence spectra were recorded for three different compositions of Eu–Gd NPs at λex = 365 nm and λem = 450 nm. Quantum yields (Φs) of sample solution was estimated using fluorescein as a standard.
Two-photon excited fluorescence intensity and lifetime imaging was performed using the 740 nm mode-locked femtosecond pulse train of a Spectra-Physics MaiTai Ti/S laser on a Leica TCS SP8 MP multiphoton laser-scanning confocal microscope (Leica Microsystems, Germany) equipped with a Leica HC PL APO CS2 40×/1.3 NA oil immersion objective. Blue and yellow nondescanned fluorescence were separated using a 500 nm long-pass dichroic mirror and isolated with HQ 460/80-2p and HQ 580/60-2p band-pass filters (Chroma Technology, Bellows Falls, VT), respectively, and detected with high-sensitivity super HyD detectors and a time-correlated single-photon counting module (830 SPC, Becker and Hickl, Berlin, Germany). The imaging field of view was 276 × 276 μm2 (128 ×128 pixels) with a line scan rate of 400 Hz (dwell time of 19.5 μs pixel–1), a line average of 3, and a total acquisition time of 120 s.
4.5. R1 and R2 Relaxivities and R1 and R2 Map Imaging Measurements
Both R1 and R2 map images, as well as both T1 and T2 relaxation times, were measured using a 9.4 T MRI instrument (Varian 9.4 T) equipped with a 4 cm Millipede RF imaging probe with triple-axis gradients (100 G cm–1 max). A series of five aqueous solutions of different concentrations (0.5, 0.25, 0.125, 0.0625, and 0.03125 mM Ln) were prepared by diluting each MRI solution with PBS. Then, both map images and relaxation times were measured using these solutions. The R1 and R2 relaxivities were then estimated from the slopes in the plots of 1/T1 and 1/T2 versus NP concentration, respectively. The measurement parameters for the fast spin-echo T1 mapping sequence were as follows: the external MR field (H) = 9.4 T, the temperature = 22 °C, the number of acquisition (NEX) = 1, the field of view (FOV) = 25 × 25 mm2, the matrix size = 128 × 128 voxels, echo train length = 16, echo spacing = 8.1 ms, slice thickness = 2 mm, seven repetition times (TRs) used in linear increments from 200–2000 ms, and the echo time (TE) = 32.42 ms. The signal was fit to the following equation using MATLAB to find T1
where S is signal for a given voxel and S0 is the signal of that voxel at saturation. T2 mapping was carried out using a multi-echo scan with the same parameters as the T1 scan with the following exceptions: the number of echoes = 10, 10 TEs linearly spaced from 10–100 ms, and TR = 3000 ms. T2 mapping was performed using Osirix and linear fitting followed in MATLAB.
4.6. Controlled Cortical Impact (CCI) Mouse Model of TBI
All animal work was approved by and done in accordance with the University of Nebraska—Lincoln IACUC. We utilized a severe controlled cortical impact (CCI) mouse model of TBI.50 Six-week-old female C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were acclimated for 1 week prior to any procedures. Anesthesia was induced with 3% isoflurane gas via inhalation, and mice were affixed to a stereotaxic frame (David Kopf Instruments, Tujunga, CA) using ear and bite bars and anesthesia was maintained at ∼1.5%. The hair of the scalp was removed with Nair (Church and Dwight Co., Inc., Princeton, NJ), and the scalp was disinfected with a betadine scrub and isopropanol wipes. Lidocaine (0.05 mL of 5 mg mL–1) and bupivacaine (0.05 mL of 0.3 mg mL–1) were applied to the scalp, and buprenorphine SR (60 μL of 0.5 mg mL–1) was injected subcutaneously. A midline incision (approximately 1 cm) was made on the scalp over bregma. Using a surgical drill, an approximately 2 mm burr hole was made in the skull over the left frontoparietal cortex (2 mm anterior and 2 mm left of lambda). A controlled cortical impactor (Hatteras Instruments, Cary, NC) attached to the stereotaxic frame was used to impact the brain normal to the dura surface at a depth of 1.5 mm and a velocity of 4 m s–1 with a dwell time of 80 ms using a 2 mm convex tip. Bleeding was controlled, and incisions were closed using tissue adhesive. Mice were placed in a recovery cage on a heating pad and monitored until awake prior to transfer to the imaging room. A total of three mice were used for these experiments.
4.7. Ktrans MRI Method for In Vivo Imaging
Animal imaging was performed using the same MRI system used for R1 and R2 assessments. All imaging studies were also approved by the University of Nebraska—Lincoln IACUC. Mice were induced and maintained using approximately 1.5% isoflurane with adjustments made to maintain the breathing rate between 40 and 80 breaths per minute. Ktrans imaging to determine NP accumulation and retention in TBI was performed similarly to our previously published work.22 Precontrast gradient echo scans were performed with flip angles (FAs) of 10 and 30° with the following parameters: TR = 54.24 ms, TE = 2.73 ms, data matrix = 128 × 128, 10 slices each with a field of view = 20 × 20 × 1 mm3, and 5 averages for a scan time (temporal resolution) of 34 s. Mice were injected with 100 μL of PBS containing 0.5 mM Ln via tail vein catheter, followed by 100 μL of PBS flush to assure the administration of all contrast into the animal. Postcontrast gradient echo scans all used a FA of 30° with the same parameters as listed above. The serial acquisition was performed for 1 h following the injection resulting in about 100 postcontrast scans per animal. R1 mapping was performed using the variable flip angle method based on the following equation51
where SSPGR is signal intensity, α is FA, E1 is exp(TR/T1), and M0 is a proportionality factor related to longitudinal magnetization. E1 describes the linear relationship between the two signal intensity ratios, taking the slope, m, of that line enables calculation of T1 as
Concentration maps were then generated by the comparison of baseline R1 maps with postcontrast R1 maps using the following equation
where C is the concentration at time t, R1(t) is the postcontrast R1value at t, R1(t0) is baseline R1, and r1 is the relaxivity of the contrast agent. Ktrans, the contrast extravasation rate constant, mapping was then performed using a reference region (RR) model derived from the extended Tofts’ model52
 |
where CTOI and CRR are concentrations in the tissue of interest (TOI) and the RR, respectively, Ktrans,TOI and Ktrans, RR are Ktrans in the TOI and RR, respectively, and ve,TOI and ve,RR are extravascular–extracellular volume fractions for the TOI and RR, respectively. Muscle tissue from within the FOV was used in all animals. MATLAB was used to execute a least-squares curve fitting routine to calculate Ktrans for each voxel in the brain. Pharmacokinetic parameters were determined from blood concentration profiles determined from the MR images used for Ktrans mapping following our previously reported procedures.22 Briefly, regions of interest (ROIs) were selected in either carotid artery of concentration map images and used to calculate blood half-life and elimination constants from the 1 h series of imaging.
4.8. Fluorescence Imaging
A fluorescence microscope (Leica DMI3000B, Leica Biosystems) was used to take the fluorescence image of mice brain. Mice were euthanized immediately following the MR imaging protocol via 4% paraformaldehyde perfusion under anesthesia, and the brain tissue was collected, trimmed, and postfixed in 4% paraformaldehyde in Dulbecco’s phosphate-buffered saline (DPBS, Thermo Fisher Scientific, Waltham, MA) for 24 h followed by 30% sucrose in DPBS for 3 days at 4 °C. The brains were then embedded in an optimal cutting temperature (OCT, Fisher Scientific, Waltham, MA) compound, frozen on dry ice, and stored at −80 °C. The brains were sliced coronally at a thickness of 15 μm with a cryotome (Leica Biosystems, Wetzlar, Germany). Sections were washed with DPBS to remove excess OCT. The brain slices were blocked with 3% normal donkey serum, 0.3% Triton X-100, and 0.1% sodium azide in DPBS. The primary and secondary antibodies (Ab) were diluted in the blocking buffer. The brain sections were incubated with rat primary Ab against CD31 (1:250, ab56299, Abcam, Cambridge, U.K.) for 2 days at 4 °C then washed with blocking buffer three times for 5 min each before being incubated with a 1:250 dilution of goat secondary Ab against rat AF488 (ab150158, Abcam) for 2 h at room temperature. The brain sections were again washed with the blocking buffer three times for 5 min each before being stained with 1:1000 dilution of propidium iodide for 15 min, washed with DPBS two times for 5 min each, rinsed with DI water, and preserved with ProLong Gold Antifade Mountant (Thermo Fisher Scientific). The excitation and emission wavelength were used to observe NPs at 360 ± 20 and 470 ± 20 nm, respectively. CD31 and propidium iodide were observed under 488 and 555 nm filters, respectively. The objective magnifications used for the imaging were 20× and 40×.
Acknowledgments
F.M.K. acknowledges support from an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20GM103480), grant funding from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (R01NS109488), and the Nebraska Settlement Biomedical Research Development Funds. Part of this research was conducted at the Integrative Biological Imaging Facility at Creighton University, Omaha, NE. This facility, supported by the C.U. Medical School, was constructed with support from grants from the National Center for Research Resources (5P20RR016469) and the National Institute for General Medical Science (NIGMS) (8P20GM103427), a component of the National Institutes of Health (NIH). This investigation is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS, NINDS, or NIH. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under the grant no. DGE-1610400 to H.A.M. We thank Brandon McDonald for his assistance in surgeries to generate CCI mice.
The authors declare no competing financial interest.
References
- Centers for Disease Control and Prevention. Surveillance Report of Traumatic Brain Injury-Related Emergency Department Visits,Hospitalizations, and Deaths—United States, 2014. 2019, https://www.cdc.gov/traumaticbraininjury/pdf/TBI-Surveillance-Report-FINAL_508.pdf.
- Gardner R. C.; Burke J. F.; Nettiksimmons J.; Kaup A.; Barnes D. E.; Yaffe K. Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol. 2014, 71, 1490–1497. 10.1001/jamaneurol.2014.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordström P.; Michaëlsson K.; Gustafson Y.; Nordström A. Traumatic brain injury and young onset dementia: a nationwide cohort study. Ann. Neurol. 2014, 75, 374–381. 10.1002/ana.24101. [DOI] [PubMed] [Google Scholar]
- Shively S.; Scher A. I.; Perl D. P.; Diaz-Arrastia R. Dementia resulting from traumatic brain injury: what is the pathology?. Arch. Neurol. 2012, 69, 1245–1251. 10.1001/archneurol.2011.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982, 239, 57–69. 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- Kontos H. A.; Povlishock J. T. Oxygen radicals in brain injury. Cent. Nerv. Syst. Trauma 1986, 3, 257–263. 10.1089/cns.1986.3.257. [DOI] [PubMed] [Google Scholar]
- Hall E. D.; Andrus P. K.; Yonkers P. A. Brain hydroxyl radical generation in acute experimental head injury. J. Neurochem. 1993, 60, 588–594. 10.1111/j.1471-4159.1993.tb03189.x. [DOI] [PubMed] [Google Scholar]
- Johnson V. E.; Stewart J. E.; Begbie F. D.; Trojanowski J. Q.; Smith D. H.; Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Yoshioka H.; Kim G. S.; Jung J. E.; Okami N.; Sakata H.; Maier C. M.; Narasimhan P.; Goeders C. E.; Chan P. H. Oxidative Stress in Ischemic Brain Damage: Mechanisms of Cell Death and Potential Molecular Targets for Neuroprotection. Antioxid. Redox Signaling 2011, 14, 1505–1517. 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan P. G.; Rabchevsky A. G.; Hicks R. R.; Gibson T. R.; Fletcher-Turner A.; Scheff S. W. Dose-response curve and optimal dosing regimen of cyclosporin A after traumatic brain injury in rats. Neuroscience 2000, 101, 289–295. 10.1016/S0306-4522(00)00380-8. [DOI] [PubMed] [Google Scholar]
- Hall E. D. Beneficial effects of acute intravenous ibuprofen on neurologic recovery of head-injured mice: comparison of cyclooxygenase inhibition with inhibition of thromboxane A2 synthetase or 5-lipoxygenase. Cent. Nerv. Syst. Trauma 1985, 2, 75–83. 10.1089/cns.1985.2.75. [DOI] [PubMed] [Google Scholar]
- Bony B. A.; Kievit F. M. A Role for Nanoparticles in Treating Traumatic Brain Injury. Pharmaceutics 2019, 11, 473 10.3390/pharmaceutics11090473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj V. N.; Nguyen D. T.; Kodibagkar V. D.; Stabenfeldt S. E. Nanoparticle-Based Therapeutics for Brain Injury. Adv. Healthcare Mater. 2018, 7, 1700668 10.1002/adhm.201700668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelaz B.; Alexiou C.; Alvarez-Puebla R. A.; Alves F.; Andrews A. M.; Ashraf S.; Balogh L. P.; Ballerini L.; Bestetti A.; Brendel C.; Bosi S.; Carril M.; Chan W. C. W.; Chen C.; Chen X.; Chen X.; Cheng Z.; Cui D.; Du J.; Dullin C.; Escudero A.; Feliu N.; Gao M.; George M.; Gogotsi Y.; Grünweller A.; Gu Z.; Halas N. J.; Hampp N.; Hartmann R. K.; Hersam M. C.; Hunziker P.; Jian J.; Jiang X.; Jungebluth P.; Kadhiresan P.; Kataoka K.; Khademhosseini A.; Kopeček J.; Kotov N. A.; Krug H. F.; Lee D. S.; Lehr C.-M.; Leong K. W.; Liang X.-J.; Ling Lim M.; Liz-Marzán L. M.; Ma X.; Macchiarini P.; Meng H.; Möhwald H.; Mulvaney P.; Nel A. E.; Nie S.; Nordlander P.; Okano T.; Oliveira J.; Park T. H.; Penner R. M.; Prato M.; Puntes V.; Rotello V. M.; Samarakoon A.; Schaak R. E.; Shen Y.; Sjöqvist S.; Skirtach A. G.; Soliman M. G.; Stevens M. M.; Sung H.-W.; Tang B. Z.; Tietze R.; Udugama B. N.; VanEpps J. S.; Weil T.; Weiss P. S.; Willner I.; Wu Y.; Yang L.; Yue Z.; Zhang Q.; Zhang Q.; Zhang X.-E.; Zhao Y.; Zhou X.; Parak W. J. Diverse Applications of Nanomedicine. ACS Nano 2017, 11, 2313–2381. 10.1021/acsnano.6b06040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo D.; Magsam A. W.; Kelly A. M.; Stayton P. S.; Kievit F. M.; Convertine A. J. Core-Cross-Linked Nanoparticles Reduce Neuroinflammation and Improve Outcome in a Mouse Model of Traumatic Brain Injury. ACS Nano 2017, 11, 8600–8611. 10.1021/acsnano.7b03426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitner B. R.; Marcano D. C.; Berlin J. M.; Fabian R. H.; Cherian L.; Culver J. C.; Dickinson M. E.; Robertson C. S.; Pautler R. G.; Kent T. A.; Tour J. M. Antioxidant Carbon Particles Improve Cerebrovascular Dysfunction Following Traumatic Brain Injury. ACS Nano 2012, 6, 8007–8014. 10.1021/nn302615f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy M. K.; Wu L.; Kou W.; Ghorpade A.; Labhasetwar V. Superoxide Dismutase-Loaded PLGA Nanoparticles Protect Cultured Human Neurons Under Oxidative Stress. Appl. Biochem. Biotechnol. 2008, 151, 565. 10.1007/s12010-008-8232-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A.; Morris V. B.; Labhasetwar V.; Ghorpade A. Nanoparticle-mediated catalase delivery protects human neurons from oxidative stress. Cell Death Dis. 2013, 4, e903. 10.1038/cddis.2013.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. L.; Ypma M.; Chiarelli P. A.; Park J.; Ellenbogen R. G.; Stayton P. S.; Mourad P. D.; Lee D.; Convertine A. J.; Kievit F. M. Theranostic Oxygen Reactive Polymers for Treatment of Traumatic Brain Injury. Adv. Funct. Mater. 2016, 26, 4124–4133. 10.1002/adfm.201504416. [DOI] [Google Scholar]
- Marcano D. C.; Bitner B. R.; Berlin J. M.; Jarjour J.; Lee J. M.; Jacob A.; Fabian R. H.; Kent T. A.; Tour J. M. Design of poly(ethylene glycol)-functionalized hydrophilic carbon clusters for targeted therapy of cerebrovascular dysfunction in mild traumatic brain injury. J. Neurotrauma 2013, 30, 789–796. 10.1089/neu.2011.2301. [DOI] [PubMed] [Google Scholar]
- Bharadwaj V. N.; Rowe R. K.; Harrison J.; Wu C.; Anderson T. R.; Lifshitz J.; Adelson P. D.; Kodibagkar V. D.; Stabenfeldt S. E. Blood–brainbarrier disruption dictates nanoparticle accumulation following experimental brain injury. Nanomedicine 2018, 14, 2155–2166. 10.1016/j.nano.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller H. A.; Magsam A. W.; Tarudji A. W.; Romanova S.; Weber L.; Gee C. C.; Madsen G. L.; Bronich T. K.; Kievit F. M. Evaluating differential nanoparticle accumulation and retention kinetics in a mouse model of traumatic brain injury via K(trans) mapping with MRI. Sci. Rep. 2019, 9, 16099 10.1038/s41598-019-52622-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj V. N.; Lifshitz J.; Adelson P. D.; Kodibagkar V. D.; Stabenfeldt S. E. Temporal assessment of nanoparticle accumulation after experimental brain injury: Effect of particle size. Sci. Rep. 2016, 6, 29988 10.1038/srep29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd B. J.; Galle A.; Daglas M.; Rosenfeld J. V.; Medcalf R. Traumatic brain injury opens blood-brain barrier to stealth liposomes via an enhanced permeability and retention (EPR)-like effect. J. Drug Targeting 2015, 23, 847–853. 10.3109/1061186X.2015.1034280. [DOI] [PubMed] [Google Scholar]
- Xu W.; Bony B. A.; Kim C. R.; Baeck J. S.; Chang Y.; Bae J. E.; Chae K. S.; Kim T. J.; Lee G. H. Mixed lanthanide oxide nanoparticles as dual imaging agent in biomedicine. Sci. Rep. 2013, 3, 3210 10.1038/srep03210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton F. A.; Wilkinson G.. Advanced Inorganic Chemistry, 4th ed.; John Wiley & Sons: New York, 1980. [Google Scholar]
- Wakefield G.; Keron H. A.; Dobson P. J.; Hutchison J. L. Synthesis and Properties of Sub-50-nm Europium Oxide Nanoparticles. J. Colloid Interface Sci. 1999, 215, 179–182. 10.1006/jcis.1999.6225. [DOI] [PubMed] [Google Scholar]
- Flores-González M. A.; Louis C.; Bazzi R.; Ledoux G.; Lebbou K.; Roux S.; Perriat P.; Tillement O. Elaboration of nanostructured Eu3+-doped Gd2O3 phosphor fine spherical powders using polyol-mediated synthesis. Appl. Phys. A 2005, 81, 1385–1391. 10.1007/s00339-005-3215-3. [DOI] [Google Scholar]
- Yokel R. A. Nanoparticle brain delivery: a guide to verification methods. Nanomedicine 2020, 15, 409–432. 10.2217/nnm-2019-0169. [DOI] [PubMed] [Google Scholar]
- van Vlerken L. E.; Vyas T. K.; Amiji M. M. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm. Res. 2007, 24, 1405–1414. 10.1007/s11095-007-9284-6. [DOI] [PubMed] [Google Scholar]
- Suk J. S.; Xu Q.; Kim N.; Hanes J.; Ensign L. M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Delivery Rev. 2016, 99, 28–51. 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokerst J. V.; Lobovkina T.; Zare R. N.; Gambhir S. S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon G. S. Polymeric micelles for delivery of poorly water-soluble compounds. Crit. Rev. Ther. Drug Carrier Syst. 2003, 20, 357–403. 10.1615/CritRevTherDrugCarrierSyst.v20.i5.20. [DOI] [PubMed] [Google Scholar]
- Kanaras A. G.; Kamounah F. S.; Schaumburg K.; Kiely C. J.; Brust M. Thioalkylated tetraethylene glycol: a new ligand for water soluble monolayer protected gold clusters. Chem. Commun. 2002, 2294–2295. 10.1039/b207838b. [DOI] [PubMed] [Google Scholar]
- Elci S. G.; Jiang Y.; Yan B.; Kim S. T.; Saha K.; Moyano D. F.; Yesilbag Tonga G.; Jackson L. C.; Rotello V. M.; Vachet R. W. Surface Charge Controls the Suborgan Biodistributions of Gold Nanoparticles. ACS Nano 2016, 10, 5536–5542. 10.1021/acsnano.6b02086. [DOI] [PubMed] [Google Scholar]
- Kim B.; Han G.; Toley B. J.; Kim C. K.; Rotello V. M.; Forbes N. S. Tuning payload delivery in tumour cylindroids using gold nanoparticles. Nat. Nanotechnol. 2010, 5, 465–472. 10.1038/nnano.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderlind F.; Pedersen H.; Petoral R. M.; Käll P.-O.; Uvdal K. Synthesis and characterisation of Gd2O3 nanocrystals functionalised by organic acids. J. Colloid Interface Sci. 2005, 288, 140–148. 10.1016/j.jcis.2005.02.089. [DOI] [PubMed] [Google Scholar]
- Hug S. J.; Bahnemann D. Infrared spectra of oxalate, malonate and succinate adsorbed on the aqueous surface of rutile, anatase and lepidocrocite measured with in situ ATR-FTIR. J. Electron Spectrosc. Relat. Phenom. 2006, 150, 208–219. 10.1016/j.elspec.2005.05.006. [DOI] [Google Scholar]
- Gao J.; Li Q.; Wang C.; Tan H. Ratiometric detection of hydroxy radicals based on functionalized europium(III) coordination polymers. Mikrochim. Acta 2017, 185, 9 10.1007/s00604-017-2581-9. [DOI] [PubMed] [Google Scholar]
- George M. R.; Golden C. A.; Grossel M. C.; Curry R. J. Modified dipicolinic acid ligands for sensitization of europium(III) luminescence. Inorg. Chem. 2006, 45, 1739–1744. 10.1021/ic051461u. [DOI] [PubMed] [Google Scholar]
- Dong H.; Du S. R.; Zheng X. Y.; Lyu G. M.; Sun L. D.; Li L. D.; Zhang P. Z.; Zhang C.; Yan C. H. Lanthanide Nanoparticles: From Design toward Bioimaging and Therapy. Chem. Rev. 2015, 115, 10725–10815. 10.1021/acs.chemrev.5b00091. [DOI] [PubMed] [Google Scholar]
- Mutas M.; Strelow C.; Kipp T.; Mews A. Specific binding and internalization: an investigation of fluorescent aptamer-gold nanoclusters and cells with fluorescence lifetime imaging microscopy. Nanoscale 2018, 10, 20453–20461. 10.1039/C8NR06639F. [DOI] [PubMed] [Google Scholar]
- Estandarte A. K.; Botchway S.; Lynch C.; Yusuf M.; Robinson I. The use of DAPI fluorescence lifetime imaging for investigating chromatin condensation in human chromosomes. Sci. Rep. 2016, 6, 31417 10.1038/srep31417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahav G.; Hirshberg A.; Salomon O.; Amariglio N.; Trakhtenbrot L.; Fixler D. Fluorescence lifetime imaging of DAPI-stained nuclei as a novel diagnostic tool for the detection and classification of B-cell chronic lymphocytic leukemia. Cytometry, Part A 2016, 89, 644–652. 10.1002/cyto.a.22890. [DOI] [PubMed] [Google Scholar]
- Badieirostami M.; Carpenter C.; Pratx G.; Xing L.; Sun C. Upconversion Luminescence Imaging of Tumors with EGFR-Affibody Conjugated Nanophosphors. MRS Adv. 2019, 4, 2461–2470. 10.1557/adv.2019.242. [DOI] [Google Scholar]
- Naczynski D. J.; Sun C.; Türkcan S.; Jenkins C.; Koh A. L.; Ikeda D.; Pratx G.; Xing L. X-ray-Induced Shortwave Infrared Biomedical Imaging Using Rare-Earth Nanoprobes. Nano Lett. 2015, 15, 96–102. 10.1021/nl504123r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y.; Mahmood A.; Chopp M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon D. W.; McGeachy M. J.; Bayir H.; Clark R. S.; Loane D. J.; Kochanek P. M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. 10.1038/nrneurol.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R. S.; Schiding J. K.; Kaczorowski S. L.; Marion D. W.; Kochanek P. M. Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models. J. Neurotrauma 1994, 11, 499–506. 10.1089/neu.1994.11.499. [DOI] [PubMed] [Google Scholar]
- Yu S.; Kaneko Y.; Bae E.; Stahl C. E.; Wang Y.; van Loveren H.; Sanberg P. R.; Borlongan C. V. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009, 1287, 157–163. 10.1016/j.brainres.2009.06.067. [DOI] [PubMed] [Google Scholar]
- Deoni S. C.; Peters T. M.; Rutt B. K. High-resolution T1 and T2 mapping of the brain in a clinically acceptable time with DESPOT1 and DESPOT2. Magn. Reson. Med. 2005, 53, 237–241. 10.1002/mrm.20314. [DOI] [PubMed] [Google Scholar]
- Tofts P. S.; Brix G.; Buckley D. L.; Evelhoch J. L.; Henderson E.; Knopp M. V.; Larsson H. B.; Lee T. Y.; Mayr N. A.; Parker G. J.; Port R. E.; Taylor J.; Weisskoff R. M. Estimating kinetic parameters from dynamic contrast-enhanced T(1)-weighted MRI of a diffusable tracer: standardized quantities and symbols. J. Magn. Reson. Imaging 1999, 10, 223–232. . [DOI] [PubMed] [Google Scholar]