

Abstract

Separation techniques hyphenated to high-resolution mass spectrometry are essential in untargeted metabolomic analyses. Due to the complexity and size of the resulting data, analysts rely on computer-assisted tools to mine for features that may represent a chromatographic signal. However, this step remains problematic, and a high number of false positives are often obtained. This work reports a novel approach where each step is carefully controlled to decrease the likelihood of errors. Datasets are first corrected for baseline drift and background noise before the MS scans are converted from profile to centroid. A new alignment strategy that includes purity control is introduced, and features are quantified using the original data with scans recorded as profile, not the extracted features. All the algorithms used in this work are part of the Finnee Matlab toolbox that is freely available. The approach was validated using metabolites in exhaled breath condensates to differentiate individuals diagnosed with asthma from patients with chronic obstructive pulmonary disease. With this new pipeline, twice as many markers were found with Finnee in comparison to XCMS-online, and nearly 50% more than with MS-Dial, two of the most popular freeware for untargeted metabolomics analysis.
Introduction
Liquid chromatography hyphenated to high-resolution mass spectrometry (LC-HRMS) is a fast-developing technique for untargeted proteomic and metabolomic analyses.1−5 Modern instruments provide unique separation power, high mass resolution, and accurate mass measurements, allowing the separation, detection, and quantification of hundreds of compounds, with concentrations spanning many orders of magnitude, in a single run.6 The datasets resulting from a single LC-HRMS run are a collection of MS spectra gathered along the time and can range from hundreds of MB to a few GB. Due to the wealth of information that is contained in the dataset, computer-assisted MS analysis tools are nowadays central to the analytical scheme.7 The main tasks of these tools are to separate chromatographic-like information from the noise (mining or feature extraction), to recognize features from the same compound in the different datasets (peak alignment), and to analyze all the gathered information (chemometrics or machine learning).1,5,8−12 The underlying assumption is that the variations observed in the data are related to chemical variations in the samples and thus to the metabolomic activities. However, this assumption is not always correct, and many observed variations are the result of processing errors, false positives, and misalignment.13,14 For example, Myers and co-workers have recently demonstrated that 30 to 60% false positives,15 depending on the experimental conditions, were detected by two of the most used packages (XCMS and mzMine).16 Such a high number of false positives are worrying as these will be carried over throughout the whole analytical pipeline, potentially resulting in false compound identification.17 This issue is not specific to free software; indeed, Li and co-workers compared two commercial software (MarkerView and Compound Discoverer) with three free alternatives (MS-Dial, MZmine 2, XCMS), and similar performances in the detection of correct features derived from compounds in the mixtures were observed.18
Different controls have been proposed to improve the reliability of the features mined in metabolomics.13,19 One of the most relevant concepts introduced in untargeted analyses was the quality control sample (QC).13,19−21 QCs should be representative of the composition and matrix of all samples. They are generally made of small aliquots of all target samples, and they are injected between samples at regular intervals. Features are mined and aligned between all samples and QC. Features present in all QC samples with a low quantitative coefficient of variation are likely to be well processed; only these features should be used for the sample. With complex samples, more than 50% features are often discarded after QC evaluation.22 This number will increase if minor peaks are considered. While QCs allow removal of many false-positive features (noise or baseline recognized as a peak), correct features can also be discarded, and this is principally due to errors during the different computerized steps. Maximizing extracted aligned features while decreasing false positives is one of the main challenges in untargeted metabolomics,7 and improving and validating the algorithms used during untargeted data mining will contribute to this goal.
Finnee is a Matlab toolbox designed for analysis in hyphenated datasets. Previous works describe a data cleaning approach, with algorithms to correct baseline drift and background noise. The corrections were made with MS scans recorded as profile, and it has been demonstrated that this step allows reduction in the number of false positives, even with peaks with low intensities.23,24 This manuscript presents the next steps with feature extraction, QC alignment methodologies using a constrained clustering approach, and targeted reanalysis using the original data. Algorithms are described and validated by comparing the metabolomic signatures of exhaled breath condensates (EBCs) of asthmatic individuals with chronic obstructive pulmonary disease (COPD) patients.
EBCs are obtained by condensation of gases and droplets released during exhalation.25−28 This sampling procedure is suitable for patients of all age groups, irrespective of the disease severity, as it is safe, noninvasive, easy, and straightforward. Traditionally, pulmonologists focus their attention on nonspecific characteristics of the EBC, such as the pH that correlates with airway inflammation,29 but it has also been demonstrated that EBCs are a source of biomarkers and can elucidate the inflammation process of the lungs, such as asthma or COPD.30−34 However, these biomarkers are often present at low concentrations, which complicates the analysis.35 Results obtained with Finnee are compared with XCMS-online36,37 and MS-Dial.38
Results and Discussion
Workflow of the Computerized Approach for Finnee
The workflow developed in this work is separated into three steps; steps 1 and 2 are illustrated in Scheme 1.
Scheme 1. Schematic Illustration of the Workflow of the Computerized Finnee Approach Used in This Study.

Step 1: Extracting features of interest – This step is similar to classical pipelines and aims to find and align features along all QCs. Briefly, after the acquisition, datasets resulting from each QC sample are converted from their proprietary format to the open mzML format.39 For this work, msConvert,40 supported by ProteoWizard,41 was used. MS scans should be obtained as profile (or continuous) spectra rather than centroid (or discrete) spectra.42 The QCs are cleaned from baseline drift and then from background noise using Finnee23,24 (see Data Cleaning) before converting the MS scans from profile to centroid spectra. QCs can now be mined for chromatographic-like features (see Peak Mining). Figures of merit for all peaks from each QC are summarized in a peak list. In this work, a constrained clustering approach was developed to align features present in the QC (see Peak List Alignment). At the end of this step, a series of features present in one or more QCs are obtained. These are characterized by their coordinate (accurate mass, mac, and centroid time, tc) and peak area. Only features present in more than 50% QCs are retained.
Step 2: Optimization and validation – Features, previously selected, are reanalyzed from the original data using the region of interest (ROI) (see Region of Interest (ROI) and Quantification). Quantification is performed in two steps. First, the peak limits (times at peak start and peak end) are obtained for each feature in every QC. Then, for each feature, consolidated peak limits are obtained. All features are analyzed for a third time using these common limits. Features are filtered to only retain the ones common to all QCs with a relative standard deviation of their area below a set threshold (in this work, 20%).
Step 3: Sample analysis – Samples are analyzed sequentially using the original data with the ROI and the consolidated limits. Only features previously selected are mined for. This approach not only allows analysis of the target features with high reliability but also avoids the need to align hundreds of peak lists with the corresponding computer limitation.
Data Cleaning
Data cleaning is used to remove needless information in datasets. The procedure has been modified from previous works.23,24Scheme 2 details the different steps used in the data cleaning process.
Scheme 2. Schematic Illustration of the Data Cleaning Workflow Used in This Study.

Briefly, for each run, the dataset, converted to mzML, is opened using the Finnee toolbox.43 The master mzAxis (Mmz) is calculated using the most intense spectrum. All MS spectra are then aligned to the Mmz using a linear interpolation algorithm. This simple transformation allows use of a common m/z axis for all spectra. Data can easily be read at a given time to obtain the MS spectrum or at a given m/z value. The profile obtained at a specific m/z value is named a single m/z profile (SMP). It should be emphasized that this approach is different from the binning techniques,9. Extracted ion profiles (EIPs) can be obtained summing nearby SMPs.
The SMP that suffers from the influence of background ions can be detected due to the low number of nonzero intensities.24 These can be corrected for baseline drift using, for example, the asymmetrically reweighted penalized least squares (arPLS) algorithms.44 The noise at each m/z is estimated, and points, whose neighbor intensities are lower than 10 times the value of the noise, are considered as background noise, and their intensities are set to zero.24
Peak Mining
MS spectra are converted from profile to centroid,23 and chromatographic-like features are recognized and extracted as data points, in successive scans, whose centroid m/z does not differ by more than a set value.23 For each feature, figures of merit (FOMs) are calculated (including the chromatographic statistical moments23 and accurate masses42) and are summarized in peak tables. Because the datasets have already been corrected for baseline drift and background noise, no deconvolution or baseline correction algorithms are used. A detected feature may be a single ion, but it can also be multiple, nonbaseline-separated, isobaric ions.
Peak List Alignment
Scheme 3 presents the algorithms used to align the features shared in different datasets. One of the peak lists is randomly selected as the seeding list, and each peak within the list is assigned to a single cluster. The following list is randomly chosen, and for each feature within this list, the Euclidian distance, d, to all clusters are computed as
| 1 |
where d(i,n) is the Euclidian distance between peak i and cluster n, mac,i and tc,i are the accurate mass and centroid time of peak i, respectively, and m̅ac, n and t̅c, n are the average accurate masses and centroid times of all features within the cluster n. Wr is a correcting factor; e.g., a Wr 100 means that a difference of 0.001 in the m/z will have the same importance as a difference of 0.1 min in the time (see Optimization of Finnee Peak Alignment with QC Samples).
Scheme 3. Schematic Illustration of the Constrained Clustering Approach.

A peak will be assigned to its closest cluster if and only if mac and tc differences are below predefined values; otherwise, the feature will start a new cluster. Once all peaks are arranged into clusters, the clusters’ figures of merit are calculated. These figures include the number of elements, the average accurate mass, and the associated standard deviation.
Overlapping clusters are then detected using the following rule:
| 2 |
where Δm̅a and Δt̅c are the average accurate mass difference and the average centroid time difference between two clusters, respectively, and sma and stc are the within-cluster standard deviations of the accurate masses and of the centroid times, respectively. Two clusters, detected as overlapping, are merged, and the process is repeated until no overlapping clusters remain.
Each final cluster is checked for purity using the Pearson correlation coefficient ρi(n) using eq 3.
| 3 |
Y(i,n) is the profile of the feature i in the cluster, represented as a two-dimensional array, time, and intensity; Y̅(n) is calculated as the average of all profiles within the cluster n. cov is the covariance, σY(i, n) is the standard deviation of Y(i, n), and σY̅(n) is the standard deviation of Y̅(n). The Pearson correlation coefficient is an efficient way of comparing two distributions independent of their intensities. A cluster is assumed pure if all profiles correlated to the average profile result in ρi higher than a set value (typically 0.7). This makes sure that all profiles within a cluster are similar.
Region of Interest (ROI) and Quantification
In this work, following the determination of putative features, defined by their accurate masses and centroid time, a targeted approach was implemented to extract an ROI in the original data for each feature. The ROI is an m × n matrix that contains all points from the original dataset, whose time and m/z are within a set interval. For this work, the time interval corresponded to the centroid time of the targeted feature ±1 min, and the m/z interval spanned 11 increments in the Mmz centered around the m/z closest to the accurate mass of the target features. An example of an ROI with tc = 1.95 min and mac = 97.0760 is shown in Figure 1.
Figure 1.

(A) ROI corresponding to t = 1.95 ± 1.00 min and m/z = 97.0760 ± 5 increments. (B) Corresponding profiles with, in black, the peak areas as a function of time and, in red, the mass centroid. Black arrows correspond to the local maxima; red arrows correspond to the peaks’ limits.
The ROI analysis is done, first, by calculating at each time the area and mass centroid along the m/z axis. In Figure 1B, the black profile corresponds to the variation of the mass peak area A(t) as a function of time, and the scattered red dots are the corresponding mass centroid values m/z(t). In the area profile, data were corrected for baseline drift as in Data Cleaning only if more than 95% of the data are non-null.
Peak limits are calculated in two ways. First, local maxima are determined using a moving window of size 2*wdz + 1, with, typically, wdz = 5. The window scans all the points in the profile. The central point is recognized as a local maximum if it is the highest value within the window, and there are no null values within the window. Peak start, tstart, and peak end, tend, are measured independently for every feature as the time of the first and last points before and after each local maximum with intensity null or negative. Second, consolidated limits are calculated by pooling the limits from the same features from all QCs. Outliers are detected and removed using the MAD algorithms,45 and consolidated peak start, Ctstart, and peak end, Ctend, are calculated as
| 4 |
| 5 |
where 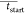 and
and  are the means of the
peak starts and the
peak ends and σtstart and σtend are the associated
standard deviations, respectively.
are the means of the
peak starts and the
peak ends and σtstart and σtend are the associated
standard deviations, respectively.
For each peak, the volume, V, time centroid, tc, and accurate mass, ma, are calculated using46
| 6 |
| 7 |
| 8 |
In the case of multiple peaks, only the peak whose peak centroid and accurate mass are closest to the target values is conserved.
Optimization of Finnee Peak Alignment with QC Samples
Following the mining step, an average of 13000 features, with maximum intensities ranging from 25000 to 109, was identified. The alignment was performed as described in the Experimental Section and Computational Methods, where the optimization of three parameters is indicated (Δm̅a, Δtc®, and Wr). Δm̅a and Δtc® correspond to the constrained accurate mass and centroid time differences, respectively, that control if the feature is arranged in an existing or in a new cluster, and Wr is a weight factor. The influence of these factors was assessed by the number of features common to all QCs with an RSD of the peak area below 20%. At this stage, figures of merit are calculated using the mined features, not the ROI (Table 1).
Table 1. Alignment Parameters and Number of Obtained Features.
| Wr | 170 | 135 | 135 | 135 | 135 |
| Δm̅a | 0.005 | 0.005 | 0.002 | 0.01 | 0.02 |
| Δtc® | 0.5 | 0.675 | 0.27 | 1.35 | 271 |
| number of featuresa | 1781 | 1781 | 1776 | 1782 | 1779 |
Number of aligned features present in all QCs, with an RSD of the peak area below 20%.
Comparison with MS-Dial and XCMS Online
Parameters for MS-Dial and XCMS were also optimized using the QC samples as described in the Supporting Information. Table 2 summarizes the results obtained by MS-Dial, XCMS, and Finnee. For Finnee, QCs were quantified in two different ways, either using the extracted features as performed classically or using the ROI, as described in the Experimental Section and Computational Methods. Each software was evaluated using the number of aligned features that are common to all QCs and with an RSD of the peak areas below 20%.
Table 2. Comparison between MS-Dial, XCMS, and Finnee.
QCs were quantified with the mined features.
QCs were quantified with the ROI and consolidated peak limits.
Number of aligned features present in all QCs, with an RSD of the peak area below 20%.
Finnee obtained, when using the mined features, 11% more markers than XCMS and 18% more markers than MS-Dial. However, performance is significantly improved when the markers are reanalyzed using a targeted approach with ROI with 28% more markers found. It is believed that reanalyzing the markers using a targeted approach allows mitigation of the errors generated by the successful transformation. The Venn diagram in Figure 2 illustrates the repartition of the markers.
Figure 2.

Venn diagram showing the number of features found by Finnee (red), MS-Dial (blue), and XCMS (green).
656 common features were found by all approaches, and 992 were found by two approaches. Interestingly, 81% of the features found by XCMS were also found by Finnee, and 66% of the features found by MS-Dial were also found by Finnee. However, few features found by MS-Dial and XCMS were not found by Finnee.
Differential Analysis of Asthma and COPD
As final validation of the Finnee pipeline, EBC samples from three groups (five controls, five patients diagnosed with COPD, and five patients diagnosed with asthma as described in the Experimental Section and Computational Methods, all run in triplicate) were analyzed by XCMS, MS-Dial, and Finnee. For XCMS, three pair jobs were performed with the optimized parameters: asthma vs COPD, asthma vs control, and COPD vs control. After completion, results were exported, and the features were aligned with the QC samples (see the Supporting Information). Only features aligned with QC features with the previous constraints were selected. Triplicates were pooled, features were aligned, and those not present in all triplicates were discarded. For each feature, the average area was calculated.
Welch’s t-test (t-test assuming unequal variance) was used to select features in which the mean of the peak areas is significantly different between two groups (p < 0.05, n = 5 in each group). The same approach was used for MS-Dial. The areas were not corrected for instrumental variation with the QC samples. However, pooling the replicates that were injected at random positions within the sample sequence allows the effect of this bias to be diminished. For Finnee, the samples were analyzed using the ROI with consolidated peak limits, optimized using the QC samples. Previously, data in triplicate were averaged, and Welch’s t-test was used to retain features where the assumption of equal means at the 5% significance level (h > 0.05) was not verified.
Figure 3 shows the Venn diagrams for the markers found by Finnee, MS-Dial, and XCMS when pairing (A) asthma with control, (B) COPD with control, and (C) asthma with COPD groups. The profiles of the three pair jobs are similar. In all cases, XCMS was found to have the lowest number of markers followed by MS-Dial, with on average 28% more markers than XCMS, and then Finnee with 43% more markers than MS-Dial. The high number of markers, found with the three software, for the pair job asthma vs COPD should be noted, which is 10 times more than with the pair job asthma vs control and COPD vs control. As the sequence of injections, including replicates, were random, bias due to instrumental variations can be ruled out. However, the sample size is small, and it should be increased to validate this preliminary work.
Figure 3.

Venn diagram showing the number of discriminating markers between (A) asthma and control, (B) COPD and control, and (C) asthma and COPD found by Finnee (red), MS-Dial (blue), and XCMS (green).
Finally, each marker was visually inspected to assess its reliability in discriminating two groups. For each marker, the extracted ion profiles (EIPs) were obtained in each sample. The EIPs were aligned to a common time axis, and EIPs from each classification were averaged. An example is shown in Figure 4 where asthma and control groups are compared. The plain dark lines are the average of all EIPs with m/z = 249.148 ± 0.005 (spanning 11 points in the m/z axis), and the dotted red lines are the means plus or minus one standard deviation. All figures are in a shared folder https://drive.google.com/drive/folders/1mkQyDcGbSG3KP_VpfATuXNv6D69D_wWJ?usp=sharing, together with the Excel files “Comprehensive results.xlsx” that summarize the results of the classification.
Figure 4.

Averaged (black trace) and corresponding standard deviation (red trace) of the 15 extracted ion profiles with m/z = 249.148 ± 0.005, for asthma and control groups.
Markers were classified in three groups in the Excel file (green: figures that corroborate the predicted markers; yellow: figures that display a peak at the predicted time but with a mean height similar in both groups; red: no chromatographic peaks observed at the predicted time or m/z). Multiple coeluting peaks are not treated differently compared to single baseline-separated peaks. On average, less than 5% of all markers were classified as false positives (red: absence of peaks) with each software. 18% of the markers were classified as yellow (presence of a peak but doubting the ability of this peak to differentiate the two groups) with MS-Dial, 25% with Finnee, and 29% with XCMS. This represents excellent predictive values with all software with a clear advantage to Finnee with those data due to the higher number of markers predicted. It is particularly notable that many markers predicted by Finnee cover markers predicted by one of the two other software, but not both; however, very few markers predicted by the two other software were not predicted by Finnee (see Figure 2), highlighting the reliability of the pipeline.
Conclusions
The different tools developed in the Finnee toolbox aim to process hyphenated MS datasets with thoroughness. In this work, the algorithms have been used to recognize peaks, at low intensities, in EBC samples. With this data, Finnee outperformed XCMS with 82% more markers discovered and MS-Dial with 43% more markers. This result should not be extrapolated to other studies; however, this demonstrated the validity of the pipeline. It should be emphasized that the detected features should not be considered yet as markers of asthma and COPD diseases. The samples analyzed were few, and the markers should also be identified to determine if they are endogenous metabolites or exogenous chemicals. The aim of this work was to design tools that will allow extraction of markers at low intensities. The pipeline will be used in a large-scale metabolomics project aiming at obtaining reliable markers for COPD, asthma, and asthma COPD overlap in EBC samples.
Experimental Section and Computational Methods
EBC Samples and Clinical Assessment
EBC samples were collected from 15 individuals randomly selected (five controls, five with asthma medical diagnosis, and five with COPD medical diagnosis, as assessed by the OLDER Study – Obstructive Lung Diseases in Elders). The Ethics Committee of Nova Medical School approved this study.
Asthma was assigned when the patient reported respiratory symptoms, was a nonsmoker, and presented a positive reversibility test (FEV1 > 12% and 200 mL). COPD disease was attributed to those who also reported being current smokers, had a post-bronchodilator FEV1/FVC < 0.70, and had a negative reversibility test.
LC–MS Analysis
Samples were analyzed in triplicate by LC–MS using an Orbitrap Q Exactive Focus (Thermo Scientific) coupled to an Ultimate 3000 UHPLC (Thermo Scientific). A pooled quality control (QC) sample was used to compensate for any possible time-dependent batch effects. The QC samples were created using a small aliquot from each sample. The QC samples were reinjected at regular intervals to bracket the samples. The separation was performed using a Waters XBridge column C18 (2.1 × 150 mm, 3.5 μm particle size, P/N 186003023). The mobile phase A was water with 0.1% formic acid (v/v), and mobile phase B was acetonitrile with 0.1% formic acid (v/v) (Optima LC–MS Grade, Fisher Scientific). The gradient program was as follows: 1 min at 1% B; 1–13 min from 1 to 99% B, 13–15 min at 99% B, 15–16 min from 99 to 1% B, and 4 min at 1% B. The column temperature was maintained at 30 °C, and a flow rate of 400 μL/min was used.
The Q Exactive Focus MS method consisted of several cycles of full MS scan (R = 70000) followed by three ddMS2 scans (R = 17500), with a (N)CE of 30 and in positive mode. External calibration was performed using LTQ ESI Positive Ion Calibration Solution (Thermo Scientific) and the lock mass enabled internal calibration. Data were obtained using the Xcalibur software v.4.0.27.19 (Thermo Scientific). The raw MS files, as recorded by the instrument, are available from the corresponding author upon reasonable request.
Programming
Finnee was developed using Matlab R2019a (Mathworks). The toolbox is open-access and freely accessible, including the new functions that were designed for this manuscript. The Matlab code used in this study is available in Zenodo with the identifier doi: 10.5281/zenodo.3581436.43 The code source can also be downloaded from GitHub (https://github.com/glerny/finnee2016/). For this work, functions were programmed and run using a PC equipped with an Intel Core i7 CPU (2.80 GHz) and 18.0 GB RAM.
XCMS online (https://xcmsonline.scripps.edu) was used as control. The following parameters were used (description of the optimization process can be found in the Supporting Information):
Feature detection: method centWave (with ppm: 100; minimum peak width: 5 s; maximum peak width: 60s)
Retention time correction: method none
Alignment: default (bw: 5; mnfrac: 0.5; mzwid: 0.015)
Statistics: unpaired parametric t-test
MS-Dial (ver. 3.98) was used with the following parameters:
Data collection: MS1 tolerance: 0.002 m/z
Peak detection: minimum peak height: 100 amu, mass slice width: 0.005 m/z
Alignment: retention time tolerance: 0.05 min, MS1 tolerance: 0.005 m/z
Acknowledgments
This work was financially supported by the projects: (i) UID/EQU/00511/2019 - Laboratory for Process Engineering, Environment, Biotechnology and Energy – LEPABE funded by national funds through FCT/MCTES (PIDDAC); (ii) POCI-01-0145-FEDER-029702 and POCI-01-0145-FEDER-031297 funded by FEDER funds through COMPETE2020 – Programa Operacional Competitividade e Internacionalização (POCI) and by national funds (PIDDAC) through FCT/MCTES; (iii) AstraZeneca – Projecto OLDER (CEDOC/2015/59); (iv) iNOVA4Health - UID/Multi/04462/2013, financially supported by FCT/Ministério da Educação e Ciência, and co-funded by FEDER under the PT2020 Partnership Agreement.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01610.
Description of XCMS and MS-Dial optimization (PDF)
Author Contributions
G.L.E, N.N., P.C.-M., and P.G.-A. designed the research. G.L.E. developed the Finnee program. P.G.-A., A.C.L.G., and R.G. analyzed the samples. N.N., P.C.-M., and J.G.M. recovered the EBC samples and were responsible for the clinical assessment. G.L.E. took the lead in writing the manuscript. All authors discussed the results and contributed to the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Zang X.; Monge M. E.; Fernández F. M. Mass Spectrometry-Based Non-Targeted Metabolic Profiling for Disease Detection: Recent Developments. TrAC, Trends Anal. Chem. 2019, 158–169. 10.1016/j.trac.2019.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gika H.; Virgiliou C.; Theodoridis G.; Plumb R. S.; Wilson I. D. Untargeted LC/MS-Based Metabolic Phenotyping (Metabonomics/Metabolomics): The State of the Art. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2019, 1117, 136–147. 10.1016/j.jchromb.2019.04.009. [DOI] [PubMed] [Google Scholar]
- Finney G. L.; Blackler A. R.; Hoopmann M. R.; Canterbury J. D.; Wu C. C.; MacCoss M. J. Label-Free Comparative Analysis of Proteomics Mixtures Using Chromatographic Alignment of High-Resolution ΜLC-MS Data. Anal. Chem. 2008, 80, 961–971. 10.1021/ac701649e. [DOI] [PubMed] [Google Scholar]
- Wang X.; Shen S.; Rasam S. S.; Qu J. MS1 Ion Current-Based Quantitative Proteomics: A Promising Solution for Reliable Analysis of Large Biological Cohorts. Mass Spectrom. Rev. 2019, 38, 461–482. 10.1002/mas.21595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrochategui E.; Jaumot J.; Lacorte S.; Tauler R. Data Analysis Strategies for Targeted and Untargeted LC-MS Metabolomic Studies: Overview and Workflow. TrAC, Trends Anal. Chem. 2016, 82, 425–442. 10.1016/j.trac.2016.07.004. [DOI] [Google Scholar]
- Krug S.; Kastenmüller G.; Stückler F.; Rist M. J.; Skurk T.; Sailer M.; Raffler J.; Römisch-Margl W.; Adamski J.; Prehn C.; et al. The Dynamic Range of the Human Metabolome Revealed by Challenges. FASEB J. 2012, 26, 2607–2619. 10.1096/fj.11-198093. [DOI] [PubMed] [Google Scholar]
- Johnson C. H.; Ivanisevic J.; Benton H. P.; Siuzdak G. Bioinformatics: The next Frontier of Metabolomics. Anal. Chem. 2015, 87, 147–156. 10.1021/ac5040693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccard J.; Veuthey J. L.; Rudaz S. Knowledge Discovery in Metabolomics: An Overview of MS Data Handling. J. Sep. Sci. 2010, 290–304. 10.1002/jssc.200900609. [DOI] [PubMed] [Google Scholar]
- Katajamaa M.; Orešič M. Data Processing for Mass Spectrometry-Based Metabolomics. J. Chromatogr. A 2007, 1158, 318–328. 10.1016/j.chroma.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Cajka T.; Fiehn O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 524–545. 10.1021/acs.analchem.5b04491. [DOI] [PubMed] [Google Scholar]
- Wei X.; Shi X.; Kim S.; Zhang L.; Patrick J. S.; Binkley J.; McClain C.; Zhang X. Data Preprocessing Method for Liquid Chromatography-Mass Spectrometry Based Metabolomics. Anal. Chem. 2012, 84, 7963–7971. 10.1021/ac3016856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccard J.; Rudaz S. Harnessing the Complexity of Metabolomic Data with Chemometrics. J. Chemom. 2014, 28, 1–9. 10.1002/cem.2567. [DOI] [Google Scholar]
- Quintás G.; Sánchez-Illana Á.; Piñeiro-Ramos J. D.; Kuligowski J. Data Quality Assessment in Untargeted LC-MS Metabolomics. Compr. Anal. Chem. 2018, 82, 137–164. 10.1016/bs.coac.2018.06.002. [DOI] [Google Scholar]
- Baran R. Untargeted Metabolomics Suffers from Incomplete Raw Data Processing. Metabolomics 2017, 13, 107. 10.1007/s11306-017-1246-3. [DOI] [Google Scholar]
- Myers O. D.; Sumner S. J.; Li S.; Barnes S.; Du X. Detailed Investigation and Comparison of the XCMS and MZmine 2 Chromatogram Construction and Chromatographic Peak Detection Methods for Preprocessing Mass Spectrometry Metabolomics Data. Anal. Chem. 2017, 89, 8689–8695. 10.1021/acs.analchem.7b01069. [DOI] [PubMed] [Google Scholar]
- Coble J. B.; Fraga C. G. Comparative Evaluation of Preprocessing Freeware on Chromatography/Mass Spectrometry Data for Signature Discovery. J. Chromatogr. A 2014, 1358, 155–164. 10.1016/j.chroma.2014.06.100. [DOI] [PubMed] [Google Scholar]
- Myers O. D.; Sumner S. J.; Li S.; Barnes S.; Du X. One Step Forward for Reducing False Positive and False Negative Compound Identifications from Mass Spectrometry Metabolomics Data: New Algorithms for Constructing Extracted Ion Chromatograms and Detecting Chromatographic Peaks. Anal. Chem. 2017, 89, 8696–8703. 10.1021/acs.analchem.7b00947. [DOI] [PubMed] [Google Scholar]
- Li Z.; Lu Y.; Guo Y.; Cao H.; Wang Q.; Shui W. Comprehensive Evaluation of Untargeted Metabolomics Data Processing Software in Feature Detection, Quantification and Discriminating Marker Selection. Anal. Chim. Acta 2018, 1029, 50–57. 10.1016/j.aca.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Dudzik D.; Barbas-Bernardos C.; García A.; Barbas C. Quality Assurance Procedures for Mass Spectrometry Untargeted Metabolomics. a Review. J. Pharm. Biomed. Anal. 2018, 147, 149–173. 10.1016/j.jpba.2017.07.044. [DOI] [PubMed] [Google Scholar]
- Sangster T.; Major H.; Plumb R.; Wilson A. J.; Wilson I. D. A Pragmatic and Readily Implemented Quality Control Strategy for HPLC-MS and GC-MS-Based Metabonomic Analysis. Analyst 2006, 1075–1078. 10.1039/b604498k. [DOI] [PubMed] [Google Scholar]
- Beger R. D.; Dunn W. B.; Bandukwala A.; Bethan B.; Broadhurst D.; Clish C. B.; Dasari S.; Derr L.; Evans A.; Fischer S.; et al. Towards Quality Assurance and Quality Control in Untargeted Metabolomics Studies. Metabolomics 2019, 15, 4. 10.1007/s11306-018-1460-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker E. S.; Patti G. J. Perspectives on Data Analysis in Metabolomics: Points of Agreement and Disagreement from the 2018 ASMS Fall Workshop. J. Am. Soc. Mass Spectrom. 2019, 30, 2031–2036. 10.1007/s13361-019-02295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erny G. L.; Acunha T.; Simó C.; Cifuentes A.; Alves A. Finnee - A Matlab Toolbox for Separation Techniques Hyphenated High Resolution Mass Spectrometry Dataset. Chemom. Intell. Lab. Syst. 2016, 155, 138–144. 10.1016/j.chemolab.2016.04.013. [DOI] [Google Scholar]
- Erny G. L.; Acunha T.; Simó C.; Cifuentes A.; Alves A. Background Correction in Separation Techniques Hyphenated to High-Resolution Mass Spectrometry – Thorough Correction with Mass Spectrometry Scans Recorded as Profile Spectra. J. Chromatogr. A 2017, 1492, 98–105. 10.1016/j.chroma.2017.02.052. [DOI] [PubMed] [Google Scholar]
- Davis M. D.; Montpetit A.; Hunt J. Exhaled Breath Condensate. An Overview. Immunol. Allergy Clin. North Am. 2012, 32, 363–375. 10.1016/j.iac.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth I. Exhaled Breath Condensate in Disease Monitoring. Clin. Pulm. Med. 2003, 10, 195–200. 10.1097/01.cpm.0000059379.82525.4a. [DOI] [Google Scholar]
- Horváth I.; Hunt J.; Barnes P. J. Exhaled Breath Condensate: Methodological Recommendations and Unresolved Questions. Eur. Respir. J. 2005, 26, 523–548. 10.1183/09031936.05.00029705. [DOI] [PubMed] [Google Scholar]
- Hunt J. Exhaled Breath Condensate: An Evolving Tool for Noninvasive Evaluation of Lung Disease. J. Allergy Clin. Immunol. 2002, 110, 28–34. 10.1067/mai.2002.124966. [DOI] [PubMed] [Google Scholar]
- Martins P. C.; Valente J.; Papoila A. L.; Caires I.; Araújo-Martins J.; Matae P.; Lopes M.; Torres S.; Rosado-Pinto J.; Borrego C.; et al. Airways Changes Related to Air Pollution Exposure in Wheezing Children. Eur. Respir. J. 2012, 39, 246–253. 10.1183/09031936.00025111. [DOI] [PubMed] [Google Scholar]
- van Mastrigt E.; de Jongste J. C.; Pijnenburg M. W. The Analysis of Volatile Organic Compounds in Exhaled Breath and Biomarkers in Exhaled Breath Condensate in Children - Clinical Tools or Scientific Toys?. Clin. Exp. Allergy 2015, 45, 1170–1188. 10.1111/cea.12454. [DOI] [PubMed] [Google Scholar]
- Kubáň P.; Foret F. Exhaled Breath Condensate: Determination of Non-Volatile Compounds and Their Potential for Clinical Diagnosis and Monitoring. A Review. Anal. Chim. Acta 2013, 805, 1–18. 10.1016/j.aca.2013.07.049. [DOI] [PubMed] [Google Scholar]
- Borrill Z. L.; Roy K.; Singh D. Exhaled Breath Condensate Biomarkers in COPD. Eur. Respir. J. 2008, 32, 472–486. 10.1183/09031936.00116107. [DOI] [PubMed] [Google Scholar]
- Carraro S.; Rezzi S.; Reniero F.; Héberger K.; Giordano G.; Zanconato S.; Guillou C.; Baraldi E. Metabolomics Applied to Exhaled Breath Condensate in Childhood Asthma. Am. J. Respir. Crit. Care Med. 2007, 175, 986–990. 10.1164/rccm.200606-769OC. [DOI] [PubMed] [Google Scholar]
- Montuschi P.; Barnes P. J. Analysis of Exhaled Breath Condensate for Monitoring Airway Inflammation. Trends Pharmacol. Sci. 2002, 23, 232–237. 10.1016/S0165-6147(02)02020-5. [DOI] [PubMed] [Google Scholar]
- Effros R. M.; Hoagland K. W.; Bosbous M.; Castillo D.; Foss B.; Dunning M.; Gare M.; Lin W.; Sun F. Dilution of Respiratory Solutes in Exhaled Condensates. Am. J. Respir. Crit. Care Med. 2002, 165, 663–669. 10.1164/ajrccm.165.5.2101018. [DOI] [PubMed] [Google Scholar]
- Tautenhahn R.; Patti G. J.; Rinehart D.; Siuzdak G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda H.; Ivanisevic J.; Johnson C. H.; Kurczy M. E.; Benton H. P.; Rinehart D.; Nguyen T.; Ray J.; Kuehl J.; Arevalo B.; et al. Interactive XCMS Online: Simplifying Advanced Metabolomic Data Processing and Subsequent Statistical Analyses. Anal. Chem. 2014, 86, 6931–6939. 10.1021/ac500734c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugawa H.; Cajka T.; Kind T.; Ma Y.; Higgins B.; Ikeda K.; Kanazawa M.; Vandergheynst J.; Fiehn O.; Arita M. MS-DIAL: Data-Independent MS/MS Deconvolution for Comprehensive Metabolome Analysis. Nat. Methods 2015, 12, 523–526. 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens L.; Chambers M.; Sturm M.; Kessner D.; Levander F.; Shofstahl J.; Tang W. H.; Römpp A.; Neumann S.; Pizarro A. D.; et al. MzML—A Community Standard for Mass Spectrometry Data. Mol. Cell. Proteomics 2011, R110.000133. 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adusumilli R.; Mallick P.. Data Conversion with ProteoWizard MsConvert. In Methods in Molecular Biology; 2017; Vol. 1550, pp 339–368, 10.1007/978-1-4939-6747-6_23. [DOI] [PubMed] [Google Scholar]
- Chambers M. C.; MacLean B.; Burke R.; Amodei D.; Ruderman D. L.; Neumann S.; Gatto L.; Fischer B.; Pratt B.; Egertson J.; et al. A Cross-Platform Toolkit for Mass Spectrometry and Proteomics. Nat. Biotechnol. 2012, 30, 918–920. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray K. K.; Boyd R. K.; Eberlin M. N.; Langley G. J.; Li L.; Naito Y. Definitions of Terms Relating to Mass Spectrometry (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1515–1609. 10.1351/PAC-REC-06-04-06. [DOI] [Google Scholar]
- Erny G.Glerny/Finnee2016: Master Mz. Zenodo January 1, 2017, 10.5281/zenodo.831658. [DOI] [Google Scholar]
- Baek S.-J.; Park A.; Ahn Y.-J.; Choo J. Baseline Correction Using Asymmetrically Reweighted Penalized Least Squares Smoothing. Analyst 2015, 140, 250–257. 10.1039/c4an01061b. [DOI] [PubMed] [Google Scholar]
- Leys C.; Ley C.; Klein O.; Bernard P.; Licata L. Detecting Outliers: Do Not Use Standard Deviation around the Mean, Use Absolute Deviation around the Median. J. Exp. Soc. Psychol. 2013, 49, 764–766. 10.1016/j.jesp.2013.03.013. [DOI] [Google Scholar]
- Misra S.; Wahab M. F.; Patel D. C.; Armstrong D. W. The Utility of Statistical Moments in Chromatography Using Trapezoidal and Simpson’s Rules of Peak Integration. J. Sep. Sci. 2019, 42, 1644–1657. 10.1002/jssc.201801131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.